
Anticancer Activity and Molecular Mechanisms of an Ursodeoxycholic Acid Methyl Ester-Dihydroartemisinin Hybrid via a Triazole Linkage in Hepatocellular Carcinoma Cells

 , ,
, ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
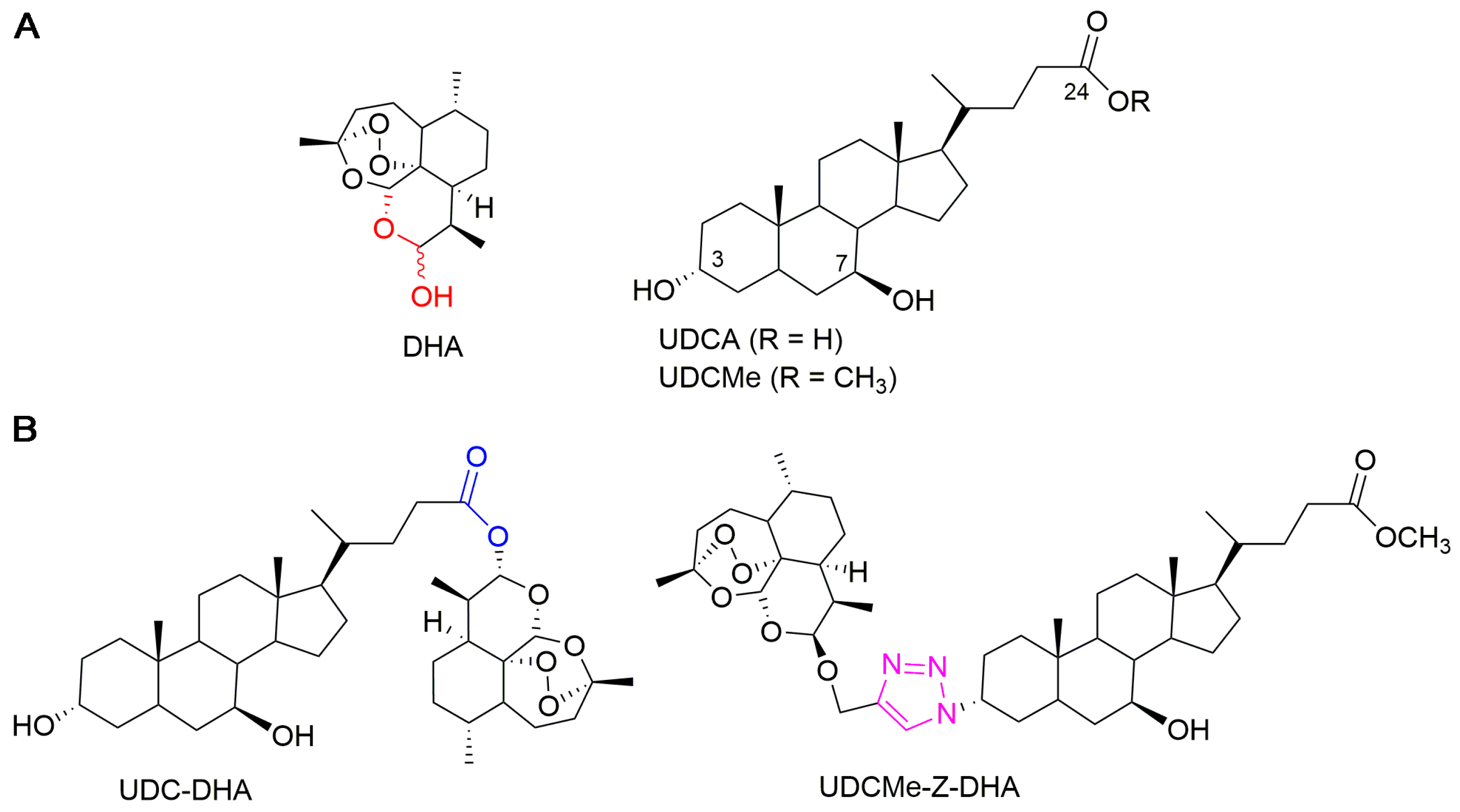
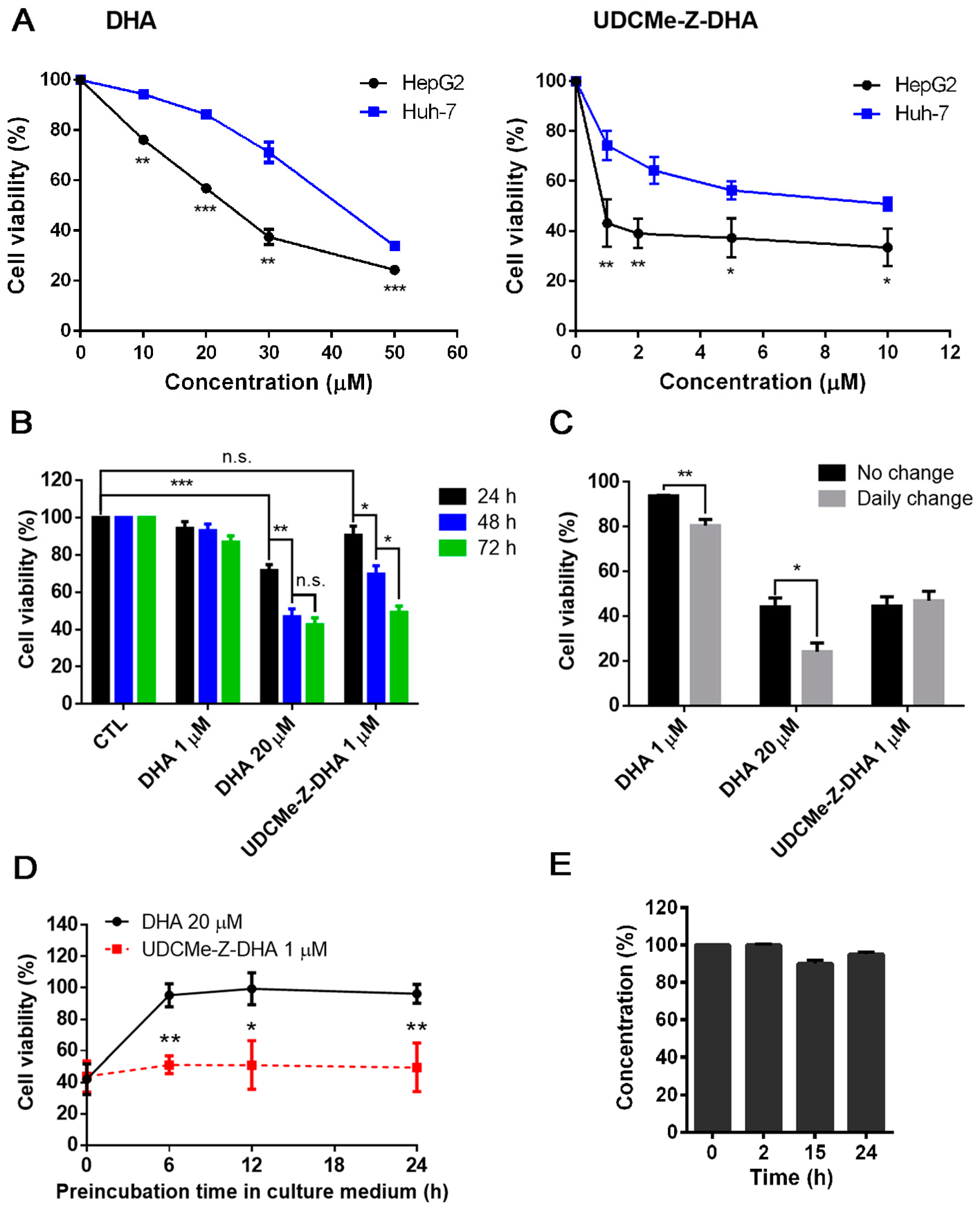
2.1. The Anticancer Effect of UDCMe-Z-DHA Is More Potent Than DHA in HCC Cells
2.2. UDCMe-Z-DHA Is More Stable Than DHA
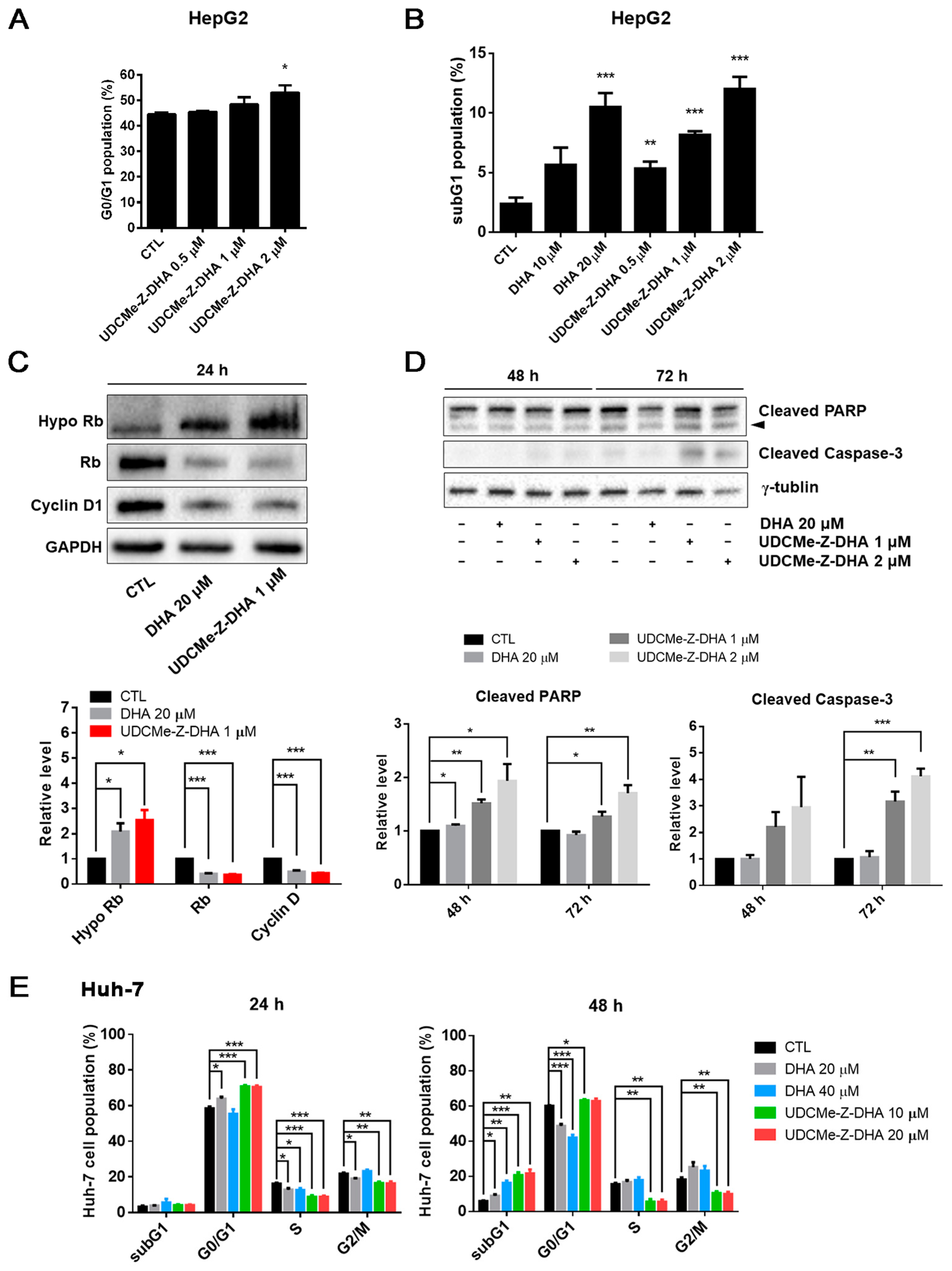
2.3. The Effect of DHA and UDCMe-Z-DHA on Cell Cycle Progression and Apoptosis in HCC Cells
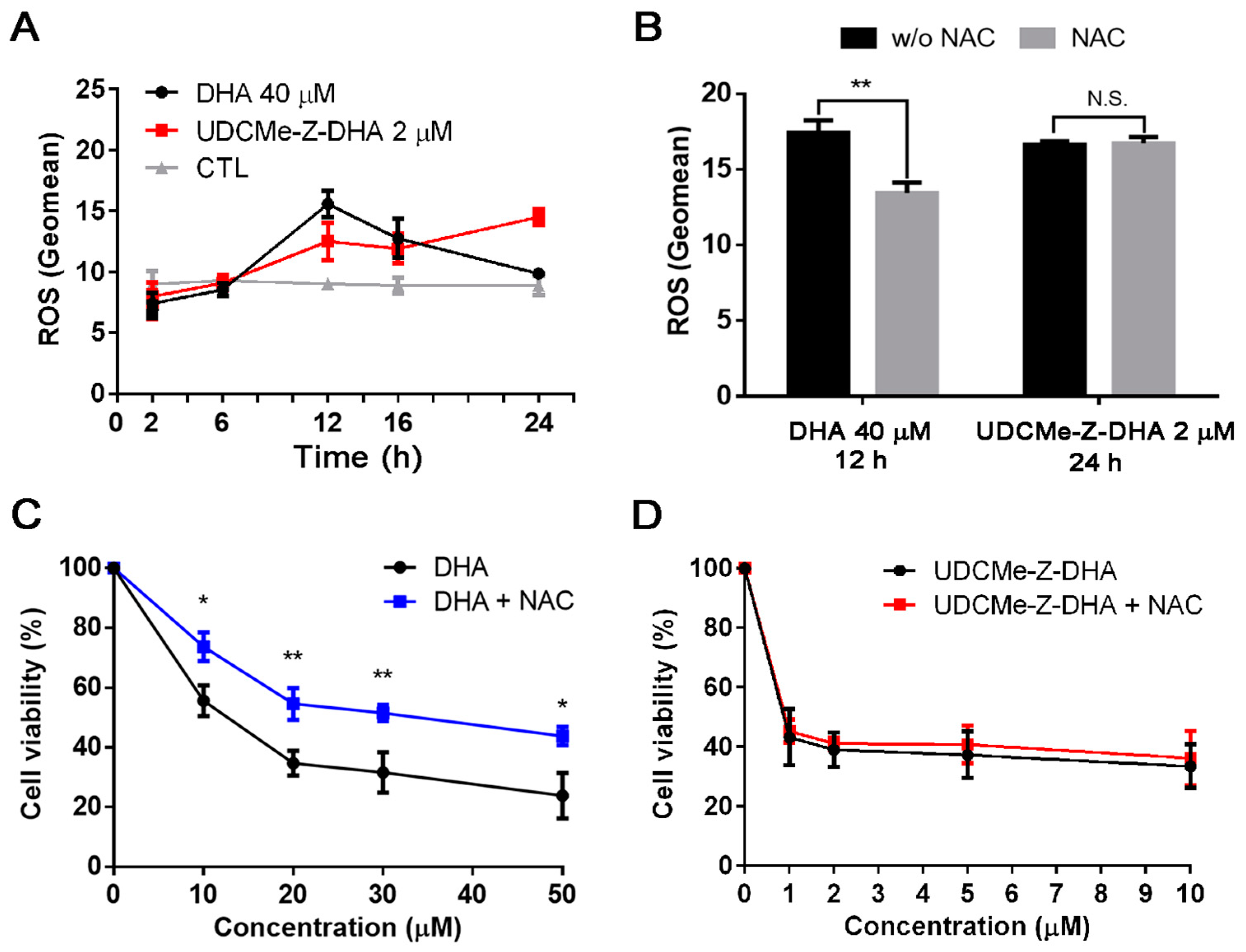
2.4. The Effect of DHA and UDCMe-Z-DHA on ROS Production in HepG2 Cells
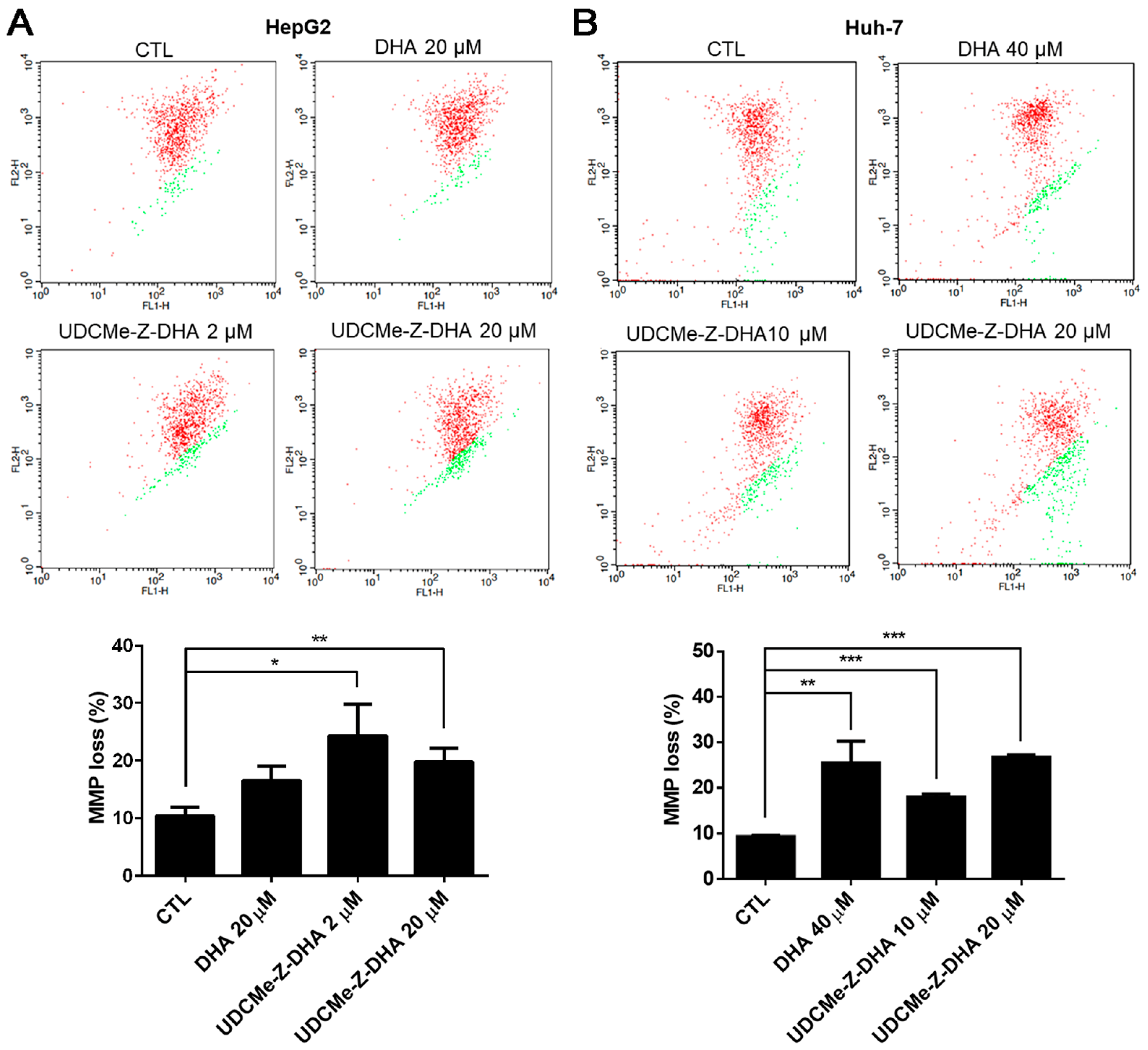
2.5. The Effect of DHA and UDCMe-Z-DHA on Depolarization of MMP in HCC Cells
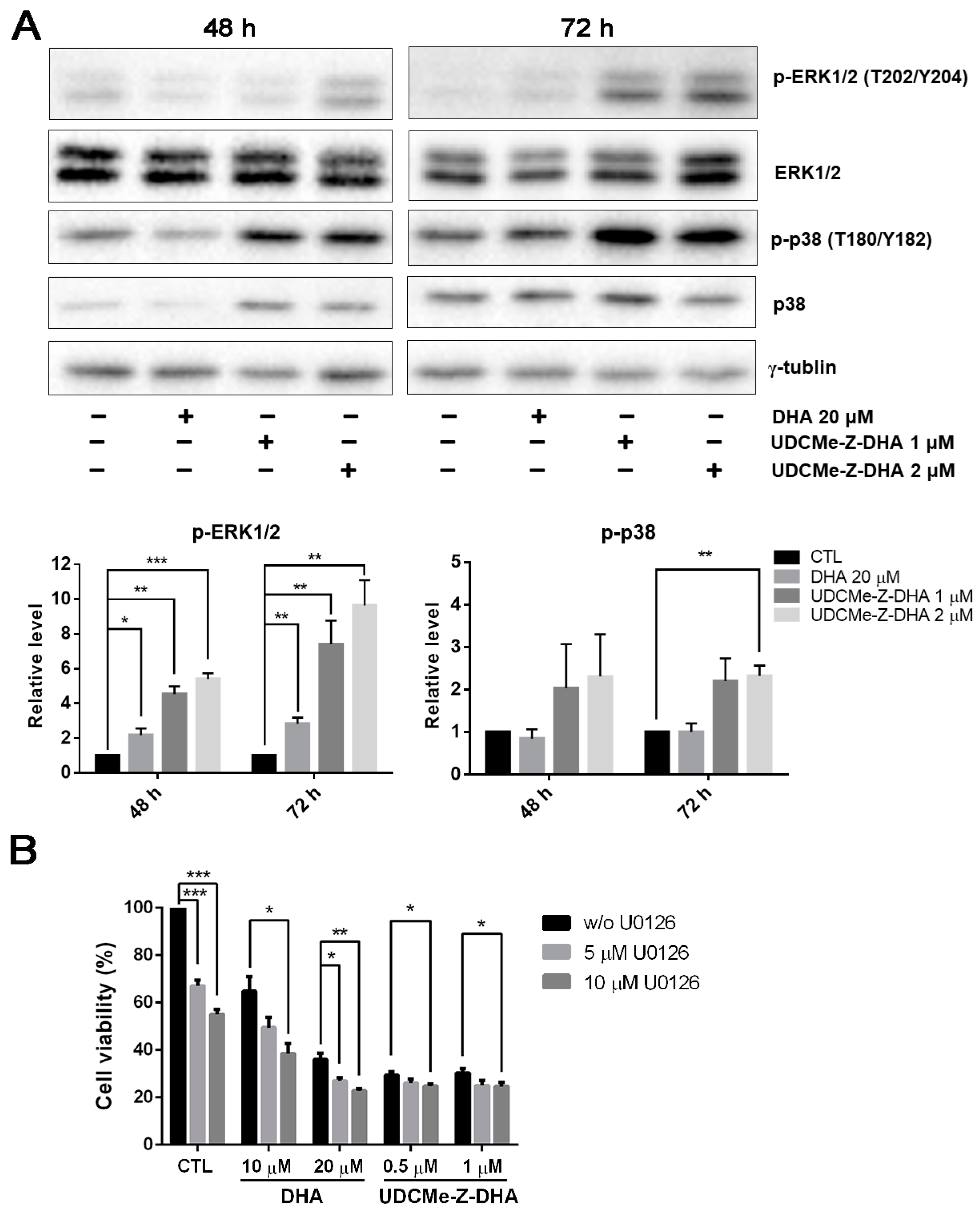
2.6. The Effect of DHA and UDCMe-Z-DHA on the MAPK Signaling Pathway in HepG2 Cells
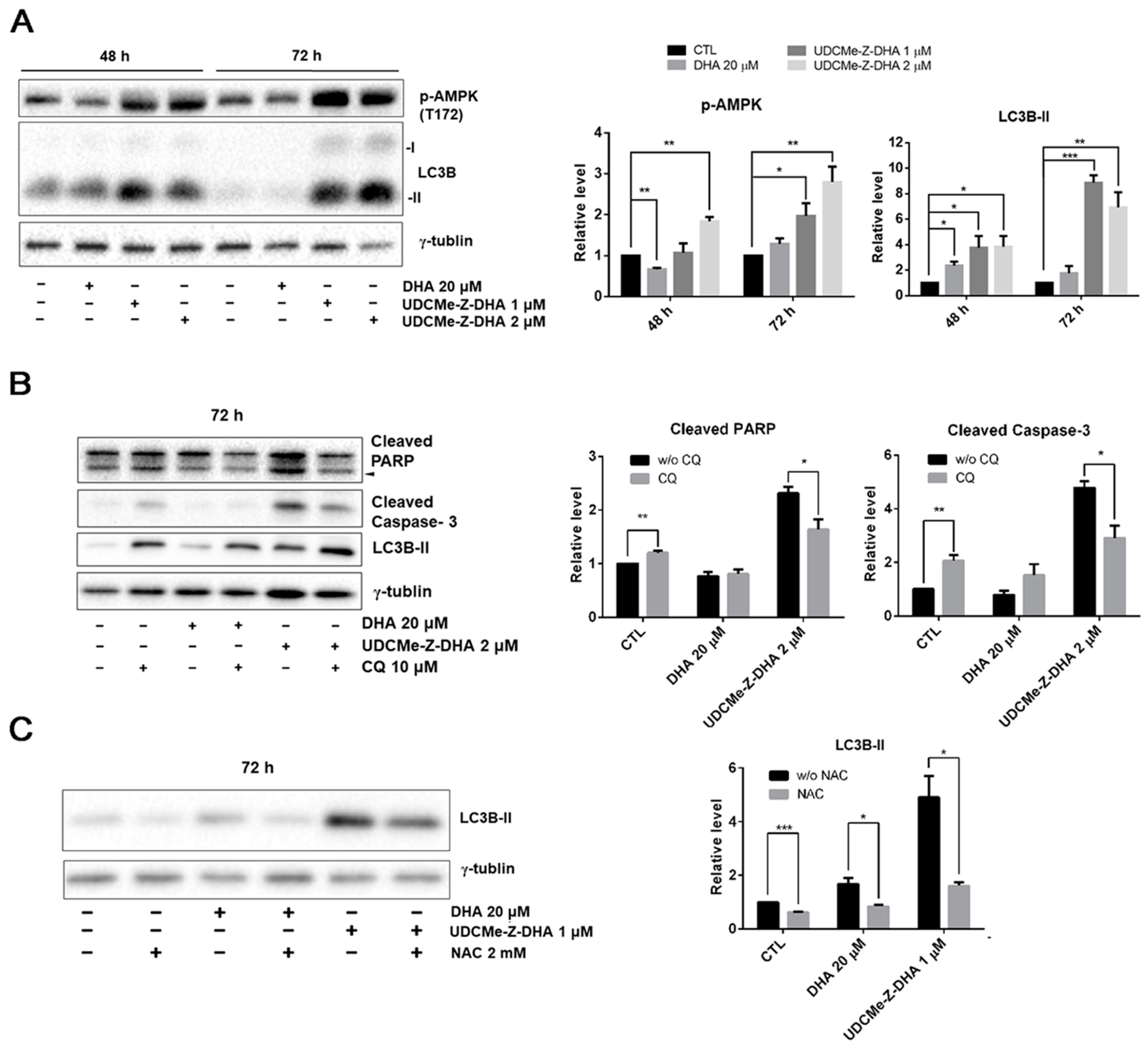
2.7. The Effect of DHA and UDCMe-Z-DHA on the AMPK/Autophagy Signaling Pathway in HepG2 Cells
2.8. Autophagy Induced by UDCMe-Z-DHA May Be Associated with Apoptosis in HepG2 Cells
2.9. ROS Induced by UDCMe-Z-DHA May Cause Autophagy in HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines and Cell Culture
4.3. Cell Viability Assay
4.4. HPLC-MS/MS Analysis
4.5. Cell Cycle Analysis
4.6. Western Blot Analysis
4.7. DCFH-DA Assay
4.8. JC-1 Assay
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma. Hepatology 2021, 73, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Balogh, J.; Victor, D., 3rd; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinima: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef]
- Ozakyol, A. Global epidemiology of hepatocellular carcinoma (HCC epidemiology). J. Gastrointest. Cancer 2017, 48, 238–240. [Google Scholar] [CrossRef]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, H.; Mu, L.; Yang, X. Artemisinins as anticancer drugs: Novel therapeutic approaches, molecular mechanisms, and clinical trials. Front. Pharmacol. 2020, 11, 529881. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, X.; Liu, J.; Gao, N.; Yang, R.; Wang, Q.; Ji, J.; Ma, L.; He, Q. Dihydroartemisinin inhibits the tumorigenesis and metastasis of breast cancer via downregulating CIZ1 expression associated with TGF-β1 signaling. Life Sci. 2020, 248, 117454. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhong, B.; Li, Y.; Liu, X. Dihydroartemisinin increases apoptosis of colon cancer cells through targeting Janus kinase 2/signal transducer and activator of transcription 3 signaling. Oncol. Lett. 2018, 15, 1949–1954. [Google Scholar] [CrossRef]
- Jiang, J.; Geng, G.; Yu, X.; Liu, H.; Gao, J.; An, H.; Cai, C.; Li, N.; Shen, D.; Wu, X.; et al. Repurposing the anti-malarial drug dihydroartemisinin suppresses metastasis of non-small-cell lung cancer via inhibiting NF-kB/GLUT1 axis. Oncotarget 2016, 7, 87271–87283. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wang, D.; Zhang, R.; Wang, H. Experimental therapy of hepatoma with artemisinin and its derivatives: In vitro and in vivo activity, chemosensitization, and mechanisms of action. Clin. Cancer Res. 2008, 14, 5519–5530. [Google Scholar] [CrossRef]
- Faustino, C.; Serafim, C.; Rijo, P.; Reis, C.P. Bile acid and bile acid derivatives: Use in drug delivery systems and as therapeutic agents. Expert Opin. Drug Deliv. 2016, 13, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, N.S.; Hua, J.; Powell, M.A.; Gibb, R.K.; Mutch, D.G.; Herzog, T.J. Novel cytotoxic agents from an unexpected source: Bile acids and ovarian tumor apoptosis. Gynecol. Oncol. 2007, 107, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Navacchia, M.L.; Marchesi, E.; Perrone, D. Bile acid conjugates with anticancer activity: Most recent research. Molecules 2021, 26, 25. [Google Scholar] [CrossRef] [PubMed]
- Sreekanth, V.; Bansal, S.; Motiani, R.K.; Kundu, S.; Muppu, S.K.; Majumdar, T.D.; Panjamurthy, K.; Sengupta, S.; Bajaj, A. Design, synthesis, and mechanistic investigations of bile acid−tamoxifen conjugates for breast cancer therapy. Bioconjug. Chem. 2013, 24, 1468–1484. [Google Scholar] [CrossRef]
- Zhang, D.; Li, D.; Shang, L.; Hea, Z.; Suna, J. Transporter-targeted cholic acid-cytarabine conjugates for improved oral absorption. Int. J. Pharm. 2016, 511, 161–169. [Google Scholar] [CrossRef]
- Melloni, E.; Marchesi, E.; Preti, L.; Casciano, F.; Rimondi, E.; Romani, A.; Secchiero, P.; Navacchia, M.L.; Perrone, D. Synthesis and biological investigation of bile acid-paclitaxel hybrids. Molecules 2022, 27, 471. [Google Scholar] [CrossRef]
- Agarwal, D.S.; Anantaraju, H.S.; Sriram, D.; Yogeeswari, P.; Nanjegowda, S.H.; Mallu, P.; Sakhuja, R. Synthesis, characterization and biological evaluation of bile acid aromatic/heteroaromatic amides linked via amino acids as anti-cancer agents. Steroids 2016, 107, 87–97. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Mari, L.; Chinaglia, N.; Gallerani, E.; Gavioli, R.; Capobianco, M.L.; Perrone, D. Rational design of nucleoside–bile acid conjugates incorporating a triazole moiety for anticancer evaluation and SAR exploration. Molecules 2017, 22, 1710. [Google Scholar] [CrossRef]
- Marchesi, E.; Chinaglia, N.; Capobianco, M.L.; Marchetti, P.; Huang, T.E.; Weng, H.C.; Guh, J.H.; Hsu, L.C.; Perrone, D.; Navacchia, M.L. Dihydroartemisinin-bile acid hybridization as an effective approach to enhance dihydroartemisinin anticancer activity. ChemMedChem 2019, 14, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.E.; Deng, Y.N.; Hsu, J.L.; Leu, W.J.; Marchesi, E.; Capobianco, M.L.; Marchetti, P.; Navacchia, M.L.; Guh, J.H.; Perrone, D.; et al. Evaluation of the anticancer activity of a bile acid-dihydroartemisinin hybrid ursodeoxycholic-dihydroartemisinin in hepatocellular carcinoma cells. Front. Pharmacol. 2020, 11, 599067. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; Marchi, E.D.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 329635. [Google Scholar] [CrossRef] [PubMed]
- Cobb, M.H. MAP kinase pathways. Prog. Biophys. Mol. Biol. 1999, 71, 479–500. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.J.; Meng, L.H.; Cai, Y.J.; Chen, Q.; Tong, L.J.; Lin, L.P.; Ding, J. Dihydroartemisinin induces apoptosis in HL-60 leukemia cells dependent of iron and p38 mitogen-activated protein kinase activation but independent of reactive oxygen species. Cancer Biol. Ther. 2008, 7, 1017–1023. [Google Scholar] [CrossRef]
- Rubinfeld, H.; Seger, R. The ERK cascade: A prototype of MAPK signaling. Mol. Biotechnol. 2005, 31, 151–174. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef]
- Parapini, S.; Olliaro, P.; Navaratnam, V.; Taramelli, D.; Basilico, N. Stability of the antimalarial drug dihydroartemisinin under physiologically relevant conditions: Implications for clinical treatment and pharmacokinetic and in vitro assays. Antimicrob. Agents Chemother. 2015, 59, 4046–4052. [Google Scholar] [CrossRef]
- Satoh, T.; Hosokawa, M. The mammalian carboxyesterases: From molecules to functions. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 257–288. [Google Scholar] [CrossRef]
- Fukami, T.; Yokoi, T. The emerging role of human esterases. Drug Metab. Pharmacokinet. 2012, 27, 466–477. [Google Scholar] [CrossRef]
- Zhou, C.H.; Wang, Y. Recent researches in triazole compounds as medicinal drugs. Curr. Med. Chem. 2012, 19, 239–280. [Google Scholar] [CrossRef]
- Dysko, A.; Baker, Y.R.; McClorey, G.; Wood, M.J.; Fenner, S.; Williams, G.; El-Sagheer, A.; Brown, T. Covalently attached intercalators restore duplex stability and splice-switching activity to triazole-modified oligonucleotides. RSC Chem. Biol. 2022, 3, 765–772. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Zhang, H.; Yun, J.; Chen, G.G.; Lai, P.B.S. Dihydroartemisinin exhibits antitumor activity toward hepatocellular carcinoma in vitro and invivo. Biochem. Pharmacol. 2012, 83, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Zhitkovich, A. N-acetylsysteine: Antioxidant, aldehyde scavenger, and more. Chem. Res. Toxicol. 2019, 32, 1318–1319. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Yao, X.; Li, X.; Zhang, D.; Xie, Y.; Sun, B.; Li, H.; Sun, L.; Zhang, X. B-cell lymphoma 2 inhibitor ABT-737 induces Beclin1-and reactive oxygen species-dependent autophagy in Adriamycin-resistant human hepatocellular carcinoma cells. Tumour Biol. 2017, 39, 1010428317695965. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Fan, M.; Shen, C.; Dai, W.; Bao, Y.; Liu, J.H.; Yu, B.Y. Novel dihydroartemisinin derivative DHA-37 induces autophagic cell death through upregulation of HMGB1 in A549 cells. Cell Death Dis. 2018, 9, 1048. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Zhang, E.; Chen, J.; Huang, X.; Yan, H.; Cao, W.; Qu, J.; Gu, H.; Xu, R. Dihydroartemisinin modulates apoptosis and autophagy in multiple myeloma through the P38/MAPK and Wnt/β-catenin signaling pathways. Oxid. Med. Cell. Longev. 2020, 2020, 6096391. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Azad, M.; Gibson, S. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Ureshino, R.P.; Rocha, K.K.; Lopes, G.S.; Bincoletto, C.; Smaili, S.S. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal. 2014, 21, 123–137. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and autophagy: Interactions and molecular regulatory mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Ni, Z.; Dai, X.; Wang, B.; Ding, W.; Rae Smith, A.; Xu, L.; Wu, D.; He, F.; Lian, J. The novel BH-3 mimetic apogossypolone induces Beclin-1-and ROS-mediated autophagy in human hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e589. [Google Scholar]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.A.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., 2nd; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Jiang, L.; Zhang, H.; Shimoda, L.A.; DeBerardinis, R.J.; Semenza, G.L. Analysis of hypoxia-induced metabolic reprogramming. Methods Enzymol. 2014, 542, 425–455. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HepG2 | Huh-7 | ||
|---|---|---|---|---|
| IC50 (μM) 1 | Ratio 2 | IC50 (μM) 1 | Ratio 2 | |
| DHA | 22.7 ± 0.39 | 40.0 ± 1.34 | ||
| UDCMe-Z-DHA | 1.00 ± 0.20 | 22.7 | 10.6 ± 2.28 | 3.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, Y.-F.; Kung, F.-L.; Huang, T.-E.; Deng, Y.-N.; Guh, J.-H.; Marchetti, P.; Marchesi, E.; Perrone, D.; Navacchia, M.L.; Hsu, L.-C. Anticancer Activity and Molecular Mechanisms of an Ursodeoxycholic Acid Methyl Ester-Dihydroartemisinin Hybrid via a Triazole Linkage in Hepatocellular Carcinoma Cells. Molecules 2023, 28, 2358. https://doi.org/10.3390/molecules28052358
Hsu Y-F, Kung F-L, Huang T-E, Deng Y-N, Guh J-H, Marchetti P, Marchesi E, Perrone D, Navacchia ML, Hsu L-C. Anticancer Activity and Molecular Mechanisms of an Ursodeoxycholic Acid Methyl Ester-Dihydroartemisinin Hybrid via a Triazole Linkage in Hepatocellular Carcinoma Cells. Molecules. 2023; 28(5):2358. https://doi.org/10.3390/molecules28052358
Chicago/Turabian StyleHsu, Ya-Fen, Fan-Lu Kung, Tzu-En Huang, Yi-Ning Deng, Jih-Hwa Guh, Paolo Marchetti, Elena Marchesi, Daniela Perrone, Maria Luisa Navacchia, and Lih-Ching Hsu. 2023. "Anticancer Activity and Molecular Mechanisms of an Ursodeoxycholic Acid Methyl Ester-Dihydroartemisinin Hybrid via a Triazole Linkage in Hepatocellular Carcinoma Cells" Molecules 28, no. 5: 2358. https://doi.org/10.3390/molecules28052358
APA StyleHsu, Y.-F., Kung, F.-L., Huang, T.-E., Deng, Y.-N., Guh, J.-H., Marchetti, P., Marchesi, E., Perrone, D., Navacchia, M. L., & Hsu, L.-C. (2023). Anticancer Activity and Molecular Mechanisms of an Ursodeoxycholic Acid Methyl Ester-Dihydroartemisinin Hybrid via a Triazole Linkage in Hepatocellular Carcinoma Cells. Molecules, 28(5), 2358. https://doi.org/10.3390/molecules28052358