
Total Synthesis of Lineaflavones A, C, D, and Analogues

Abstract
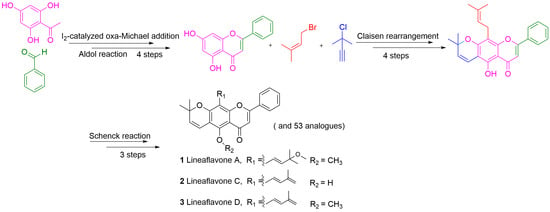
1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Materials
3.3. Procedure for the Synthesis of Lineaflavones A, C, D
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Al-Khayri, J.; Sahana, G.; Nagella, P.; Joseph, B.; Alessa, F.; Al-Mssallem, M. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef] [PubMed]
- Maleki, S.; Crespo, J.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food. Chem. 2019, 299, 125124. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.; Zhu, X.; Chan, K.; Liu, Z.; Chan, C.; Chow, T.; Chong, T.; Law, M.; Cui, J.; Chow, L.; et al. Flavonoid Monomers as Potent, Nontoxic, and Selective Modulators of the Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2021, 64, 14311–14331. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.; Diwan, A.; Chandra, S. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Juca, M.; Filho, F.C.; de Almeida, J.; Mesquita, D.; Barriga, J.; Dias, K.; Barbosa, T.; Vasconcelos, L.; Leal, L.; Ribeiro, J.; et al. Flavonoids: Biological activities and therapeutic potential. Nat. Prod. Res. 2020, 34, 692–705. [Google Scholar] [CrossRef]
- Raffa, D.; Maggio, B.; Raimondi, M.; Plescia, F.; Daidone, G. Recent discoveries of anticancer flavonoids. Eur. J. Med. Chem. 2017, 142, 213–228. [Google Scholar] [CrossRef]
- Deng, M.; Jia, X.; Dong, L.; Liu, L.; Huang, F.; Chi, J.; Ma, Q.; Zhao, D.; Zhang, M.; Zhang, R. Structural elucidation of flavonoids from Shatianyu (Citrus grandis L. Osbeck) pulp and screening of key antioxidant components. Food Chem. 2022, 366, 130605. [Google Scholar] [CrossRef] [PubMed]
- Owor, R.; Bedane, K.; Zuhlke, S.; Derese, S.; Ong’amo, G.; Ndakala, A.; Spiteller, M. Anti-inflammatory Flavanones and Flavones from Tephrosia linearis. J. Nat. Prod. 2020, 83, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Jung, E.; Lee, Y.; Seo, J.; Ahn, S.; Koh, D.; Lim, Y.; Shin, S. A Novel Synthetic Compound(E)-5-((4-oxo-4H-chromen-3-yl)methyleneamino)-1-phenyl-1H-pyrazole-4-carbonitrile Inhibits TNFalpha-Induced MMP9 Expression via EGR-1 Downregulation in MDA-MB-231 Human Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5080. [Google Scholar] [CrossRef]
- Helesbeux, J.-J.; Duval, O.; Guilet, D.; Séraphin, D.; Rondeau, D.; Richomme, P. Regioselectivity in the ene reaction of singlet oxygen with ortho-prenylphenol derivatives. Tetrahedron 2003, 59, 5091–5104. [Google Scholar] [CrossRef]
- Jian, J.; Fan, J.; Yang, H.; Lan, P.; Li, M.; Liu, P.; Gao, H.; Sun, P. Total Synthesis of the Flavonoid Natural Product Houttuynoid A. J. Nat. Prod. 2018, 81, 371–377. [Google Scholar] [CrossRef]
- Liu, M.; Wilairat, P.; Go, M.-L. Antimalarial Alkoxylated and Hydroxylated Chalones: Structure-Activity Relationship Analysis. J. Med. Chem. 2001, 44, 4443–4452. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Xiao, Z.; Qiu, Y.; Shu, S.; Chen, X.; Chen, X.; Zhuang, F.; Zhao, Y.; Liang, G.; Liu, Z. Synthesis and Anti-inflammatory Evaluation of (R)−, (S)−, and (+/−)-Sanjuanolide Isolated from Dalea frutescens. J. Nat. Prod. 2019, 82, 748–755. [Google Scholar] [CrossRef]
- Yenjai, C.; Wanich, S. Cytotoxicity against KB and NCI-H187 cell lines of modified flavonoids from Kaempferia parviflora. Bioorg Med. Chem. Lett. 2010, 20, 2821–2823. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Xu, F.; Wang, G.; Xie, S.; Li, W.; Yao, H.; Ma, C.; Zhu, Z.; Xu, J.; Xu, S. Design, synthesis, and biological evaluation of truncated deguelin derivatives as Hsp90 inhibitors. Eur. J. Med. Chem. 2019, 167, 485–498. [Google Scholar] [CrossRef]
- Farmer, R.; Scheidt, K. A Concise Enantioselective Synthesis and Cytotoxic Evaluation of the Anticancer Rotenoid Deguelin Enabled by a Tandem Knoevenagel/Conjugate Addition/Decarboxylation Sequence. Chem. Sci. 2013, 4, 3304–3309. [Google Scholar] [CrossRef]
- Ollevier, T.; Mwene-Mbeja, T. Bismuth Triflate Catalyzed [1,3] Rearrangement of Aryl 3-Methylbut-2-enyl Ethers. Synthesis 2006, 2006, 3963–3966. [Google Scholar] [CrossRef]
- Han, J.; Li, X.; Guan, Y.; Zhao, W.; Wulff, W.; Lei, X. Enantioselective biomimetic total syntheses of kuwanons I and J and brosimones A and B. Angew. Chem. Int. Ed. 2014, 53, 9257–9261. [Google Scholar] [CrossRef]
- Monaco, M.; Fazzi, D.; Tsuji, N.; Leutzsch, M.; Liao, S.; Thiel, W.; List, B. The Activation of Carboxylic Acids via Self-Assembly Asymmetric Organocatalysis: A Combined Experimental and Computational Investigation. J. Am. Chem. Soc. 2016, 138, 14740–14749. [Google Scholar] [CrossRef]
- Qiu, H.; Shuai, B.; Wang, Y.; Liu, D.; Chen, Y.; Gao, P.; Ma, H.; Chen, S.; Mei, T. Enantioselective Ni-Catalyzed Electrochemical Synthesis of Biaryl Atropisomers. J. Am. Chem. Soc. 2020, 142, 9872–9878. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lai, J.; Kong, W.; Li, S. Asymmetric Synthesis of Sakuranetin-Relevant Flavanones for the Identification of New Chiral Antifungal Leads. J. Agric. Food. Chem. 2022, 70, 3409–3419. [Google Scholar] [CrossRef] [PubMed]
- Zore, M.; Gilbert-Girard, S.; Reigada, I.; Patel, J.; Savijoki, K.; Fallarero, A.; Yli-Kauhaluoma, J. Synthesis and Biological Evaluation of Fingolimod Derivatives as Antibacterial Agents. ACS Omega 2021, 6, 18465–18486. [Google Scholar] [CrossRef]
- Shaffer, C.; Cai, S.; Peng, J.; Robles, A.; Hartley, R.; Powell, D.; Du, L.; Cichewicz, R.; Mooberry, S. Texas Native Plants Yield Compounds with Cytotoxic Activities against Prostate Cancer Cells. J. Nat. Prod. 2016, 79, 531–540. [Google Scholar] [CrossRef]
- Ito, C.; Itoigawa, M.; Kumagaya, M.; Okamoto, Y.; Ueda, K.; Nishihara, T.; Kojima, N.; Furukawa, H. Isoflavonoids with Antiestrogenic Activity from Millettia pachycarpa. J. Nat. Prod. 2006, 69, 138–141. [Google Scholar] [CrossRef]
- Thanh, V.T.T.; Mai, H.D.T.; Pham, V.; Litaudon, M.; Dumontet, V.; Gueritte, F.; Nguyen, V.; Chau, V. Acetylcholinesterase inhibitors from the leaves of Macaranga kurzii. J. Nat. Prod. 2012, 75, 2012–2015. [Google Scholar] [CrossRef] [PubMed]
- Polbuppha, I.; Suthiphasilp, V.; Maneerat, T.; Charoensup, R.; Limtharakul, T.; Cheenpracha, S.; Pyne, S.; Laphookhieo, S. Macluracochinones A-E, antimicrobial flavonoids from Maclura cochinchinensis (Lour.) Corner. Phytochemistry 2021, 187, 112773. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Schrader, M.; Kim, D.; Baik, M.; Park, Y.; Chang, S. Harnessing Secondary Coordination Sphere Interactions That Enable the Selective Amidation of Benzylic C-H Bonds. J. Am. Chem. Soc. 2019, 141, 15356–15366. [Google Scholar] [CrossRef] [PubMed]
- Daskiewicz, J.-B.; Depeint, F.; Viornery, L.; Bayet, C.; Comte-Sarrazin, G.; Comte, G.; Gee, J.M.; Johnson, I.T.; Ndjoko, K.; Hostettmann, K.; et al. Effects of Flavonoids on Cell Proliferation and Caspase Activation in a Human Colonic Cell Line HT29: An SAR Study. J. Med. Chem. 2005, 48, 2790–2804. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yuan, H.; Men, Y.; Liu, Q.; Zhang, J.; Xu, X. Cross-Cycloaddition of Two Different Isocyanides: Chemoselective Heterodimerization and [3+2]-Cyclization of 1,4-Diazabutatriene. Angew. Chem. Int. Ed. 2016, 55, 7077–7080. [Google Scholar] [CrossRef]
- Zheng, S.-Y.; Shen, Z.-W. Total synthesis of Hirtellanine A. Tetrahedron Lett. 2010, 51, 2883–2887. [Google Scholar] [CrossRef]
- Guo, C.; Fleige, M.; Janssen-Müller, D.; Daniliuc, C.G.; Glorius, F. Cooperative N-Heterocyclic Carbene/Palladium-Catalyzed Enantioselective Umpolung Annulations. J. Am. Chem. Soc. 2016, 138, 7840–7843. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Conditions | Yield(%) a | 1/3 b |
| 1 | H2SO4:MeOH = 1:10 | 60 | 0:1 |
| 2 | HCl:MeOH = 1:10 | 78 | 0:1 |
| 3 | 3N HCl:MeOH = 1:5 | 76 | 1:1 |
| 4 | 3N HCl:MeOH = 1:10 | 85 | 2.2:1 |
| 5 | 3N HCl:MeOH = 1:15 | 77 | 1.8:1 |
| 6 | 2N HCl:MeOH = 1:15 | 70 | 1.5:1 |
| 7 | 1N HCl:MeOH = 1:15 | 62 | 1:3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Fu, Y.; Ma, R.; Jin, H.; Zhao, W. Total Synthesis of Lineaflavones A, C, D, and Analogues. Molecules 2023, 28, 2373. https://doi.org/10.3390/molecules28052373
Wang R, Fu Y, Ma R, Jin H, Zhao W. Total Synthesis of Lineaflavones A, C, D, and Analogues. Molecules. 2023; 28(5):2373. https://doi.org/10.3390/molecules28052373
Chicago/Turabian StyleWang, Rui, Yu Fu, Ran Ma, Hongzhen Jin, and Wei Zhao. 2023. "Total Synthesis of Lineaflavones A, C, D, and Analogues" Molecules 28, no. 5: 2373. https://doi.org/10.3390/molecules28052373
APA StyleWang, R., Fu, Y., Ma, R., Jin, H., & Zhao, W. (2023). Total Synthesis of Lineaflavones A, C, D, and Analogues. Molecules, 28(5), 2373. https://doi.org/10.3390/molecules28052373