

Decoding the Conformational Selective Mechanism of FGFR Isoforms: A Comparative Molecular Dynamics Simulation

 , and
, and
Abstract
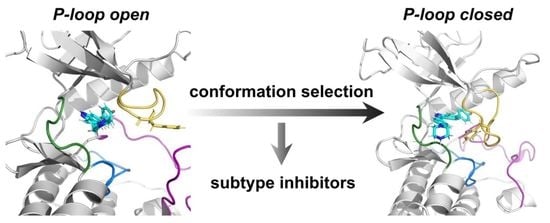
1. Introduction
2. Results
2.1. Compound 38 Loading Enhanced Systems Stability
2.2. Compound 38 Binding Enhanced Local Conformational Dynamics in FGFR2
2.3. Compound 38 Binding Induced the Approaching Conformations of the N- and C-lobes in FGFR2
2.4. Compound 38 Binding Induced a Conformational Transition from the Open to the Closed Conformation of the P-loop in FGFR2
2.5. The Hydrophobic Channel by the Closed P-loop Was a Major Mechanism for Selective Inhibition
2.6. The Interaction Network Reveals the Driving Force of P-loop Folding
3. Discussion
4. Materials and Methods
4.1. Preparation of Stimulation Systems
4.2. Molecular Dynamics Simulations
4.3. Dynamic Cross-Correlation Matrix (DCCM) Analysis
4.4. Principal Component Analysis and Free Energy Landscapes
4.5. Binding Free Energy
4.6. Markov State Model Construction and Validation
4.7. Community Network Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target Ther. 2020, 5, 181–218. [Google Scholar] [CrossRef]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; Andre, F.; Soria, J.C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Adjei, A.A. FGFR Signaling as a Target for Lung Cancer Therapy. J. Thorac. Oncol. 2016, 11, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef]
- Yang, W.; Yao, Y.W.; Zeng, J.L.; Liang, W.J.; Wang, L.; Bai, C.Q.; Liu, C.H.; Song, Y. Prognostic value of FGFR1 gene copy number in patients with non-small cell lung cancer: A meta-analysis. J. Thorac. Dis. 2014, 6, 803–809. [Google Scholar]
- Matsumoto, K.; Arao, T.; Hamaguchi, T.; Shimada, Y.; Kato, K.; Oda, I.; Taniguchi, H.; Koizumi, F.; Yanagihara, K.; Sasaki, H.; et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br. J. Cancer 2012, 106, 727–732. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.G.; Yu, Y.; Eathiraj, S.; Wang, Y.X.; Savage, R.E.; Lapierre, J.M.; Schwartz, B.; Abbadessa, G. Preclinical Activity of ARQ 087, a Novel Inhibitor Targeting FGFR Dysregulation. PLoS ONE 2016, 11, e0162594. [Google Scholar] [CrossRef] [PubMed]
- Perera, T.; Jovcheva, E.; Vialard, J.; Verhulst, T.; Esser, N.; Wroblowski, B.; Gilissen, R.; Freyne, E.; King, P.; Platero, S.; et al. JNJ-42756493 is an inhibitor of FGFR-1, 2, 3 and 4 with nanomolar affinity for targeted therapy. Cancer Res. 2014, 74, 1738. [Google Scholar] [CrossRef]
- Kalyukina, M.; Yosaatmadja, Y.; Middleditch, M.J.; Patterson, A.V.; Smaill, J.B.; Squire, C.J. TAS-120 Cancer Target Binding: Defining Reactivity and Revealing the First Fibroblast Growth Factor Receptor 1 (FGFR1) Irreversible Structure. Chemmedchem 2019, 14, 494–500. [Google Scholar] [CrossRef]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef]
- Zhao, G.; Li, W.Y.; Chen, D.; Henry, J.R.; Li, H.Y.; Chen, Z.; Zia-Ebrahimi, M.; Bloem, L.; Zhai, Y.; Huss, K.; et al. A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Mol. Cancer Ther. 2011, 10, 2200–2210. [Google Scholar] [CrossRef]
- Hoy, S.M. Pemigatinib: First Approval. Drugs 2020, 80, 923–929. [Google Scholar] [CrossRef]
- Heroult, M.; Ellinghaus, P.; Sieg, C.; Brohm, D.; Gruenewald, S.; Collin, M.P.; Boemer, U.; Lobell, M.; Huebsch, W.; Ocker, M.; et al. Preclinical profile of BAY 1163877-a selective pan-FGFR inhibitor in phase 1 clinical trial. Cancer Res. 2014, 74, 1739. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Akiyama, N.; Tsukaguchi, T.; Fujii, T.; Sakata, K.; Sase, H.; Isobe, T.; Morikami, K.; Shindoh, H.; Mio, T.; et al. The Fibroblast Growth Factor Receptor Genetic Status as a Potential Predictor of the Sensitivity to CH5183284/Debio 1347, a Novel Selective FGFR Inhibitor. Mol. Cancer Ther. 2014, 13, 2547–2558. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, A.; Tella, S.H.; Kommalapati, A.; Yu, J.; Kim, R. Prevention and treatment of FGFR inhibitor-associated toxicities. Crit. Rev. Oncol. Hemat. 2020, 155, 103091–103097. [Google Scholar] [CrossRef]
- Casaletto, J.; Maglic, D.; Toure, B.B.; Taylor, A.; Schoenherr, H.; Hudson, B.; Bruderek, K.; Zhao, S.P.; O’Hearn, P.; Gerami-Moayed, N.; et al. RLY-4008, a novel precision therapy for FGFR2-driven cancers designed to potently and selectively inhibit FGFR2 and FGFR2 resistance mutations. Cancer Res. 2021, 81, 1455. [Google Scholar] [CrossRef]
- Hagel, M.; Miduturu, C.; Sheets, M.; Rubin, N.; Weng, W.F.; Stransky, N.; Bifulco, N.; Kim, J.L.; Hodous, B.; Brooijmans, N.; et al. First Selective Small Molecule Inhibitor of FGFR4 for the Treatment of Hepatocellular Carcinomas with an Activated FGFR4 Signaling Pathway. Cancer Discov. 2015, 5, 424–437. [Google Scholar] [CrossRef]
- Turner, L.D.; Trinh, C.H.; Hubball, R.A.; Orritt, K.M.; Lin, C.C.; Burns, J.E.; Knowles, M.A.; Fishwick, C.W.G. From Fragment to Lead: De Novo Design and Development toward a Selective FGFR2 Inhibitor. J. Med. Chem. 2022, 65, 1481–1504. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.C.; He, X.H.; Rehman, A.U.; Li, X.Y.; Qiu, Y.R.; Pu, J.; Lu, S.Y.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2021, 12, 464–476. [Google Scholar] [CrossRef]
- Lu, S.Y.; Chen, Y.Y.; Wei, J.C.; Zhao, M.Z.; Ni, D.; He, X.H.; Zhang, J. Mechanism of allosteric activation of SIRT6 revealed by the action of rationally designed activators. Acta Pharm. Sin. B. 2021, 11, 1355–1361. [Google Scholar] [CrossRef]
- Lu, S.Y.; He, X.H.; Yang, Z.; Chai, Z.T.; Zhou, S.H.; Wang, J.Y.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.P.; et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 2021, 12, 4721–4735. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Ji, D.; Lei, C.Y.; Chen, Y.F.; Qiu, Y.R.; Li, X.Y.; Li, M.Y.; Ni, D.; Pu, J.; Zhang, J.; et al. Mechanistic insights into the effect of phosphorylation on Ras conformational dynamics and its interactions with cell signaling proteins. Comput. Struct. Biotec. J. 2021, 19, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, C.R.W.; Rai, B.K.; Munchhof, M.J.; Liu, S.P.; Wang, J.; Bhattacharya, S.K.; Buckbinder, L. Understanding the Impact of the P-loop Conformation on Kinase Selectivity. J. Chem. Inf. Model. 2011, 51, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Tacconi, E.M.C.; Zimmer, J.; Liang, Y.K.; Gray, N.S.; Tarsounas, M.; Knapp, S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014, 10, 853–860. [Google Scholar] [CrossRef] [PubMed]
- McClendon, C.L.; Kornev, A.P.; Gilson, M.K.; Taylor, S.S. Dynamic architecture of a protein kinase (vol 111, pg E4623, 2014). Proc. Natl. Acad. Sci. USA 2014, 111, 16973. [Google Scholar] [CrossRef]
- Liu, C.; Li, Z.Z.; Liu, Z.H.; Yang, S.Y.; Wang, Q.; Chai, Z.T. Understanding the P-Loop Conformation in the Determination of Inhibitor Selectivity Toward the Hepatocellular Carcinoma-Associated Dark Kinase STK17B. Front. Mol. Biosci. 2022, 9, 478–488. [Google Scholar] [CrossRef]
- Counago, R.M.; Allerston, C.K.; Savitsky, P.; Azevedo, H.; Godoi, P.H.; Wells, C.I.; Mascarello, A.; Gama, F.H.D.; Massirer, K.B.; Zuercher, W.J.; et al. Structural characterization of human Vaccinia-Related Kinases (VRK) bound to small-molecule inhibitors identifies different P-loop conformations. Sci. Rep. 2017, 7, 7501–7512. [Google Scholar] [CrossRef]
- Mohammadi, M.; McMahon, G.; Sun, L.; Tang, C.; Hirth, P.; Yeh, B.K.; Hubbard, S.R.; Schlessinger, J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997, 276, 955–960. [Google Scholar] [CrossRef]
- Collie, G.W.; Michaelides, I.N.; Embrey, K.; Stubbs, C.J.; Borjesson, U.; Dale, I.L.; Snijder, A.; Barlind, L.; Song, K.; Khurana, P.; et al. Structural Basis for Targeting the Folded P-Loop Conformation of c-MET. ACS Med. Chem. Lett. 2021, 12, 162–167. [Google Scholar] [CrossRef]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Perez-Hernandez, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.H.; Noe, F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef]
- Chandel, T.I.; Zaman, M.; Khan, M.V.; Ali, M.; Rabbani, G.; Ishtikhar, M.; Khan, R.H. A mechanistic insight into protein-ligand interaction, folding, misfolding, aggregation and inhibition of protein aggregates: An overview. Int. J. Biol. Macromol. 2018, 106, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Dis. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.; Hulme, C.; Hurley, L.H. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: A structural analysis of the binding interactions of Gleevec®, Nexavar®, and BIRB-796. Bioorgan. Med. Chem. 2010, 18, 5738–5748. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.C.; Peterson, J.R.; Dunbrack, R.L.; Levy, R.M. Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef]
- Millan, D.S.; Bunnage, M.E.; Burrows, J.L.; Butcher, K.J.; Dodd, P.G.; Evans, T.J.; Fairman, D.A.; Hughes, S.J.; Kilty, I.C.; Lemaitre, A.; et al. Design and Synthesis of Inhaled p38 Inhibitors for the Treatment of Chronic Obstructive Pulmonary Disease. J. Med. Chem. 2011, 54, 7797–7814. [Google Scholar] [CrossRef]
- Palmieri, L.; Rastelli, G. alpha C helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discov. Today 2013, 18, 407–414. [Google Scholar] [CrossRef]
- Herbert, C.; Schieborr, U.; Saxena, K.; Juraszek, J.; De Smet, F.; Alcouffe, C.; Bianciotto, M.; Saladino, G.; Sibrac, D.; Kudlinzki, D.; et al. Molecular Mechanism of SSR128129E, an Extracellularly Acting, Small-Molecule, Allosteric Inhibitor of FGF Receptor Signaling. Cancer Cell 2013, 23, 489–501. [Google Scholar] [CrossRef]
- Wang, J.M.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald-an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints-Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Li, M.Y.; Wang, Y.H.; Fan, J.G.; Zhuang, H.M.; Liu, Y.Q.; Ji, D.; Lu, S.Y. Mechanistic Insights into the Long-range Allosteric Regulation of KRAS Via Neurofibromatosis Type 1 (NF1) Scaffold Upon SPRED1 Loading. J. Mol. Biol. 2022, 434, 167730–167748. [Google Scholar] [CrossRef]
- Li, X.Y.; Wang, C.X.; Peng, T.; Chai, Z.T.; Ni, D.; Liu, Y.Q.; Zhang, J.; Chen, T.; Lu, S.Y. Atomic-scale insights into allosteric inhibition and evolutional rescue mechanism of Streptococcus thermophilus Cas9 by the anti-CRISPR protein AcrIIA6. Comput. Struct. Biotechnol. J. 2021, 19, 6108–6124. [Google Scholar] [CrossRef]
- Zhang, H.; Ni, D.; Fan, J.G.; Li, M.Y.; Zhang, J.; Hua, C.; Nussinov, R.; Lu, S.Y. Markov State Models and Molecular Dynamics Simulations Reveal the Conformational Transition of the Intrinsically Disordered Hypervariable Region of K-Ras4B to the Ordered Conformation. J. Chem. Inf. Model. 2022, 62, 4222–4231. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential Dynamics of Proteins. Proteins-Struct. Funct. Genet. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Wang, E.C.; Sun, H.Y.; Wang, J.M.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T.J. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Sun, H.Y.; Duan, L.L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.C.; Zhu, F.; Zhang, J.Z.H.; Hou, T.J. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef] [PubMed]
- Husic, B.E.; Pande, V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Prinz, J.H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M.; Held, M.; Chodera, J.D.; Schutte, C.; Noe, F. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011, 134, 174105–174127. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernandez, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Eargle, J.; Luthey-Schulten, Z. NetworkView: 3D display and analysis of protein center dot RNA interaction networks. Bioinformatics 2012, 28, 3000–3001. [Google Scholar] [CrossRef]
- Zhang, H.; Chu, G.; Wang, G.; Yao, M.; Lu, S.; Chen, T. Mechanistic Understanding of the Palmitoylation of G(o) Protein in the Allosteric Regulation of Adhesion Receptor GPR97. Pharmaceutics 2022, 14, 1856. [Google Scholar] [CrossRef]
- Zhuang, H.M.; Fan, X.H.; Ji, D.; Wang, Y.H.; Fan, J.G.; Li, M.Y.; Ni, D.; Lu, S.Y.; Li, X.L.; Chai, Z.T. Elucidation of the conformational dynamics and assembly of Argonaute-RNA complexes by distinct yet coordinated actions of the supplementary microRNA. Comput. Struct. Biotechnol. J. 2022, 20, 1352–1365. [Google Scholar] [CrossRef]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Targets | Disease Type | Reference |
|---|---|---|---|
| AZD4547 | FGFR1–3 | ER+ breast cancer | [12] |
| Derazantinib (ARQ-087) | FGFR1–4 | Advanced intrahepatic cholangiocarcinoma with FGFR2 gene aberrations | [13] |
| Erdafitinib (JNJ42756493) | FGFR1–4 | Locally advanced or metastatic bladder cancer, etc. | [14] |
| Futibatinib (TAS-120) | FGFR1–4 | Metastatic breast cancer with FGFR2 amplification | [15] |
| Infigratinib (BGJ398) | FGFR1–3 | Advanced or metastatic cholangiocarcinoma | [16] |
| LY2874455 | FGFR1–4 | Advanced cancer | [17] |
| Pemigatinib (INCB054828) | FGFR1–3 | Unresectable or metastatic cholangiocarcinoma | [18] |
| Rogaratinib (BAY1163877) | FGFR1–3 | Squamous non-small cell lung cancer | [19] |
| Zoligratinib (Debio-1347) | FGFR1–3 | Advanced solid tumors | [20] |
| Complex | ΔEele | ΔEvdw | ΔGpol | ΔGnp | ΔGbinding |
|---|---|---|---|---|---|
| FGFR1-38 | −47.72 ± 3.00 | −18.60 ± 7.85 | 41.28 ± 6.18 | −5.82 ± 0.25 | −30.86 ± 3.73 |
| FGFR2-38 | −40.92 ± 3.09 | −46.93 ± 9.89 | 56.48 ± 8.03 | −5.27 ± 0.29 | −36.64 ± 4.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Yasen, M.; Lu, S.; Ma, D.-N.; Chai, Z. Decoding the Conformational Selective Mechanism of FGFR Isoforms: A Comparative Molecular Dynamics Simulation. Molecules 2023, 28, 2709. https://doi.org/10.3390/molecules28062709
Zhang M, Yasen M, Lu S, Ma D-N, Chai Z. Decoding the Conformational Selective Mechanism of FGFR Isoforms: A Comparative Molecular Dynamics Simulation. Molecules. 2023; 28(6):2709. https://doi.org/10.3390/molecules28062709
Chicago/Turabian StyleZhang, Mingyang, Miersalijiang Yasen, Shaoyong Lu, De-Ning Ma, and Zongtao Chai. 2023. "Decoding the Conformational Selective Mechanism of FGFR Isoforms: A Comparative Molecular Dynamics Simulation" Molecules 28, no. 6: 2709. https://doi.org/10.3390/molecules28062709
APA StyleZhang, M., Yasen, M., Lu, S., Ma, D.-N., & Chai, Z. (2023). Decoding the Conformational Selective Mechanism of FGFR Isoforms: A Comparative Molecular Dynamics Simulation. Molecules, 28(6), 2709. https://doi.org/10.3390/molecules28062709