
Papaverinol-N-Oxide: A Microbial Biotransformation Product of Papaverine with Potential Antidiabetic and Antiobesity Activity Unveiled with In Silico Screening

 ,
,  ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
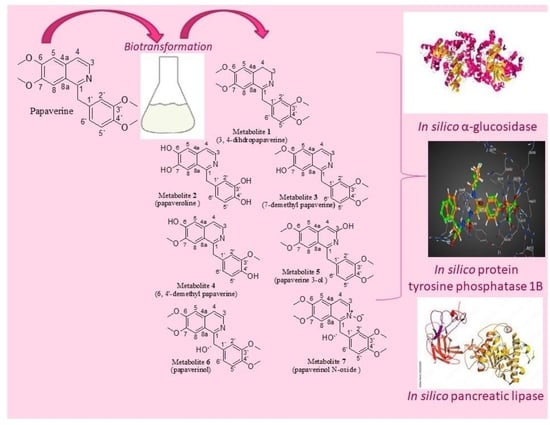
1. Introduction
2. Results and Discussion
2.1. Identification of the Biotransformation Products
2.2. ADMET/Pharmacokinetic Properties and Drug-Likeness Predictions of Papaverine and Its Metabolites (1–7)
2.3. Docking Studies of Metabolites on the Active Sites of PTP1B (1G7F) and α-Glucosidase (3A4A)
2.4. Docking Studies of Metabolites on the Active Site of Lipase (PDB: 1LPB)
2.5. MD Simulation
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Microorganism
3.3. Large-Scale Fermentation and Isolation of the Metabolites
3.3.1. Papaverine Transformation via Cunninghamella Elegans NRRL 2310
3.3.2. Papaverine Transformation via Rhodotorula Rubra NRRL y1592
3.3.3. Papaverine Transformation via Cunninghamella Blackesleeana NRRL 1369
3.3.4. Papaverine Transformation via Penicillium Chrysogeneum ATCC 10002
3.4. ADMET/Pharmacokinetic Properties and Drug-Likeness Predictions
3.5. Molecular Docking Studies
3.6. MD Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smitha, S.; Singh, S.; Singh, R. Microbial biotransformation: A process for chemical alterations. J. Bacteriol. Mycol. Open Access 2017, 4, 47–51. [Google Scholar]
- Ghasemi, Y.; Rasoul-Amini, S.; Fotooh-Abadi, E. The biotransformation, biodegradation, and bioremediation of organic compounds by microalgae1. J. Phycol. 2011, 47, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, Z.Y.; Cui, J.L.; Wang, M.L.; Wang, J.H. Biotransformation ability of endophytic fungi: From species evolution to industrial applications. Appl. Microbiol. Biotechnol. 2021, 105, 7095–7113. [Google Scholar] [CrossRef] [PubMed]
- Thawabteh, A.; Juma, S.; Bader, M.; Karaman, D.; Scrano, L.; Bufo, S.A.; Karaman, R. The biological activity of natural alkaloids against herbivores, cancerous cells and pathogens. Toxins 2019, 11, 656. [Google Scholar] [CrossRef]
- Daley, S.K.; Cordell, G.A. Alkaloids in contemporary drug discovery to meet global disease needs. Molecules 2021, 26, 3800. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, D.; Deborah, A.; Neil, C. Microbial transformation of alkaloids. Curr. Opin. Microbiol. 2002, 5, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Eliwa, D.; Albadry, M.A.; Ibrahim, A.S.; Kabbash, A.; Meepagala, K.; Khan, I.A.; El-Aasr, M.; Ross, S.A. Biotransformation of papaverine and in silico docking studies of the metabolites on human phosphodiesterase 10a. Phytochemistry 2021, 183, 112598. [Google Scholar] [CrossRef]
- Verdeil, J.; Bister-Miel, F.; Guignard, J.; Viel, C. Papaverine biotransformation by Silene alba cell suspension. Planta Med. 1986, 52, 1–3. [Google Scholar] [CrossRef]
- Dorisse, P.; Gleye, J.; Loiseau, P.; Puig, P.; Edy, M.; Henry, M. Papaverine biotransformation in plant cell suspension cultures. J. Nat. Prod. 1988, 51, 532–536. [Google Scholar] [CrossRef]
- El Sayed, K. Microbial transformation of papaveraldine. Phytochemistry 2000, 53, 675–678. [Google Scholar] [CrossRef]
- Belpaire, F.; Bogaert, M.; Rosseel, M.; Anteunis, M. Metabolism of papaverine. I. Identification of metabolites in rat bile. Xenobiotica 1975, 5, 413–420. [Google Scholar] [CrossRef]
- Bustanji, Y.; Taha, M.O.; Al-Masri, I.M.; Mohammad, M.K. Docking simulations and in vitro assay unveil potent inhibitory action of papaverine against protein tyrosine phosphatase 1B. Biol. Pharm. Bull. 2009, 32, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Thareja, S.; Aggarwal, S.; Bhardwaj, T.R.; Kumar, M. Protein tyrosine phosphatase 1B inhibitors: A molecular level legitimate approach for the management of diabetes mellitus. Med. Res. Rev. 2012, 32, 459–517. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.I.; Ketsawatsomkron, P.; de Chantemele, E.J.B.; Mintz, J.D.; Muta, K.; Salet, C.; Black, S.M.; Tremblay, M.L.; Fulton, D.J.; Marrero, M.B.; et al. Deletion of protein tyrosine phosphatase 1B improves peripheral insulin resistance and vascular function in obese, leptin-resistant mice via reduced oxidant tone. Circ. Res. 2009, 105, 1013–1022. [Google Scholar] [CrossRef]
- Lebovitz, H.E. Alpha-Glucosidase Inhibitors. Endocrinol. Metab. Clin. N. Am. 1997, 26, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.J. The role of anti-obesity medication in prevention of diabetes and its complications. J. Obes. Metab. Syndr. 2019, 28, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Almasri, I. Pancreatic lipase inhibition by papaverine: Investigation by simulated molecular docking and subsequent in vitro evaluation. Jordan J. Pharm. Sci. 2013, 6, 271–279. [Google Scholar] [CrossRef]
- Salter, R.; Beshore, D.; Colletti, S.; Evans, L.; Gong, Y.; Helmy, R.; Liu, Y.; Maciolek, C.; Martin, G.; Pajkovic, N.; et al. Microbial biotransformation—An important tool for the study of drug metabolism. Xenobiotica 2019, 49, 877–886. [Google Scholar] [CrossRef]
- Sebek, O. Fungal transformations as a useful method for the synthesis of organic compounds. Mycologia 1983, 75, 383–394. [Google Scholar] [CrossRef]
- Galat, A. Synthesis of Papaverine and Some Related Compounds. J. Am. Chem. Soc. 1951, 73, 3654–3656. [Google Scholar] [CrossRef]
- Brochmann-Hanssen, E.; Hirai, K. Opium alkaloids. VII. Isolation of a new benzylisoquinoline alkaloid. Synthesis and NMR studies of papaveroline trimethyl ethers. J. Pharm. Sci. 1968, 57, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Hermann, T.W.; Girreser, U.; Michalski, P.; Piotrowska, K. Oxidation and degradation products of papaverine. Part I: Gadamer and Schulemann’s papaverinol synthesis revisited. Arch. Pharm. 2002, 335, 167–169. [Google Scholar] [CrossRef]
- de Montellano, P.R.O. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Nafie, M.S.; Shaldam, M.A.; Elmaaty, A.A.; Antar, S.A.; El-Hamaky, A.A.; Saleh, M.A.; Elkamhawy, A.; Tawfik, H.O. Ligand-Based Design on the Dog-Bone-Shaped BIBR1532 Pharmacophoric Features and Synthesis of Novel Analogues as Promising Telomerase Inhibitors with in vitro and in vivo Evaluations. J. Med. Chem. 2023, 66, 777–792. [Google Scholar] [CrossRef]
- Hassan, S.S.U.; Abbas, S.Q.; Ali, F.; Ishaq, M.; Bano, I.; Hassan, M.; Jin, H.-Z.; Bungau, S.G. A Comprehensive In Silico Exploration of Pharmacological Properties, Bioactivities, Molecular Docking, and Anticancer Potential of Vieloplain F from Xylopia vielana Targeting B-Raf Kinase. Molecules 2022, 27, 917. [Google Scholar] [CrossRef]
- Olasupo, S.B.; Uzairu, A.; Shallangwa, G.A.; Uba, S. Unveiling novel inhibitors of dopamine transporter via in silico drug design, molecular docking, and bioavailability predictions as potential antischizophrenic agents. Future J. Pharm. Sci. 2021, 7, 63. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Bellmann, L.; Klein, R.; Rarey, M. Calculating and Optimizing Physicochemical Property Distributions of Large Combinatorial Fragment Spaces. J. Chem. Inf. Model. 2022, 62, 2800–2810. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Buğday, N.; Rehman, A.U.; Özdemir, İ. Synthesis, molecular docking, and biological evaluation of 5-alkyl(aryl)-2-isobutylthiazole derivatives: As α-amylase, α-glucosidase, and protein kinase inhibitors. Appl. Organomet. Chem. 2022, 36, e6641. [Google Scholar] [CrossRef]
- Tawfik, H.O.; El-Hamaky, A.A.; El-Bastawissy, E.A.; Shcherbakov, K.A.; Veselovsky, A.V.; Gladilina, Y.A.; Zhdanov, D.D.; El-Hamamsy, M.H. New Genetic Bomb Trigger: Design, Synthesis, Molecular Dynamics Simulation, and Biological Evaluation of Novel BIBR1532-Related Analogs Targeting Telomerase against Non-Small Cell Lung Cancer. Pharmaceuticals 2022, 15, 481. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tian, Y.; Gao, Y.; Wu, F.; Luo, X.; Ju, X.; Liu, G. In silico design of novel HIV-1 NNRTIs based on combined modeling studies of dihydrofuro [3,4-d] pyrimidines. Front. Chem. 2020, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.V.; Serafim, R.B.; da Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Neto, M.F.A.; Leite, F.H.A.; Taft, C.A.; da Silva, C.H.T.P.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef] [PubMed]
- Tamilarasan, R.; Ganesan, K.; Subramani, A.; Benazir, L.; Ali, M.M.; Mohammed, A.A. Synthesis, Characterization, Pharmacogenomics, and Molecular Simulation of Pyridinium Type of Ionic Liquids and Their Applications. ACS Omega 2023, 8, 4146–4155. [Google Scholar] [CrossRef] [PubMed]
- Dulsat, J.; López-Nieto, B.; Estrada-Tejedor, R.; Borrell, J.I. Evaluation of Free Online ADMET Tools for Academic or Small Biotech Environments. Molecules 2023, 28, 776. [Google Scholar] [CrossRef] [PubMed]
- Ugwu-Korie, N.; Quaye, O.; Wright, E.; Languon, S.; Agyapong, O.; Broni, E.; Gupta, Y.; Kempaiah, P.; Kwofie, S.K. Structure-Based Identification of Natural-Product-Derived Compounds with Potential to Inhibit HIV-1 Entry. Molecules 2023, 28, 474. [Google Scholar] [CrossRef]
- Kara, G.; Calin, G.A.; Ozpolat, B. RNAi-based therapeutics and tumor targeted delivery in cancer. Adv. Drug Deliv. Rev. 2022, 182, 114113. [Google Scholar] [CrossRef]
- Krishna, S.; Borrel, A.; Huang, R.; Zhao, J.; Xia, M.; Kleinstreuer, N. High-throughput chemical screening and structure-based models to predict hERG inhibition. Biology 2022, 11, 209. [Google Scholar] [CrossRef]
- Tamrakar, A.K.; Maurya, C.K.; Rai, A.K. PTP1B inhibitors for type 2 diabetes treatment: A patent review (2011–2014). Expert Opin. Ther. Pat. 2014, 24, 1101–1115. [Google Scholar] [CrossRef]
- Bleasdale, J.E.; Ogg, D.; Palazuk, B.J.; Jacob, C.S.; Swanson, M.L.; Wang, X.-Y.; Thompson, D.P.; Conradi, R.A.; Mathews, W.R.; Laborde, A.L. Small molecule peptidomimetics containing a novel phosphotyrosinebioisostere inhibit protein tyrosine phosphatase 1B and augment insulin action. Biochemistry 2001, 40, 5642–5654. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef]
- Murugesu, S.; Ibrahim, Z.; Ahmed, Q.U.; Yusoff, N.I.N.; Uzir, B.F.; Perumal, V.; Abas, F.; Saari, K.; El-Seedi, H.; Khatib, A. Characterization of α-glucosidase inhibitors from Clinacanthus nutans Lindau leaves by gas chromatography-mass spectrometry-based metabolomics and molecular docking simulation. Molecules 2018, 23, 2402. [Google Scholar] [CrossRef]
- Askarzadeh, M.; Azizian, H.; Adib, M.; Mohammadi-Khanaposhtani, M.; Mojtabavi, S.; Faramarzi, M.A.; Sajjadi-Jazi, S.M.; Larijani, B.; Hamedifar, H.; Mahdavi, M. Design, synthesis, in vitro α-glucosidase inhibition, docking, and molecular dynamics of new phthalimide-benzenesulfonamide hybrids for targeting type 2 diabetes. Sci. Rep. 2022, 12, 10569. [Google Scholar] [CrossRef]
- Egloff, M.P.; Marguet, F.; Buono, G.; Verger, R.; Cambillau, C.; Van Tilbeurgh, H. The 2.46 A resolution structure of the pancreatic lipase-colipase complex inhibited by a C11 alkyl phosphonate. Biochemistry 1995, 34, 2751–2762. [Google Scholar] [CrossRef] [PubMed]
- Veeramachaneni, G.K.; Raj, K.K.; Chalasani, L.M.; Annamraju, S.K.; Talluri, V.R. Shape based virtual screening and molecular docking towards designing novel pancreatic lipase inhibitors. Bioinformation 2015, 11, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.; Al-Masri, I.M.; Issa, A. Inhibition of pancreatic lipase by berberine and dihydroberberine: An investigation by docking simulation and experimental validation. Med. Chem. Res. 2013, 22, 2273–2278. [Google Scholar] [CrossRef]
- Noor, Z.I.; Ahmed, D.; Rehman, H.M.; Qamar, M.T.; Froeyen, M.; Ahmad, S.; Mirza, M.U. In vitro antidiabetic, anti-obesity and antioxidant analysis of Ocimum basilicum aerial biomass and in silico molecular docking simulations with alpha-amylase and lipase enzymes. Biology 2019, 8, 92. [Google Scholar] [CrossRef]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.L.; Zhao, Z.; Wang, Y. The Lid Domain in Lipases: Structural and Functional Determinant of Enzymatic Properties. Front. Bioeng. Biotechnol. 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Croitoru, A.; Park, S.-J.; Kumar, A.; Lee, J.; Im, W.; MacKerell, A.D.; Aleksandrov, A. Additive CHARMM36 Force Field for Nonstandard Amino Acids. J. Chem. Theory Comput. 2021, 17, 3554–3570. [Google Scholar] [CrossRef]
- Silva, T.F.D.; Vila-Viçosa, D.; Reis, P.; Victor, B.; Diem, M.; Oostenbrink, C.; Machuqueiro, M. The Impact of Using Single Atomistic Long-Range Cutoff Schemes with the GROMOS 54A7 Force Field. J. Chem. Theory Comput. 2018, 14, 5823–5833. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Yasuoka, K.; Takahashi, K.Z. Critical test of isotropic periodic sum techniques with group-based cut-off schemes. Sci. Rep. 2018, 8, 4185. [Google Scholar] [CrossRef]
- Aboukhatwa, S.M.; Ibrahim, A.O.; Aoyama, H.; Al-Behery, A.S.; Shaldam, M.A.; El-Ashmawy, G.; Tawfik, H.O. Nicotinonitrile-derived apoptotic inducers: Design, synthesis, X-ray crystal structure and Pim kinase inhibition. Bioorg. Chem. 2022, 129, 106126. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH (Multiplicities, J in Hz) | |||||||
|---|---|---|---|---|---|---|---|---|
| Papaverine | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| 3 | 8.33, d (5.6) | 3.47, t (7.2) | 8.31, d (5.7) | 8.29, d (5.2) | 8.36, d (5.6) | - | 8.35, d (5.6) | 8.41, d (5.2) |
| 4 | 7.53, d (5.7) | 3.07, t (7.2) | 7.50, d (5.6) | 7.52, d (5.2) | 7.41, d (5.6) | 6.99, s | 7.52, d (5.6) | 7.51, d (5.2) |
| 5 | 7.01, s | 7.06, s | 7.14, s | 7.07, s | 7.34, s | 7.05, s | 7.02, s | 7.06, s |
| 8 | 7.41, s | 7.38, s | 7.45, s | 7.54, s | 7.40, s | 7.33, s | 7.07, s | 7.12, s |
| 2′ | 6.94, d (1.8) | 6.79, d (1.6) | 6.93, d (1.6) | 6.82, d (1.2) | 6.92, d (1.2) | 6.61, d (1.2) | 6.79, d (1.2) | 6.82, d (1.6) |
| 5′ | 6.74, brs | 6.87, d (7.6) | 6.77, d (8.4) | 6.70, d (8.4) | 6.75, d (8.0) | 6.76, d (8.4) | 6.75, d (8.4) | 6.78, d (8.4) |
| 6′ | 6.82, m | 6.81, dd (7.6, 1.6) | 6.82, dd (8.4, 1.6) | 6.73, dd (8.4, 1.2) | 6.88, dd (8.0, 1.2) | 6.83, dd (8.4, 1.2) | 6.89, dd (8.4, 1.2) | 6.93, dd (8.4, 1.6) |
| α | 4.56, s | 4.54, s | 4.45, s | 4.45, s | 4.55, s | 4.43, s | 6.08, s | 6.15, s |
| 6-Ome | 4.02, s | 3.99, s | - | 4.02, s | - | 3.98, s | 3.95, s | 4.03, s |
| 7-Ome | 3.93, s | 3.90, s | - | - | 3.91, s | 3.88, s | 3.71, s | 3.90, s |
| 3′-Ome | 3.78, s | 3.76, s | - | 3.72, s | 3.76, s | 3.76, s | 3.76, s | 3.76, s |
| 4′-Ome | 3.80, s | 3.81, s | - | 3.77, s | - | 3.82, s | 3.81, s | 3.82, s |
| 6-OH | - | - | 5.06, s | - | 6.03, br s | - | - | - |
| 7-OH | - | - | 5.15, s | 6.02, br s | - | - | - | - |
| 3′-OH | - | - | 5.33, s | - | - | - | - | - |
| 4′-OH | - | - | 5.64, s | - | 5.94, br s | - | - | - |
| 3-OH | - | - | __ | - | - | 2.36, br s | - | - |
| α-OH | - | - | __ | - | - | - | 6.38, br s | 6.50, br s |
| Position | δH (Multiplicities, J) | |||||||
|---|---|---|---|---|---|---|---|---|
| Papaverine | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| 1 | 156.9, C | 156.5, C | 157.5, C | 156.4, C | 157.3, C | 158.9, C | 157.0, C | 155.6, C |
| 3 | 139.0, CH | 46.5, CH2 | 141.7, CH | 139.9, CH | 141.2, CH | 165.9, C | 138.6, CH | 138.0, CH |
| 4 | 119.6, CH | 26.2, CH2 | 119.0, CH | 119.3, CH | 117.7, CH | 98.3, CH | 119.5, CH | 119.8, CH |
| 4a | 134.6, C | 134.1, C | 133.5, C | 132.0, C | 133.6, C | 134.0, C | 132.8, C | 133.5, C |
| 5 | 105.6, CH | 104.8, CH | 105.7, CH | 105.3, CH | 103.2, CH | 108.7, CH | 105.0, CH | 105.2, CH |
| 6 | 150.7, C | 153.4, C | 150.5, C | 150.1, C | 150.8, C | 151.7, C | 152.2, C | 151.9, C |
| 7 | 149.3, C | 150.4, C | 148.4, C | 147.8, C | 147.2, C | 149.4, C | 149.5, C | 149.7, C |
| 8 | 104.5, CH | 103.3, CH | 104.1, CH | 102.2, CH | 103.7, CH | 105.1, CH | 102.6, CH | 103.6, CH |
| 8a | 122.9, C | 121.5, C | 122.1, C | 122.1, C | 121.9, C | 118.1, C | 120.9, C | 120.9, C |
| 1′ | 131.6, C | 133.2, C | 133.0, C | 134.3, C | 133.2, C | 131.9, C | 135.6, C | 136.1, C |
| 2′ | 111.3, CH | 111.1, CH | 112.0, CH | 112.1, CH | 111.5, CH | 110.6, CH | 111.3, CH | 110.9, CH |
| 3′ | 147.9, C | 148.8, C | 146.1, C | 149.1, C | 148.0, C | 149.7, C | 148.0, C | 148.4, C |
| 4′ | 149.3, C | 149.3, C | 145.0, C | 148.1, C | 146.2, C | 146.8, C | 150.2, C | 149.0, C |
| 5′ | 112.2, CH | 109.2, CH | 110.1, CH | 110.4, CH | 110.3, CH | 112.3, CH | 110.2, CH | 110.3, CH |
| 6′ | 120.7, CH | 120.6, CH | 120.8, CH | 120.6, CH | 120.3, CH | 122.3, CH | 120.0, CH | 120.0, CH |
| α | 40.6, CH2 | 47.0, CH2 | 43.4, CH2 | 44.6, CH2 | 42.4, CH2 | 42.4, CH2 | 72.5, CH | 72.4, CH |
| 6-Ome | 55.9, CH3 | 55.5, CH3 | - | 56.3, CH3 | - | 56.3, CH3 | 55.9, CH3 | 55.9, CH3 |
| 7-Ome | 56.10, CH3 | 55.6, CH3 | - | - | 55.3, CH3 | 56.1, CH3 | 55.7, CH3 | 55.7, CH3 |
| 3′-Ome | 56.12, CH3 | 55.7, CH3 | - | 56.2, CH3 | 55.5, CH3 | 56.0, CH3 | 55.6, CH3 | 55.6, CH3 |
| 4′-Ome | 56.4, CH3 | 55.8, CH3 | - | 55.8, CH3 | - | 55.3, CH3 | 55.6, CH3 | 55.6, CH3 |
| Papaverine | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| MW a | 339.39 | 341.40 | 283.28 | 325.36 | 311.33 | 355.39 | 355.39 | 355.39 |
| Rot. Bond b | 6 | 6 | 2 | 5 | 4 | 6 | 6 | 6 |
| HBA b | 5 | 5 | 5 | 5 | 5 | 6 | 6 | 5 |
| HBD c | 0 | 0 | 4 | 1 | 2 | 1 | 1 | 0 |
| TPSA d | 49.83 | 49.30 | 93.80 | 60.82 | 71.82 | 70.06 | 70.06 | 62.40 |
| Log Po/w | 3.86 | 3.30 | 2.64 | 3.55 | 3.25 | 3.56 | 3.35 | 3.09 |
| Papaverine | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| Absorption | ||||||||
| Water Solubility | −4.79 | −4.94 | −3.23 | −4.47 | −4.01 | −4.21 | −3.70 | −4.17 |
| GI | 100 | 100 | 79.85 | 98.62 | 95.14 | 95.89 | 96.54 | 98.24 |
| Log Kp | −2.72 | −2.72 | −2.76 | −2.64 | −2.77 | −2.73 | −2.73 | −2.66 |
| Distribution | ||||||||
| BBB | 0.37 | 0.04 | −1.10 | 0.18 | −0.47 | −0.47 | −0.49 | 0.13 |
| Log PS | −2.41 | −2.36 | −2.46 | −2.37 | −2.39 | −3.19 | −3.31 | −2.43 |
| VD | 0.03 | 0.13 | −0.05 | 0.13 | 0.06 | -0.13 | 0.03 | 0.64 |
| Metabolism | ||||||||
| CYP1A2 Inhibitor | √ | √ | √ | √ | √ | √ | √ | √ |
| CYP2C9 Inhibitor | √ | √ | X | √ | √ | √ | √ | X |
| CYP2C19 Inhibitor | √ | √ | X | √ | √ | √ | √ | X |
| CYP3A4 Inhibitor | X | X | X | X | X | X | X | X |
| CYP2D6 Inhibitor | X | X | X | X | X | √ | √ | X |
| Excretion | ||||||||
| Total Clearance | 0.37 | 0.51 | 0.14 | 0.33 | 0.25 | 0.71 | 0.44 | 1.00 |
| Renal OCT2 Sub. | X | X | X | X | X | X | X | X |
| Papaverine | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | Metabolite | |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| Ames Toxicity | X | X | X | X | X | X | X | X |
| hERG I Inhibitor | X | X | X | X | X | X | X | X |
| hERG II Inhibitor | √ | X | √ | √ | √ | √ | √ | √ |
| Oral Toxicity | 2202 | 2000 | 1930 | 2100 | 2064 | 2074 | 2029 | 2374 |
| Oral Toxicity Classification | V | IV | IV | V | V | V | V | V |
| Hepatotoxicity | √ | X | X | √ | √ | √ | X | X |
| Skin Sensitivity | X | X | X | X | X | X | X | X |
| Isolate | Binding Energy (BE) (kcal.mol−1) | |
|---|---|---|
| PTP1B (PDB: 1G7F) | α-Glucosidase (PDB: 3A4A) | |
| Papaverine | −6.18 | −6.97 |
| Metabolite 1 | −6.08 | −4.80 |
| Metabolite 2 | −5.74 | −5.78 |
| Metabolite 3 | −6.22 | −5.28 |
| Metabolite 4 | −6.01 | −6.26 |
| Metabolite 5 | −6.57 | −4.86 |
| Metabolite 6 | −6.72 | −5.12 |
| Metabolite 7 | −6.86 | −7.77 |
| Redocked Ligand | −7.78 | −5.77 |
| Acarbose | −7.90 | −6.57 |
| Isolate | Binding Energy (BE) (kcal.mol−1) Lipase (PDB: 1LPB) |
|---|---|
| Papaverine | −5.60 |
| Metabolite 1 | −5.83 |
| Metabolite 2 | −5.19 |
| Metabolite 3 | −5.81 |
| Metabolite 4 | −5.69 |
| Metabolite 5 | −5.82 |
| Metabolite 6 | −5.66 |
| Metabolite 7 | −6.05 |
| Redocked Ligand | −5.67 |
| Orlistat | −6.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eliwa, D.; Kabbash, A.; El-Aasr, M.; Tawfik, H.O.; Batiha, G.E.-S.; Mahmoud, M.H.; De Waard, M.; Eldehna, W.M.; Ibrahim, A.-R.S. Papaverinol-N-Oxide: A Microbial Biotransformation Product of Papaverine with Potential Antidiabetic and Antiobesity Activity Unveiled with In Silico Screening. Molecules 2023, 28, 1583. https://doi.org/10.3390/molecules28041583
Eliwa D, Kabbash A, El-Aasr M, Tawfik HO, Batiha GE-S, Mahmoud MH, De Waard M, Eldehna WM, Ibrahim A-RS. Papaverinol-N-Oxide: A Microbial Biotransformation Product of Papaverine with Potential Antidiabetic and Antiobesity Activity Unveiled with In Silico Screening. Molecules. 2023; 28(4):1583. https://doi.org/10.3390/molecules28041583
Chicago/Turabian StyleEliwa, Duaa, Amal Kabbash, Mona El-Aasr, Haytham O. Tawfik, Gaber El-Saber Batiha, Mohamed H. Mahmoud, Michel De Waard, Wagdy M. Eldehna, and Abdel-Rahim S. Ibrahim. 2023. "Papaverinol-N-Oxide: A Microbial Biotransformation Product of Papaverine with Potential Antidiabetic and Antiobesity Activity Unveiled with In Silico Screening" Molecules 28, no. 4: 1583. https://doi.org/10.3390/molecules28041583
APA StyleEliwa, D., Kabbash, A., El-Aasr, M., Tawfik, H. O., Batiha, G. E.-S., Mahmoud, M. H., De Waard, M., Eldehna, W. M., & Ibrahim, A.-R. S. (2023). Papaverinol-N-Oxide: A Microbial Biotransformation Product of Papaverine with Potential Antidiabetic and Antiobesity Activity Unveiled with In Silico Screening. Molecules, 28(4), 1583. https://doi.org/10.3390/molecules28041583