
Diastereoselective Synthesis of cis-2,6-Disubstituted Dihydropyrane Derivatives through a Competitive Silyl-Prins Cyclization versus Alternative Reaction Pathways



Abstract
1. Introduction
- (a)
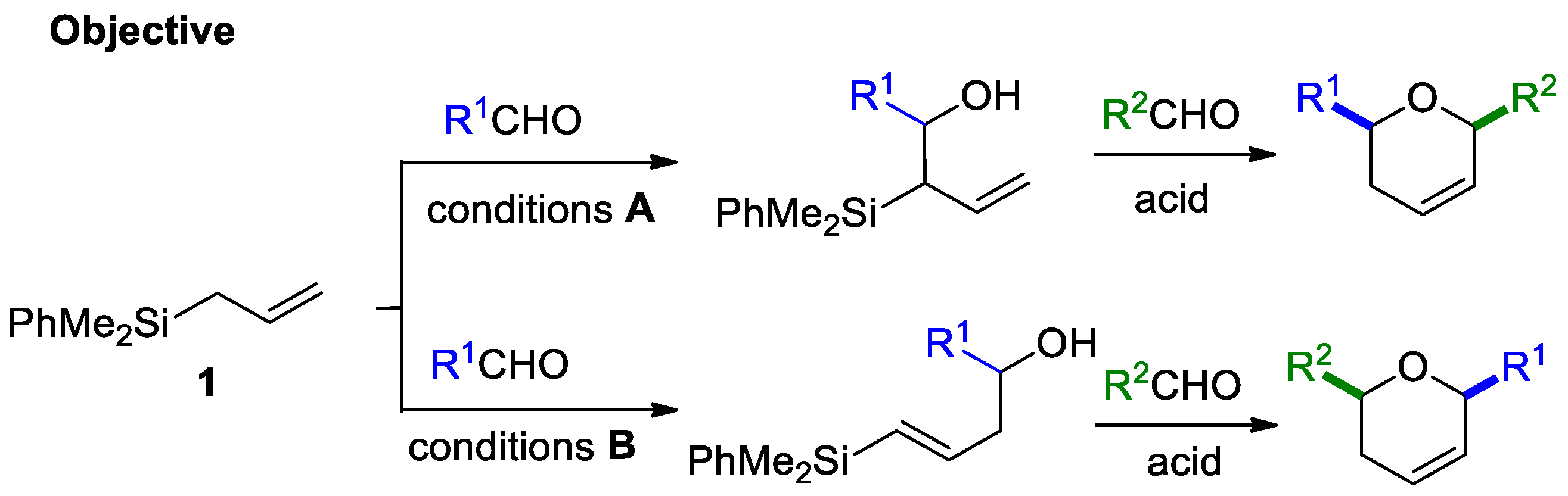
- Selective methodologies are needed to obtain exclusively allyl- or vinyl silylalcohols from a common allylsilane precursor (conditions A and B, Scheme 1).
- (b)
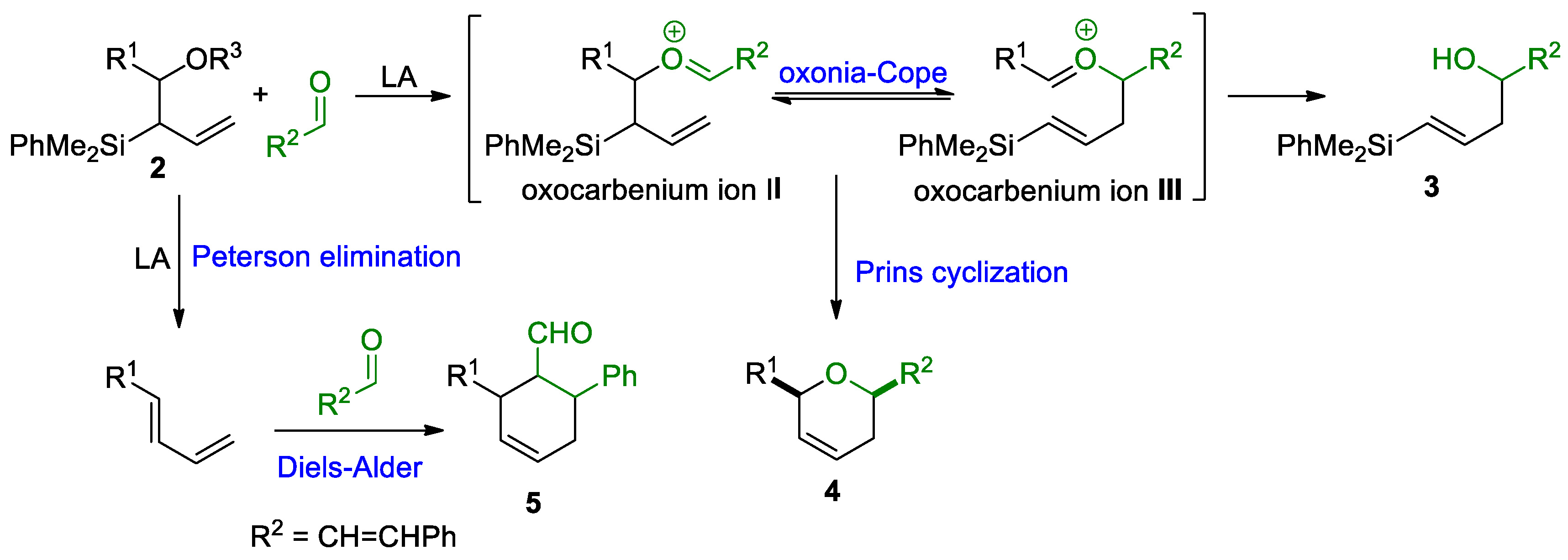
- Alternative reaction pathways may interfere in the silyl-Prins cyclization reaction outcome [19], generating complex mixtures due to the formation of highly reactive intermediates.
2. Results
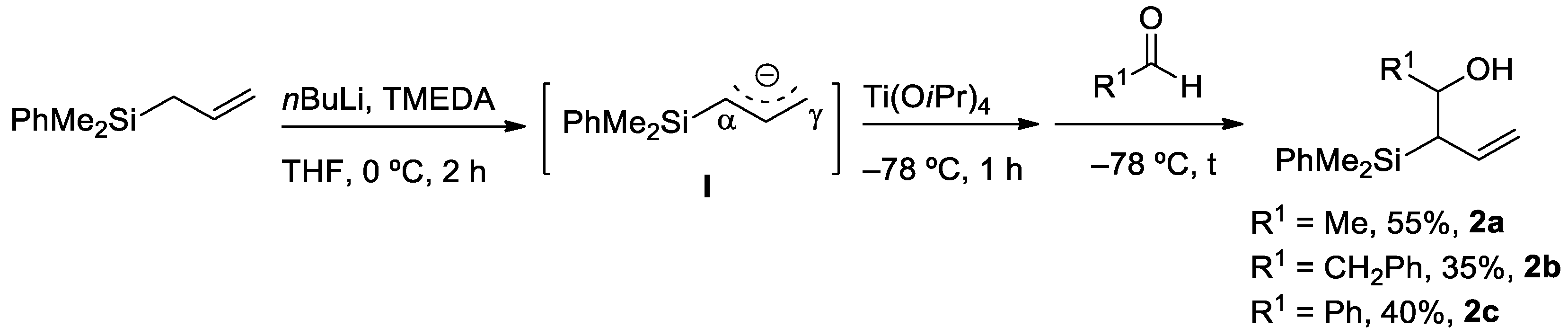
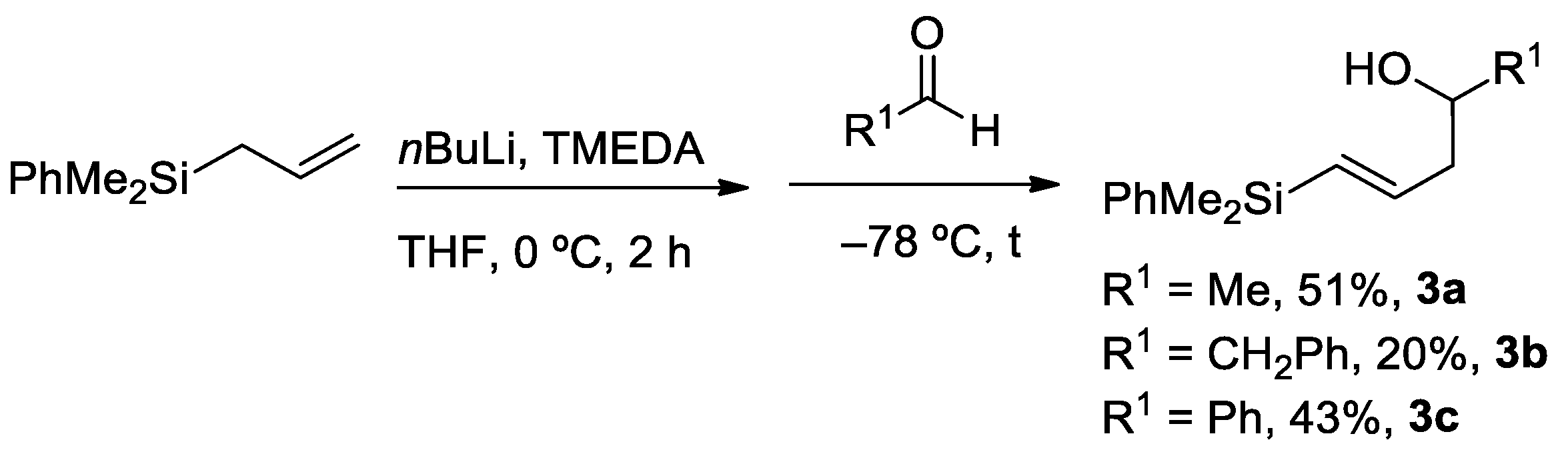
2.1. Synthesis of Allyl- and Vinylsilyl Alcohols
2.2. Study of the Silyl-Prins Cyclization with Allylsilyl Alcohols 2
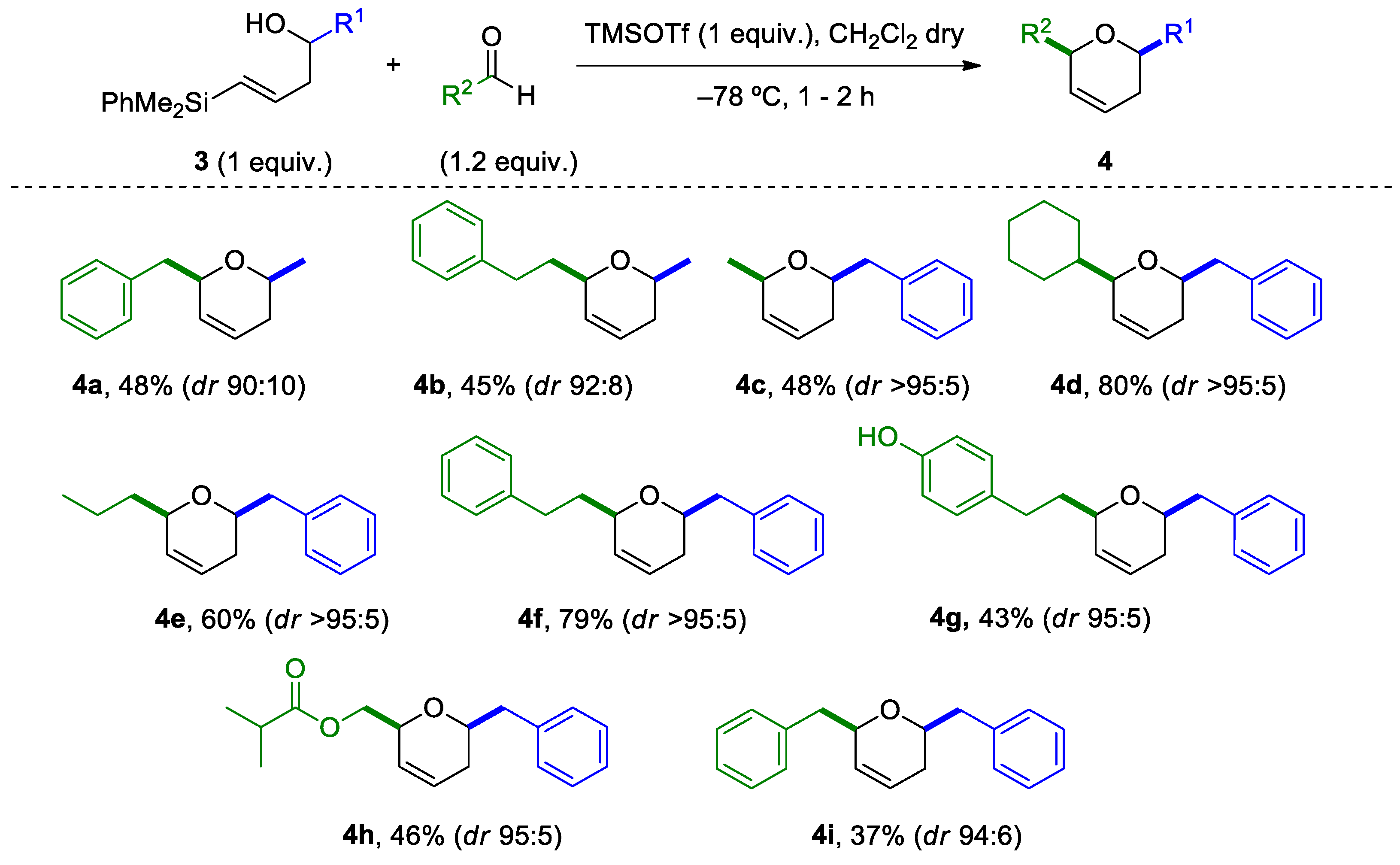
2.3. Silyl-Prins Cyclization Study of Vinylsilyl Alcohols 3
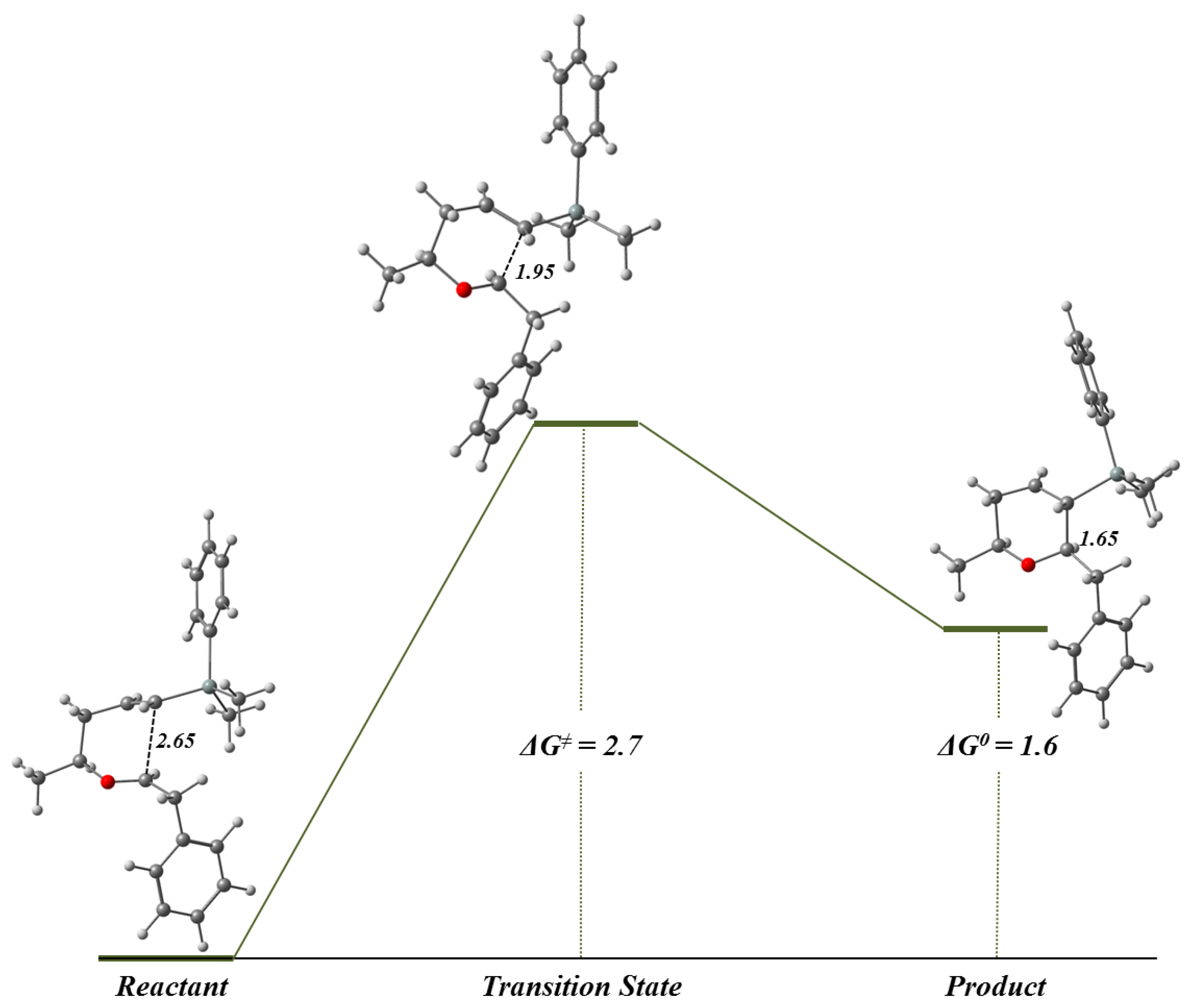
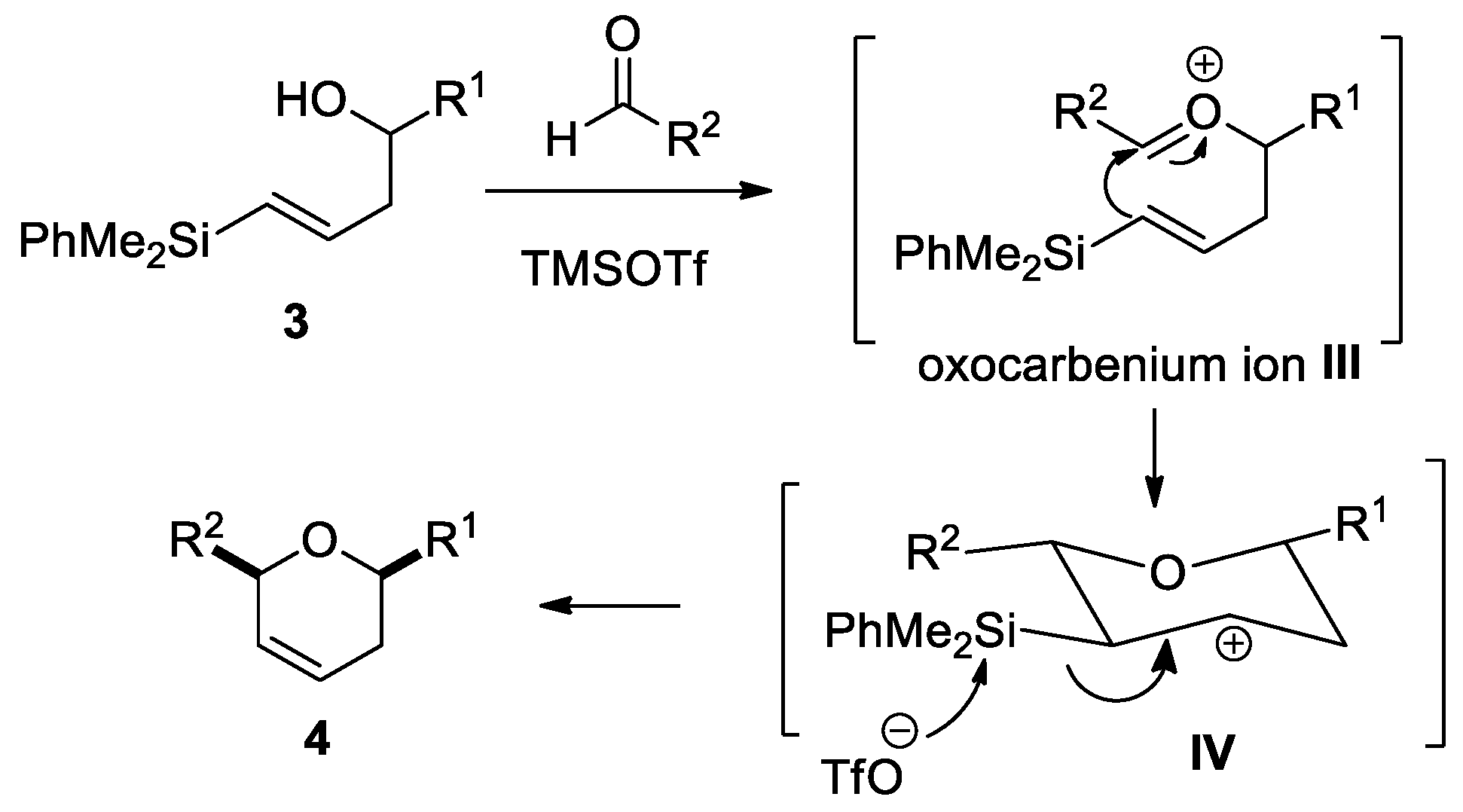
2.4. Reaction Mechanism Study
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of Allyl(diphenyl)silane 1
3.3. Synthesis of Allylsilyl Alcohols 2
3.4. Synthesis of Vinylsilyl Alcohols 3
3.5. TMSOTf-Promoted Cyclization Reaction to Afford Dihydropyranes 4
3.6. BF3·OEt2-Promoted Diels Alder Reaction to Afford Derivatives 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nasir, N.M.; Ermanis, K.; Clarke, P.A. Strategies for the construction of tetrahydropyran rings in the synthesis of natural products. Org. Biomol. Chem. 2014, 12, 3323–3335. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H. Contemporary Strategies for the Synthesis of Tetrahydropyran Derivatives: Application to Total Synthesis of Neopeltolide, a Marine Macrolide Natural Product. Mar. Drugs 2016, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Reyes, E.; Prieto, L.; Uria, U.; Carrillo, L.; Vicario, J.L. Recent Advances in the Prins Reaction. ACS Omega 2022, 7, 31621–31627. [Google Scholar] [CrossRef]
- Díez-Poza, C.; Barbero, A. Synthesis of O− And N− Heterocycles by Silyl-Prins Cyclization of Allylsilanes. Eur. J. Org. Chem. 2017, 32, 4651–4665. [Google Scholar] [CrossRef]
- Pastor, I.M.; Yus, M. Focused Update on the Prins Reaction and the Prins Cyclization. Curr. Org. Chem. 2012, 16, 1277–1312. [Google Scholar] [CrossRef]
- Díez-Poza, C.; Barbero, H.; Díez-Varga, A.; Barbero, A. The Silyl-Prins Reaction as an Emerging Method for the Synthesis of Heterocycles in Progress in Heterocyclic Chemistry. In Progress of Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 30, pp. 13–41. [Google Scholar]
- Su, Q.; Panek, J.S. Total Synthesis of (-)-Apicularen A. J. Am. Chem. Soc. 2004, 126, 2425–2430. [Google Scholar] [CrossRef]
- Kjellgren, J.; Szabó, K.J. Synthesis of stereodefined vinyl-tetrahydropyran and vinyl-octahydrochromene derivatives via acetization-cyclization of allylsilanes with aldehydes. Origin of the high stereoselectivity. Tetrahedron Lett. 2002, 43, 1123–1126. [Google Scholar] [CrossRef]
- Keck, G.E.; Kraft, M.B.; Truong, A.P.; Li, W.; Sánchez, C.C.; Kedei, N.; Lewin, N.E.; Blumberg, P.M. Convergent Assembly of Highly Potent Analogues of Bryostatin 1 via Pyran Annulation: Bryostatin Look-Alikes that Mimic Phorbol Ester Function. J. Am. Chem. Soc. 2008, 130, 6660–6661. [Google Scholar] [CrossRef]
- Sánchez, C.C.; Keck, G.E. Total Synthesis of (+)-Dactylolide. Org. Lett. 2005, 7, 3053–3056. [Google Scholar] [CrossRef]
- Overman, L.E.; Castaneda, A.; Blumenkopf, T.A. Acetal-initiated cyclizations of vinylsilanes: A general synthesis of allylically unsaturated oxacyclics. J. Am. Chem. Soc. 1986, 108, 1303–1304. [Google Scholar]
- Semeyn, C.; Blaauw, R.H.; Hiemstra, H.; Speckamp, W.N. Stereoselective Synthesis of Dihydropyrans via Vinylsilane-Terminated Cyclizations of Ester-Substituted Oxycarbenium Ion Intermediates. J. Org. Chem. 1997, 62, 3426–3427. [Google Scholar] [CrossRef]
- Dobbs, A.P.; Martinović, S. The Silyl-Prins reaction: A novel method for the synthesis of dihydropyrans. Tetrahedron Lett. 2002, 43, 7055–7057. [Google Scholar] [CrossRef]
- Lian, Y.; Hinkle, R.J. BiBr3-Initiated Tandem Addition/Silyl-Prins Reactions to 2,6-disubstituted Dihydropyrans. J. Org. Chem. 2006, 71, 7071–7074. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.; Wang, K.; Gao, L.; Song, Z. Chrial crotyl geminal bis(silane): A useful reagent for asymmetric Sakurai allylation by selective desilylation-enabled chirality transfer. Chem. Commun. 2017, 53, 3078–3081. [Google Scholar]
- Barbero, A.; Diez-Varga, A.; Pulido, F.J.; González-Ortega, A. Synthesis of Azepane Derivatives by Silyl-aza-Prins Cyclization of Allylsilyl Amines: Influence of the Catalyst in the Outcome of the Reaction. Org. Lett. 2016, 18, 1972–1975. [Google Scholar] [CrossRef]
- Barbero, A.; Díez-Varga, A.; Herrero, M.; Pulido, F.J. From Silylated Trishomoallylic Alcohols to Dioxaspiroundecades or Oxocanes: Catalyst and Substitution Influence. J. Org. Chem. 2016, 81, 2704–2712. [Google Scholar] [CrossRef]
- Barbero, A.; Díez-Poza, C. Unexpected Dominio Silyl-Prins/Aryl Migration Process from Geminal Vinylsilyl alcohols. Org. Lett. 2021, 23, 8385–8389. [Google Scholar]
- Alder, R.W.; Carta, F.; Reed, C.A.; Stoyanova, I.; Willis, C.L. Searching for intermediates in Silyl Prins cyclizations: The 2-oxa—5-adamantyl carbocation. Org. Biomol. Chem. 2010, 8, 1551–1559. [Google Scholar] [CrossRef]
- Ramesh, R.; Reddy, D.S. Zinc mediated allylations of chlorosilanes promoted by ultrasound: Synthesis of novel constrained sila aminoacids. Org. Biomol. Chem. 2014, 12, 4093–4097. [Google Scholar] [CrossRef]
- Chen, M.; Roush, W.R. Enantioselective synthesis of syn- and anti-β-hydroxiallylsilanes via allenes hydroboration-aldehyde allylboration reactions. Org. Lett. 2011, 13, 1992–1995. [Google Scholar] [CrossRef]
- Karimi, B.; Golshani, B. Mild and Highly Efficient Method for the silylation of Alcohols Using Hexamethyldisilazane Catalyzed by Iodine Under Nearly Neutral Reaction Conditions. J. Org. Chem. 2000, 65, 7228–7230. [Google Scholar] [CrossRef] [PubMed]
- Pulido, F.J.; Barbero, A.; Val, P.; Díez, A.; González-Ortega, A. Efficiency of Acid- and Mercury-Catalyzed Cyclization Reactions in the Synthesis of Tetrahydrofurans from Allylsilyl Alcohols. Eur. J. Org. Chem. 2012, 2012, 5350–5356. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; van Gisbergen, S.J.A.; Fonseca Guerra, C.; Baerends, E.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF2013. SCM, Theoretical Chemistry. Available online: http://www.scm.com (accessed on 29 January 2023).
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.A. Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Chemcraft. Available online: http://www.chemcraftprog.com (accessed on 29 January 2023).
- Markó, I.E.; Dobbs, A.P.; Scheirmann, V.; Chellé, F.; Bayston, D.J. Concise and Stereocontrolled Assembly of Substituted Dihydropyrans: Synthetic Studies Towards the trans-dioxadecalin Subunit of Okadaic Acid. Tetrahedron Lett. 1997, 38, 2899–2902. [Google Scholar]
- Viswanathan, G.S.; Yang, J.; Li, C.-J. A Novel Stereoselective Cyclization to Functionalized Dihydropyrans. Org. Lett. 1999, 1, 993–995. [Google Scholar] [CrossRef]
- Jasti, R.; Rychnovsky, S.D. Racemization in Prins Cyclization Reactions. J. Am. Chem. Soc. 2006, 128, 13640–13648. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R1 | R2 | R3 | LA (1 equiv.) | T (°C) | NMR Analysis (Isolated Yield) |
|---|---|---|---|---|---|---|
| 1 | CH3 | CH2CH2Ph | H | TMSOTf | −78 | 3 |
| 2 | CH3 | CH2CH2Ph | H | TMSOTf | 0 | Complex mixture |
| 3 a | CH3 | CH2CH2Ph | H | TMSOTf | −78 | Complex mixture |
| 4 | CH3 | CH2CH2Ph | H | BF3·OEt2 | −78 | Complex mixture |
| 5 | CH3 | CH=CHPh | H | TMSOTf | −20 | Aldehyde |
| 6 a | CH2Ph | CH3 | H | TMSOTf | −78 | 3a + 4a |
| 7 a | CH2Ph | CH2Ph | H | TMSOTf | −78 | 4i |
| 8 a | Ph | CH3 | H | TMSOTf | −78 | Complex mixture |
| 9 | Ph | CH2CH2Ph | H | TMSOTf | −78 | Complex mixture |
| 10 | CH3 | CH=CHPh | TMS | TMSOTf | −78 | 2a |
| 11 | CH3 | CH=CHPh | TMS | BF3·OEt2 | −78 | Complex mixture |
| 12 | CH3 | CH=CHPh | TMS | BF3·OEt2 | 0 | 5 (29%) |

| Entry | LA (equiv.) | Temperature (°C) | Isolated Yield |
|---|---|---|---|
| 1 a | BF3·OEt2 (1.0) | 0 | Complex mixture |
| 2 | BF3·OEt2 (1.2) | 0 | Complex mixture |
| 3 | BF3·OEt2 (0.5) | 0 | Complex mixture |
| 4 | TMSCl (1.5) | 0 to rt b | nr |
| 5 a | TMSOTf (1.0) | −78 | 35% |
| 6 | TMSOTf (1.0) | −78 | 48% |
| 7 | TMSOTf (0.5) | −78 | 25% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña, L.F.; López, E.; Sánchez-González, Á.; Barbero, A. Diastereoselective Synthesis of cis-2,6-Disubstituted Dihydropyrane Derivatives through a Competitive Silyl-Prins Cyclization versus Alternative Reaction Pathways. Molecules 2023, 28, 3080. https://doi.org/10.3390/molecules28073080
Peña LF, López E, Sánchez-González Á, Barbero A. Diastereoselective Synthesis of cis-2,6-Disubstituted Dihydropyrane Derivatives through a Competitive Silyl-Prins Cyclization versus Alternative Reaction Pathways. Molecules. 2023; 28(7):3080. https://doi.org/10.3390/molecules28073080
Chicago/Turabian StylePeña, Laura F., Enol López, Ángel Sánchez-González, and Asunción Barbero. 2023. "Diastereoselective Synthesis of cis-2,6-Disubstituted Dihydropyrane Derivatives through a Competitive Silyl-Prins Cyclization versus Alternative Reaction Pathways" Molecules 28, no. 7: 3080. https://doi.org/10.3390/molecules28073080
APA StylePeña, L. F., López, E., Sánchez-González, Á., & Barbero, A. (2023). Diastereoselective Synthesis of cis-2,6-Disubstituted Dihydropyrane Derivatives through a Competitive Silyl-Prins Cyclization versus Alternative Reaction Pathways. Molecules, 28(7), 3080. https://doi.org/10.3390/molecules28073080