
Design, Synthesis, In Silico and POM Studies for the Identification of the Pharmacophore Sites of Benzylidene Derivatives


 , , , ,
, , , ,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Results
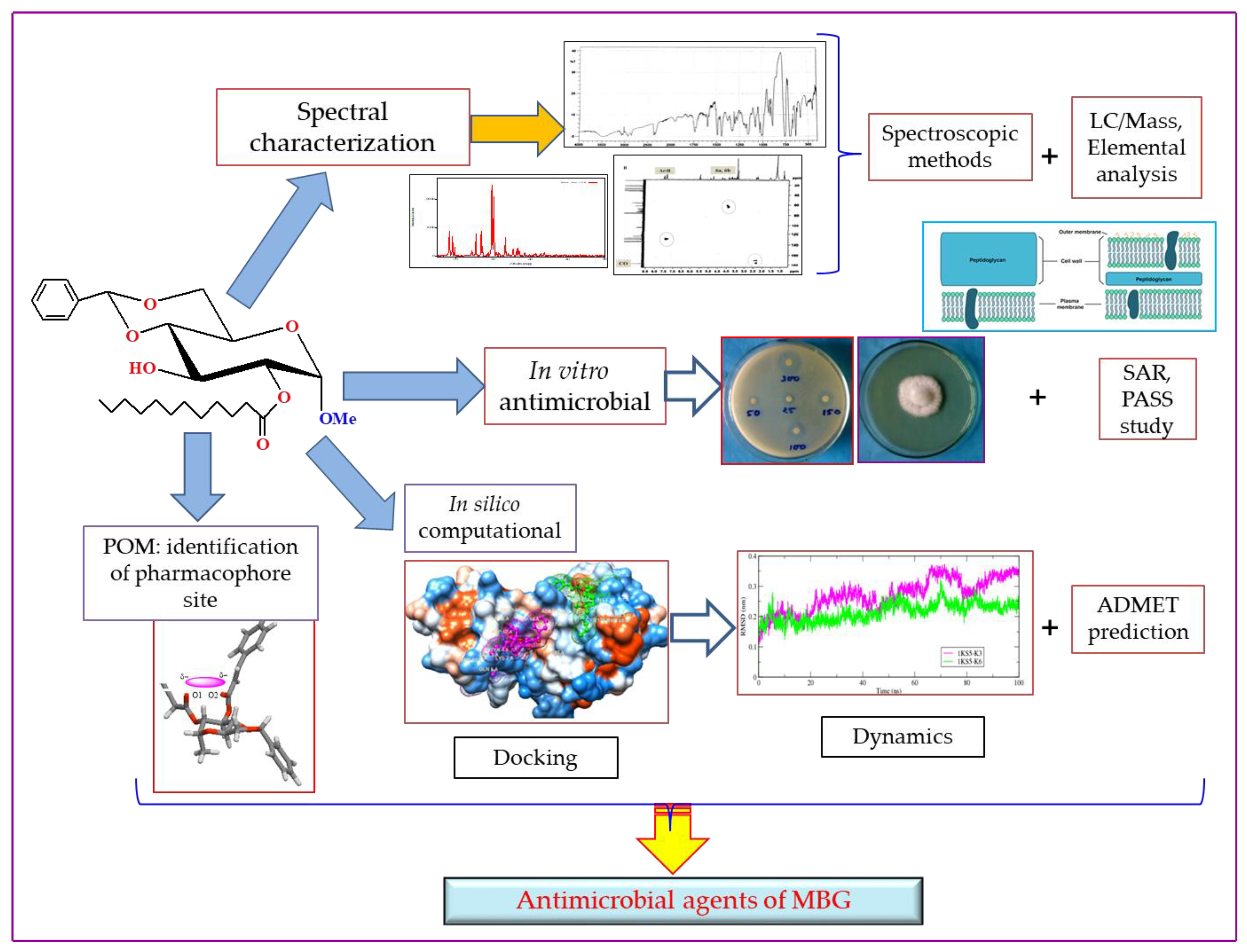
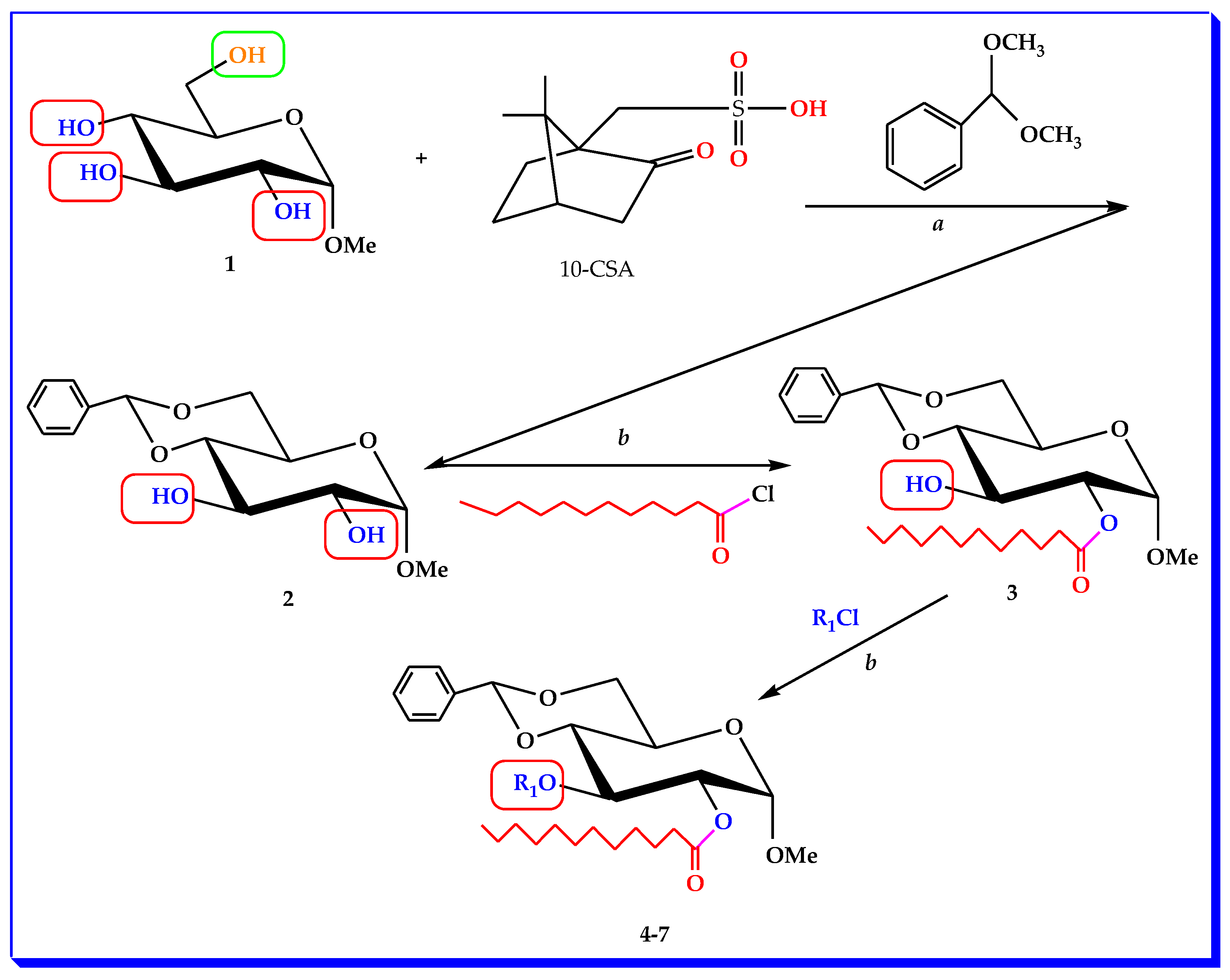
2.1. Chemistry and Characterization

| Compounds | 4 | 5 | 6 | 7 |
| R1 | CH3(CH2)12CO- | CH3(CH2)14CO- | (C6H5)3C- | C6H5CH=CHCO- |
2.2. Antibacterial Activity
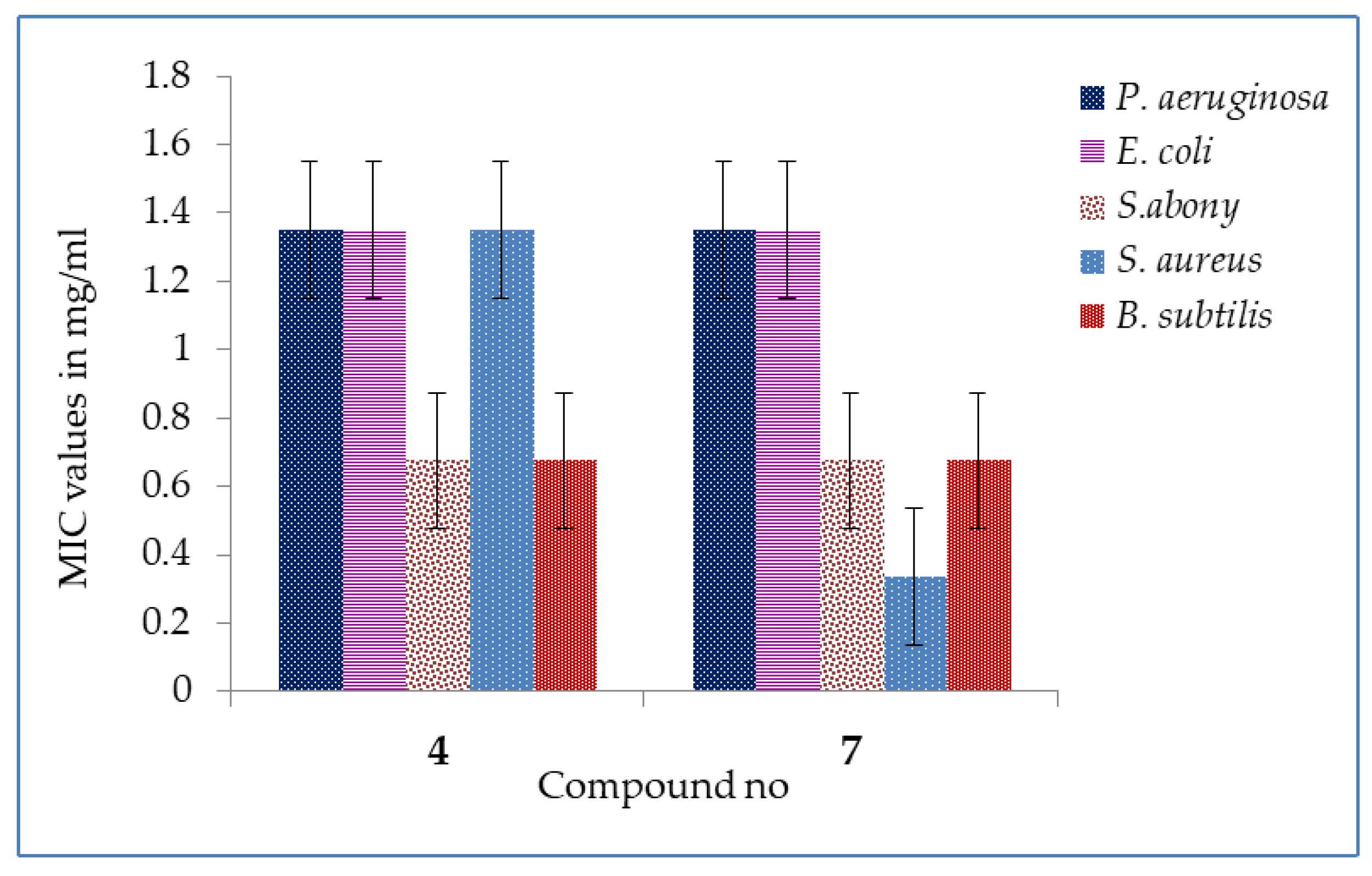
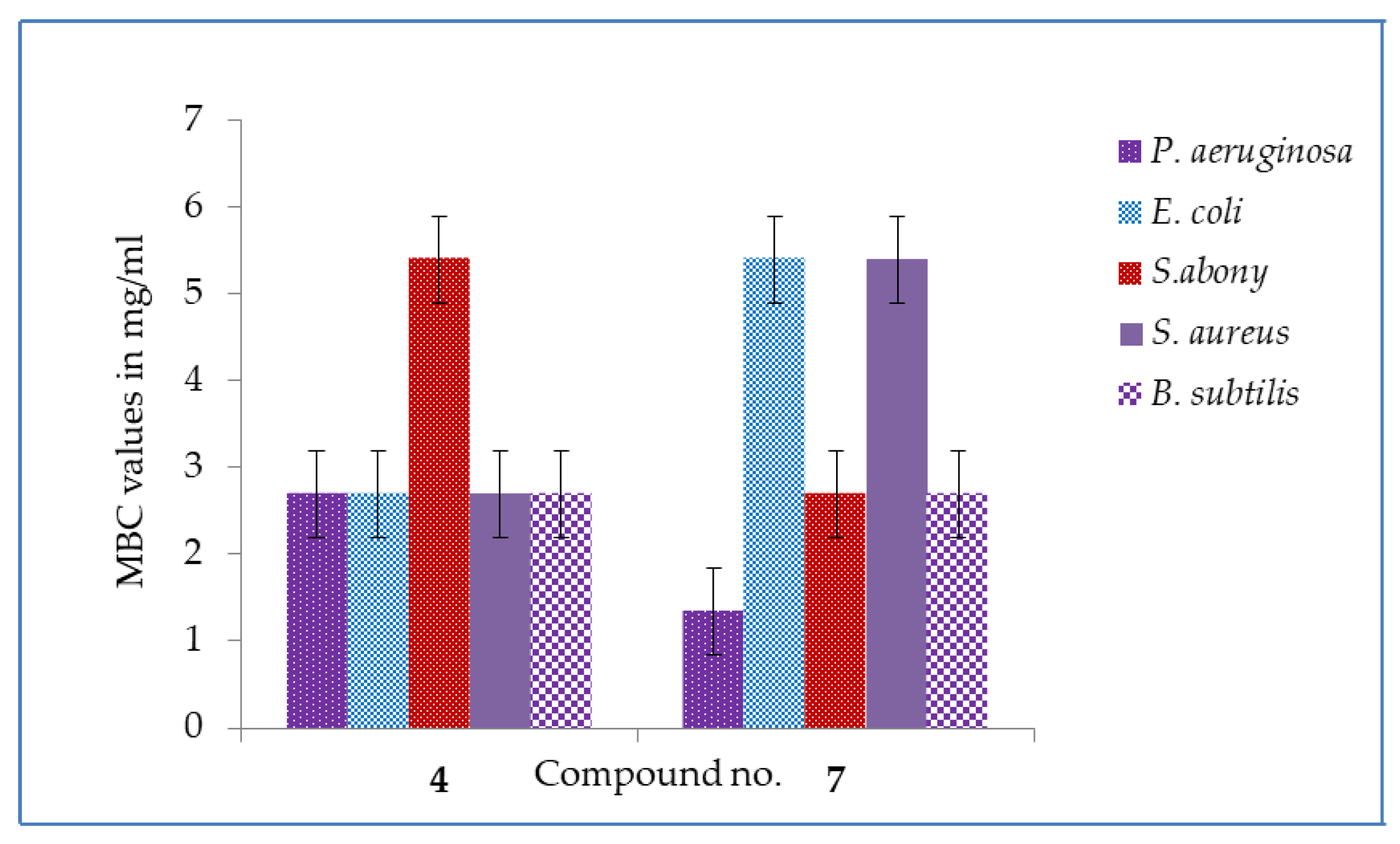
2.3. MIC and MBC Values
2.4. Antifungal Susceptibility
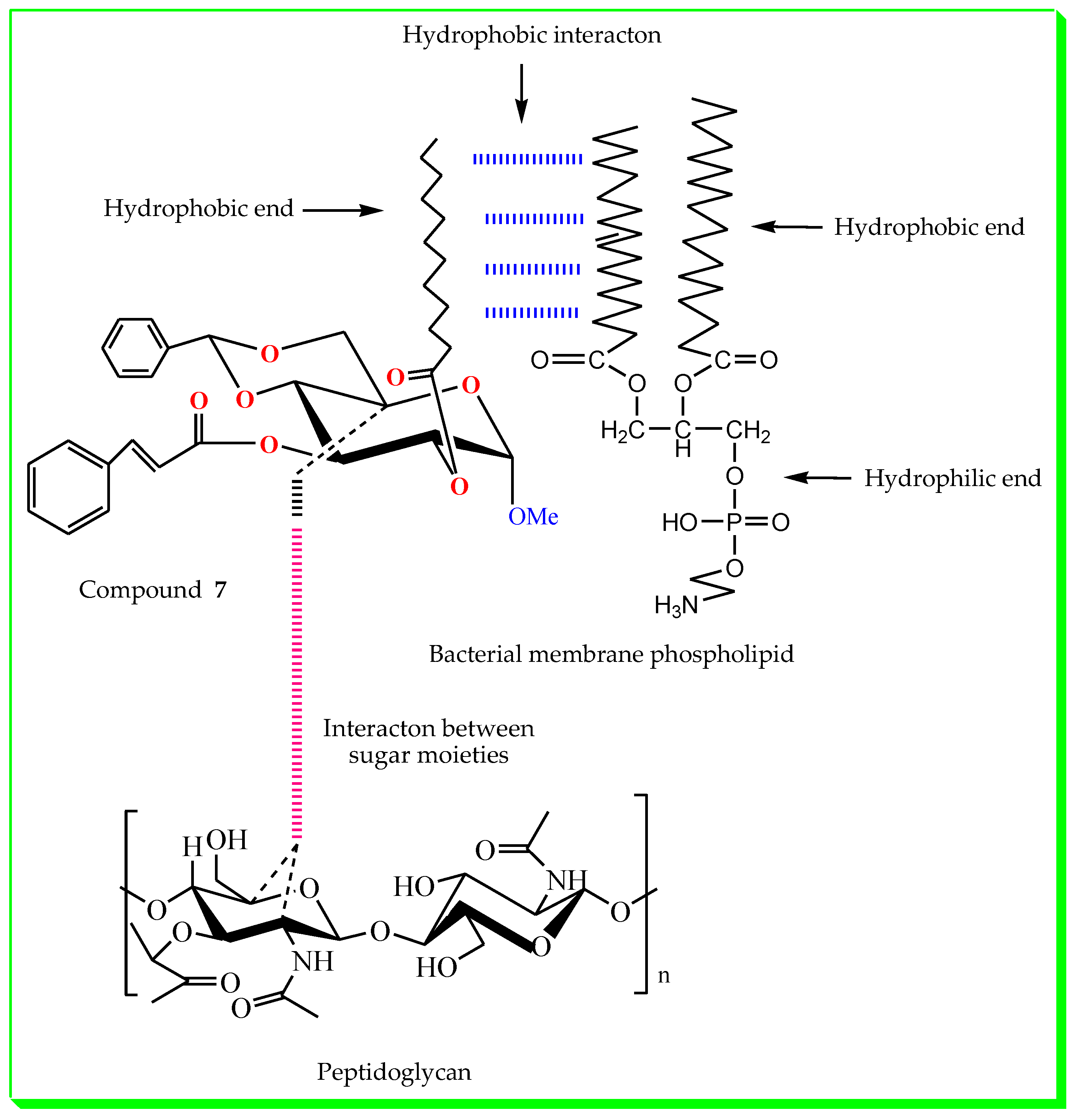
2.5. Structure–Activity Relationship
2.6. PASS Prediction
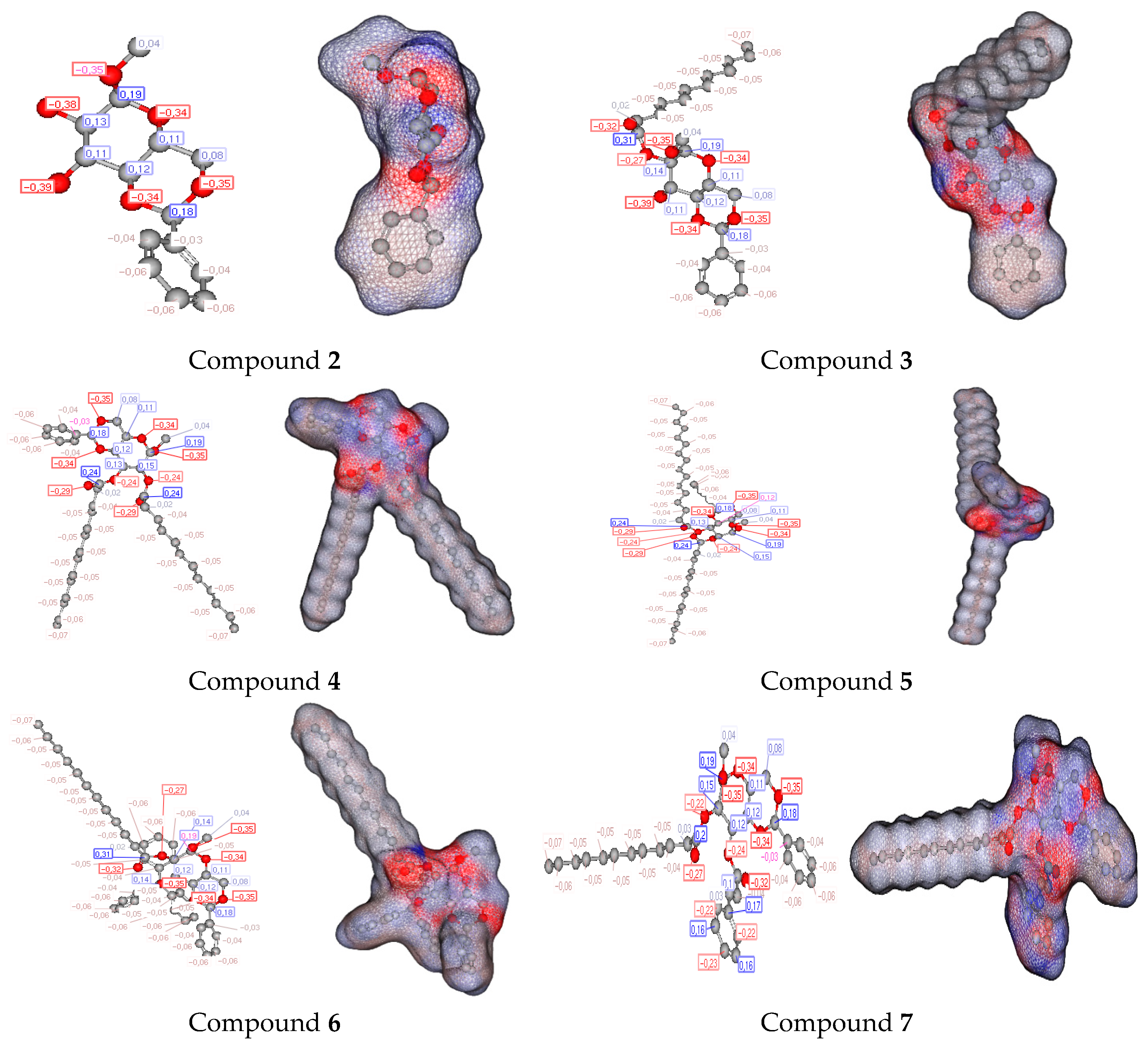
2.7. DFT
2.8. Biological Validation
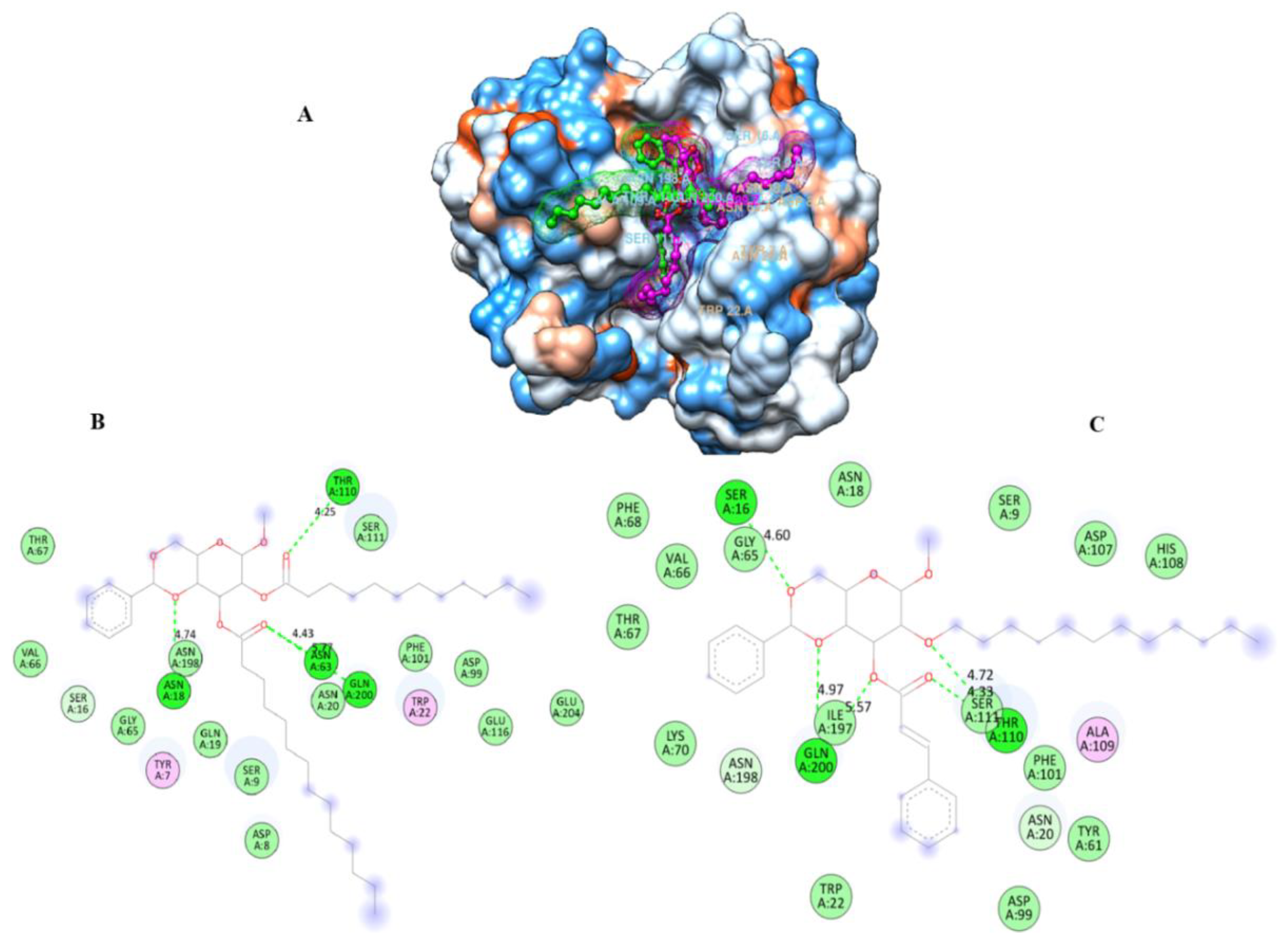
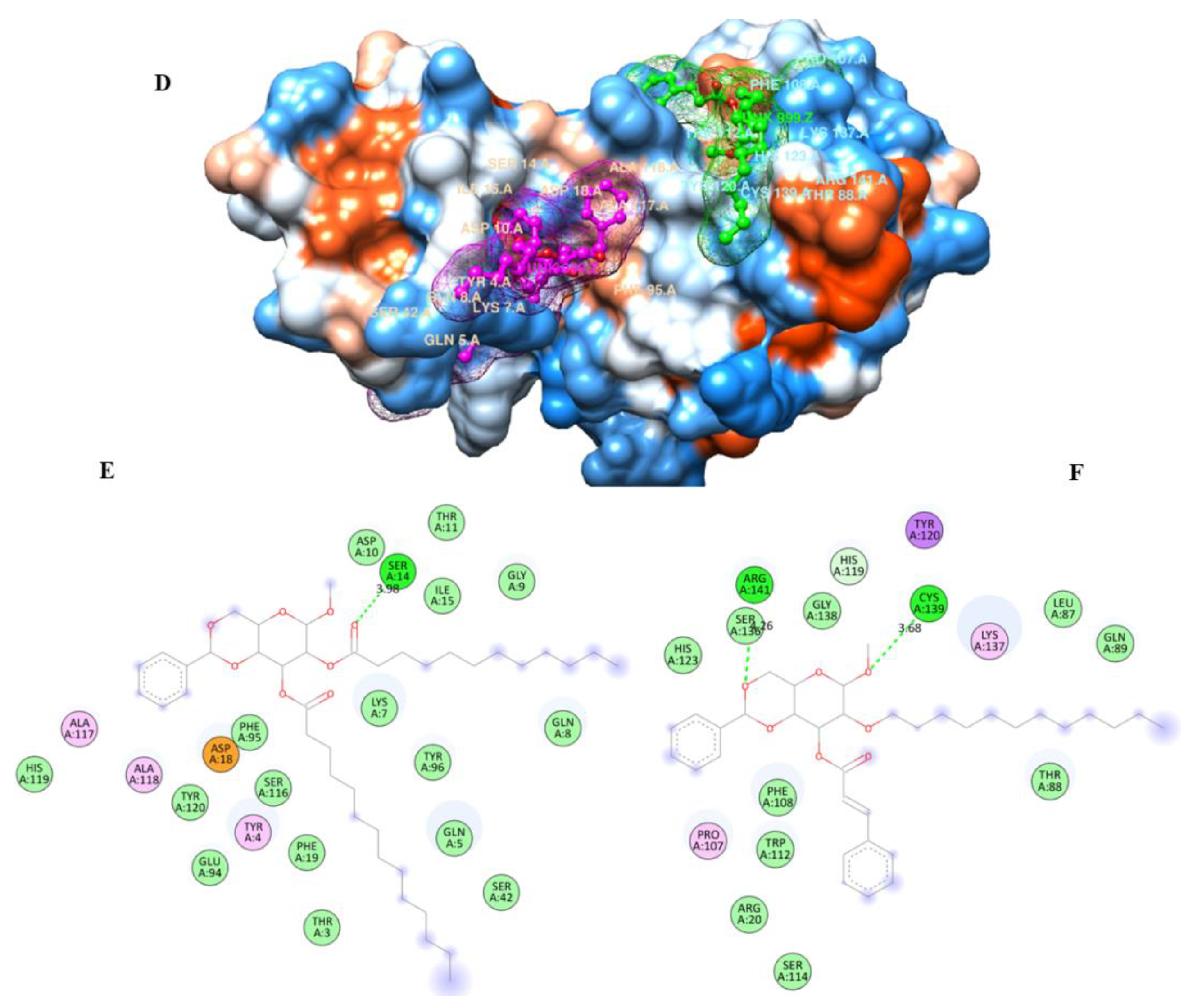
2.9. Molecular Docking
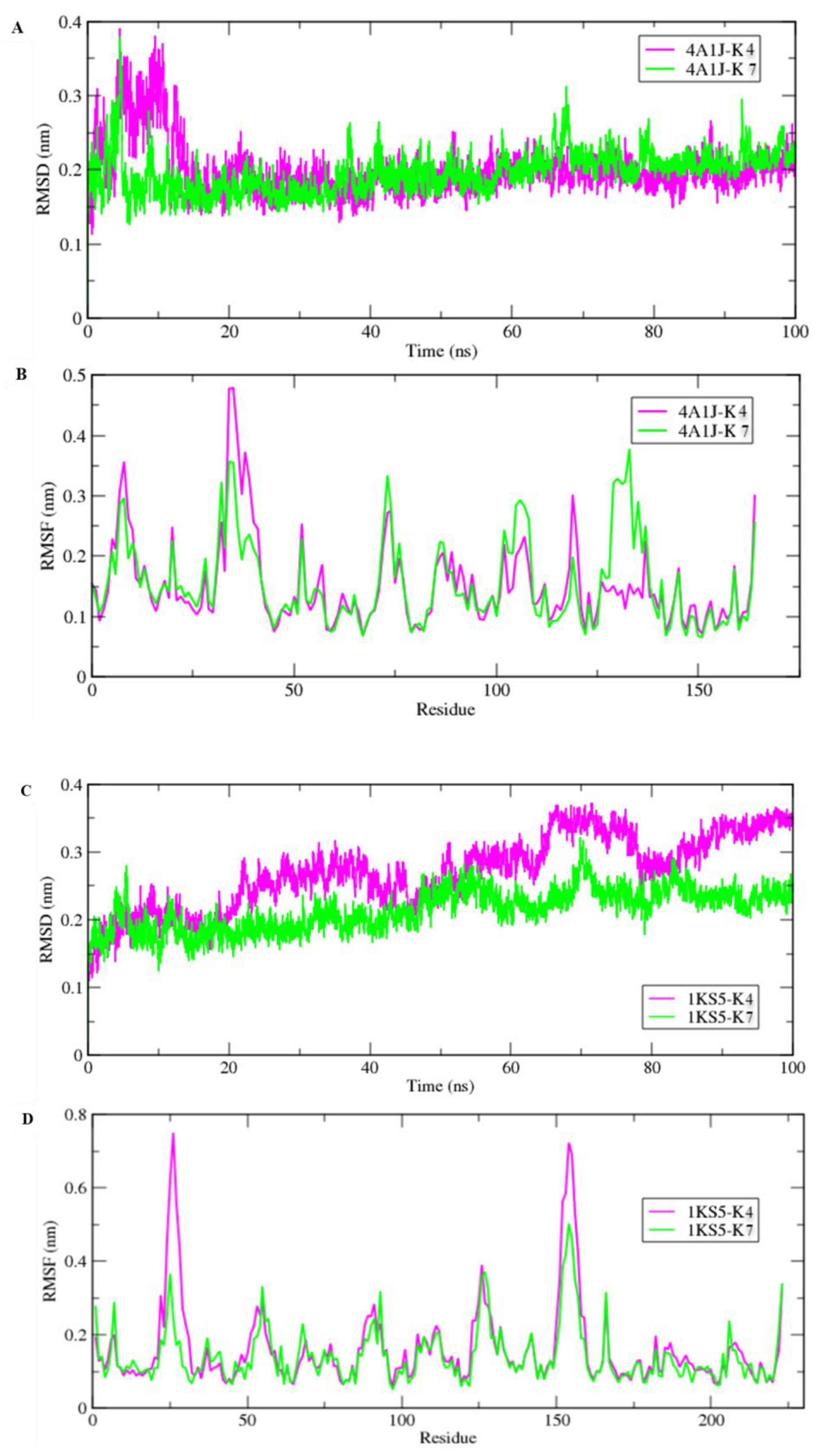
2.10. Molecular Dynamic Simulations
2.11. MM/PBSA
2.12. Pharmacokinetics Properties
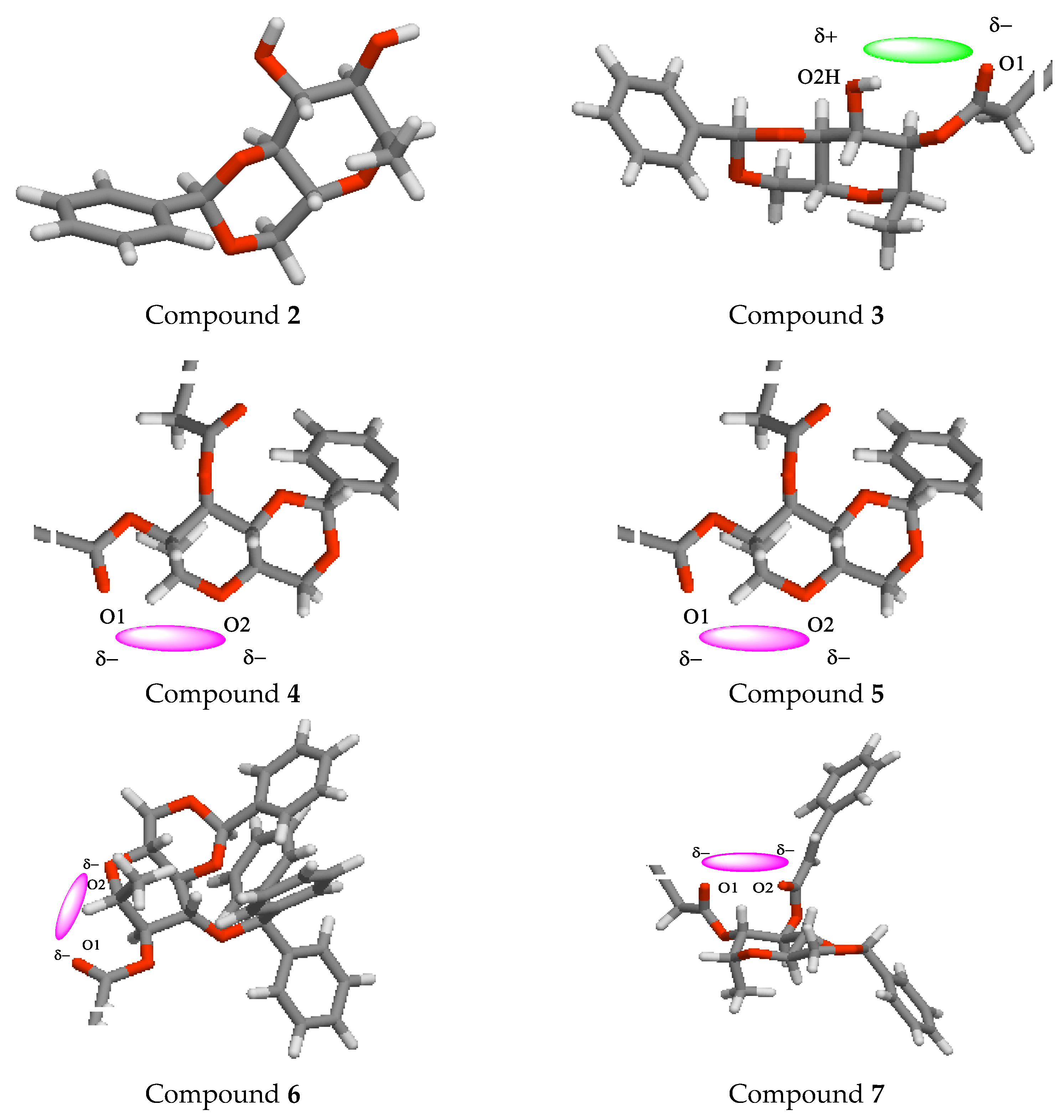
2.13. POM Analyses: Identification of the Pharmacophore Sites
3. Materials and Methods
3.1. General Information
3.2. Synthesis of MBG Derivatives
3.2.1. Methyl 4,6-O-benzylidene-α-d-glucopyranoside (2)
3.2.2. Methyl 4,6-O-benzylidine-2-O-lauroyl-α-d-glucopyranoside (3)
3.2.3. General Procedure for the Synthesis of 2-O-lauroyl Derivatives (4–7)
3.2.4. Methyl 4,6-O-benzylidine-2-O-lauroyl-3-O-myristoyl-α-d-glucopyranoside (4)
3.2.5. Methyl 4,6-O-benzylidine-2-O-lauroyl-3-O-palmitoyl-α-d-glucopyranoside (5)
3.2.6. Methyl 4,6-O-benzylidine-2-O-lauroyl-3-O-(triphenylmethyl)-α-d-glucopyranoside (6)
3.2.7. Methyl 4,6-O-benzylidine-3-O-cinnamoyl-2-O-lauroyl-α-d-glucopyranoside (7)
3.3. Microbial Strains
3.4. In Vitro Antibacterial Activity
3.5. In Vitro Antifungal Activity
3.6. MIC and MBC Tests
3.7. Structure–Activity Relationship
3.8. Quantum Chemical Calculations
3.9. Prediction of the Activity Spectra
3.10. Biological Validation
3.11. Molecular Docking
3.12. Molecular Dynamic Simulations
3.13. Prediction of the Pharmacokinetic and ADME Properties
3.14. POM Analyses
4. Future Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bertozzi, C.R.; Kiessling, L.L. Chemical Glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef]
- Chen, S.; Fukuda, M. Cell Type-specific Roles of Carbohydrates in Tumor Metastasis. Meth. Enzymol. 2006, 416, 371–380. [Google Scholar]
- Varki, A. Biological Roles of Oligosaccharides: All of the Theories are Correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Werz, D.B. Synthesis and Medical Applications of Oligosaccharides. Nature 2007, 446, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Koulenti, D.; Song, A.; Ellingboe, A.; Abdul-Aziz, M.H.; Harris, P.; Gavey, E.; Lipman, J. Infections by Multidrug-Resistant Gram-Negative Bacteria: What’s New in Our Arsenal and What’s in the Pipeline? Int. J. Antimicrob. Agents 2019, 53, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Geddes-McAlister, J.; Shapiro, R.S. New Pathogens, New Tricks: Emerging, Drug-Resistant Fungal Pathogens and Future Prospects for Antifungal Therapeutics. Ann. N. Y. Acad. Sci. 2019, 1435, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, E.M. Plants: An Alternative Source for Antimicrobials. J. Appl. Pharm. Sci. 2011, 1, 16–20. [Google Scholar]
- Wang, L.; Hu, C.; Shao, L. The Antimicrobial Activity of Nanoparticles: Present Situation and Prospects for the Future. Int. J. Nanomed. 2017, 12, 1227–1249. [Google Scholar] [CrossRef] [PubMed]
- Farag, N.A.; El-Tayeb, W. Design, Synthesis and Docking Studies of New Furobenzopyranones and Pyranobenzopyranones as Photoreagent Towards DNA and as Antimicrobial Agents. Eur. J. Med. Chem. 2010, 45, 317–325. [Google Scholar] [CrossRef]
- Lu, H.; Wei, T.; Lou, H.; Shu, X.; Chen, Q. A Critical Review on Communication Mechanism Within Plant-Endophytic Fungi Interactions to Cope with Biotic and Abiotic Stresses. J. Fungi 2021, 7, 719. [Google Scholar] [CrossRef]
- Wing-Fu, L. Non-Conjugated Polymers with Intrinsic Luminescence for Drug Delivery. J. Drug Deliv. Sci. Technol. 2020, 59, 101916. [Google Scholar]
- Kawsar, S.M.A.; Islam, M.; Jesmin, S.; Manchur, M.A.; Hasan, I.; Rajia, S. Evaluation of the Antimicrobial Activity and Cytotoxic Effect of Some Uridine Dderivatives. Int. J. Biosci. 2018, 12, 211–219. [Google Scholar]
- Kawsar, S.M.A.; Hamida, A.A.; Sheikh, A.U.; Hossain, M.K.; Shagir, A.C.; Sanaullah, A.F.M.; Manchur, M.A.; Hasan, I.; Ogawa, Y.; Fujii, Y.; et al. Chemically Modified Uridine Molecules Incorporating Acyl Residues to Enhance Antibacterial and Cytotoxic Activities. Int. J. Org. Chem. 2015, 5, 232–245. [Google Scholar] [CrossRef]
- Shagir, A.C.; Bhuiyan, M.M.R.; Ozeki, Y.; Kawsar, S.M.A. Simple and Rapid Synthesis of Some Nucleoside Derivatives: Structural and Spectral Characterization. Curr. Chem. Lett. 2016, 5, 83–92. [Google Scholar]
- Rana, K.M.; Ferdous, J.; Hosen, A.; Kawsar, S.M.A. Ribose Moieties Acylation and Characterization of Some Cytidine Analogs. J. Sib. Fed. Univ. Chem. 2020, 13, 465–478. [Google Scholar] [CrossRef]
- Bulbul, M.Z.H.; Chowdhury, T.S.; Misbah, M.M.H.; Ferdous, J.; Dey, S.; Kawsar, S.M.A. Synthesis of New Series of Pyrimidine Nucleoside Derivatives Bearing the Acyl Moieties as Potential Antimicrobial Agents. Pharmacia 2021, 68, 23–34. [Google Scholar] [CrossRef]
- Arifuzzaman, M.; Islam, M.M.; Rahman, M.M.; Mohammad, A.R.; Kawsar, S.M.A. An Efficient Approach to the Synthesis of Thymidine Derivatives Containing Various Acyl Groups: Characterization and Antibacterial Activities. ACTA Pharm. Sci. 2018, 56, 7. [Google Scholar] [CrossRef]
- Maowa, J.; Alam, A.; Rana, K.M.; Dey, S.; Hosen, A.; Fujii, Y.; Hasan, I.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, Characterization, Synergistic Antimicrobial Properties and Molecular Docking of Sugar Modified Uridine Derivatives. Ovidius Univ. Ann. Chem. 2021, 32, 6–21. [Google Scholar] [CrossRef]
- Alam, A.; Hosen, M.A.; Hosen, A.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, Characterization, and Molecular Docking against a Receptor Protein FimH of Escherichia coli (4XO8) of Thymidine Derivatives. J. Mex. Chem. Soc. 2021, 65, 256–276. [Google Scholar] [CrossRef]
- Rana, K.M.; Maowa, J.; Alam, A.; Dey, S.; Hosen, A.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. In Silico DFT Study, Molecular Docking, and ADMET Predictions of Cytidine Analogs with Antimicrobial and Anticancer Pproperties. In Silico Pharmacol. 2021, 9, 42. [Google Scholar] [CrossRef]
- Farhana, Y.; Amin, M.R.; Hosen, A.; Kawsar, S.M.A. Bromobenzoylation of Methyl α-D-Mannopyranoside: Synthesis and Spectral Characterization. J. Sib. Fed. Univ. Chem. 2021, 14, 171–183. [Google Scholar]
- Devi, S.R.; Jesmin, S.; Rahman, M.; Manchur, M.A.; Fujii, Y.; Kanaly, R.A.; Ozeki, Y.; Kawsar, S.M.A. Microbial Efficacy and Two Step Synthesis of Uridine Derivatives with Spectral Characterization. Acta Pharm. Sci. 2019, 57, 47–68. [Google Scholar] [CrossRef]
- Alam, A.; Hosen, M.A.; Islam, M.; Ferdous, J.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, Antibacterial and Cytotoxicity Assessment of Modified Uridine Molecules. Curr. Adv. Chem. Biochem. 2021, 6, 114–129. [Google Scholar]
- Kawsar, S.M.A.; Kumar, A. Computational Investigation of Methyl α-D-Glucopyranoside Derivatives as Inhibitor Against Bacteria, Fungi and COVID-19 (SARS-2). J. Chil. Chem. Soc. 2021, 66, 5206–5214. [Google Scholar] [CrossRef]
- Mirajul, M.I.; Arifuzzaman, M.; Monjur, M.R.; Atiar, M.R.; Kawsar, S.M.A. Novel Methyl 4,6-O-Benzylidene-α-D-Glucopyranoside Derivatives: Synthesis, Structural Characterization and Evaluation of Antibacterial Activities. Hacet. J. Biol. Chem. 2019, 47, 153–164. [Google Scholar]
- Kabir, A.K.M.S.; Kawsar, S.M.A.; Bhuiyan, M.M.R.; Rahman, M.S.; Chowdhury, M.E. Antimicrobial Screening Studies of Some Derivatives of Methyl α-D-Glucopyranoside. Pak. J. Sci. Ind. Res. 2009, 52, 138–142. [Google Scholar]
- Kawsar, S.M.A.; Kabir, A.K.M.S.; Bhuiyan, M.M.R.; Siddiqa, A.; Anwar, M.N. Synthesis, Spectral and Antimicrobial Screening Studies of Some Acylated D-Glucose Derivatives. Rajiv. Gandhi. Univ. Health Sci. J. Pharm. Sci. 2012, 2, 107–115. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Hosen, M.A.; Fujii, Y.; Ozeki, Y. Thermochemical, DFT, Molecular Docking and Pharmacokinetic Studies of Methyl β-D-Galactopyranoside Esters. J. Comput. Chem. Mol. Model. 2020, 4, 452–462. [Google Scholar] [CrossRef]
- Kabir, A.K.M.S.; Kawsar, S.M.A.; Bhuiyan, M.M.R.; Rahman, M.S.; Banu, B. Biological Ealuation of Some Octanoyl Derivatives of Methyl 4,6-O-Cyclohexylidene-α-D-gGlucopyranoside. Chittagong Univ. J. Biol. Sci. 2008, 3, 53–64. [Google Scholar]
- Rahman, M.; Islam, M.; Arifuzzaman, M.; Ferdous, J.; Rahman, M.A.; Hasan, I.; Asaduzzaman, A.K.M.; Kawsar, S.M.A. Two Steps Synthesis of Uracil-1-β-D-Ribofuranoside Esters: Characterization, Antibacterial and Anticancer Activities. J. Bang. Chem. Soc. 2019, 30, 46. [Google Scholar]
- Misbah, M.M.H.; Ferdous, J.; Bulbul, M.Z.H.; Chowdhury, T.S.; Dey, S.; Hasan, I.; Kawsar, S.M.A. Evaluation of MIC, MBC, MFC and Anticancer Activities of Acylated Methyl β-D-Galactopyranoside Esters. Int. J. Biosci. 2020, 16, 299–309. [Google Scholar]
- Kawsar, S.M.A.; Matsumoto, R.; Fujii, Y.; Matsuoka, H.; Masuda, N.; Iwahara, C.; Yasumitsu, H.; Kanaly, R.A.; Sugawara, S.; Hosono, M.; et al. Cytotoxicity and Glycan-Binding Profile of α-D-Galactose-Binding Lectin from the Eggs of a Japanese Sea Hare (Aplysia kurodai). Protein J. 2011, 30, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Maowa, J.; Hosen, M.A.; Alam, A.; Rana, K.M.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Pharmacokinetics and Molecular Docking Studies of Uridine Derivatives as SARS-COV-2 Mpro Inhibitors. Phys. Chem. Res. 2021, 9, 385–412. [Google Scholar]
- Farhana, Y.; Amin, M.R.; Hosen, M.A.; Bulbul, M.Z.H.; Dey, S.; Kawsar, S.M.A. Monosaccharide Derivatives: Synthesis, Antimicrobial, PASS, Antiviral, and Molecular Docking studies Against SARS-COV-2 Mpro Inhibitors. J. Cellul. Chem. Technol. 2021, 55, 477–499. [Google Scholar]
- Bulbul, M.Z.H.; Hosen, M.A.; Ferdous, J.; Misbah, M.M.H.; Kawsar, S.M.A. Thermochemical, DFT Study, Physicochemical, Molecular Docking and ADMET Predictions of Some Modified Uridine Derivatives. Int. J. New Chem. 2021, 8, 88–110. [Google Scholar]
- Chowdhury, T.S.; Ferdous, J.; Bulbul, M.Z.H.; Misbah, M.M.H.; Dey, S.; Hasan, I.; Kawsar, S.M.A. Antimicrobial and Anticancer Activities of some Partial Acylated Thymidine Derivatives. J. Bio. Sci. 2021, 29, 11–22. [Google Scholar]
- Kawsar, S.M.A.; Kabir, A.K.M.S.; Manik, M.M.; Anwar, M.N. Selective Synthesis of Some New Carbohydrate Derivatives: Antimicrobial Screening Studies Against Human and Phytopathogens. Chem. Sci. J. 2012, 73, CSJ-73. [Google Scholar]
- Mohammadi, S.; Esmail, D.; Chaleshtori, A.R.C.; Mohammed, E.; Zamani, F.; Ayoub, E. A Computational Study at Blocking Probability of the SARS-CoV-2 Spike Protein Through the Binding of Cellular Receptors. Euras. Chem. Commun. 2021, 3, 369–382. [Google Scholar]
- Kawsar, S.M.A.; Hosen, M.A.; Ahmad, S.; El Bakri, Y.; Laaroussi, H.; Hadda, T.B.; Almalki, F.A.; Ozeki, Y.; Goumri-Said, S. Potential SARS-CoV-2 RdRp Inhibitors of Cytidine Derivatives: Molecular Docking, Molecular Dynamic Simulations, ADMET, and POM Analyses for the Identification of Pharmacophore Sites. PLoS ONE 2022, 17, e0273256. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Almalki, F.A.; Hadda, T.B.; Laaroussi, H.; Khan, M.A.R.; Hosen, M.A.; Mahmud, S.; Aounti, A.; Maideen, N.M.P.; Heidarizadeh, F.; et al. Potential Antifungal Activity of Novel Carbohydrate Derivatives Validated by POM, Molecular Docking and Molecular Dynamic Ssimulations Analyses. Mol. Simul. 2023, 49, 60–75. [Google Scholar] [CrossRef]
- Mahmoud, M.; Hossein, R.A.; Saedi, A. HOMO-LUMO Photosensitization Analyses of Coronene-Cytosine Complexes. Main Group Chem. 2021, 20, 565–573. [Google Scholar]
- Ahmmed, F.; Islam, A.U.; Mukhrish, Y.E.; Bakri, Y.E.; Ahmad, S.; Ozeki, Y.; Kawsar, S.M.A. Efficient Antibacterial/Antifungal Activities: Synthesis, Molecular Docking, Molecular Dynamics, Pharmacokinetic and Binding Free Energy of Galactopyranoside Derivatives. Molecules 2023, 28, 219. [Google Scholar] [CrossRef]
- Kun, H.; Narjes, H.; Mahmoud, M.; Elham, S. Quantum Processing of Cytidine Derivatives and Evaluating their In Silico Interactions with the COVID-19 Main Protease. Main Group Chem. 2022, 21, 263–270. [Google Scholar]
- Uddin, K.M.; Hosen, M.A.; Khan, M.F.; Ozeki, Y.; Kawsar, S.M.A. Investigation of Structural, Physicochemical, Pharmacokinetics, PASS Prediction, and Molecular Docking Analysis of SARS-CoV-2 against Methyl 6-O-Myristoyl-α-D-Glucopyranoside Derivatives. Philipp. J. Sci. 2022, 151, 2215–2231. [Google Scholar] [CrossRef]
- Richtmeyer, N.K. Academic Press Inc., New York, Meth. Carbohydr. Chem. 1962, 1, 107. [Google Scholar]
- Munia, N.S.; Hosen, M.A.; Azzam, K.M.A.; Al-Ghorbani, M.; Baashen, M.; Hossain, M.K.; Ali, F.; Mahmud, S.; Shimu, M.S.S.; Almalk, F.A.; et al. Synthesis, Antimicrobial, SAR, PASS, Molecular Docking, Molecular Dynamics and Pharmacokinetics Studies of 5´-O-Uridine Derivatives Bearing Acyl Moieties: POM Study and Identification of the Pharmacophore Sites. Nucleos. Nucleot. Nucl. Acids 2022, 41, 1036–1083. [Google Scholar] [CrossRef] [PubMed]
- Howse, G.L.; Bovill, R.A.; Stephens, P.J.; Osborn, H.M. Synthesis and Antibacterial Profiles of Targeted Triclosan Derivatives. Europ. J. Med. Chem. 2019, 162, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Craig, W. Pharmacodynamics of Antimicrobial Agents as a Basis for Determining Dosage Regimens. Europ. J. Clin. Microb. Infect. Dis. 1993, 12, S6–S8. [Google Scholar] [CrossRef]
- Amin, M.R.; Yasmin, F.; Hosen, M.A.; Dey, S.; Mahmud, S.; Saleh, M.A.; Hasan, I.; Fujii, Y.; Yamada, M.; Ozeki, Y.; et al. Synthesis, Antimicrobial, Anticancer, PASS, Molecular Docking, Molecular Dynamic Simulations and Pharmacokinetic Predictions of Some Methyl β-D-Galactopyranoside Analogs. Molecules 2021, 26, 7016. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Hosen, M.A.; Ahmad, S.; ul Qamar, M.T.; Dey, S.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, Antimicrobial, Anticancer Activities, PASS Prediction, Molecular Docking, Molecular Dynamics and Pharmacokinetic Studies of Designed Methyl α-D-Glucopyranoside Esters. J. Mol. Struct. 2022, 1260, 132761. [Google Scholar] [CrossRef]
- Liu, B.L.; Hu, X.; He, H.L.; Qiu, L.; Li, Y.Z.; Ding, W.B. A New Epicatechin Glucopyranoside Derivative From Styrax suberifolius. Nat. Prod. Res. 2020, 34, 1977–1983. [Google Scholar] [CrossRef]
- Celik, I.; Erol, M.; Kuyucuklu, G. Molecular Modeling, Density Functional Theory, ADME Prediction and Antimicrobial Activity Studies of 2-(substituted)oxazolo[4,5-b]pyridine Derivatives. New J. Chem. 2021, 45, 11108–11118. [Google Scholar] [CrossRef]
- Mary, Y.S.S.; Yalcin, G.; Mary, Y.S.; Resmi, K.S.; Thomas, R.; Onkol, T.; Kasap, E.N.; Yldiz, I. Spectroscopic, Quantum Mechanical Studies, Ligand Protein Interactions and Photovoltaic Efficiency Modeling of Some Bioactive Benzothiazolinone Acetamide Analogs. Chem. Papers 2020, 74, 1957–1964. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Hosen, M.A.; Kawsar, S.M.A. An Optimization and Pharmacokinetic Studies of some Thymidine Derivatives. Turk. Comput. Theoret. Chem. 2020, 4, 59–66. [Google Scholar]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.W.; Kirby, W.M.M.; Sherris, J.C.; Turck, M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef]
- Grover, R.K.; Moore, J.D. In-Vitro Efficacy of Certain Essential Oils and Plant Extracts Against Three Major Pathogens of Jatropha curcas L. Phytopathology 1962, 52, 876–879. [Google Scholar]
- Melvin, P.W.; Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Disk Susceptibility Tests, 23rd Informational Supplement M100-S23; Clinical and Laboratory Standards Institute (CLSI): Wayne, NJ, USA, 2013. [Google Scholar]
- Hunt, W.A. The Effects of Aliphatic Alcohols on the Biophysical and Biochemical Correlates of Membrane Function. Adv. Exp. Med. Biol. 1975, 56, 195–210. [Google Scholar]
- Kim, Y.M.; Farrah, S.; Baney, R.H. Structure–Antimicrobial Activity Relationship for Silanols, a New Class of Disinfectants, Compared with Alcohols and Phenols. Int. J. Antimicrob. Agents 2007, 29, 217–222. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, M.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. Gauss View, Version 5; Semichem Inc., Shawnee Mission: Irvine, CA, USA, 2009. [Google Scholar]
- Kawsar, S.M.A.; Kabir, A.K.M.S.; Manik, M.M.; Hossain, M.K.; Anwar, M.N. Antibacterial and Mycelial Growth Inhibition of Some Acylated Derivatives of D-Glucopyranoside. Int. J. Biosc. 2012, 2, 66–73. [Google Scholar]
- Onodera, K.; Satou, K.; Hirota, H. Evaluations of Molecular Docking Programs for Virtual Screening. J. Chem. Info. Model. 2007, 47, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Chen, C.C. GEMDOCK: A Generic Evolutionary Method for Molecular Docking. Proteins Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B. GROMACS: High Performance Molecular Simulations Through Multi-level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; van Aalten, D.M. PRODRG: A Tool for High-Throughput Crystallography of Protein-Ligand Complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Gunsteren, W.F.V. A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling Through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Amin, M.R.; Yasmin, F.; Dey, S.; Mahmud, S.; Saleh, M.A.; Emran, T.B.; Hasan, I.; Rajia, S.; Ogawa, Y.; Fujii, Y.; et al. Methyl β-D-Galactopyranoside Esters as Potential Inhibitors for SARS-CoV-2 Protease Enzyme: Synthesis, Antimicrobial, PASS, Molecular Docking, Molecular Dynamics Simulations and Quantum Computations. Glycoconj. J. 2021, 39, 261–290. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R. Open Source Drug Discovery Consortium, Lynn A. g_mmpbsa--a GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-Like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Lakhrissi, Y.; Rbaa, M.; Tuzun, B.; Hichar, A.; Anouar, E.H.; Ounnie, K.; Almalki, F.; Hadda, T.B.; Zarrouk, A.; Lakhrissi, B. Synthesis, Structural Confirmation, Antibacterial Properties and Bio-Informatics Computational Analyses of New Pyrrole Based on 8-Hydroxyquinoline. J. Mol. Struct. 2022, 1259, 132683. [Google Scholar] [CrossRef]
- Fujii, Y.; Kawsar, S.M.A.; Matsumoto, R.; Yasumitsu, H.; Naoto, I.; Dohgasaki, C.; Hosono, M.; Nitta, K.; Hamako, J.; Matsui, T.; et al. A-D-Galactose-Binding Lectin Purified from Coronate Moon Turban, Turbo (Lunella) coreensis, with a Unique Amino Acid Sequence and the Ability to Recognize Lacto-Series Glycophingolipids. Comp. Biochem. Physiol. 2011, 158B, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Hadda, T.B.; Ali, M.A.; Masand, V.; Gharby, S.; Fergoug, T.; Warad, I. Tautomeric Origin of Dual Effects of N1-Nicotinoyl-3-(4’-Hydroxy-3’-Methylphenyl)-5-[(sub)Phenyl]-2-Pyrazolines on Bacterial and Viral Strains: POM Analyses as New Efficient Bioinformatics’ Platform to Predict and Optimize Bioactivity of Drugs. Med. Chem. Res. 2013, 22, 1438–1449. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Hosen, M.A.; Bakri, Y.E.; Ahmad, S.; Affi, S.T.; Goumri-Said, S. In Silico Approach for Potential Antimicrobial Agents through Antiviral, Molecular Docking, Molecular Dynamics, Pharmacokinetic and Bioactivity Predictions of Galactopyranoside Derivatives. Arab J. Basic Appl. Sci. 2022, 29, 99–112. [Google Scholar] [CrossRef]
- Berredjem, M.; Bouzina, A.; Bahadi, R.; Bouacida, S.; Rastija, V.; Djouad, S.-E.; Sothea, T.O.; Almalki, F.A.; Hadda, T.B.; Aissaoui, M. Antitumor Activity, X-Ray Crystallography, In Silico Study of Some-Sulfamido-Phosphonates. Identification of Pharmacophore Sites. J. Mol. Struct. 2022, 1250, 131886. [Google Scholar] [CrossRef]
- Hadda, T.B.; Sezer, S.F.; Orhan, I.E.; Zgou, H.; Rauf, A.; Mabkhot, Y.N.; Bennani, B.; Emam, D.R.; Asayari, N.A.K.A.; Muhsinah, A.B.; et al. Spiro Heterocyclic Compounds as Potential Anti-Alzheimer Agents (Part 2): Their Metal Chelation Capacity, POM Analyses and DFT Sudies. Med. Chem. 2020, 16, 834–843. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diameter of Inhibition Zones (in mm) | |||||
|---|---|---|---|---|---|
| Entry | B. subtilis (G + ve) | S. aureus (G + ve) | E. coli (G − ve ) | S. abony (G − ve) | P. aeruginosa (G − ve) |
| 2 | NI | NI | 10.0 ± 0.1 | 10.0 ± 0.1 | NI |
| 3 | 5.0 ± 0.1 | NI | 8.0 ± 0.1 | NI | NI |
| 4 | ** 21.0 ± 0.3 | * 16.0 ± 0.2 | 7.0 ± 0.1 | ** 24.0 ± 0.3 | * 15.0 ± 0.2 |
| 5 | NI | 10.0 ± 0.1 | 5.0 ± 0.1 | ** 20 ± 0.3 | NI |
| 6 | NI | 6.0 ± 0.1 | 11.0 ± 0.1 | NI | 10.0 ± 0.1 |
| 7 | ** 21.0 ± 0.3 | ** 31.0 ± 0.4 | * 18.0 ± 0.2 | ** 26.5 ± 0.4 | ** 20.0 ± 0.3 |
| Azithromycin | ** 19.0 ± 0.2 | ** 18.0 ± 0.2 | ** 17.0 ± 0.2 | ** 19.0 ± 0.2 | ** 17.0 ± 0.2 |
| Entry | Percentage (%) of Inhibition | |
|---|---|---|
| Aspergillus niger | Aspergillusflavus | |
| 2 | 25.45 ± 0.04 | NI |
| 3 | 32.67 ± 0.05 | 28.48 ± 0.03 |
| 4 | * 85.55 ± 0.05 | * 64.44 ± 0.05 |
| 5 6 | * 74.88 ± 0.05 | 40.00 ± 0.05 |
| * 70.22 ± 0.05 | NI | |
| 7 | * 73.33 ± 0.05 | 38.88 ± 0.05 |
| Nystatin | ** 66.4 ± 0.05 | ** 63.1 ± 0.05 |
| Diameter of Inhibition Zone in Mm | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Antibacterial | Antifungal | Antioxidant | Anticarcinogenic | ||||
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| 2 | 0.342 | 0.011 | 0.521 | 0.012 | 0.403 | 0.041 | 0.731 | 0.008 |
| 3 | 0.455 | 0.014 | 0.681 | 0.010 | 0.637 | 0.005 | 0.843 | 0.005 |
| 4 | 0.566 | 0.011 | 0.761 | 0.010 | 0.661 | 0.004 | 0.776 | 0.006 |
| 5 | 0.501 | 0.013 | 0.743 | 0.008 | 0.585 | 0.005 | 0.709 | 0.007 |
| 6 | 0.421 | 0.013 | 0.743 | 0.008 | 0.585 | 0.005 | 0.709 | 0.007 |
| 7 | 0.581 | 0.011 | 0.759 | 0.005 | 0.601 | 0.004 | 0.787 | 0.006 |
| Energy | Fungal Target Proteins | Bacterial Target Proteins | ||
|---|---|---|---|---|
| 1KS5-4 | 1KS5-7 | 4A1J-4 | 4A1J-7 | |
| Van der Waals | −173.798 ± 25.063 | −191.792 ± 27.019 | −186.159 ± 22.931 | −144.754 ± 18.180 |
| Electrostatic | −8.833 ± 10.975 | −0.483 ± 5.877 | −24.110 ± 16.763 | −0.121 ± 4.572 |
| Polar solvation | 67.371 ± 19.038 | 56.234 ± 21.781 | 67.151 ± 22.032 | 46.781 ± 29.273 |
| SASA | −17.994 +/−2.694 | −20.052 ± 2.824 | −19.244 ± 2.657 | −14.770 ± 1.985 |
| Binding | −133.254 ± 20.879 | −156.093 ± 23.130 | −162.362 ± 22.027 | −112.863 ± 30.839 |
| Entry | MW (g/mole) | Toxicity Risks [a] | Drug Score Calculations [b] | ||||||
|---|---|---|---|---|---|---|---|---|---|
| MUT | TUM | IRRI | REP | cLogP | cLogS | DL | DS | ||
| 2 | 282.29 |  | | | | −0.39 | −1.49 | −5.08 | 0.48 |
| 3 | 464.60 | | | | | 4.64 | −4.6 | −26.7 | 0.26 |
| 4 | 674.96 | | | | | 10.57 | −8.25 | −27.22 | 0.07 |
| 5 | 703.01 | | | | | 11.48 | −8.79 | −27.22 | 0.07 |
| 6 | 706.92 | | | | | 9.14 | −7.71 | −28.06 | 0.07 |
| 7 | 594.75 | | |  | | 6.89 | −6.55 | −30.14 | 0.06 |
); highly toxic, (); REP, reproductive effective; IRRIT, irritant; TUM; tumorigenic; MUT; mutagenic. [b] DS, drug score; DL, drug likeness.| Entry | Lipinski Parameters Calculations [a] | Drug Likeness [b] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TPSA | NONH | NV | VOL | GPCRL | ICM | KI | NRL | PI | EI | |
| 2 | 77 | 2 | 0 | 249 | −0.25 | −0.05 | −0.30 | −0.57 | −0.09 | 0.49 |
| 3 | 84 | 1 | 1 | 453 | −0.06 | −0.11 | −0.28 | −0.27 | 0.09 | 0.35 |
| 4 | 90 | 0 | 2 | 691 | −0.51 | −1.23 | −0.95 | −1.02 | −0.22 | −0.53 |
| 5 | 90 | 0 | 2 | 725 | −0.27 | −1.55 | −1.24 | −1.32 | −0.37 | −0.80 |
| 6 | 73 | 0 | 2 | 685 | −1.01 | −2.01 | −1.60 | −1.60 | −0.56 | −1.02 |
| 7 | 90 | 0 | 2 | 572 | −0.18 | −0.65 | −0.47 | −0.45 | −0.04 | −0.02 |
| Compounds (δH Values) | |||||
|---|---|---|---|---|---|
| Protons | 3 | 4 | 5 | 6 | 7 |
| Ar-H | 7.51 (2H, m), 7.37 (3H, m) | 7.44 (2H, m), 7.35 (3H, m) | 7.44 (2H, m, Ar-H), 7.35 (3H, m) | 8.21 (2H, m), 8.12 (3H, m), 7.28 (6H, m), 7.01 (9H, m) | 7.55 (2H, m), 7.53 (2H, m), 7.39 (3H, m), 7.28 (3H, m) |
| PhCH- | 5.56 (1H, s) | 5.51 (1H, s) | 5.50 (1H, s) | 5.09 (1H, s) | 5.34 (1H, s) |
| PhCH=CHCO- | 7.76 (1H, d, J = 12.1 Hz) | ||||
| PhCH=CHCO- | 6.49 (1H, d, J = 12.1 Hz) | ||||
| H-1 | 4.99 (1H, d, J = 4.2 Hz) | 5.34 (1H, d, J = 3.7 Hz) | 4.97 (1H, d, J = 3.7 Hz) | 4.96 (1H, d, J = 3.7 Hz) | 5.11 (1H, d, J = 3.6 Hz) |
| H-2 | 4.84 (1H, dd, J = 3.7 Hz, 9.7 Hz) | 4.95 (1H, dd, J = 3.6 Hz, 9.8 Hz) | 4.89 (1H, dd, J = 3.6 Hz, 9.8 Hz) | 4.32 (1H, dd, J = 3.7 Hz, 9.7 Hz) | 5.09 (1H, dd, J = 3.6 Hz, and 9.8 Hz, H-2) |
| H-3 | 4.24 (1H, t, J = 9.7 Hz) | 5.62 (1H, t, J = 9.6 Hz) | 5.61 (1H, t, J = 9.6 Hz) | 5.36 (1H, t, J = 9.7 Hz) | 5.82 (1H, t, J = 9.8 Hz) |
| H-6a | 3.98 (1H, dd, J = 4.7, 10.1 Hz | 4.90 (1H, dd, J = 4.7, 10.1 Hz) | 4.33 (1H, dd, J = 4.7, 10.1 Hz) | 4.29 (1H, dd, J = 4.7, 10.1 Hz) | 4.40 (1H, dd, J = 4.7, 10.1 Hz) |
| H-5 | 3.81 (1H, ddd, J = 4.7, 9.7, 14.1 Hz) | 4.33 (1H, ddd, J = 4.7, 9.7, 14.1 Hz) | 3.94 (1H, ddd, J = 4.7, 9.7, 14.1 Hz) | 4.16 (1H, ddd, J = 4.7, 9.7, 14.1 Hz) | 4.17 (1H, ddd, J = 4.8, 9.8, 14.2 Hz) |
| H-6b | 3.74 (1H, t, J = 10.1 Hz) | 3.87 (1H, t, J = 10.2 Hz) | 3.86 (1H, t, J = 10.2 Hz) | 4.14 (1H, t, J = 10.2 Hz) | 4.13 (1H, t, J = 10.2 Hz) |
| H-4 | 3.57 (1H, t, J = 9.7 Hz) | 3.68 (1H, t, J = 9.8 Hz) | 3.57 (1H, t, J = 9.8 Hz) | 3.71 (1H, t, J = 9.8 Hz) | 4.12 (1H, t, J = 9.8 Hz) |
| 1-OCH3 | 3.50 (3H, s) | 3.46 (3H, s) | 3.41 (3H, s) | 3.68 (3H, s) | 3.68 (3H, s) |
| {CH3(CH2)9CH2CO-} | 2.38 (2H, m) | 2.35 (2H, m) | 2.32 (2H, m) | 2.35 (2H, m) | 2.36 (2H, m) |
| {CH3(CH2)8CH2CH2CO-} | 1.66 (2H, m) | 1.60 (2H, m) | 1.61 (2H, m) | 1.64 (2H, m) | 1.66 (2H, m) |
| {CH3(CH2)8CH2CH2CO-} | 1.28 {16H, m) | 1.35 (16H, m) | 1.27 (16H, m) | 1.28 (16H, m) | 1.30 (16H, m) |
| {CH3(CH2)10CO-} | 0.90 (3H, m) | 0.90 (3H, m) | 0.90 (3H, m) | 0.89 (3H, m) | 0.90 (3H, m) |
| {CH3(CH2)11CH2CO-} | 2.40 (2H, m) | ||||
| CH3(CH2)10CH2CH2CO- | 1.64 (2H, m) | ||||
| {CH3(CH2)10CH2CH2CO-} | 1.28 (20H, br, m) | ||||
| {CH3(CH2)12CO-} | 0.88 (3H, t, J = 6.8 Hz) | ||||
| {CH3(CH2)13CH2CO-} | 2.37 (2H, m) | ||||
| {CH3(CH2)13CH2CO-} | 1.66 (26H, m) | ||||
| {CH3(CH2)14CO-} | 0.90 (3H, m) | ||||
| Compounds (δC Values) | |||||
|---|---|---|---|---|---|
| Protons | 3 | 4 | 5 | 6 | 7 |
| {CH3(CH2)10CO-} | 177.90 | 177.87 | 177.11 | 177.12 | 177.28 |
| (C6H5CH-) | 136.20, 129.10, 128.42 (×2), 126.34 (×2) | 136.43, 129.45, 128.01 (×2), 126.31 (×2) | 137.22, 128.13 (×3), 126.31 (×2) | 137.11, 129.10, 128.30 (×2), 126.37 (×2) | |
| (C6H5CH-) | 125.31 | 125.21 | 125.21 | 125.42 | |
| Glucose ring | 105.09 (C-1), 73.95 (C-2), 76.09 (C-4), 74.25 (C-3), 69.35 (C-5), 62.05 (C-6) | 105.11 (C-1), 73.21 (C-2), 76.42 (C-4), 74.23 (C-3), 69.11 (C-5), 62.31 (C-6) | 105.11 (C-1), 73.43 (C-2), 76.54 (C-4), 74.65 (C-3), 69.22 (C-5), 62.08 (C-6) | 105.05 (C-1), 73.55 (C-2), 76.32 (C-4), 74.26 (C-3), 69.63 (C-5), 61.32 (C-6) | 105.55 (C-1), 73.54 (C-2), 76.44 (C-4), 74.53 (C-3), 69.53 (C-5), 62.43 (C-6) |
| (1-OCH3) | 58.06 | 58.44 | 58.44 | 58.24 | 58.43 |
| {CH3(CH2) 10CO-} | 34.18, 31.94, 29.64 (×2), 29.50, 29.31 (×2), 25.22, 24.97, 22.73 | 34.32, 31.39, 29.32 (×2), 29.11, 29.01 (×2), 25.21, 24.33, 22.42 | 34.28, 31.33, 29.61 (×2), 25.23, 24.21, 22.33 | 34.16, 31.43, 29.45 (×2), 29.43, 29.73 (×2), 25.53, 24.53, 22.63 | 34.54, 31.54, 29.22 (×2), 29.52, 29.12 (×2), 25.23, 24.52, 22.63 |
| {CH3(CH2)10CO-} | 14.17 | 14.12 | 14.12 | 14.17 | 14.43 |
| {CH3(CH2)12CO-} | 172.54 | ||||
| {CH3(CH2)12CO-} | 34.38, 34.36, 31.92, 31.90, 29.15, 25.01 (×2), 24.96, 22.67, 21.72, 21.69, 20.09 | ||||
| {CH3(CH2)12CO-} | 14.01 | ||||
| {CH3(CH2)14CO-} | 172.21 | ||||
| {CH3(CH2)14CO-} | 34.43, 34.38, 34.12, 31.95, 31.91, 29.52, 29.31, 25.11 (×2), 24.77, 22.61, 21.65, 21.54, 20.01 | ||||
| {CH3(CH2)14CO-} | 14.02 | ||||
| [(C6H5)3C-] | 145.20 (×3), 129.61 (×6), 127.84 (×3), 127.64 (×6) | 136.54, 129.55, 128.45 (×2), 126.55 (×2) | |||
| [(C6H5)3C-] | 81.31 | 125.35 | |||
| (C6H5CH=CHCO-) | 165.84 | ||||
| (C6H5CH=CHCO-) | 150.58 | ||||
| (C6H5CH=CHCO-) | 136.95, 132.01, 129.20 (×2), 129.06 (×2) | ||||
| (C6H5CH=CHCO-) | 122.26 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosen, M.I.; Mukhrish, Y.E.; Jawhari, A.H.; Celik, I.; Erol, M.; Abdallah, E.M.; Al-Ghorbani, M.; Baashen, M.; Almalki, F.A.; Laaroussi, H.; et al. Design, Synthesis, In Silico and POM Studies for the Identification of the Pharmacophore Sites of Benzylidene Derivatives. Molecules 2023, 28, 2613. https://doi.org/10.3390/molecules28062613
Hosen MI, Mukhrish YE, Jawhari AH, Celik I, Erol M, Abdallah EM, Al-Ghorbani M, Baashen M, Almalki FA, Laaroussi H, et al. Design, Synthesis, In Silico and POM Studies for the Identification of the Pharmacophore Sites of Benzylidene Derivatives. Molecules. 2023; 28(6):2613. https://doi.org/10.3390/molecules28062613
Chicago/Turabian StyleHosen, Mohammad I., Yousef E. Mukhrish, Ahmed Hussain Jawhari, Ismail Celik, Meryem Erol, Emad M. Abdallah, Mohammed Al-Ghorbani, Mohammed Baashen, Faisal A. Almalki, Hamid Laaroussi, and et al. 2023. "Design, Synthesis, In Silico and POM Studies for the Identification of the Pharmacophore Sites of Benzylidene Derivatives" Molecules 28, no. 6: 2613. https://doi.org/10.3390/molecules28062613
APA StyleHosen, M. I., Mukhrish, Y. E., Jawhari, A. H., Celik, I., Erol, M., Abdallah, E. M., Al-Ghorbani, M., Baashen, M., Almalki, F. A., Laaroussi, H., Hadda, T. B., & Kawsar, S. M. A. (2023). Design, Synthesis, In Silico and POM Studies for the Identification of the Pharmacophore Sites of Benzylidene Derivatives. Molecules, 28(6), 2613. https://doi.org/10.3390/molecules28062613