
A KNIME Workflow to Assist the Analogue Identification for Read-Across, Applied to Aromatase Activity

 , ,
, , 
Abstract
1. Introduction
1.1. The Process of Category Formation and Read-Across
- Decision context (also called problem formulation and scenario definition): This serves to define the purpose and thus the scenario of the read-across prediction: for prioritization, or hazard or risk assessment. The scope and decision context determine the level of uncertainty that can be tolerated [1,5].
- Analogue evaluation: The most suitable candidates should have enough data, especially for the endpoint of interest. The evaluation should consider similarities with a focus on different features. The data quality and availability are decisive at this point [7]. Once this step is performed, a category is defined, and it should be evaluated for consistency [4].Each step is somehow dependent and linked to the other. For example, the identification and evaluation of the SCs are interconnected and can become an iterative process, which at the same time leads to an enrichment of the initial hypothesis.
- Data gap filling: Once a category has been obtained, the read-across can be performed. This step is called data gap filling and, in some cases, it may be a simple interpolation of source data. Three main strategies can be followed: (1) a conservative strategy based on a worst-case, that considers the TC toxic as the most toxic SC; (2) in trend analysis, if a clear trend between the activity and the structure/property of the SCs is identified; (3) a nearest neighbor approach, meaning that only the most similar SCs are used to infer the toxicity of the TC. For qualitative read-across, a strategy could be a simple majority vote approach [1,2,4,5].
- Uncertainty evaluation: some guiding documents exist on uncertainty assessment, e.g., the weight of evidence [7,8]. The RAAF also proposes a complementary strategy to address the different sources of uncertainties [6]. Finally, when a read-across is done for regulatory acceptance, all processes related to the prediction should be properly documented.
1.2. The Process of Category Formation and Read-Across
1.3. Adapted Scheme for the Present Work
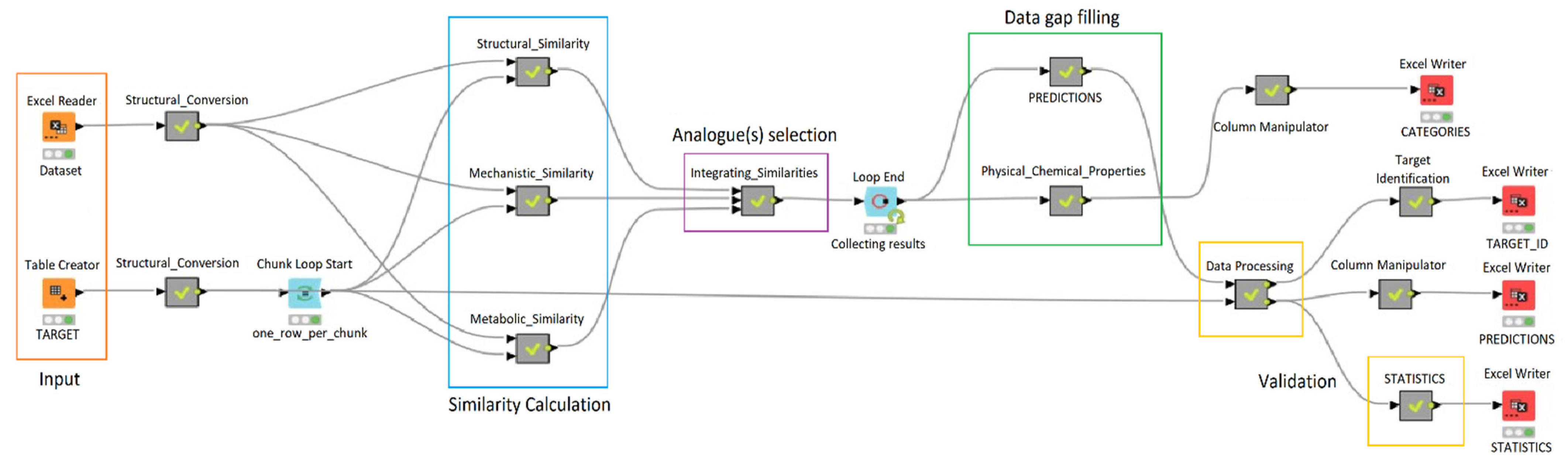
1.3.1. Constructing Read-Across Workflow
1.3.2. Structural Similarity
1.3.3. Mechanistic Similarity
1.3.4. Metabolic Similarity
1.3.5. Integrating Similarities
2. Results and Discussion
2.1. Workflow Performance
2.2. The Read-Across Case Studies
3. Materials and Methods
3.1. The Dataset on Aromatase
- 82 monoazoles compounds of which 61 were inactive and 21 active.
- 198 diazoles of which 148 were inactive and 50 active.
- 46 triazoles which contained 26 inactive and 20 active.
3.2. Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Escher, S.E.; Kamp, H.; Bennekou, S.H.; Bitsch, A.; Fisher, C.; Graepel, R.; Hengstler, J.G.; Herzler, M.; Knight, D.; Leist, M.; et al. Towards grouping concepts based on new approach methodologies in chemical hazard assessment: The read-across approach of the EU-ToxRisk project. Arch. Toxicol. 2019, 93, 3643–3667. [Google Scholar] [CrossRef]
- Schultz, T.; Amcoff, P.; Berggren, E.; Gautier, F.; Klaric, M.; Knight, D.; Mahony, C.; Schwarz, M.; White, A.; Cronin, M. A strategy for structuring and reporting a read-across prediction of toxicity. Regul. Toxicol. Pharmacol. 2015, 72, 586–601. [Google Scholar] [CrossRef] [PubMed]
- Patlewicz, G.; Ball, N.; Booth, E.D.; Hulzebos, E.; Zvinavashe, E.; Hennes, C. Use of category approaches, read-across and (Q) SAR: General considerations. Regul. Toxicol. Pharmacol. 2013, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M. An introduction to chemical grouping, categories and read-across to predict toxicity. In Chemical Toxicity Prediction, 2nd ed.; Cronin, M., Madden, J., Enoch, S., Roberts, D., Eds.; Royal Society of Chemistry: London, UK, 2013; pp. 1–29. [Google Scholar]
- Patlewicz, G.; Helman, G.; Pradeep, P.; Shah, I. Navigating through the minefield of read-across tools: A review of in silico tools for grouping. Comput. Toxicol. 2017, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Read-Across Assessment Framework (RAAF). Available online: https://echa.europa.eu/documents/10162/13628/raaf_en.pdf/614e5d61-891d-4154-8a47-87efebd1851a (accessed on 5 February 2023).
- Madden, J. Tools for grouping chemicals and forming categories. In Chemical Toxicity Prediction: Category Formation and Read-Across, 2nd ed.; Mark, C., Judith, M., Steven, E., David, R., Eds.; Royal Society of Chemistry: London, UK, 2013; pp. 72–97. [Google Scholar]
- Alfonso, A.Y.C.; Lagares, L.M.; Novic, M.; Benfenati, E.; Kumar, A. Exploration of structural requirements for azole chemicals towards human aromatase CYP19A1 activity: Classification modeling, structure-activity relationships and read-across study. Toxicol. Vitr. 2022, 81, 105332. [Google Scholar] [CrossRef]
- Gadaleta, D.; Bakhtyari, A.G.; Lavado, G.J.; Roncaglioni, A.; Benfenati, E. Automated integration of structural, biological and metabolic similarities to improve read-across. ALTEX-Altern. Anim. Exp. 2020, 37, 469–481. [Google Scholar]
- Caballero, A.Y.; Toma, C.; Gadaleta, D.; Perez, Y.; Benfenati, E. Assessment of a framework to identify analogues for read-across: Case study. In Toxicology Letters. 2019. Elsevier Ireland Ltd Elsevier House, Brookvale Plaza, East Park Shannon, Co, Clare, 00000; Elsevier Ireland Ltd.: Dublin, Ireland, 2019. [Google Scholar]
- Information, N.C.f.B. PubChem Substructure Fingerprint, in PubChem Data Specification Directory. 2009. Available online: https://web.cse.ohio-state.edu/~zhang.10631/bak/drugreposition/list_fingerprints.pdf (accessed on 13 February 2023).
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Chertó, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2. 0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminform. 2017, 9, 33. [Google Scholar]
- Gortari, E.F.-D.; García-Jacas, C.R.; Martinez-Mayorga, K.; Medina-Franco, J.L. Database fingerprint (DFP): An approach to represent molecular databases. J. Cheminform. 2017, 9, 9. [Google Scholar]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef]
- Bender, A.; Glen, R. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef]
- Webster, F.; Gagné, M.; Patlewicz, G.; Pradeep, P.; Trefiak, N.; Judson, R.S.; Barton-Maclaren, T.S. Predicting estrogen receptor activation by a group of substituted phenols: An integrated approach to testing and assessment case study. Regul. Toxicol. Pharmacol. 2019, 106, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, J.; Wu, Z.; Li, W.; Liu, G.; Tang, Y. Evaluation of different methods for identification of structural alerts using chemical ames mutagenicity data set as a benchmark. Chem. Res. Toxicol. 2017, 30, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, M.; Enoch, S.J.; Madden, J.C.; Przybylak, K.R.; Cronin, M.T.D. Hepatotoxicity: A scheme for generating chemical categories for read-across, structural alerts and insights into mechanism(s) of action. Crit. Rev. Toxicol. 2013, 43, 537–558. [Google Scholar] [CrossRef] [PubMed]
- Rostkowski, M.; Spjuth, O.; Rydberg, P. WhichCyp: Prediction of cytochromes P450 inhibition. Bioinformatics 2013, 29, 2051–2052. [Google Scholar] [CrossRef]
- Caballero-Alfonso, A.Y.; Cruz-Monteagudo, M.; Tejera, E.; Benfenati, E.; Borges, F.; Cordeiro, M.N.D.; Armijos-Jaramillo, V.; Perez-Castillo, Y. Ensemble-based modeling of chemical compounds with antimalarial activity. Curr. Top. Med. Chem. 2019, 19, 957–969. [Google Scholar] [CrossRef]
- Viganò, E.L.; Colombo, E.; Raitano, G.; Manganaro, A.; Sommovigo, A.; CM Dorne, J.L.; Benfenati, E. Virtual Extensive Read-Across: A New Open-Access Software for Chemical Read-Across and Its Application to the Carcinogenicity Assessment of Botanicals. Molecules 2022, 27, 6605. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Bubalo, M.C.; Radošević, K.; Redovniković, I.R.; Slivac, I.; Srček, V.G. Toxicity mechanisms of ionic liquids. Arh. Za Hig. Rada I Toksikol. 2017, 68, 171–179. [Google Scholar] [CrossRef]
- Cruz-Monteagudo, M.; Ancede-Gallardo, E.; Jorge, M.; Dias Soeiro Cordeiro, M.N. Chemoinformatics profiling of ionic liquids—Automatic and chemically interpretable cytotoxicity profiling, virtual screening, and cytotoxicophore identification. Toxicol. Sci. 2013, 136, 548–565. [Google Scholar] [CrossRef]
- Ranke, J.; Mölter, K.; Stock, F.; Bottin-Weber, U.; Poczobutt, J.; Hoffmann, J.; Ondruschka, B.; Filser, J.; Jastorff, B. Biological effects of imidazolium ionic liquids with varying chain lengths in acute Vibrio fischeri and WST-1 cell viability assays. Ecotoxicol. Environ. Saf. 2004, 58, 396–404. [Google Scholar] [CrossRef]
- Ranke, J.; Müller, A.; Bottin-Weber, U.; Stock, F.; Stolte, S.; Arning, J.; Störmann, R.; Jastorff, B. Lipophilicity parameters for ionic liquid cations and their correlation to in vitro cytotoxicity. Ecotoxicol. Environ. Saf. 2007, 67, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Fan, Y.; Zhang, H.; Zhong, Y.; Yang, Y.; Miao, J.; Hua, S. Inhibitory effects of ionic liquids on the lactic dehydrogenase activity. Int. J. Biol. Macromol. 2016, 86, 155–161. [Google Scholar] [CrossRef]
- Na, L.I.; Weiyan, D.U.; HUANG, Z.; Wei, Z.H.A.O.; Shoujiang, W.A.N.G. Effect of imidazolium ionic liquids on the hydrolytic activity of lipase. Chin. J. Catal. 2013, 34, 769–780. [Google Scholar]
- Agency, U.S.E.P.A. Standard Laboratory Protocol for Tox21 Assays. 2018. Available online: https://gaftp.epa.gov/COMPTOX/High_Throughput_Screening_Data/Standard_Lab_Protocol_Tox21_Assays/Tox21Assay_SLPs%20and%20Descriptions_2016.zip (accessed on 28 May 2018).
- Gadaleta, D.; Lombardo, A.; Toma, C.; Benfenati, E. A new semi-automated workflow for chemical data retrieval and quality checking for modeling applications. J. Cheminform. 2018, 10, 60. [Google Scholar] [CrossRef]
- Achar, P.N.; Aubert, A.-M. Springer correspondences for dihedral groups. Transform. Groups 2008, 13, 1–24. [Google Scholar] [CrossRef]
- Tharwat, A. Classification assessment methods. Appl. Comput. Inform. 2021, 17, 168–192. [Google Scholar] [CrossRef]
- Vian, M.; Raitano, G.; Roncaglioni, A.; Benfenati, E. In silico model for mutagenicity (Ames test), taking into account metabolism. Mutagenesis 2019, 34, 41–48. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Structural Alert_ID | SMARTS | Associated Toxicity |
|---|---|---|
| SA_1 | n1c(N)sc2cccc(c12) | Toxic * |
| SA_2 | c1c[n+](cn1CCCCCCCC)C | Toxic |
| SA_3 | N#C | Toxic |
| SA_4 | c1c(cccc1Cl)Cl | Toxic |
| SA_5 | c1ccc(cc1)c1nc(N)sc1 | Toxic |
| SA_6 | n1ccsc1C | Non_Toxic |
| SA_7 | O=c1c2c(ncn2C)n(c(=O)n1)C | Non_Toxic |
| SA_8 | O=c1ccnc([nH]1) | Non_Toxic |
| SA_9 | N=C(N)N | Non_Toxic |
| SA_10 | O=C(O)C | Non_Toxic |
| SA_11 | c1ccnn1 | Non_Toxic |
| SA_12 | S(=O)c1ccccc1 | Non_Toxic |
| SA_13 | O=C(NC)C | Non_Toxic |
| SA_14 | c1nc[nH]c1C | Non_Toxic |
| SA_15 | n1c(nnc1C)C | Toxic |
| SA_16 | C(Cn1ncnc1)C(C)(C) | Toxic |
| SA_17 | O=S(=O)(N) | Non_Toxic |
| SA_18 | n1csc(c1C) | Non_Toxic |
| SA_19 | c1cnc(n1C)C | Non_Toxic |
| SA_20 | O=C(OC)c1cncn1 | Non_Toxic |
| SA_21 | N(C)C | Non_Toxic |
| Structural Similarity | Integrated Similarities | ||||
|---|---|---|---|---|---|
| Predicted | |||||
| Positive | Negative | Positive | Negative | ||
| Experimental | Positive | 49 | 35 | 34 | 3 |
| Negative | 23 | 178 | 5 | 27 | |
| Sensitivity | 0.58 | 0.92 | |||
| Specificity | 0.89 | 0.84 | |||
| Accuracy | 0.80 | 0.88 | |||
| Error_rate | 0.20 | 0.12 | |||
| Unpredicted rate | 0.04 | 0.79 | |||
| MCC | 0.49 | 0.77 | |||
| Number of SCs Matching the Target Activity | Number of SCs not Matching the Target Activity | Ratio of SCs not Matching the Target Activity | |||
|---|---|---|---|---|---|
| StrS | 2050 | 944 | 0.46 | ||
| active | inactive | active | inactive | ||
| 500 | 1550 | 472 | 472 | ||
| IntS | 211 | 26 | 0.12 | ||
| active | inactive | active | inactive | ||
| 44 | 167 | 13 | 13 | ||
| Name | Structure * | Rank | Activity | StrS | Metabolic Similarity | Common Mechanistic Structural Alerts |
|---|---|---|---|---|---|---|
| 1-Hexadecyl-3-methylimidazolium |  | Target | Active | - | - | - |
| 1,3-Didecyl-2-methylimidazolium |  | IntS StrS | Active | 0.98 | 1 | SA2 |
| 1-Methyl-3-octadecylimidazolium hexafluorophosphate |  | IntS StrS | Active | 1 | 1 | SA2 |
| 1-Methyl-3-tetradecylimidazolium chloride |  | IntS StrS | Active | 1 | 1 | SA2 |
| 1-Hexyl-3-methylimidazolium chloride |  | StrS | Inactive | 0.95 | - | - |
| 1A2 | 2C9 | 2C19 | 2D6 | 3A4 | |
|---|---|---|---|---|---|
| 1-Hexadecyl-3-methylimidazolium chloride (target) |  |  |  |  |  |
| 0 | 0 | 0 | 1 | 0 | |
| 1-Hexyl-3-methylimidazolium chloride |  |  |  |  |  |
| 0 | 0 | 0 | 0 | 0 | |
| 1-Methyl-3-tetradecylimidazolium chloride |  |  |  |  |  |
| 0 | 0 | 0 | 1 | 0 |
| Name | Structure * | Rank | Activity | StrS | Metabolic Similarity | Common Mechanistic Structural Alerts |
|---|---|---|---|---|---|---|
| 2-Amino-6-ethoxybenzothiazole |  | Target | Active | - | - | - |
| Methabenzthiazuron |  | IntS StrS | Active | 0.73 | 1 | SA1 |
| Riluzole |  | IntS StrS | Active | 0.94 | 1 | SA1 |
| Tioxidazole |  | IntS StrS | Active | 0.94 | 1 | SA1 |
| 2-Amino-4-methoxybenzothiazole |  | StrS | Inactive | 0.90 | - | - |
| 1A2 | 2C9 | 2C19 | 2D6 | 3A4 | |
|---|---|---|---|---|---|
| 2-Amino-6- ethoxybenzothiazole (target) | 1 | 0 | 1 | 0 | 0 |
 |  |  |  |  | |
| Methabenzthiazuron | 1 | 0 | 1 | 0 | 0 |
 |  |  |  |  | |
| Riluzole | 1 | 0 | 1 | 0 | 0 |
 |  |  |  |  | |
| Tioxidazole | 1 | 0 | 1 | 0 | 0 |
 |  |  |  |  | |
| 2-Amino-4- methoxybenzothiazole | 1 | 0 | 0 | 0 | 0 |
 |  |  |  |  |
| Predicted Positive | Predicted Negative | |
|---|---|---|
| Experimental Positive | “True Positive” | “False Negative” |
| Experimental Negative | “False Positive” | “True Negative” |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caballero Alfonso, A.Y.; Chayawan, C.; Gadaleta, D.; Roncaglioni, A.; Benfenati, E. A KNIME Workflow to Assist the Analogue Identification for Read-Across, Applied to Aromatase Activity. Molecules 2023, 28, 1832. https://doi.org/10.3390/molecules28041832
Caballero Alfonso AY, Chayawan C, Gadaleta D, Roncaglioni A, Benfenati E. A KNIME Workflow to Assist the Analogue Identification for Read-Across, Applied to Aromatase Activity. Molecules. 2023; 28(4):1832. https://doi.org/10.3390/molecules28041832
Chicago/Turabian StyleCaballero Alfonso, Ana Yisel, Chayawan Chayawan, Domenico Gadaleta, Alessandra Roncaglioni, and Emilio Benfenati. 2023. "A KNIME Workflow to Assist the Analogue Identification for Read-Across, Applied to Aromatase Activity" Molecules 28, no. 4: 1832. https://doi.org/10.3390/molecules28041832
APA StyleCaballero Alfonso, A. Y., Chayawan, C., Gadaleta, D., Roncaglioni, A., & Benfenati, E. (2023). A KNIME Workflow to Assist the Analogue Identification for Read-Across, Applied to Aromatase Activity. Molecules, 28(4), 1832. https://doi.org/10.3390/molecules28041832