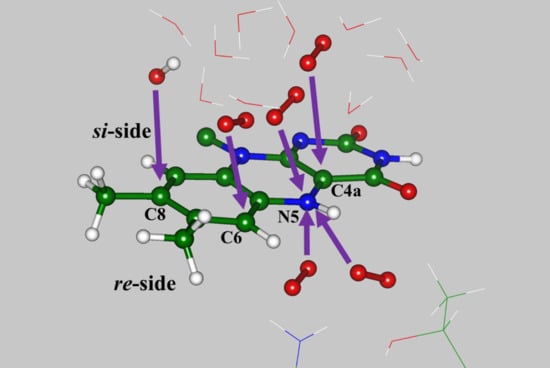
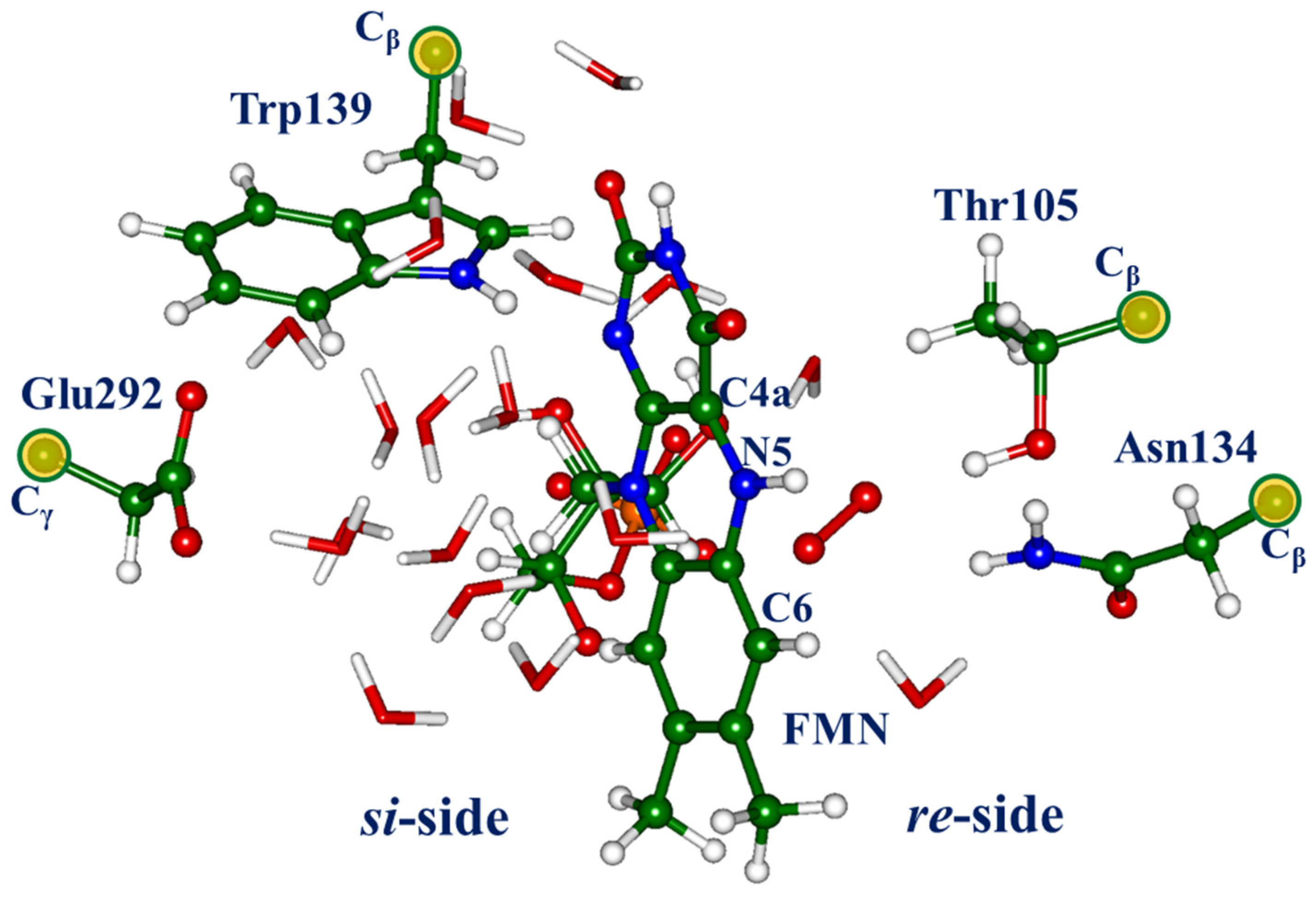
There are several important observations related to the computationally derived model systems containing the flavin-oxygen complexes. First, the optimized positions of dioxygen are located on both sides (
re-side and
si-side) of the isoalloxazine ring (see
Figure 1). The sites at the
re-side correspond to the oxygen pocket-1 noted in [
12], as well as the position of oxygen in the X-ray structure PDB ID 6SGG [
6]. The sites on the
si-side, which correspond to the oxygen pocket-5 noted in [
12], describe the oxygen molecule inside the shell of water molecules. Second, in each located oxygen-flavin complex, the dioxygen moiety should be identified as a superoxide O
2− anion rather than a neutral triplet oxygen molecule. Third, the energies of the triplet and singlet states in these flavin-oxygen complexes are almost degenerate. To support the second and third conclusions, we analyze the results of the complete active space self-consistent field (CASSCF) calculations of the complexes (for details, see
Section 3 below). Thus, assuming a switch from the triplet state potential energy surface (PES) to the singlet state PES, modeling a triplet-singlet intersystem crossing, several pathways to the reaction products arise, including the formation of the flavin-oxygen adducts. We describe these pathways in the following subsections using the QM(DFT)/MM calculations and characterize the excitation energies of the products using the XMCQDPT2 calculations.
2.1. Pathway-1: Formation of the C(4a)-peroxyflavin Species from the Si-Side Oxygen Pocket
As described in the Introduction, the formation of the transient C(4a)-(hydro)peroxyflavin intermediates, Fl
C4aOOH or Fl
C4aOO−, is commonly accepted in the chemistry of flavoenzymes [
3,
7], even though the C(4a)-peroxyflavin species in water is unstable and dissociates to hydrogen peroxide (H
2O
2) and the oxidized flavin [
3]. Previous simulations [
5,
10] described the reaction pathways from reduced lumiflavin (Fl
red) and O
2 to Fl
C4aOO− and Fl
C4aOOH using the continuum solvent models. In both calculations, the transition from the initial triplet state model system to the singlet state system occurs after electron transfer in the configuration of the radical pair superoxide—Fl
SQ. In both cases, low activation barriers of several kcal/mol were estimated.
According to the results of molecular dynamics simulations with the QM(DFT)/MM potentials for the RutA-O
2-uracil model system [
12], the C(4a)-peroxyflavin species was spontaneously formed after switching from the triplet state to the singlet state system at the trajectory frames that showed the occurrence of both dioxygen and uracil near the isoalloxazine ring at the
si-side. Complexes of RutA with dioxygen located at the
si-side pocket are located in this work using the QM/MM method (see
Section 3 below for technical details).
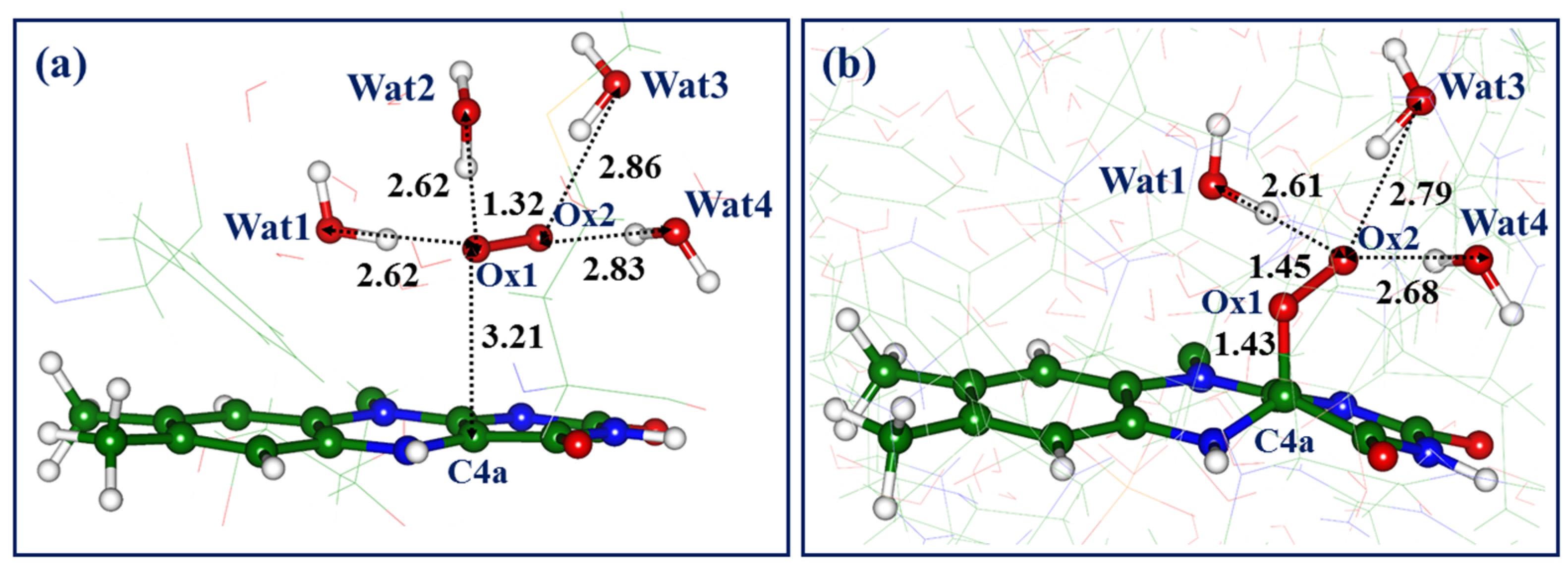
Figure 2a shows the triplet state minimum energy structure of the complex, which is denoted here as Complex-1. In this and other figures, we show critical distances (in Å) between heavy atoms and pay attention to the hydrogen-bond patterns. The water molecules, which are the components of hydrogen-bond networks, are numbered sequentially as they appear in the figures.
For Complex-1, we note that the Ox1-Ox2 distance in dioxygen increases from the value of about 1.20 Å in the gas-phase triplet state O2 molecule to 1.32 Å, thus favoring the formation of the superoxide anion. The latter is hydrogen bonded to the four nearest water molecules, with typical intermolecular distances between the oxygen atoms (2.6–2.9 Å). The C4a atom is the closest atom of the isoalloxazine ring to the dioxygen atoms, with a C4a-Ox1 distance 3.21 Å; the N5-Ox1 distance is 3.89 Å.
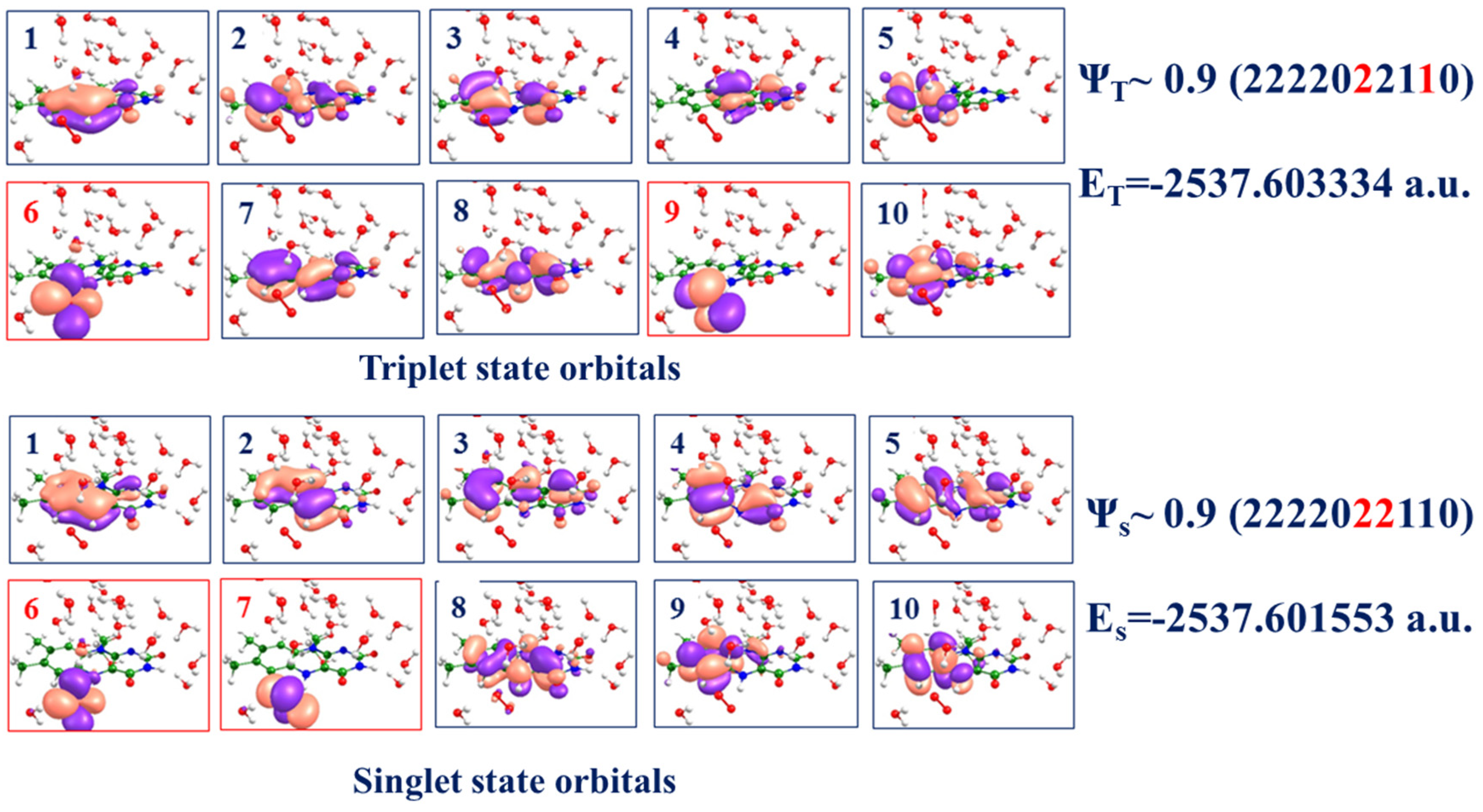
To characterize the electronic structure in Complex-1 in the triplet and singlet states, we carried out CASSCF(14/10)/cc-pVDZ calculations with the distribution of 14 electrons over 10 molecular orbitals. The latter include the orbitals located at the isoalloxazine ring and dioxygen (
Figure 3). Orbitals 5–7 in both triplet and singlet states include notable contributions from dioxygen. Two dominant electronic configurations in the triplet Ψ
T and singlet Ψ
S wavefunctions are shown on the right side of
Figure 3, indicating that the weight of the superoxide structure is considerable in the triplet state and dominant in the singlet state. Therefore, we conclude that Complex-1 bears the features of the Fl
SQ–O
2− radical pair. Notably, the CASSCF energies of the triplet and singlet states (see the right side of
Figure 3) are practically degenerate at this geometry since the energy gap is less than 0.1 kcal/mol. Therefore, we may assume that the switch from the triplet state PES to the singlet state PES in this model system occurs in the immediate vicinity of the geometry configuration of Complex-1.
Taking the structure of Complex-1 as a starting point for geometry optimization at the QM/MM level on the singlet state potential energy surface, we arrive at the structure shown in
Figure 2b. In this system, the oxygen atom, Ox1, is covalently bound to C4a, leading to the distorted isoalloxazine ring. Another oxygen atom, Ox2, is hydrogen bonded to three water molecules; one water molecule from the initial structure, Wat2 in
Figure 2a, drifted away from the dioxygen moiety. In agreement with the previous QM/MM studies of the reactions of the superoxide with the conjugated organic molecules inside proteins, e.g., with the chromophore of the green fluorescent protein [
21,
22], no potential energy barrier is detected on the pathway leading to the C(4a)-peroxyflavin species. The energy of the RutA structure with the C(4a)-peroxyflavin (
Figure 2b) is 16.8 kcal/mol lower than the level of the reactants shown in
Figure 2a.
It is instructive to compare these findings with the previously reported quantum chemistry computational results [
5,
10]. For the reaction of the reduced lumiflavin with the oxygen molecule, which starts at the triplet state PES and switches to the singlet state PES after an intersystem crossing (ISC) point, [
10] reports the structure of the ISC point optimized at the CASSCF(12/9) level. The Ox1-Ox2 distance varies from the initial value of 1.20 Å for the reactants to the value of 1.31 Å near the ISC point to be compared to our 1.32 Å in Complex-1. The charge of the dioxygen moiety at this point is almost -1, as in the present work. The Ox1-Ox2 distance in the singlet state C(4a)-peroxyflavin adduct is 1.46 Å (versus 1.45 Å in the present work). The C4a-Ox1 distance varies from 3.14 Å (versus 3.21 Å in the present work) for the triplet state reactants to 1.41 Å (versus 1.36 Å in the present work) for the singlet state C(4a)-peroxyflavin adduct. The reaction energy profile shows the barrier of about 10 kcal/mol on the way from the initial triplet state structure of Fl
−…O
2 to the ISC point (the corresponding reaction segment is unavailable in our simulations). The reported potential energy difference between the ISC point and the singlet state C(4a)-peroxyflavin adduct (see Table S4 in [
10]) for a water solution is 14.1 kcal/mol, to be compared with the value of 16.8 kcal/mol computed for the protein environment in the present work. We conclude that Complex-1 resembles the ISC point identified in [
10]. In [
5], the authors report in their
supplementary materials the computed potential energy difference of 8.1 kcal/mol between the C(4a)-peroxyflavin adduct and the reactants (Fl
−…O
2) for the water solution, using the B3LYP-D/6-311++G(2d,2p) calculations. The computed energy profile shows the energy barriers on both reaction steps: from (Fl
−…O
2) to Fl
SQ…O
2− and then to the Fl
C4aOO− adduct.
With respect to the S
0 → S
1 excitation, our XMCQDPT2 calculations (see details in
Section 3 below) report the excitation energy of the model system (
Figure 2b) at 3.47 eV (357 nm, 0.57 o.s.), in excellent agreement with the experimental absorption band known for the C(4a)-peroxyflavin intermediate [
3].
To conclude this subsection, we emphasize that the formation of the C(4a)-peroxyflavin intermediate is a commonly accepted route in monooxygenase activity, as reproduced in our calculations. We report an interesting finding that the oxygen molecule approaches the C4a position from the
si-side pocket, i.e., from the site, presumably occupied by a uracil substrate in RutA [
6]. Simulations described in [
12] show that location of both the substrate and dioxygen at the same site of the isoalloxazine ring does not prevent the formation of Fl
C4aOO−; this intermediate may occur and attack the substrate. Few theoretical papers report calculations of the functionalized flavins at the position C4a, e.g., refs. [
23,
24].
2.2. Pathway-2: Oxygen-N5 Interaction from the Si-Side Oxygen Pocket
Another triplet-state complex of the reduced flavin with dioxygen located at the
si-side pocket is shown in
Figure 4a. The QM/MM energy of this Complex-2 is 1.5 kcal/mol lower than the energy of Complex-1, indicating that these structures may be almost equally populated. Complex-2 shares the same features with Complex-1 in the sense that the superoxide describes the dioxygen moiety in this environment—the Ox1-Ox2 distance is 1.30 Å, and the charge on dioxygen is almost -1. However, in Complex-2, dioxygen stays closer to the isoalloxazine ring—the Ox2-N5 distance (2.58 Å) is about 0.6 Å shorter than the Ox1-C4a distance of 3.21 Å in Complex-1. Complex-2 also shows different hydrogen-bonding patterns as compared to Complex-1 (cf.
Figure 2a and
Figure 4a)—only two water molecules, Wat4 and Wat5 in
Figure 4a, are within the hydrogen-bond distances from the dioxygen atoms; the water molecule Wat6 stays further from the active site, located on the way to bulk solvent.
Analysis of the electronic structure in Complex-2 in the triplet and singlet states using CASSCF(14/10) calculations was carried out similarly to that described in the preceding subsection for Complex-1. Like the previous case, the dominant electronic configuration corresponds to the superoxide structure, and the energy gap between the triplet and singlet states in Complex-2 is small (about 1.6 kcal/mol).
The system shown in
Figure 4b corresponds to the minimum energy structure at the singlet state PES obtained in unconstrained QM/MM minimization initiated from the structure of Complex-2. The hydroperoxide anion HO
2− (hydrogen-bonded to water molecules Wat4, Wat5) is obtained as a result of a barrier-less transfer of the hydrogen atom transfer from N5. The QM/MM energy of the model system shown in
Figure 4b is 7.8 lower than the energy of Complex-2.
Subsequent molecular events may develop along different scenarios. First, if a substrate is present in the protein cavity, the hydroperoxide anion may serve as an oxidative agent. Second, a proton from the neighboring water molecules (here, Wat5 or Wat4) may be transferred to the HO
2− species, leading to the hydrogen peroxide molecule (see
Figure 4c for the scenario with Wat5 as a proton donor). The obtained hydroxyl anion (here, in the vicinity of the isoalloxazine ring) may serve as the oxidative agent if a substrate is located nearby. Third, a more extended chain of proton transfer events may take place over proton wires, borrowing a proton from bulk, which finally removes the charged species away from the oxidized FMN. Such proton wires involving a number of water molecules are present in this system (see
Figure 1). Proton transfer events over the aligned chains of water molecules are characterized by low energy barriers within 10 kcal/mol [
25]; thus, these pathways are feasible.
The S
0 → S
1 and S
0 → S
2 excitation energies computed at the XMCQDPT2 level at the configuration shown in
Figure 4b are 2.46 eV (503 nm, 0.20 o.s.) and 3.60 eV (344 nm, 0.13 o.s.). The absorption band maxima are close to those expected for the oxidized flavin in the protein environment. We conclude that pathway-2 describes the mechanism of flavin oxidation Fl
red− + O
2 (+H
2O) → Fl
ox + H
2O
2 +OH
− in RutA in the absence of the substrate via the occurrence of the transient hydroperoxyl species.
2.3. Pathway-3: Oxygen-N5 Interaction from the Re-Side Oxygen Pocket
The scenario described in the preceding subsection (pathway-2) is also an option when starting from one more triplet-state flavin-oxygen complex, but this time, the oxygen molecule is located at the
re-side pocket. The occurrence of the oxygen molecule at the
re-side is detected in the X-ray structure PDB ID 6SGG [
6]. According to the results of the crystallography analysis, the distance between the N5 atom of flavin and the nearest oxygen atom of O
2 is unusually short—2.1 Å, whereas the other oxygen atom is coordinated by the nitrogen atom of the Asn134 side chain (the distance is 2.6 Å) and the oxygen atom of the Thr105 side chain (the distance is 2.7 Å) [
6]. The oxygen-binding pocket at the
re-side was also characterized in our previous simulations [
11,
12], although no such short nitrogen-oxygen distance as 2.1 Å was observed in these simulations. Another important result of [
12] is that the switch from the triplet state PES to the singlet state PES at some MD trajectory frame of the flavin-oxygen complex led to the barrier-less formation of the hydroperoxide anion HO
2−, similar to what we describe in the preceding subsection of this paper.
The structure of the triplet state flavin-oxygen complex with O
2 from the
re-side as obtained in QM/MM optimization in the present work (Complex-3) is shown in
Figure 5a. We note that the dioxygen moiety again should be assigned to a superoxide O
2−; the Ox1-Ox2 distance is 1.31 Å and the charge of the dioxygen is close to -1. The geometry configuration differs from the structure PDB ID 6SGG [
6]—the dioxygen is hydrogen-bonded to flavin, Thr105, and water, whereas the Ox1-N(Asn134) distance is considerably longer (3.3 Å vs. 2.6 Å in the crystal), as well as the Ox1-N5 distance (2.6 Å vs. 2.1 Å in the crystal).
The results of the CASSCF(14/10) analysis of the electronic structure of Complex-3 (see
Figure 6) show that the dominant electronic configuration in the triplet state multiconfigurational function corresponds to the superoxide structure of dioxygen, whereas the singlet state configuration features an even higher electron population in the dioxygen orbitals. The energy gap between the triplet and singlet states in Complex-3 computed at the CASSCF level is small (1.1 kcal/mol), similar to all flavin-oxygen complexes in the active site of RutA.
At least two minimum energy structures can be located on the singlet state PES using the QM/MM calculations. The structure shown in
Figure 5b is almost isoenergetic with the triplet state Complex-3. Consistent with pathway-2 (see
Figure 4b), the dioxygen borrows a hydrogen atom from N5 to Ox1. In pathway-3, the increased electron population on dioxygen leads to the spontaneous formation of hydrogen peroxide at the expense of a nearby water molecule, Wat7. The structure shown in
Figure 5c is 6.8 kcal/mol lower in energy than the initial Complex-3. The chain of water molecules provides a suitable proton wire to move the negative charge of the hydroxyl farther away from the isoalloxazine ring.
The S
0 → S
1 and S
0 → S
2 excitation energies computed at the XMCQDPT2 level for the system shown in
Figure 5b are 2.46 eV (503 nm, 0.20 o.s.) and 3.64 eV (344 nm, 0.20 o.s.). They are almost the same as the reaction products at pathway-2 (see
Figure 4b) and close to those of the oxidized flavin in the protein environment. We conclude that pathway-3 (as well as pathway-2) describes the mechanism of flavin oxidation Fl
red− + O
2 (+H
2O) → Fl
ox + H
2O
2 + OH
− in RutA in the absence of a substrate.
2.4. Pathway-4: Formation of the N(5)-oxide Adducts
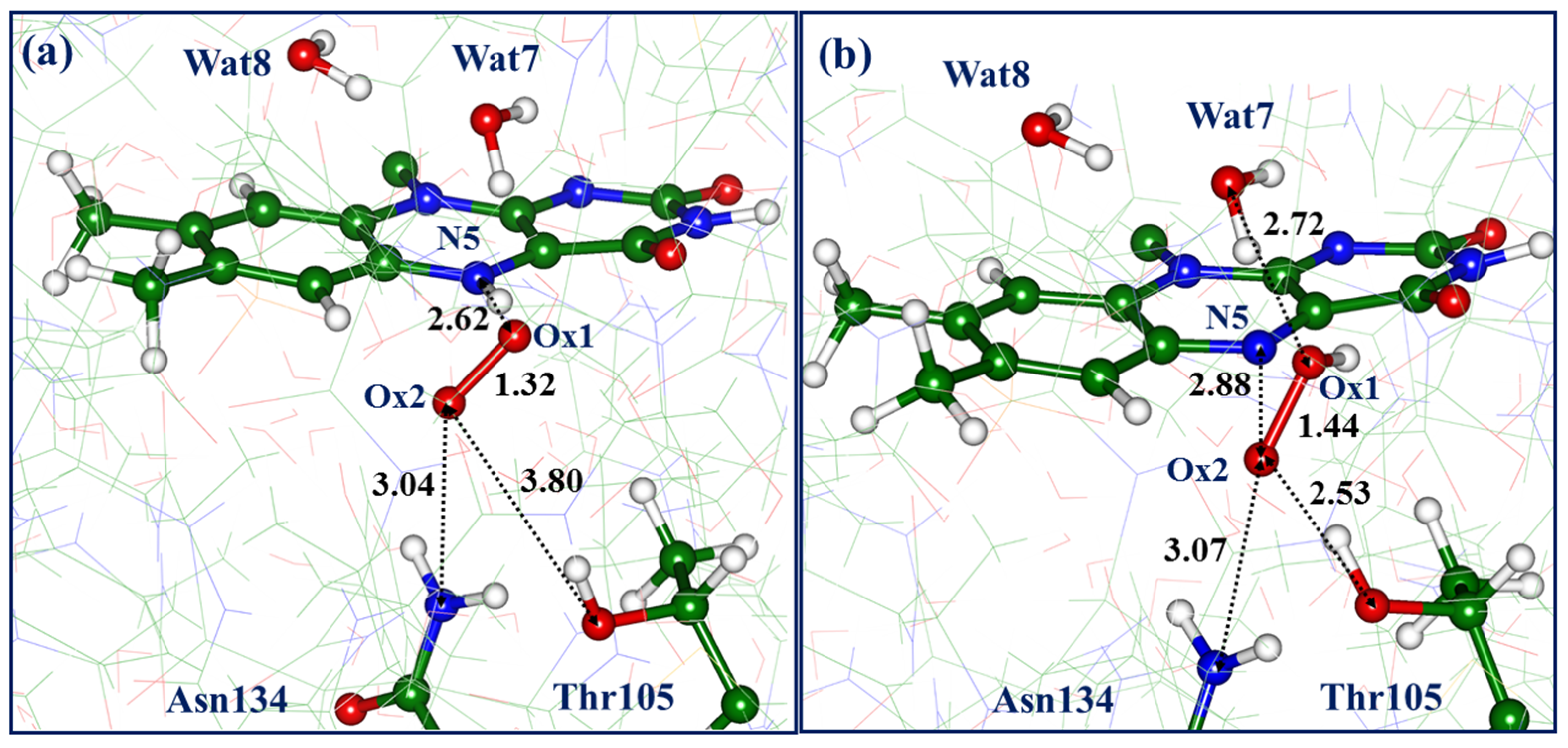
An intriguing pathway-4 also starts from the triplet state flavin-oxygen complex (Complex-4) at the
re-side, although it should be noted that its QM/MM energy is 5.6 kcal/mol higher than the energy of Complex-3. The structure shown in
Figure 7a differs from the previously described Complex-3 by the arrangement of the dioxygen—the Ox1-N5 distances are the same (2.62 Å) in both cases, but the Ox2 atom points towards Wat7 in Complex-3 and towards Asn134 in Complex-4. The latter structure shows poor coordination of dioxygen by the Thr105 side chain—the distance of 3.80 Å between Ox1 and the hydroxyl oxygen of Thr105 is long enough. Like all previously described flavin-oxygen complexes, Complex-4 accommodates the superoxide anion in the protein cavity—the Ox1-Ox2-distance is 1.32 Å; the charge of dioxygen is close to -1; and the energy gap between the triplet and singlet states is small.
Switching to the singlet-state PES at this point and subsequent unconstrained geometry optimization led to the structure (
Figure 7b) with the hydroperoxyl and Ox1-Ox2 distance of 1.44 Å. The energy of this structure is 12.8 kcal/mol lower than the level of the initial Complex-4. It is important to note the following distances: Ox2-N5 (2.88 Å) and Ox1-O(Wat7) (2.72 Å). We note in passing that all QM/MM optimized structures in both triplet and singlet states of model systems do not reveal a short oxygen-nitrogen distance of 2.09 Å between any of the dioxygen atoms and the N5 flavin atom reported in the crystal structure PDB ID 6SGG [
6]; no values less than 2.5 Å were obtained in simulations.
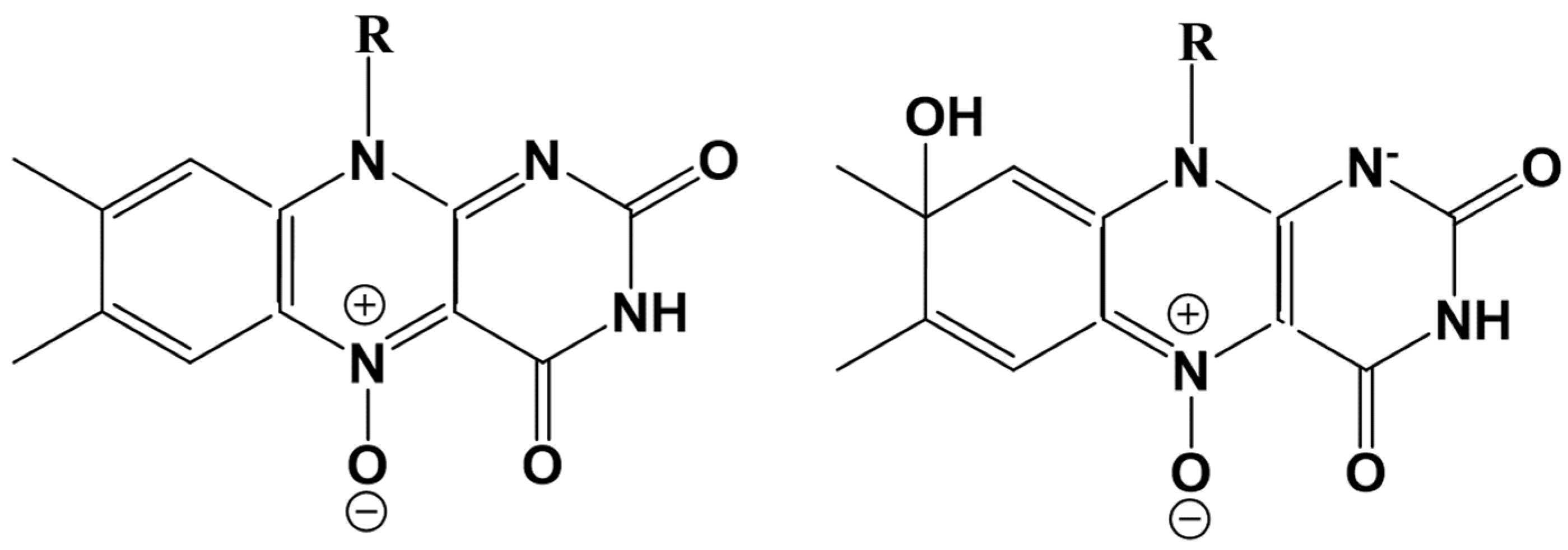
A gradual decrease of the Ox2-N5 distance results in the formation of the N-O covalent bond with a simultaneous break of the Ox1-Ox2 bond and finally leads to the formation of the flavin-N(5)-oxide, as shown in
Figure 8a. This N5-O adduct is responsible for flavin functionalization in the flavoenzyme EncM [
5,
6]. According to the present QM/MM calculations, its energy is 28.5 kcal/mol lower than the energy of Complex-4. This agrees with the results of quantum chemistry gas-phase calculations of the reaction of the reduced lumiflavin with the oxygen molecule leading to the lumiflavin-N(5)-oxide described in [
5]—the computed reaction energy is about −23 kcal/mol. Moreover, the reaction scheme depicted in
Figure 4 in [
5] seems consistent with the mechanism revealed in the present QM/MM simulations of the protein. As shown in
Figure 7b, the hydroxyl Ox1-H
− formed after the cleavage of the Ox1-Ox2 bond interacts with the water molecule Wat7, leading to a newly formed water molecule Wat9 and a hydroxyl anion.
The S
0 → S
1 and S
0 → S
2 excitation energies computed at the XMCQDPT2 level at the configuration shown in
Figure 8a are 2.44 eV (508 nm, 0.12 o.s.) and 3.25 eV (381 nm, 0.16 o.s.). These values may be compared to the absorption band maxima of 463 nm (2.69 eV) and 360–380 nm (3.26–3.44 eV) of the flavin-N(5)-oxide reported for the EncM protein (see Figure S5 in [
5]). Taking into account possible errors in calculations of excitation energies in complex systems [
26], the agreement is reasonable; moreover, the presence of the charged species OH
− near the chromophore in our model system (
Figure 8a) may cause some shifts in the computed excitation energies.
As shown in
Figure 8a,b, the N(5)-oxide undergoes a further transformation, namely, the hydroxyl emerged from Wat7 (
Figure 7b) attaches a proton from Wat8, stimulating the newly derived hydroxyl (from Wat8) to attack flavin at the C8 position to form a new covalent bond. This adduct refers to the C(8)-hydrated N(5)-oxide compound shown in
Figure 8b. These two structures are almost degenerate in energy. The hydration reaction of a chromophore in photoactive proteins upon light illumination is known, e.g., in the Dreiklang protein [
27,
28,
29]; here, this reaction may occur at the ground singlet state PES.
The S
0 → S
1 and S
0 → S
2 excitation energies computed at the XMCQDPT2 level for the system shown in
Figure 8b are 2.73 eV (454 nm, 0.38 o.s.) and 3.54 eV (350 nm, 0.12 o.s.). We see a considerable change in the S
0 → S
1 band (from 508 nm to 454 nm) due to chromophore hydration. This shift is clear as the electronic structure of flavin is closer to the Fl
ox for the flavin-N(5)-oxide and closer to the Fl
red for the flavin- C(8)-hydrated N(5)-oxide. The chemical formulas of both species are given in
Scheme 2. We note a change in the conjugation patterns consistent with the equilibrium geometry structures (see, e.g., the distances between N5 and carbon atoms in
Figure 8).
2.5. Pathway-5: Formation of the C(6)-hydroperoxyflavin Species from the Si-Side Oxygen Pocket
Finally, we describe an uncommon pathway in flavin functionalization that explores the C6 position of the isoalloxazine ring. Spontaneous covalent binding of the hydroperoxide at the C6 atom was noted in the molecular dynamics simulations with the QM/MM potentials for the RutA-O2-uracil model system [
12]. The occurrence of the reaction intermediates with the C6-O bond formation was discussed in the recent study [
30].
Figure 9a shows a fragment of the triplet-state model system (Complex-5) formed from the
si-side oxygen pocket. The energy of this structure is 5.4 kcal/mol higher than that of Complex-1, which initiates the formation of the flavin-C(4a) adduct, and is 6.9 kcal/mol higher as compared to Complex-2, which gives rise to the oxidized flavin and hydrogen peroxide.
Complex-5 shows the features common to all flavin-oxygen complexes considered in the present work: the Ox1-Ox2 distance is 1.31 Å; the charge on dioxygen is close to -1; the triplet-singlet gap is small; and the dioxygen moiety is hydrogen bonded to the nearest molecular groups, in this case, to four water molecules: Wat6, Wat7, Wat8, and Wat11. The shortest distance to the atoms of the isoalloxazine ring refers to the Ox1-C6 separation—3.02 Å.
Starting from Complex-5, a structure shown in
Figure 9b with the flavin-C(6)-hydroperoxide adduct (noted in the previous QM/MM MD simulations [
12]) is readily obtained after switching from the triplet state PES to the singlet state PES as a result of the forming covalent bond C6-Ox1 coupled with proton transfer from N5 to Ox2. The energy of this structure is 6.5 kcal/mol lower than that of Complex-5. Principally, this pathway may proceed further; a potential barrier of about 14 kcal/mol is required to break the Ox1-Ox2 bond and to arrive at the structure shown in
Figure 9c with the C(6)-C(7)-epoxide. Such cyclic patterns were noted earlier in the modeling reactions of oxygen with the chromophore of the green fluorescent protein [
22]. The energy of the thus obtained structure (
Figure 9c) is 30.1 kcal/mol lower than that of Complex-5, and this adduct may occur as a possible reaction intermediate. Its S
0 → S
1 excitation energies computed at the XMCQDPT2 level is 2.57 eV (483 nm, 0.06 o.s.).
2.6. Summary and Concluding Remarks
In this work, we apply QM/MM and quantum chemistry methods to characterize minimum energy points on the triplet state and singlet state potential energy surfaces along the possible reaction pathways initiated by the interaction of the oxygen molecule with the molecular groups in the protein cavities of the flavoenzyme RutA.
Table 1 summarizes the most important results of these simulations, grouped as the oxygen activation pathways, starting from the oxygen-containing pockets at the
si-side and
re-side of the flavin’s isoalloxazine ring.
In
Table 1, we compare the energies of the initial triplet state flavin-oxygen complexes (column 2) and of the corresponding reaction products (column 4) differently for the structures emerging from either the
si-side or
re-side oxygen pockets, despite the fact that these model systems were created for the same QM-MM partitioning scheme described below in
Section 3. As clarified in
Figure 1, the
re-side region is deeper in the protein macromolecule, whereas the
si-side is entirely in the solvent-accessible cavity. It is practically impossible to provide fully equivalent conditions for the QM/MM geometry optimization in these two regions because of the different impacts of the large amount of water molecules in the MM subsystem. For instance, the total QM/MM energies for the
re-side initial flavin-oxygen systems (Complexes-3 and 4) are about 15 kcal/mol lower than the energies of the
re-side initial complexes.
Analysis of the data collected in
Table 1 allows us to conclude that pathways 1, 2, and 3 may refer to the most probable reaction routes leading to oxygen activation in RutA if the estimated energy barriers are considered. Pathways 2 and 3 are practically similar despite the different starting positions of the oxygen molecule—from the protein interior or from the solvent-accessible cavity. These routes describe the formation of the oxidized flavin, hydrogen peroxide, and the hydroxyl anion with a reaction energy of 7–8 kcal/mol. The computed absorption band maxima (the last column in
Table 1) are the same in the two pathways showing characteristic features of the oxidized flavin. Pathway-1 leading to the formation of the flavin-C(4a)-peroxide well-known from experimental studies is also highly likely, as validated by the computed relative energies and absorption spectra.
Pathways 4 and 5 are initiated from the structures, which may be less populated considering the computed energies (6–7 kcal/mol above the level of the lowest-energy structures on the respective side) of Complex-4 and Complex-5. However, the products of the corresponding reactions, whose energies are low enough relative to the level reactants, are very interesting and describe the known [
3,
4,
5,
6,
7] and newly characterized covalent flavin-oxygen adducts.
The computed energies of the reaction products (column 4 in
Table 1) are large enough to expect that the corresponding species may be detected experimentally. The computed excitation energies and oscillator strengths are reported for every reaction product, providing useful data to analyze transient absorption bands in prospective spectral studies.
Next, we emphasize the essential role of water molecules and of proton transfer along hydrogen bond networks involving water molecules in the solvent-accessible protein cavities of RutA. This refers to all pathways studied here, and it is important to note that proton transfer over the aligned chains of water molecules is characterized by low energy barriers—within 10 kcal/mol, e.g., according to [
25].
Finally, we comment that the energy profiles connecting these minimum energy points are roughly estimated in the present QM/MM calculations. We point out that there are unavoidable errors in the estimates of the activation barriers on reaction routes calculated by the QM/MM method with the modern quantum chemistry software packages, amounting to several kcal/mol [
31].
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}