
Organometallic Chemistry within the Structured Environment Provided by the Macrocyclic Cores of Carbaporphyrins and Related Systems

Abstract
1. Introduction
2. Synthetic Routes to Carbaporphyrinoid Systems
3. Organometallic Chemistry of N-Confused Porphyrins
4. X-Confused Heteroporphyrins
5. Organometallic Chemistry of True Carbaporphyrins
6. Organometallic Chemistry of Azuliporphyrins
7. Organometallic Chemistry of Benziporphyrins, Naphthiporphyrins and Related Systems
8. Oxybenziporphyrins, Oxynaphthiporphyrins and Related Systems
9. Miscellaneous Monocarbaporphyrinoids
10. Dicarbaporphyrinoid Systems
11. Tri- and Tetracarbaporphyrins
12. Contracted Carbaporphyrinoids
13. Expanded Carbaporphyrinoids
14. Related Systems
15. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mironov, A. Handbook of Porphyrin Science—With Applications to Chemistry, Physics, Material Science, Engineering, Biology and Medicine; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing: Singapore, 2012; Volume 18, pp. 303–413. [Google Scholar]
- Thuita, D.W.; Brückner, C. Metals Complexes of Porphyrinoids Containing Nonpyrrolic Heterocycles. Chem. Rev. 2022, 122, 7990–8052. [Google Scholar] [CrossRef]
- Lash, T.D. Carbaporphyrinoid Systems. Chem. Rev. 2017, 117, 2313–2446. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D. Metal Complexes of Carbaporphyrinoid Systems. Chem. Asian J. 2014, 9, 682–705. [Google Scholar] [CrossRef] [PubMed]
- Furuta, H.; Maeda, H.; Osuka, A. Confusion, Inversion, and Creation—A New Spring from Porphyrin Chemistry. Chem. Commun. 2002, 38, 1795–1804. [Google Scholar] [CrossRef]
- Harvey, J.D.; Ziegler, C.J. Developments in the Metal Chemistry of N-Confused Porphyrin. Coord. Chem. Rev. 2003, 247, 1–19. [Google Scholar] [CrossRef]
- Srinivasan, A.; Furuta, H. Confusion Approach to Porphyrinoid Chemistry. Acc. Chem. Res. 2005, 38, 10–20. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grażyński, L. Core Modified Porphyrins—A Macrocyclic Platform for Organometallic Chemistry. Coord. Chem. Rev. 2005, 249, 2510–2533. [Google Scholar] [CrossRef]
- Maeda, H.; Furuta, H. A Dozen Years of N-Confusion: From Synthesis to Supramolecular Chemistry. Pure Appl. Chem. 2006, 78, 29–44. [Google Scholar] [CrossRef]
- Toganoh, M.; Furuta, H. Handbook of Porphyrin Science—With Applications to Chemistry, Physics, Material Science, Engineering, Biology and Medicine; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing: Singapore, 2010; Volume 2, pp. 103–192. [Google Scholar]
- Toganoh, M.; Furuta, H. Blooming of Confused Porphyrinoids—Fusion, Expansion, Contraction, and More Confusion. Chem. Commun. 2012, 48, 937–954. [Google Scholar] [CrossRef]
- Toganoh, M.; Furuta, H. Creation from Confusion to Fusion in the Porphyrin World—The Last Three Decades of N-Confused Porphyrinoid Chemistry. Chem. Rev. 2022, 122, 8313–8437. [Google Scholar] [CrossRef]
- Geier, G.R., III; Haynes, D.M.; Lindsey, J.S. An Efficient One- Flask Synthesis of N-Confused Tetraphenylporphyrin. Org. Lett. 1999, 1, 1455–1458. [Google Scholar] [CrossRef]
- Geier, G.R., III; Ciringh, Y.; Li, F.; Haynes, D.M.; Lindsey, J.S. A Survey of Acid Catalysts for Use in Two-Step, One-Flask Syntheses of meso-Substituted Porphyrinic Macrocycles. Org. Lett. 2000, 2, 1745–1748. [Google Scholar] [CrossRef]
- Lash, T.D.; Hayes, M.J. Carbaporphyrins. Angew. Chem., Int. Ed. Engl. 1997, 36, 840–842. [Google Scholar] [CrossRef]
- Lash, T.D. Out of the Blue! Azuliporphyrins and Related Carbaporphyrinoid Systems. Acc. Chem. Res. 2016, 49, 471–482. [Google Scholar] [CrossRef]
- Stępień, M.; Latos-Grażyński, L. Benziporphyrins: Exploring Arene Chemistry in Macrocyclic Environment. Acc. Chem. Res. 2005, 38, 88–98. [Google Scholar] [CrossRef]
- Lash, T.D. Benziporphyrins, a Unique Platform for Exploring the Aromatic Characteristics of Porphyrinoid Systems. Org. Biomol. Chem. 2015, 13, 7846–7878. [Google Scholar] [CrossRef]
- Lash, T.D. Oxybenziporphyrin, an Aromatic Semiquinone Porphyrin Analog with Pathways for 18π-Electron Delocalization. Angew. Chem., Int. Ed. Engl. 1995, 34, 2533–2535. [Google Scholar] [CrossRef]
- Bergman, K.M.; Ferrence, G.M.; Lash, T.D. Tropiporphyrins, Cycloheptatrienyl Analogues of the Porphyrins: Synthesis, Spectroscopy, Chemistry and Structural Characterization of a Silver(III) Derivative. J. Org. Chem. 2004, 69, 7888–7897. [Google Scholar] [CrossRef]
- Latos-Grażyński, L. Core-Modified Heteroanalogues of Porphyrins and Metalloporphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 2, pp. 361–416. [Google Scholar]
- Lash, T.D. Syntheses of Novel Porphyrinoid Chromophores. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M.; Guilard, R., Eds.; Academic Press, San Diego, CA, USA, 2000; pp. 125–199.
- Lash, T.D. Carbaporphyrinoids: Taking the Heterocycle Out of Nature’s [18]Annulene. Synlett 2000, 2000, 279–295. [Google Scholar]
- Lash, T.D. Recent Advances on the Synthesis and Chemistry of Carbaporphyrins and Related Porphyrinoid Systems. Eur. J. Org. Chem. 2007, 2007, 5461–5481. [Google Scholar] [CrossRef]
- Pawlicki, M.; Latos-Grażyński, L. Carbaporphyrinoids—Synthesis and Coordination Properties. In Handbook of Porphyrin Science—With Applications to Chemistry, Physics, Material Science, Engineering, Biology and Medicine; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing: Singapore, 2010; Volume 2, pp. 104–192. [Google Scholar]
- Lash, T.D. Carbaporphyrins and Related Systems. Synthesis, Characterization, Reactivity and Insights into the Nature of Porphyrinoid Aromaticity. In Handbook of Porphyrin Science—With Applications to Chemistry, Physics, Material Science, Engineering, Biology and Medicine; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing: Singapore, 2012; Volume 16, 329p. [Google Scholar]
- Szyszko, B.; Latos-Grażyński, L. Core Chemistry and Skeletal Rearrangements of Porphyrinoids and Metalloporphyrinoids. Chem. Soc. Rev. 2015, 44, 3588–3616. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Abebayehu, A.; Mulugeta, E.; Sareen, D.; Lee, C.-H. Recent Advances in meso-Alkylidenyl Carbaporphyrinoids. J. Porphyr. Phthalocyanines 2016, 20, 21–34. [Google Scholar] [CrossRef]
- Lash, T.D. Heteroporphyrins and Carbaporphyrins. In Fundamentals of Porphyrin Chemistry: A 21st Century Approach; Brothers, P.J., Senge, M.O., Eds.; Wiley: Hoboken, NJ, USA, 2022; Volume 1, pp. 385–451. [Google Scholar]
- Lash, T.D. Recent Developments in the Chemistry of Heteroporphyrins and Heterocarbaporphyrins. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Elsevier: Oxford, UK, 2022; Volume 138, pp. 243–334. [Google Scholar]
- Furuta, H.; Asano, T.; Ogawa, T. N-Confused Porphyrin: A New Isomer of Tetraphenylporphyrin. J. Am. Chem. Soc. 1994, 116, 767–768. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grażyński, L.; Rachlewicz, K.; Glowiak, T. Tetra-p-tolylporphyrin with an Inverted Pyrrole Ring: A Novel Isomer of Porphyrin. Angew. Chem. Int. Ed. Engl. 1994, 33, 779–781. [Google Scholar] [CrossRef]
- Aronoff, S.; Calvin, M. The Porphyrin-like Products of the Reaction of Pyrrole with Benzaldehyde. J. Org. Chem. 1943, 8, 205–223. [Google Scholar] [CrossRef]
- Ball, R.H.; Dorough, G.D.; Calvin, M. A Further Study of the Porphine-like Products of the Reaction of Benzaldehyde and Pyrrole. J. Am. Chem. Soc. 1946, 68, 2278–2281. [Google Scholar] [CrossRef]
- Senge, M.O. Extroverted Confusion—Linus Pauling, Melvin Calvin, and Porphyrin Isomers. Angew. Chem. Int. Ed. 2011, 50, 4272–4277. [Google Scholar] [CrossRef]
- Lash, T.D. Porphyrin Synthesis by the “3 + 1” Approach: New Applications for an Old Methodology. Chem. Eur. J. 1996, 2, 1197–1200. [Google Scholar] [CrossRef]
- Lash, T.D. What’s in a Name? The MacDonald Condensation. J. Porphyr. Phthalocyanines 2016, 20, 855–888. [Google Scholar] [CrossRef]
- Broadhurst, M.J.; Grigg, R.; Johnson, A.W. Synthesis of Porphin Analogues Containing Furan and/or Thiophen Rings. J. Chem. Soc. C 1971, 1971, 3681–3690. [Google Scholar]
- Sessler, J.L.; Johnson, M.R.; Lynch, V. Synthesis and Crystal Structure of a Novel Tripyrrane-Containing Porphyrinogen-like Macrocycle. J. Org. Chem. 1987, 52, 4394–4397. [Google Scholar] [CrossRef]
- Latham, A.N.; Lash, T.D. Synthesis and characterization of N-methylporphyrins, heteroporphyrins, carbaporphyrins and related systems. J. Org. Chem. 2020, 85, 13050–13068. [Google Scholar] [CrossRef]
- Boudif, A.; Momenteau, M. Synthesis of a Porphyrin-2, 3-diacrylic acid using a New ‘3 + 1’ Type Procedure. J. Chem. Soc. Chem. Commun. 1994, 30, 2069–2070. [Google Scholar] [CrossRef]
- Lin, Y.; Lash, T.D. Porphyrin Synthesis by the “3 + 1” Methodology: A Superior Approach for the Preparation of Porphyrins with Fused 9, 10-Phenanthroline Subunits. Tetrahedron Lett. 1995, 36, 9441–9444. [Google Scholar] [CrossRef]
- Chandrasekar, P.; Lash, T.D. Versatile “3 + 1” Syntheses of Acenaphthoporphyrins, a New Family of Highly Conjugated Tetrapyrroles. Tetrahedron Lett. 1996, 37, 4873–4876. [Google Scholar] [CrossRef]
- Boudif, A.; Momenteau, M. A New Convergent Method for Porphyrin Synthesis Based on a ‘3 + 1’ Condensation. J. Chem. Soc. Perkin Trans. 1 1996, 1996, 1235–1242. [Google Scholar] [CrossRef]
- Sessler, J.L.; Genge, J.W.; Urbach, A.; Sansom, P.A. ‘3 + 1’ Approach to Monofunctionalized Alkyl Porphyrins. Synlett 1996, 1996, 187–188. [Google Scholar] [CrossRef]
- Lash, T.D. Porphyrins with exocyclic rings. Part 9. Porphyrin synthesis by the “3 + 1” approach. J. Porphyr. Phthalocyanines 1997, 1, 29–44. [Google Scholar] [CrossRef]
- Lash, T.D.; Chandrasekar, P.; Osuma, A.T.; Chaney, S.T.; Spence, J.D. Porphyrins with Exocyclic Rings. Part 13. Synthesis and Characterization of Highly Modified Porphyrin Chromophores with Fused Acenaphthylene and Benzothiadiazole Rings. J. Org. Chem. 1998, 63, 8455–8469. [Google Scholar] [CrossRef]
- Lash, T.D.; Rauen, P.J. Extended Porphyrinoid Chromophores: Heteroporphyrins Fused to Phenanthrene and Acenaphthylene. Tetrahedron 2021, 100, 132481. [Google Scholar] [CrossRef]
- Berlin, K.; Breitmaier, E. Benziporphyrin, a Benzene-Containing, Nonaromatic Porphyrin Analogue. Angew. Chem. Int. Ed. Engl. 1994, 33, 1246–1247. [Google Scholar] [CrossRef]
- Lash, T.D.; Chaney, S.T.; Richter, D.T. Conjugated Macrocycles Related to the Porphyrins. Part 12. Oxybenzi-and Oxypyriporphyrins: Aromaticity and Conjugation in Highly Modified Porphyrinoid Structures. J. Org. Chem. 1998, 63, 9076–9088. [Google Scholar] [CrossRef]
- Lash, T.D.; Chaney, S.T. Conjugated Macrocycles Related to the Porphyrins. Part 7. Tropiporphyrin: Tropylium versus Porphyrinoid Aromaticity. Tetrahedron Lett. 1996, 37, 8825–8828. [Google Scholar] [CrossRef]
- Berlin, K.; Steinbeck, E.; Breitmaier, E. Synthesis of Carbaporphyrinoids from Tripyrranes and Unsaturated Dialdehydes. Synthesis 1996, 1996, 336–340. [Google Scholar] [CrossRef]
- Berlin, K. Carbaporphyrins. Angew. Chem. Int. Ed. Engl. 1996, 35, 1820–1822. [Google Scholar] [CrossRef]
- Lash, T.D.; Chaney, S.T. Azuliporphyrin, a Case of Borderline Porphyrinoid Aromaticity. Angew. Chem. Int. Ed. Engl. 1997, 36, 839–840. [Google Scholar] [CrossRef]
- Hayes, M.J.; Lash, T.D. Conjugated Macrocycles Related to the Porphyrins. Part 10. Carbachlorins. Chem. Eur. J. 1998, 4, 508–511. [Google Scholar] [CrossRef]
- Lash, T.D.; Richter, D.T.; Shiner, C.M. Conjugated Macrocycles Related to the Porphyrins. Part 16. Synthesis of Hexa- and Heptaalkyl Substituted Inverted or N-Confused Porphyrins by the “3 + 1” Methodology. J. Org. Chem. 1999, 64, 7973–7982. [Google Scholar] [CrossRef]
- Richter, D.T.; Lash, T.D. Conjugated Macrocycles Related to the Porphyrins. Part 18. Synthesis and Spectroscopic Characterization of Electron-rich Benzi- and Oxybenziporphyrins: Influence of Steric and Electronic Factors on Porphyrinoid Aromaticity. Tetrahedron 2001, 57, 3659–3673. [Google Scholar] [CrossRef]
- Lash, T.D.; Hayes, M.J.; Spence, J.D.; Muckey, M.A.; Ferrence, G.M.; Szczepura, L.F. Conjugated Macrocycles Related to the Porphyrins. Part 21. Synthesis, Spectroscopy, Electrochemistry and Structural Characterization of Carbaporphyrins. J. Org. Chem. 2002, 67, 4860–4874. [Google Scholar] [CrossRef]
- Jiao, W.; Lash, T.D. tert-Butyl Substituted Tripyrranes: Insights into the Steric and Conformational Factors that Influence Porphyrinoid Ring Formation in the “3 + 1” Methodology. J. Org. Chem. 2003, 68, 3896–3901. [Google Scholar] [CrossRef]
- Liu, D.; Lash, T.D. Conjugated Macrocycles Related to the Porphyrins. Part 25. 1H NMR Spectroscopic Evidence for a Preferred [18]Annulene Substructure in Carbaporphyrins from the Magnitude of Selected 4JHH CH=C-CH3 Coupling Constants. J. Org. Chem. 2003, 68, 1755–1761. [Google Scholar] [CrossRef]
- Lash, T.D.; Colby, D.A.; Graham, S.R.; Chaney, S.T. Synthesis, Spectroscopy and Reactivity of meso-Unsubstituted Azuliporphyrins and their Heteroanalogues. Oxidative Ring Contractions to Carba-, Oxacarba-, Thiacarba- and Selenacarbaporphyrins. J. Org. Chem. 2004, 69, 8851–8864. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Von Ruden, A.L. Synthesis and Reactivity of N-methyl and N-phenyl meso-Unsubstituted N-Confused Porphyrins. J. Org. Chem. 2008, 73, 9417–9425. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Young, A.M.; Von Ruden, A.L.; Ferrence, G.M. Adding to the Confusion! Synthesis and Metalation of Pyrazole Analogues of the Porphyrins. Chem. Commun. 2008, 44, 6309–6311. [Google Scholar] [CrossRef] [PubMed]
- Young, A.M.; Von Ruden, A.L.; Lash, T.D. Pyrazole Analogues of the Porphyrins and Oxophlorins. Org. Biomol. Chem. 2011, 9, 6293–6305. [Google Scholar] [CrossRef]
- Lash, T.D.; Young, A.M.; Rasmussen, J.M.; Ferrence, G.M. Naphthiporphyrins. J. Org. Chem. 2011, 76, 5636–5651. [Google Scholar] [CrossRef]
- Lash, T.D.; Bergman, K.M. Further Observations on Conformational and Substituent Effects in Acid Catalyzed “3 + 1” Cyclizations of Tripyrranes with Aromatic Dialdehydes. J. Org. Chem. 2012, 77, 9774–9783. [Google Scholar] [CrossRef]
- Li, D.; Lash, T.D. Synthesis and Reactivity of Carbachlorins and Carbaporphyrins. J. Org. Chem. 2014, 79, 7112–7121. [Google Scholar] [CrossRef]
- Gao, R.; AbuSalim, D.I.; Lash, T.D. Pyreniporphyrins, Porphyrin Analogues that Incorporate a Polycyclic Aromatic Hydrocarbon Subunit within the Macrocyclic Framework. J. Org. Chem. 2017, 82, 6680–6688. [Google Scholar] [CrossRef]
- Sahota, N.; Ferrence, G.M.; Lash, T.D. Synthesis and Properties of Carbaporphyrin and Carbachlorin Dimethyl Esters Derived from Cyclopentanedialdehydes. J. Org. Chem. 2017, 82, 9715–9730. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lash, T.D. Synthesis and Oxidation of Internally Chlorinated Carbachlorins. Eur. J. Org. Chem. 2017, 2017, 6775–6780. [Google Scholar] [CrossRef]
- Grabowski, E.Y.; AbuSalim, D.I.; Lash, T.D. Naphtho[2,3-b]carbaporphyrins. J. Org. Chem. 2018, 83, 11825–11838. [Google Scholar] [CrossRef] [PubMed]
- Cillo, C.M.; Geiger, M.A.; Lash, T.D. Exploring the Limitations of the MacDonald ‘3 + 1’ Condensation in the Preparation of Porphyrins with Fused Electron-Withdrawing Heterocyclic Rings: Synthesis of a Bis(thiadiazolo)benzoporphyrin and a Related Benzocarbaporphyrin. Tetrahedron Lett. 2020, 61, 152576. [Google Scholar] [CrossRef]
- Lash, T.D.; Chaney, S.T.; Johnson, A.L.; Weisbond, J.T.; Ferrence, G.M. Synthesis, Characterization and Metalation of 22-Hydroxybenziporphyrins. J. Porphyr. Phthalocyanines 2021, 25, 1095–1103. [Google Scholar] [CrossRef]
- Cramer, E.K.; Lash, T.D. Synthesis of a Series of Tropone-fused Porphyrinoids. J. Org. Chem. 2022, 87, 952–962. [Google Scholar] [CrossRef]
- Cramer, E.K.; AbuSalim, D.I.; Lash, T.D. Oxyquinoliziniporphyrins: Introduction of a Heterocyclic Dimension to Carbaporphyrinoid Systems. Org. Lett. 2022, 24, 5402–5406. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Lash, T.D. Synthesis, Spectroscopy and Metallation of Mixed Carbaporphyrinoid Systems. Chem. Commun. 2002, 38, 2426–2427. [Google Scholar] [CrossRef]
- Liu, D.; Ferrence, G.M.; Lash, T.D. Oxybenziporphyrins, Oxypyriporphyrins, Benzocarbaporphyrins, and their 23-Oxa- and 23-Thia Analogues: Synthesis, Spectroscopic Characterization, Metalation, and Structural Characterization of a Palladium(II) Organometallic Derivative. J. Org. Chem. 2004, 69, 6079–6093. [Google Scholar] [CrossRef]
- Graham, S.R.; Colby, D.A.; Lash, T.D. Azulene Analogues of Tripyrranes and Carbaporphyrinoids Therefrom. Angew. Chem. Int. Ed. 2002, 41, 1371–1374. [Google Scholar] [CrossRef]
- Lash, T.D.; El-Beck, J.A.; Ferrence, G.M. Syntheses and Reactivity of meso-Unsubstituted Azuliporphyrins Derived from 6-tert-Butyl- and 6-Phenylazulene. J. Org. Chem. 2007, 72, 8402–8415. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; El-Beck, J.A.; Ferrence, G.M. Synthesis, Structural Characterization and Reactivity of Heteroazuliporphyrins. Org. Biomol. Chem. 2014, 12, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Lash, T.D. Preparation of Tripyrrane Analogues from Resorcinol and 2-Methylresorcinol for Applications in the Synthesis of New Benziporphyrin Systems. Chem. Commun. 2004, 40, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Miyake, K.; Xu, L.; Ferrence, G.M. Synthesis of a Series of Aromatic Benziporphyrins and Heteroanalogues via Tripyrrane-like Intermediates Derived from Resorcinol and 2-Methylresorcinol. J. Org. Chem. 2011, 76, 6295–6308. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Toney, A.M.; Castans, K.M.; Ferrence, G.M. Synthesis of Benziporphyrins and Heterobenziporphyrins and an Assessment of the Diatropic Characteristics of the Protonated Species. J. Org. Chem. 2013, 78, 9143–9152. [Google Scholar] [CrossRef] [PubMed]
- Fosu, S.C.; Ferrence, G.M.; Lash, T.D. Synthesis and metalation of dimethoxybenziporphyrins, thiabenziporphyrins and dibenziporphyrins. J. Org. Chem. 2014, 79, 11061–11074. [Google Scholar] [CrossRef]
- Lash, T.D.; Pokharel, K.; Serling, J.M.; Yant, V.R.; Ferrence, G.M. Aromatic and Nonaromatic Pyriporphyrins. Org. Lett. 2007, 9, 2863–2866. [Google Scholar] [CrossRef]
- Liu, B.Y.; Bruckner, C.; Dolphin, D. A meso-Unsubstituted N-Confused Porphyrin Prepared by Rational Synthesis. Chem. Commun. 1996, 32, 2141–2142. [Google Scholar] [CrossRef]
- Lash, T.D.; Lammer, A.D.; Idate, A.S.; Colby, D.A.; White, K. Preparation of Azulene-derived Fulvenedialdehydes and their Application to the Synthesis of Stable adj-Dicarbaporphyrinoids. J. Org. Chem. 2012, 77, 2368–2381. [Google Scholar] [CrossRef]
- Lash, T.D.; Lammer, A.D.; Ferrence, G.M. Neo-Confused Porphyrins, a New Class of Porphyrin Isomers. Angew. Chem. Int. Ed. 2011, 50, 9718–9721. [Google Scholar] [CrossRef]
- Li, R.; Ferrence, G.M.; Lash, T.D. Synthesis of a Neo-Confused Porphyrin and an Unusual Dihydroporphyrin Derivative. Chem. Commun. 2013, 49, 7537–7539. [Google Scholar] [CrossRef]
- Li, R.; Lammer, A.D.; Ferrence, G.M.; Lash, T.D. Synthesis, Structural Characterization, Aromatic Properties and Metalation of Neo-Confused Porphyrins, a Newly Discovered Class of Porphyrin Isomers. J. Org. Chem. 2014, 79, 4078–4093. [Google Scholar]
- Stępien, M.; Latos-Grażyński, L. Tetraphenylbenziporphyrin—A Ligand for Organometallic Chemistry. Chem. Eur. J. 2001, 7, 5113–5117. [Google Scholar] [CrossRef]
- Lash, T.D.; Yant, V.R. Improved Syntheses of meso-Tetraarylbenziporphyrins and Observations of substituent Effects on the Diatropic Characteristics of these Formally Nonaromatic Carbaporphyrinoids. Tetrahedron 2009, 65, 9527–9535. [Google Scholar]
- Colby, D.A.; Lash, T.D. Adaptation of the Rothemund Reaction for Carbaporphyrin Synthesis: Preparation of meso-Tetraphenylazuliporphyrin and Related Benzocarbaporphyrins. Chem. Eur. J. 2002, 8, 5397–5402. [Google Scholar] [CrossRef]
- Lash, T.D.; Colby, D.A.; Ferrence, G.M. Further Studies on the Synthesis of meso-Tetraarylazuliporphyrins under Lindsey-Rothemund Reaction Conditions and Their Conversion into Benzocarbaporphyrins. Eur. J. Org. Chem. 2003, 2003, 4533–4548. [Google Scholar] [CrossRef]
- El-Beck, J.A.; Lash, T.D. Synthesis and Reactivity of 23-tert-Butyl- and 23-Phenyltetraarylazuliporphyrins: An Analysis of the Effect of Bulky Substituents on Oxidative Ring Contractions to Benzocarbaporphyrins. Eur. J. Org. Chem. 2007, 2007, 3981–3990. [Google Scholar] [CrossRef]
- Stępień, M.; Latos-Grażyński, L. Tetraphenyl-p-benziporphyrin: A Carbaporphyrinoid with Two Linked Carbon Atoms in the Coordination Core. J. Am. Chem. Soc. 2002, 124, 3838–3839. [Google Scholar] [PubMed]
- Colby, D.A.; Lash, T.D. Calix[4]azulene. J. Org. Chem. 2002, 67, 1031–1033. [Google Scholar] [PubMed]
- Lash, T.D.; El-Beck, J.A.; Colby, D.A. Synthesis of a Tetraazulene Porphodimethene Analogue. J. Org. Chem. 2009, 74, 8830–8833. [Google Scholar] [CrossRef] [PubMed]
- Mysliborski, R.; Latos-Grażyński, L. Carbaporphyrinoids Containing a Pyridine Moiety: 3-Aza-meta-benziporphyrin and 24- Thia-3-aza-meta-benziporphyrin. Eur. J. Org. Chem. 2005, 2005, 5039–5048. [Google Scholar] [CrossRef]
- Garbicz, M.; Latos-Grażyński, L. A meso-Tetraaryl-21-carbaporphyrin: Incorporation of a Cyclopentadiene Unit into a Porphyrin Architecture. Angew. Chem. Int. Ed. 2019, 58, 6089–6093. [Google Scholar] [CrossRef]
- Szyszko, B.; Latos-Grażyński, L. Expanded Carbaporphyrinoids. Angew. Chem. Int. Ed. 2020, 59, 16874–16901. [Google Scholar] [CrossRef] [PubMed]
- Stateman, L.M.; Lash, T.D. Syntheses of Carbaporphyrinoid Systems using a Carbatripyrrin Methodology. Org. Lett. 2015, 17, 4522–4525. [Google Scholar] [CrossRef] [PubMed]
- Smolczyk, T.J.; Lash, T.D. Alphabet Soup within a Porphyrinoid Cavity: Synthesis of Heterocarbaporphyrins with CNNO, CNOO, CNSO and CNSeO Cores from an Oxacarbatripyrrin. Chem. Commun. 2018, 54, 9003–9006. [Google Scholar] [CrossRef]
- Lash, T.D.; Mathius, M.A.; AbuSalim, D.I. Synthesis of Chrysoporphyrins and a Related Benzopyrene-fused System. J. Org. Chem. 2022, 87, 16276–16296. [Google Scholar] [CrossRef]
- Lash, T.D.; Fosu, S.C.; Smolczyk, T.J.; AbuSalim, D.I. Synthesis of Expanded Porphyrinoids with Azulene and Indene Subunuits and an opp-Dioxadicarbaporphyrin from Fulvene Carbinols and a Dioxacarbatripyrrin. J. Org. Chem. 2018, 83, 12619–12631. [Google Scholar] [CrossRef]
- Lash, T.D.; AbuSalim, D.I.; Ferrence, G.M. Synthesis of Telluracarbaporphyrins and a Related Palladium(II) Complex: Evidence for Hypervalent Interactions. Inorg. Chem. 2021, 60, 9833–9847. [Google Scholar] [CrossRef]
- Seo, W.S.; Cho, Y.J.; Yoon, S.C.; Park, J.T.; Park, Y. Synthesis and Structure of ansa-Cyclopentadienyl Pyrrolyl Titanium Complexes: [(η5-C5H4)CH3(2-C4H3N)]Ti(NMe2)2 and [1,3-{CH2(2- C4H3N)}2(η5-C5H3)]Ti(NMe2). J. Organomet. Chem. 2001, 640, 79–84. [Google Scholar] [CrossRef]
- Berlicka, A.; Dutka, P.; Szterenberg, L.; Latos-Grażyński, L. Towards True Carbaporphyrinoids: Synthesis of 21-Carba-23-thiaporphyrin. Angew. Chem. Int. Ed. 2014, 53, 4885–4889. [Google Scholar] [CrossRef] [PubMed]
- Berlicka, A.; Foryś-Martowłos, P.; Hassa, K.; Białek, M.J.; Ślepokura, K.; Latos-Grażyński, L. 21-Carba-23-oxaporphyrinoids and 21-Oxo-21-carba-23-oxaporphyrinoids: Macrocyclic π-Conjugation involving the Carbonyl Moiety. Org. Chem. Front. 2022, 9, 5440–5452. [Google Scholar] [CrossRef]
- Lash, T.D. The Azuliporphyrin-Carbaporphyrin Connection. Chem. Commun. 1998, 34, 1683–1684. [Google Scholar] [CrossRef]
- Noboa, M.A.; AbuSalim, D.I.; Lash, T.D. Azulichlorins and Benzocarbachlorins Derived Therefrom. J. Org. Chem. 2019, 84, 11649–11664. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Stateman, L.M.; AbuSalim, D.I. Synthesis of Azulitriphyrins (1.2.1) and Related Benzocarbatriphyrins. J. Org. Chem. 2019, 84, 14733–14744. [Google Scholar] [CrossRef]
- Furuta, H.; Ishizuka, T.; Osuka, A.; Dejima, H.; Nakagawa, H.; Ishikawa, Y. NH Tautomerism of N-Confused Porphyrin. J. Am. Chem. Soc. 2001, 123, 6207–6208. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, P.J.; Latos-Grażyński, L. N-Methyltetraphenylporphyrin with an Inverted N-Methylpyrrole Ring: The First Isomer of N-Methyltetraphenylporphyrin. J. Chem. Soc. Perkin Trans. 2 1995, 1995, 503–509. [Google Scholar] [CrossRef]
- Furuta, H.; Kubo, N.; Maeda, H.; Ishizuka, T.; Osuka, A.; Nanami, H.; Ogawa, T. N-Confused Double-Decker Porphyrins. Inorg. Chem. 2000, 39, 5424–5425. [Google Scholar] [CrossRef]
- Furuta, H.; Youfu, K.; Maeda, H.; Osuka, A. Facile Formation of N-Confused Porphyrin Dimers by Platinum(II) Coordination to the Outer-Nitrogen Atoms. Angew. Chem. Int. Ed. 2003, 42, 2186–2188. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Schmidt, I. Diatereoselective Assembling of 21-C-Alkylated Nickel(II) Complexes of Inverted Porphyrin on a Platinum(II) Template. Inorg. Chem. 2004, 43, 1885–1894. [Google Scholar] [CrossRef]
- Won, D.-H.; Toganoh, M.; Uno, H.; Furuta, H. Pt(II) N-Confused Porphyrin: An Expanded Pyrrole That Affords a Stable π-Anion. Dalton Trans. 2009, 39, 6151–6158. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grażyński, L.; Schmidt, I. Copper(II) Complexes of Inverted Porphyrin and Its Methylated Derivatives. Inorg. Chem. 2000, 39, 5475–5482. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Ishikawa, Y.; Matsuda, T.; Osuka, A.; Furuta, H. Control of Cu(II) and Cu(III) States in N-Confused Porphyrin by Protonation/Deprotonation at the Peripheral Nitrogen. J. Am. Chem. Soc. 2003, 125, 11822–11823. [Google Scholar] [CrossRef] [PubMed]
- Bohle, D.S.; Chen, W.-C.; Hung, C.-H. Metal Oxidation Promoted C-H Activation in Manganese Complexes of N-Confused Porphyrin. Inorg. Chem. 2002, 41, 3334–3336. [Google Scholar] [CrossRef]
- Harvey, J.D.; Ziegler, C.J. Dianionic and Trianionic Macrocycles in Cobalt N-Confused Porphyrin Complexes. Chem. Commun. 2004, 40, 1666–1667. [Google Scholar] [CrossRef] [PubMed]
- Niino, T.; Toganoh, M.; Andrioletti, B.; Furuta, H. Rhodium N-Confused Porphyrin-Catalyzed Alkene Cyclopropanation. Chem. Commun. 2006, 42, 4335–4337. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, A.; Toganoh, M.; Niino, T.; Osuka, A.; Furuta, H. Synthesis of N-Confused Tetraphenylporphyrin Rhodium Complexes Having Versatile Metal Oxidation States. Inorg. Chem. 2008, 47, 11305–11313. [Google Scholar] [CrossRef]
- Harvey, J.D.; Shaw, J.L.; Herrick, R.S.; Ziegler, C.J. The Synthesis of Isostructural Mo2+ Porphyrin and N-Confused Porphyrin Complexes. Chem. Commun. 2005, 41, 4663–4665. [Google Scholar] [CrossRef]
- He, H.; Ye, Z.; Shimizu, D.; Sumra, I.; Zhang, Y.; Liang, Z.; Zeng, Y.; Xu, L.; Osuka, A.; Ke, Z.; et al. Formation of Stable NiIII N-Confused Porphyrins Aided by a 3-Ethoxy Group. Chem. Eur. J. 2022, 28, e202103272. [Google Scholar] [CrossRef]
- Furuta, H.; Ogawa, T.; Uwatoko, Y.; Araki, K. N-Confused Tetraphenylporphyrin-Silver(III) Complex. Inorg. Chem. 1999, 38, 2676–2682. [Google Scholar] [CrossRef]
- Brückner, C. The Silver Complexes of Porphyrins, Corroles, and Carbaporphyrins: Silver in the Oxidation States II and III. J. Chem. Ed. 2004, 81, 1665–1669. [Google Scholar] [CrossRef]
- Toganoh, M.; Niino, T.; Furuta, H. Luminescent Au(III) Organometallic Complex of N-Confused Tetraphenylporphyrin. Chem. Commun. 2008, 44, 4070–4072. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzek, N.; Pawlicki, M.; Latos-Grażyński, L. Regioselective Amination of Carbaporpholactone and N-Confused Porphyrin. J. Org. Chem. 2009, 74, 8547–8553. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzek, N.; Latos-Grażyński, L.; Szterenberg, L. Regioselective phosphorylation and Thiophosphorylation of N-Confused Porphyrin: A Route to Hybrid Carbaporphyrinoids. Org. Biomol. Chem. 2012, 10, 8064–8075. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Zhang, T.; Yu, D.; Yang, X.; Jiang, H.-W.; Xiao, J.-C.; Chen, Q.-Y. Porphyriynes: 18-π-Conjugated Macrocycles Incorporating a Triple Bond. Org. Lett. 2022, 24, 1716–1721. [Google Scholar] [CrossRef]
- Liu, J.-C.; Ishizuka, T.; Osuka, A.; Furuta, H. Modulation of Axial Coordination in N-Confused Porphyrin-Antimony(V) Dibromide Complex by Proton Stimulus. Chem. Commun. 2003, 39, 1908–1909. [Google Scholar] [CrossRef]
- Furuta, H.; Ishizuka, T.; Osuka, A. Flexible Inner and Outer Coordination of Zn(II) N-Confused Porphyrin Complex. J. Am. Chem. Soc. 2002, 124, 5622–5623. [Google Scholar] [CrossRef]
- Chen, W.-C.; Hung, C.-H. Synthesis and Characterization of Iron N-Confused Porphyrins: Structural Evidences of Agostic Interaction. Inorg. Chem. 2001, 40, 5070–5071. [Google Scholar] [CrossRef]
- Rachlewicz, K.; Wang, S.-L.; Ko, J.-L.; Hung, C.-H.; Latos-Grażyński, L. Oxidation and Oxygenation of Iron Complexes of 2-Aza-21-carbaporphyrin. J. Am. Chem. Soc. 2004, 126, 4420–4431. [Google Scholar] [CrossRef]
- Hung, C.-H.; Wang, S.-L.; Ko, J.-L.; Peng, C.-H.; Hu, C.-H.; Lee, M.-T. Demetalation of the Regioselective Oxygenation Product of an N-Confused Porphyrin Complex. Org. Lett. 2004, 6, 1393–1396. [Google Scholar] [CrossRef]
- Pawlicki, M.; Latos-Grażyński, L. O-Confusion Approach in Construction of Carbaporphyrinoids. Chem. Rec. 2006, 6, 64–78. [Google Scholar] [CrossRef]
- Pawlicki, M.; Latos-Grażyński, L. Pyrrole-Appended Derivatives of O-Confused Oxaporphyrins and Their Complexes with Nickel(II), Palladium(II), and Silver(III). Chem. Eur. J. 2003, 9, 4650–4660. [Google Scholar] [CrossRef]
- Pawlicki, M.; Kanska, I.; Latos-Grażyński, L. Copper(II) and Copper(III) Complexes of Pyrrole-Appended Oxacarbaporphyrin. Inorg. Chem. 2007, 46, 6575–6584. [Google Scholar] [CrossRef]
- Pawlicki, M.; Latos-Grażyński, L. O-Confused Oxaporphyrin-An Intermediate in Formation of 3-Substituted 2-Oxa-21-Carbaporphyrins. J. Org. Chem. 2005, 70, 9123–9130. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzek, N.; Pawlicki, M.; Szterenberg, L.; Latos-Grażyński, L. Organocopper(II) Complex of 21-Diphenylphosphoryl-Carbaporpholactone Hybrid: A Side-On Coordination Mode of Copper(II). J. Am. Chem. Soc. 2009, 131, 7224–7225. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.J.; Pawlicki, M.; Sprutta, N.; Szterenberg, L.; Latos-Grażyński, L. Cadmium(II) and Zinc(II) Complexes of S-Confused Thiaporphyrin. Inorg. Chem. 2006, 45, 8664–8671. [Google Scholar] [CrossRef] [PubMed]
- Muckey, M.A.; Szczepura, L.F.; Ferrence, G.M.; Lash, T.D. Silver(III) Carbaporphyrins: The First Organometallic Complexes of True Carbaporphyrins. Inorg. Chem. 2002, 41, 4840–4842. [Google Scholar] [CrossRef]
- Clezy, P.S.; Fookes, C.J.R.; Mirza, A.H. The chemistry of pyrrolic compounds. XXXVII. Monobenzoporphyrins: The rhodoporphyrin of petroleum deposits. Aust. J. Chem. 1977, 30, 1337–1347. [Google Scholar] [CrossRef]
- Lash, T.D. Geochemical Origins of Sedimentary Benzoporphyrins and Tetrahydrobenzoporphyrins. Energy Fuels 1993, 7, 166–171. [Google Scholar] [CrossRef]
- Hayes, M.J.; Spence, J.D.; Lash, T.D. Facile Oxidation of a Carbaporphyrin at the Internal Carbon Atom: Synthesis of Novel Benzo[18]annulene Ketals. Chem. Commun. 1998, 34, 2409–2410. [Google Scholar] [CrossRef]
- Lash, T.D.; Muckey, M.A.; Hayes, M.J.; Liu, D.; Spence, J.D.; Ferrence, G.M. Regioselective Oxidation of Benzocarbaporphyrins with Ferric Chloride: A Facile Synthesis of Bridged [18]Annulene Ketals with Strong Absorptions in the Far Red and an Unexpected Halogenation Reaction at the Interior Carbon Atom. J. Org. Chem. 2003, 68, 8558–8570. [Google Scholar] [CrossRef]
- Morgenthaler, J.B.; Peters, S.J.; Cedeño, D.L.; Constantino, M.H.; Edwards, K.A.; Kamowski, E.M.; Passini, J.C.; Butkus, B.E.; Young, A.M.; Lash, T.D.; et al. Carbaporphyrin Ketals as Potential Agents for a New Photodynamic Therapy Treatment of Leishmaniasis. Bioorg. Med. Chem 2008, 16, 7033–7038. [Google Scholar] [CrossRef]
- Taylor, V.M.; Cedeño, D.L.; Muñoz, D.L.; Jones, M.A.; Lash, T.D.; Young, A.M.; Constantino, M.H.; Esposito, N.; Vélez, I.D.; Robledo, S.M. In Vivo and In Vitro Studies of the Utility of Dimethyl and Diethyl Carbaporphyrin Ketals in the Treatment of Cutaneous Leishmaniasis. Antimicr. Agents Chemother. 2011, 55, 4755–4764. [Google Scholar] [CrossRef]
- Lash, T.D.; Colby, D.A.; Szczepura, T.D. New Riches in Carbaporphyrin Chemistry: Silver and Gold Organometallic Complexes of Benzocarbaporphyrins. Inorg. Chem. 2004, 43, 5258–5267. [Google Scholar] [CrossRef]
- Adiraju, V.A.K.; Ferrence, G.M.; Lash, T.D. Rhodium(I), Rhodium(III) and Iridium(III) Carbaporphyrins. Dalton Trans. 2016, 45, 13691–13694. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Rasmussen, J.M.; Bergman, K.M.; Colby, D.A. Carbaporphyrinoid Chemistry has a Silver Lining! Silver(III) Oxybenzi-, Oxynaphthi-, Tropi- and Benzocarbaporphyrins. Org. Lett. 2004, 6, 549–552. [Google Scholar] [CrossRef]
- Lash, T.D.; Darrow, W.T.; Latham, A.N.; Sahota, N.; Ferrence, G.M. Rhodium complexes of carbaporphyrins, carbachlorins, adj-dicarbaporphyrins and an adj-dicarbachlorin. Inorg. Chem. 2019, 58, 7511–7526. [Google Scholar] [CrossRef] [PubMed]
- Alemayehu, A.B.; Vasquez-Lima, H.; Teat, S.J.; Ghosh, A. Unexpected Molecular Structure of a Putative Rhenium-Dioxo-Benzocarbaporphyrin Complex. Implications for the Highest Transition Metal Valence in a Porphyrin-Type Ligand Environment. ChemistryOpen 2019, 8, 1298–1302. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D. Unexpected Alkyl Group Migration in Palladium(II) Benzocarbaporphyrins. Org. Lett. 2011, 13, 4632–4635. [Google Scholar] [CrossRef]
- Latham, A.N.; Ferrence, G.M.; Lash, T.D. Metalation and Methyl Group Migration in 21-, 22- and 23-Methylcarbaporphyrins: Synthesis and Characterization of Palladium(II), Rhodium(I) and Rhodium(III) derivatives. Organometallics 2019, 38, 575–585. [Google Scholar] [CrossRef]
- Berlicka, A.; Latos-Grażyński, L. 21-Carbaporphyrin: A Cyclopentadiene Moiety Entrapped into a Porphyrin Scaffold. J. Porphyr. Phthalocyanines 2020, 24, 1–20. [Google Scholar] [CrossRef]
- AbuSalim, D.I.; Lash, T.D. Aromatic Character and Stability of Neo-Confused Porphyrin Tautomers and Related Compounds. Org. Biomol. Chem. 2013, 11, 8306–8323. [Google Scholar] [CrossRef] [PubMed]
- Mathius, M.A.; Chhoeun, J.M.; Kaufman, R.H.; AbuSalim, D.I.; Lash, T.D. manuscript in preparation.
- Lash, T.D.; Ferrence, G.M. Metalation and Selective Oxidation of Diphenyl-23-oxa-, thia-, and selena-21-carbaporphyrins. Inorg. Chem. 2017, 56, 11426–11434. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Ferrence, G.M.; Lash, T.D. Preparation of Furan and Thiophene-derived Fulvene Dialdehydes: Synthesis and Structural Characterization of a 22-Oxa-21-carbaporphyrin and a Related Palladium(II) Organometallic Complex. J. Org. Chem. 2010, 75, 6563–6573. [Google Scholar] [CrossRef]
- AbuSalim, D.I.; Lash, T.D. Tropylium and Porphyrinoid Character in Carbaporphyrinoid Systems. Relative Stability and Aromatic Characteristics of Azuliporphyrin and Tropiporphyrin Tautomers, Protonated Species, and Related Structures. J. Phys. Chem. A 2019, 123, 230–246. [Google Scholar] [CrossRef]
- Graham, S.R.; Ferrence, G.M.; Lash, T.D. Organometallic Chemistry of Carbaporphyrinoids: Synthesis and Characterization of Nickel(II) and Palladium(II) Azuliporphyrins. Chem. Commun. 2002, 38, 894–895. [Google Scholar] [CrossRef]
- Lash, T.D.; Colby, D.A.; Graham, S.R.; Ferrence, G.M.; Szczepura, L.F. Organometallic Chemistry of Azuliporphyrins: Synthesis, Spectroscopy, Electrochemistry and Structural Characterization of Nickel(II), Palladium(II) and Platinum(II) Complexes of Azuliporphyrins. Inorg. Chem. 2003, 42, 7326–7338. [Google Scholar] [CrossRef]
- Lash, T.D.; Pokharel, K.; Zeller, M.; Ferrence, G.M. Iridium(III) Azuliporphyrins. Chem. Commun. 2012, 48, 11793–11795. [Google Scholar] [CrossRef]
- Stateman, L.M.; Ferrence, G.M.; Lash, T.D. Rhodium(III) Azuliporphyrins. Organometallics 2015, 34, 3842–3848. [Google Scholar] [CrossRef]
- Adiraju, V.A.K.; Ferrence, G.M.; Lash, T.D. Regioselective Oxidation and Metalation of meso-Unsubstituted Azuliporphyrins. Org. Biomol. Chem. 2016, 14, 10523–10533. [Google Scholar] [CrossRef]
- Kuwabara, J.; Munezawa, G.; Okamoto, K.; Kanbara, T. Palladium(II) and Platinum(II) Complexes Bearing a κ3SCS Pincer Ligand with an Azulene Unit. Dalton Trans. 2010, 39, 6255–6261. [Google Scholar] [CrossRef]
- Colby, D.A.; Ferrence, G.M.; Lash, T.D. Oxidative Metalation of Azuliporphyrins with Copper(II) Salts: Formation of a Porphyrin Analogue System with a Unique Fully Conjugated Nonaromatic Azulene Subunit. Angew. Chem. Int. Ed. 2004, 43, 1346–1349. [Google Scholar] [CrossRef]
- Lash, T.D.; Colby, D.A.; El-Beck, J.A.; AbuSalim, D.I.; Ferrence, G.M. Preparation, Structural Characterization, Assessment of Potential Antiaromaticity and Metalation of 21-Oxyazuliporphyrins. Inorg. Chem. 2015, 54, 9174–9187. [Google Scholar] [CrossRef]
- Białek, M.J.; Chmielewski, P.J.; Latos-Grażyński, L. C-H and C-M Activation, Aromaticity Tuning, and Co···Ru Interactions Confined in the Azuliporphyrin Framework. Chem. Eur. J. 2019, 25, 14536–14545. [Google Scholar] [CrossRef]
- Białek, M.J.; Latos-Grażyński, L. Merging of Inner and Outer Ruthenium Organometallic Coordination Motifs within an Azuliporphyrin Framework. Chem. Commun. 2014, 50, 9270–9272. [Google Scholar] [CrossRef]
- Białek, M.J.; Bialonska, A.; Latos-Grażyński, L. Oxidation and Oxygenation of Carbonyl Ruthenium(II) Azuliporphyrin. Inorg. Chem. 2015, 54, 6184–6194. [Google Scholar] [CrossRef]
- Białek, M.J.; Latos-Grażyński, L. Palladium(II), Ruthenium(II), and Ruthenium(III) Complexes of 23-Thiaazuliporphyrins: The Case of Coordination-Induced Contraction. Inorg. Chem. 2016, 55, 1758–1769. [Google Scholar] [CrossRef]
- Moriones, J.S.D.; Latham, A.N.; Lash, T.D. Synthesis of Internally Alkylated Azuliporphyrins. J. Porphyr. Phthalocyanines 2020, 24, 817–829. [Google Scholar] [CrossRef]
- Yi, H.; Lei, A. Pd-Catalyzed Hydroxylation of Aryl Boronic Acids Using In Situ Generated Hydrogen Peroxide. Chem. Eur. J. 2017, 23, 10023–10027. [Google Scholar] [CrossRef]
- Lash, T.D.; Gilot, G.C.; AbuSalim, D.I. Tropone-fused Carbaporphyrins. J. Org. Chem. 2014, 79, 9704–9716. [Google Scholar] [CrossRef] [PubMed]
- AbuSalim, D.I.; Lash, T.D. Relative Stability of Benziporphyrin and Naphthiporphyrin Tautomers and the Emergence of Macrocyclic Diatropicity. Org. Biomol. Chem. 2014, 12, 8719–8736. [Google Scholar] [CrossRef]
- Stępień, M.; Latos-Grażyński, L.; Szterenberg, L.; Panek, J.; Latajka, Z. Cadmium(II) and Nickel(II) Complexes of Benziporphyrins. A Study of Weak Intramolecular Metal-Arene Interactions. J. Am. Chem. Soc. 2004, 126, 4566–4580. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-H.; Chang, F.-C.; Lin, C.-Y.; Rachlewicz, K.; Stepień, M.; Latos-Grażyński, L.; Lee, G.-H.; Peng, S.-M. Iron and Copper Complexes of Tetraphenyl-m-benziporphyrin: Reactivity of the Internal C-H Bond. Inorg. Chem. 2004, 43, 4118–4120. [Google Scholar] [CrossRef] [PubMed]
- Stępień, M.; Latos-Grażyński, L. Regioselective Pyridination of m-Benziporphyrin. Org. Lett. 2003, 5, 3379–3381. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, J.T.; Lash, T.D. Dimethoxytetraphenylbenziporphyrins. Tetrahedron Lett. 2003, 44, 8613–8616. [Google Scholar] [CrossRef]
- Lash, T.D.; Szymanski, J.T.; Ferrence, G.M. Tetraaryldimethoxybenziporphyrins. At the Edge of Carbaporphyrinoid Aromaticity. J. Org. Chem. 2007, 72, 6481–6492. [Google Scholar] [CrossRef]
- Chang, G.-F.; Wang, C.-H.; Lu, H.-C.; Kan, L.-S.; Chao, I.; Chen, W.H.; Kumar, A.; Lo, L.; de La Rosa, A.C.M.; Hung, C.-H. Factors That Regulate the Conformation of m-Benziporphodimethene Complexes: Agostic Metal–Arene Interaction, Hydrogen Bonding, and η2,π Coordination. Chem. Eur. J. 2011, 17, 11332–11343. [Google Scholar] [CrossRef]
- Hung, C.-H.; Chang, G.-F.; Kumar, A.; Lin, G.-F.; Luo, L.-Y.; Ching, W.-M.; Diau, E.W.-G. m-Benziporphodimethene: A New Porphyrin Analogue Fluorescence Zinc(II) Sensor. Chem. Commun. 2008, 44, 978–980. [Google Scholar] [CrossRef]
- Huang, C.; Li, Y.; Yang, J.; Cheng, N.; Liu, H.; Li, Y. Construction of Multidimensional Nanostructures by Self-assembly of a Porphyrin Analogue. Chem. Commun. 2010, 46, 3161–3163. [Google Scholar] [CrossRef]
- Ribas, X.; Jackson, D.A.; Donnadieu, B.; Mahía, J.; Parella, T.; Xifra, R.; Hedman, B.; Hodgson, K.O.; Llobet, A.; Stack, T.D.P. Aryl C-H Activation by CuII To Form an Organometallic Aryl-CuIII Species: A Novel Twist on Copper Disproportionation. Angew. Chem. Int. Ed. 2002, 41, 2991–2994. [Google Scholar] [CrossRef]
- Huffman, L.M.; Stahl, S.S. Carbon-Nitrogen Bond Formation Involving Well-Defined Aryl-Copper(III) Complexes. J. Am. Chem. Soc. 2008, 130, 9196–9197. [Google Scholar] [CrossRef]
- King, A.E.; Huffman, L.M.; Casitas, A.; Costas, M.; Ribas, X.; Stahl, S.S. Copper-Catalyzed Aerobic Oxidative Functionalization of an Arene C-H Bond: Evidence for an Aryl-Copper(III) Intermediate. J. Am. Chem. Soc. 2010, 132, 12068–12073. [Google Scholar] [CrossRef]
- Yao, B.; Wang, D.X.; Huang, Z.T.; Wang, M.X. Room-temperature Aerobic Formation of a Stable Aryl–Cu(III) Complex and its Reactions with Nucleophiles: Highly Efficient and Diverse Arene C-H Functionalizations of Azacalix[1]arene[3]pyridine. Chem. Commun. 2009, 45, 2899–2901. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Zhao, L.; Wang, M.-X. Regiospecific Functionalization of Azacalixaromatics through Copper-Mediated Aryl C-H Activation and C-O Bond Formation. Org. Lett. 2011, 13, 6560–6563. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Wang, Z.-L.; Zhang, H.; Wang, D.-X.; Zhao, L.; Wang, M.-X. Cu(ClO4)2-Mediated Arene C-H Bond Halogenations of Azacalixaromatics Using Alkali Metal Halides as Halogen Sources. J. Org. Chem. 2012, 77, 3336–3340. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Cu(OTf)2-Catalyzed Selective Arene C–H Bond Hydroxylation and Nitration with KNO2 as an Ambident O- and N-Nucleophile via a Cu(II)-Cu(III)-Cu(I) Mechanism. Org. Lett. 2013, 15, 3836–3839. [Google Scholar] [CrossRef]
- Hurej, K.; Pawlicki, M.; Szterenberg, L.; Latos-Grażyński, L. A Rhodium-Mediated Contraction of Benzene to Cyclopentadiene: Transformations o fRhodium(III) m-Benziporphyrin. Angew. Chem. Int. Ed. 2016, 55, 1427–1431. [Google Scholar] [CrossRef]
- Szyszko, B.; Latos-Grażyński, L.; Szterenberg, L. Towards Aceneporphyrinoids: Synthesis and Transformations of Palladium(II) meso-Anthriporphyrin. Chem. Commun. 2012, 48, 5004–5006. [Google Scholar] [CrossRef] [PubMed]
- Szyszko, B.; Latos-Grażyński, L.; Szterenberg, L. A Facile Palladium-Mediated Contraction of Benzene to Cyclopentadiene: Transformations of Palladium(II) p-Benziporphyrin. Angew. Chem. Int. Ed. 2011, 50, 6587–6591. [Google Scholar] [CrossRef]
- Szyszko, B.; Latos-Grażyński, L. Conformational Flexibility of 1,4-Naphthiporphyrin Promotes a Palladium-Mediated Contraction of Naphthalene to Isoindene. Organometallics 2011, 30, 4354–4363. [Google Scholar] [CrossRef]
- Szyszko, B.; Kupietz, K.; Szterenberg, L.; Latos-Grażyński, L. Gold(III)-Mediated Contraction of Benzene to Cyclopentadiene: From p-Benziporphyrin to Gold(III) True Tetraarylcarbaporphyrin. Chem. Eur. J. 2014, 20, 1376–1382. [Google Scholar] [CrossRef]
- Idec, A.; Szterenberg, L.; Latos-Grażyński, L. From para-Benziporphyrin to Rhodium(III) 21-Carbaporphyrins: Imprinting Rh···η2-CC, Rh···η2-CO, and Rh···η2-CH Coordination Motifs. Chem. Eur. J. 2015, 21, 12481–12487. [Google Scholar] [CrossRef] [PubMed]
- Hurej, K.; Pawlicki, M.; Latos-Grażyński, L. Gold(III) Triggered Transformation of 22-Methyl-m-benziporphyrin Involving an Effective Contraction of Benzene to Cyclopentadiene. Chem. Eur. J. 2017, 23, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Hurej, K.; Pawlicki, M.; Latos-Grażyński, L. Rhodium-Induced Reversible C-C Bond Cleavage: Transformations of Rhodium(III) 22-Alkyl-m-benziporphyrins. Chem. Eur. J. 2018, 24, 115–126, Related studies on rhodium(III) derivatives have also been reported. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Thorat, K.G.; Lee, W.-Z.; Ravikanth, M. Synthesis, Structural, Spectral, and Electrochemical Studies of Selenabenziporphyrin and Its Pd(II) Complex. Inorg. Chem. 2018, 57, 8956–8963. [Google Scholar] [CrossRef]
- Kumar, S.; Lee, W.-Z.; Ravikanth, M. Synthesis of Tellurabenziporphyrin and Its Pd(II) Complex. Org. Lett. 2018, 20, 636–639. [Google Scholar] [CrossRef]
- Abebayehu, A.; Park, D.; Hwang, S.; Dutta, R.; Lee, C.-H. Synthesis and Spectroscopic Behaviour of Metal Complexes of meso-Alkylidenyl Carbaporphyrinoids and their Expanded Analogues. Dalton Trans. 2016, 45, 3093–3101. [Google Scholar] [CrossRef]
- Park, D.; Jeong, S.D.; Ishida, M.; Lee, C.-H. Stepwise π-Extension of meso-Alkylidenyl Porphyrins through Sequential 1,3-Dipolar Cycloaddition and Redox Reactions. Chem. Commun. 2014, 50, 9277–9280. [Google Scholar] [CrossRef]
- Mysliborski, R.; Rachlewicz, K.; Latos-Grażyński, L. Iron Complexes of N-Confused Pyriporphyrin: NMR Studies. Inorg. Chem. 2006, 45, 7828–7834. [Google Scholar] [CrossRef]
- Pacholska-Dudziak, E.; Latos-Grażyński, L. NMR Studies of Paramagnetic Metallocarbaporphyrinoids. Eur. J. Inorg. Chem. 2007, 2007, 2594–2608. [Google Scholar] [CrossRef]
- Linstead, R.P. Discoveries Among Conjugated Macrocyclic Compounds. J. Chem. Soc. 1953, 1953, 2873–2884. [Google Scholar] [CrossRef]
- Clark, P.F.; Elvidge, J.A.; Linstead, R.P. Conjugated Macrocycles. Part XXV. Cross-conjugated Macrocycles with Inner Great Rings of 16, 20, and 24 Atoms. J. Chem. Soc. 1954, 1954, 2490–2497. [Google Scholar] [CrossRef]
- Baguley, M.E.; Elvidge, J.A. Heterocyclic Imines and Amines, Part VIII. Identification of “o-Cyanothiobenzamide” as 1-Imino-3-thioisoindoline, and its Conversion with Amines into Macrocycles and Intermediates. J. Chem. Soc. 1957, 1957, 709–719. [Google Scholar] [CrossRef]
- Elvidge, J.A.; Golden, J.H. Conjugated Macrocycles. Part XXVIII. Adducts from Di-iminoisoindoline and Arylene-m-diamines, and a New Type of Cross-conjugated Macrocycle with Three-quarters of the Chromophore of Phthalocyanine. J. Chem. Soc. 1957, 1957, 700–709. [Google Scholar] [CrossRef]
- Ziegler, C.J. The Hemiporphyrazines and Related Systems. In Handbook of Porphyrin Science—With Applications to Chemistry, Physics, Material Science, Engineering, Biology and Medicine; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing: Singapore, 2012; Volume 17, pp. 113–238. [Google Scholar]
- Wu, R.; Cetin, A.; Durfee, W.S.; Ziegler, C.J. Metal-Mediated C-H Bond Activation in a Carbon-Substituted Hemiporphyrazine. Angew. Chem. Int. Ed. 2006, 45, 5670–5673. [Google Scholar] [CrossRef]
- Cetin, A.; Durfee, W.S.; Ziegler, C.J. Low-Coordinate Transition-Metal Complexes of a Carbon-Substituted Hemiporphyrazine. Inorg. Chem. 2007, 46, 6239–6241. [Google Scholar] [CrossRef]
- Sripothongnak, S.; Pischera, A.M.; Espe, M.P.; Durfee, W.S.; Ziegler, C.J. Synthesis and Characterization of Lithium Hemiporphyrazines. Inorg. Chem. 2009, 48, 1293–1300. [Google Scholar] [CrossRef]
- Cetin, A.; Sripothongnak, S.; Kawa, M.; Durfee, W.S.; Ziegler, C.J. Co(II) and Co(III) Complexes of m-Benziphthalocyanine. Chem. Commun. 2007, 43, 4289–4290. [Google Scholar] [CrossRef] [PubMed]
- Sripothongnak, S.; Barone, N.; Ziegler, C.J. C-H Activation and Ring Oxidation in Nickel Carbahemiporphyrazines. Chem. Commun. 2009, 45, 4584–4586. [Google Scholar] [CrossRef] [PubMed]
- Stępień, M.; Latos-Grażyński, L.; Lash, T.D.; Szterenberg, L. Palladium(II) Complexes of Oxybenziporphyrin. Inorg. Chem. 2001, 40, 6892–6900. [Google Scholar] [CrossRef]
- Pokharel, K.; Ferrence, G.M.; Lash, T.D. Late Transition Metal Complexes of Oxypyriporphyrin and the Platinum(II) Complex of Oxybenziporphyrin. J. Porphyr. Phthalocyanines 2017, 21, 493–501. [Google Scholar] [CrossRef]
- El-Beck, J.A.; Lash, T.D. Tetraphenyloxybenziporphyrin, a New Organometallic Ligand for Silver(III) and Gold(III). Org. Lett. 2006, 8, 5263–5266. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, S.; Anand, V.G.; Pushpan, S.K.; Sankar, J.; Chandrashekar, T.K. Core Modified Oxybenziporphyrins: New Aromatic Ligands for Metal-Carbon Bond Activation. Chem. Commun. 2002, 38, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Schick, A.J.; Paul, N.B.; Durfee, W.S.; Ziegler, C.J. Hydroxybenziphthalocyanines: Non-Aromatic Phthalocyanine Analogues that Exhibit Strong UV-Visible Absorptions. New J. Chem. 2011, 35, 794–799. [Google Scholar] [CrossRef]
- Muranaka, A.; Ohira, S.; Hashizume, D.; Koshino, H.; Kyotani, F.; Hirayama, M.; Uchiyama, M. [18]/[20]π Hemiporphyrazine: A Redox-Switchable Near-Infrared Dye. J. Am. Chem. Soc. 2012, 134, 190–193. [Google Scholar] [CrossRef]
- Muranaka, A.; Kyotani, F.; Ouyang, X.; Hashizume, D.; Uchiyama, M. Synthesis and Properties of Palladium(II) Complexes of Aromatic Hemiporphyrazines. J. Porphyr. Phthalocyanines 2014, 18, 869–874. [Google Scholar] [CrossRef]
- Lash, T.D.; Rooney, J.M. Alkylation, Metalation and Ring Contraction of Tropiporphyrin. J. Porphyr. Phthalocyanines 2020, 24, 181–190. [Google Scholar] [CrossRef]
- AbuSalim, D.I.; Lash, T.D. In Pursuit of Novel Porphyrin Isomers. Aromatic Character and Relative Stability of Conjugated Tetrapyrroles with Two Neo-Confused Rings or with Mixed Neo-Confused and N-Confused Subunits. J. Phys. Chem. A 2015, 119, 11440–11453. [Google Scholar] [CrossRef]
- Almejbel, A.S.; Lash, T.D. Synthesis of 2-Bromo- and 2-Phenyl Neo-Confused Porphyrins. Oerg. Biomol. Chem. 2020, 18, 7336–7344. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.-H.; Aslam, A.S.; Ishida, M.; Mori, S.; Furuta, H.; Cho, D.-G. 2-Naphthalen-1-yl)thiophene as a New Motif for Porphyrinoids: Meso-Fused Carbaporphyrin. J. Am. Chem. Soc. 2016, 138, 4992–4995. [Google Scholar] [CrossRef]
- Aslam, A.D.; Hong, J.-H.; Shin, J.-H.; Cho, D.-G. Synthesis of a Phlorin from a Meso-Fused Anthriporphyrin by a Diels–Alder Strategy. Angew. Chem. Int. Ed. 2017, 56, 6247–16251. [Google Scholar] [CrossRef]
- Hong, J.-H.; Aslam, A.S.; Ko, M.-S.; Choi, J.; Lee, Y.; Cho, D.-G. Bond Rotation in an Aromatic Carbaporphyrin: Allyliporphyrin. Chem. Eur. J. 2018, 24, 10054–10058. [Google Scholar] [CrossRef]
- Ko, M.-S.; Hong, J.-H.; Aslam, A.S.; Rao, P.S.; Desale, P.P.; Choi, J.; Lee, Y.; Cho, D.-G. o-(2-Thienyl)vinylarene as an Alternative Synthetic Motif for Porphyrinoids: O-Arene-Connected Porphyrinoids. Org. Lett. 2022, 24, 8812–8815. [Google Scholar] [CrossRef]
- Hong, J.-H.; Aslam, A.S.; Cho, B.; Ko, M.-S.; Kim, Y.; Heo, J.; Cho, D.-G. Carbaporphyrin Dimers That Bear a Rigid Naphthalene Motif as an Internal Strap. Org.Lett. 2021, 23, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Laxman, K.; Ravikanth, M. Antiaromatic Carbaporphyrinoids: Fluorene as a Fused Motif toward the Synthesis of meso-Fused Heterobenziporphyrins. Org. Lett. 2019, 21, 8726–8730. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Smith, B.E.; Melquist, M.J.; Godfrey, B.A. Synthesis of Indenoporphyrins, Highly Modified Porphyrins with Reduced Diatropic Characteristics. J. Org. Chem. 2011, 76, 5335–5345. [Google Scholar] [CrossRef] [PubMed]
- Saegusa, Y.; Ishizuka, T.; Shiota, Y.; Yoshizawa, K.; Kojima, T. Acid-Base Properties of a Free Base Form of a Quadruply Ring-Fused Porphyrin—Stepwise Protonation Induced by Rigid Ring-Fused Structure. J. Org. Chem. 2017, 82, 322–330. [Google Scholar] [CrossRef]
- Pachola, E.; Latos-Grażyński, L.; Ciunik, Z. A Direct Link between Annulene and Porphyrin Chemistry—21-Vacataporphyrin. Chem. Eur. J. 2002, 8, 5403–5405. [Google Scholar] [CrossRef]
- Pacholska-Dudziak, E.; Skonieczny, J.; Pawlicki, M.; Szterenberg, L.; Latos-Grażyński, L. Cadmium(II), Nickel(II), and Zinc(II) Complexes of Vacataporphyrin: A Variable Annulene Conformation inside a Standard Porphyrin Frame. Inorg. Chem. 2005, 44, 8794–8803. [Google Scholar] [CrossRef]
- Pacholska-Dudziak, E.; Skonieczny, J.; Pawlicki, M.; Szterenberg, L.; Ciunik, Z.; Latos-Grażyński, L. Palladium Vacataporphyrin Reveals Conformational Rearrangements Involving Hückel and Möbius Macrocyclic Topologies. J. Am. Chem. Soc. 2008, 130, 6182–6195. [Google Scholar] [CrossRef]
- Lash, T.D.; Romanic, J.L.; Hayes, M.J.; Spence, J.D. Towards Hydrocarbon Analogues of the Porphyrins: Synthesis and Spectroscopic Characterization of the First Dicarbaporphyrin. Chem. Commun. 1999, 35, 819–820. [Google Scholar] [CrossRef]
- Xu, L.; Lash, T.D. Synthesis of Aromatic Dicarbaporphyrinoids from Resorcinol and 2-Methylresorcinol. Tetrahedron Lett. 2006, 47, 8863–8866. [Google Scholar] [CrossRef]
- Lash, T.D.; Colby, D.A.; Idate, A.S.; Davis, R.N. Fulvene Dialdehyde Strategy for adj-Dicarbaporphyrinoid Synthesis: Preparation of a 22-Carbaazuliporphyrin. J. Am. Chem. Soc. 2007, 129, 13800–13801. [Google Scholar] [CrossRef]
- Lash, T.D.; Lammer, A.D.; Ferrence, G.M. Two-step Synthesis of Stable Dioxadicarbaporphyrins from Bis(3-indenyl)methane. Angew. Chem. Int. Ed. 2012, 51, 10871–10875. [Google Scholar] [CrossRef] [PubMed]
- AbuSalim, D.I.; Merfeld, M.L.; Lash, T.D. Dicarbaporphyrinoid Systems. Synthesis of Oxo-adj-dibenziphlorins. J. Org. Chem. 2013, 78, 10360–10368. [Google Scholar] [CrossRef] [PubMed]
- AbuSalim, D.I.; Ferrence, G.M.; Lash, T.D. Synthesis of an adj-Dicarbaporphyrin and the Formation of an Unprecedented Tripalladium Sandwich Complex. J. Am. Chem. Soc. 2014, 136, 6763–6772. [Google Scholar] [CrossRef]
- Jain, P.; AbuSalim, D.I.; Lash, T.D. adj-Dicarbaporphyrinoid Systems: Synthesis, Spectroscopic Characterization and Reactivity of 23-Carbabenziporphyrins. J. Org. Chem. 2019, 84, 10237–10256. [Google Scholar] [CrossRef]
- Sprutta, N.; Swiderska, M.; Latos-Grażyński, L. Dithiadiazuliporphyrin: Facile Generation of Carbaporphyrinoid Cation Radical and Dication. J. Am. Chem. Soc. 2005, 127, 13108–13109. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Sprutta, N.; Kim, J.O.; Koo, Y.H.; Latos-Grażyński, L.; Kim, D. Retrieving Aromaticity of Dithiadiazuliporphyrin by Oxidation: Illustration by Experimental and Theoretical Investigation. RSC Adv. 2017, 7, 19502–19505. [Google Scholar] [CrossRef]
- Sprutta, N.; Siczek, M.; Latos-Grażyński, L.; Pawlicki, M.; Szterenberg, L.; Lis, T. Dioxadiazuliporphyrin: A Near-IR Redox Switchable Chromophore. J. Org. Chem. 2007, 72, 9501–9509. [Google Scholar] [CrossRef]
- Furuta, H.; Maeda, H.; Osuka, A. Doubly N-Confused Porphyrin: A New Complexing Agent Capable of Stabilizing Higher Oxidation States. J. Am. Chem. Soc. 2000, 122, 803–807. [Google Scholar] [CrossRef]
- Maeda, H.; Osuka, A.; Furuta, H. Synthesis of A2B2 Type cis-Doubly N-Confused Porphyrins from N-Confused Dipyrromethanes. Tetrahedron 2004, 60, 2427–2432. [Google Scholar] [CrossRef]
- Araki, K.; Winnischofer, H.; Toma, H.E.; Maeda, H.; Osuka, A.; Furuta, H. Acid-Base and Spectroelectrochemical Properties of Doubly N-Confused Porphyrins. Inorg. Chem. 2001, 40, 2020–2025. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Morimoto, T.; Osuka, A.; Furuta, H. Halide-Anion Binding by Singly and Doubly N-Confused Porphyrins. Chem. Asian J. 2006, 1, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Furuta, H.; Maeda, H.; Osuka, A.; Yasutake, M.; Shinmyozu, T.; Ishikawa, Y. Inner C-Arylation of a Doubly N-Confused Porphyrin-Pd Complex in Toluene—The Possibility of a Pd3+ Intermediate. Chem. Commun. 2000, 36, 1143–1144. [Google Scholar] [CrossRef]
- Maeda, H.; Osuka, A.; Furuta, H. Trans Doubly N-Confused Porphyrins: Cu(III) Complexation and Formation of Rodlike Hydrogen-Bonding Networks. J. Am. Chem. Soc. 2003, 125, 15690–15691. [Google Scholar] [CrossRef]
- Yan, J.; Takakusaki, M.; Yang, Y.; Mori, S.; Zhang, B.; Feng, Y.; Ishida, M.; Furuta, H. Doubly N-Confused Isophlorin: Synthesis, Structure and Copper Coordination. Chem. Commun. 2014, 50, 14593–14596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ferrence, G.M.; Lash, T.D. adj-Diazuliporphyrins, a New Family of Dicarbaporphyrinoids with Unprecedented Mesoionic Characteristics. Org. Lett. 2009, 11, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; AbuSalim, D.I.; Ferrence, G.M. adj-Dicarbachlorin, the First Example of a Free Base Carbaporphyrinoid System with an Internal Methylene Unit. Chem. Commun. 2015, 51, 15952–15955. [Google Scholar] [CrossRef]
- Maulbetsch, T.; Kunz, D. Carbenaporphyrins: No Longer Missing Ligands in N-Heterocyclic Carbene Chemistry. Angew. Chem. Int. Ed. 2021, 60, 2007–2012. [Google Scholar] [CrossRef]
- Lash, T.D. Handbook of Porphyrin Science—With Applications to Chemistry, Physics, Material Science, Engineering, Biology and Medicine; World Scientific Publishing: Singapore, 2012; pp. 6–8, 267–275. [Google Scholar]
- AbuSalim, D.I.; Lash, T.D. Relative Stability and Diatropic Character of Carbaporphyrin, Dicarbaporphyrin, Tricarbaporphyrin and Quatyrin Tautomers. J. Org. Chem. 2013, 78, 11535–11548. [Google Scholar] [CrossRef]
- Lash, T.D. Origin of Aromatic Character in Porphyrinoid Systems. J. Porphyr. Phthalocyanines 2011, 15, 1093–1115. [Google Scholar] [CrossRef]
- Lash, T.D. Carbaporphyrins, Porphyrin Isomers and the Legacy of Emanuel Vogel. J. Porphyr. Phthalocyanines 2012, 16, 423–433. [Google Scholar] [CrossRef]
- Vogel, E. The Porphyrins from the ‘Annulene Chemist’s Perspective. Pure Appl. Chem. 1993, 65, 143–152. [Google Scholar] [CrossRef]
- Vogel, E. Porphyrinoid Macrocycles: A Cornucopia of Novel Chromophores. Pure Appl. Chem. 1996, 68, 1355–1360. [Google Scholar] [CrossRef]
- Vogel, E.; Haas, W.; Knipp, B.; Lex, J.; Schmickler, H. Tetraoxaporphyrin Dication. Angew. Chem. Int. Ed. Engl. 1988, 27, 406–409. [Google Scholar] [CrossRef]
- Vogel, E.; Köcher, M.; Schmickler, H.; Lex, J. Porphycene—A Novel Porphyrin Isomer. Angew. Chem. Int. Ed. Engl. 1986, 25, 257–259. [Google Scholar] [CrossRef]
- Davis, R.N.; Lash, T.D. Formation of Stable Fulvene and Difulvene Aldehydes from Benzaldehydes and an Indene-derived Enamine: Preparation of Novel Indene Fused Benzodiazepines and Attempted Syntheses of Di- and Tricarbaporphyrinoid Systems. Tetrahedron 2009, 65, 9935–9943. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Q.; Wang, Z.; Yan, C.; Li, G.; Ma, J.S. Tetra-N- confused Cyclohexapyrrole: The Unusual Product Formed by Condensation of 3,3′-Dipyrromethane with Tripyrrin-aldehyde. J. Org. Chem. 2002, 67, 4997–5000. [Google Scholar] [CrossRef]
- Rahman, S.; Zein, A.; Dawe, L.N.; Shamov, G.; Thordarson, P.; Georghiou, P.E. Supramolecular Host-Guest Complexation of Lash’s Calix[4]azulene with Tetraalkylammonium Halides and Tetrafluoroborate Salts: Binding and DFT Computational Studies. RSC Adv. 2015, 5, 54848–54852. [Google Scholar] [CrossRef]
- Georghiou, P.E.; Schneider, C.; Shamov, G.; Lash, T.D.; Rahman, S.; Giddings, D.S. Mechanochemical Formation of a 1:1 C60:tert-Butylcalix[4]azulene Supramolecular Complex: Solid-State NMR and DFT Computational Studies. Supramol. Chem. 2016, 28, 396–402. [Google Scholar] [CrossRef]
- Georghiou, P.E.; Rahman, S.; Alodhayb, A.; Nishimura, H.; Lee, J.; Wakamiya, A.; Scott, L.T. Calixazulenes: Azulene-based Calixarene Analogues—An Overview and Recent Supramolecular Complexation Studies. Beilstein J. Org. Chem. 2018, 14, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Sprutta, N.; Mackowiak, S.; Kocik, M.; Szterenberg, L.; Lis, T.; Latos-Grażyński, L. Tetraazuliporphyrin Tetracation. Angew. Chem. Int. Ed. 2009, 48, 3337–3341. [Google Scholar] [CrossRef]
- Sprutta, N.; Welnic, M.; Bialek, M.J.; Lis, T.; Szterenberg, L.; Latos-Grażyński, L. Carbocations Confined in a Thiatriazuliporphyrin Frame. Chem. Eur. J. 2016, 22, 6974–6980. [Google Scholar] [CrossRef]
- Bialek, M.J.; Sprutta, N.; Latos-Grażyński, L. Coordination-Induced Molecular Tweezing: Ruthenium Clusters Docked at Azuliporphyrinogens. Inorg. Chem. 2016, 55, 12061–12073. [Google Scholar] [CrossRef]
- Chun, Y.; Singh, N.J.; Hwang, I.-C.; Lee, J.W.; Yu, S.U.; Kim, K.S. Calix[n]imidazolium as a New Class of Positively Charged Homo-calix Compounds. Nat. Commun. 2013, 4, 1797. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.E.; Langenhahn, V.; Lügger, T.; Pape, T.; Van, D.L. Template Synthesis of a Coordinated Tetracarbene Ligand with Crown Ether Topology. Angew. Chem. Int. Ed. 2005, 44, 3759–3763. [Google Scholar] [CrossRef] [PubMed]
- Bass, H.M.; Cramer, S.A.; Price, J.L.; Jenkins, D.M. 18-Atom-Ringed Macrocyclic Tetra-imidazoliums for Preparation of Monomeric Tetra-carbene Complexes. Organometallics 2010, 29, 3235–3238. [Google Scholar] [CrossRef]
- Meyer, I.; Klawitter, S.; Demeshko, E.; Bill, F.; Meyer, A. Tetracarbene–Oxoiron(IV) Complex. Angew. Chem. Int. Ed. 2013, 52, 901–905. [Google Scholar] [CrossRef]
- Bauer, E.B.; Bernd, M.A.; Schutz, M.; Oberkofler, J.; Pothig, A.; Reich, R.M.; Kuhn, F.E. Synthesis, Characterization, and Biological Studies of Multidentate Gold(I) and Gold(III) NHC Complexes. Dalton Trans. 2019, 48, 16615–16625. [Google Scholar] [CrossRef]
- Ghavami, Z.S.; Anneser, M.R.; Kaiser, F.; Altmann, P.J.; Hofmann, B.J.; Schlagintweit, J.F.; Grivani, G.; Kühn, F.E. A Bench Stable Formal Cu(III) N-heterocyclic Carbene Accessible from Simple Copper(II) Acetate. Chem. Sci. 2018, 9, 8307–8314. [Google Scholar] [CrossRef]
- Schlachta, T.P.; Schlagintweit, J.F.; Anneser, M.R.; Esslinger, E.-M.H.J.; Muhr, M.; Stefan Haslinger, S.; Kuhn, F.E. Modification of Bio-inspired Tetra-NHC Iron Complexes with Axial Nitrile Ligands. Inorg. Chim. Acta 2021, 518, 120228. [Google Scholar] [CrossRef]
- Schlagintweit, J.F.; Hintermeier, C.; Anneser, M.R.; Esslinger, E.-M.H.J.; Haslinger, S.; Kuhn, F.E. Electronic Finetuning of a Bio-inspired Iron(II) Tetra-NHC Complex by trans Axial Isocyanide Substitution. Chem Asian J. 2020, 15, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Schlachta, T.P.; Anneser, M.R.; Schlagintweit, J.F.; Jakob, C.H.G.; Hintermeier, C.; Böth, A.D.; Reich, R.M.; Kuhn, F.E. Mimicking Reactive High-valent Diiron-μ2-oxo Intermediates of Nonheme Enzymes by an Iron Tetracarbene Complex. Chem. Commun. 2021, 57, 6644–6647. [Google Scholar] [CrossRef] [PubMed]
- Anneser, M.R.; Haslinger, S.; Pothig, A.; Cokoja, M.; D’Elia, V.; Hogerl, M.P.; Basset, J.-M.; Kuhn, F.E. Binding of Molecular Oxygen by an Artificial Heme Analogue: Investigation on the Formation of an Fe-tetracarbene Superoxo Complex. Dalton Trans. 2016, 45, 6449–6455. [Google Scholar] [CrossRef] [PubMed]
- Schlagintweit, J.F.; Altmann, P.J.; Böth, A.D.; Hofmann, B.J.; Jandl, C.; Kaußler, C.; Nguyen, L.; Reich, R.M.; Pöthig, A.; Kühn, F.E. Activation of Molecular Oxygen by a Cobalt(II) Tetra-NHC Complex. Chem. Eur. J. 2021, 27, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Bernd, M.A.; Bauer, E.B.; Oberkofler, J.; Bauer, A.; Reich, R.M.; Kuhn, F.E. Macrocyclic NHC Complexes of Group 10 Elements with Enlarged Aromaticity for Biological Studies. Dalton Trans. 2020, 49, 14106–14114. [Google Scholar] [CrossRef] [PubMed]
- Bass, H.M.; Cramer, S.A.; McCullough, A.S.; Bernstein, K.J.; Murdock, C.R.; Jenkins, D.M. Employing Dianionic Macrocyclic Tetracarbenes To Synthesize Neutral Divalent Metal Complexes. Organometallics 2013, 32, 2160–2167. [Google Scholar] [CrossRef]
- Anneser, M.R.; Powers, X.B.; Peck, K.M.; Jensen, I.M.; Jenkins, D.M. One Macrocyclic Ligand, Four Oxidation States: A 16-Atom Ringed Dianionic Tetra-NHC Macrocycle and Its Cr(II) through Cr(V) Complexes. Organometallics 2019, 38, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Anneser, M.R.; Elpitiya, G.R.; Powers, X.B.; Jenkins, D.M. Toward a Porphyrin-Style NHC: A 16-Atom Ringed Dianionic Tetra-NHC Macrocycle and Its Fe(II) and Fe(III) Complexes. Organometallics 2019, 38, 981–987. [Google Scholar] [CrossRef]
- Carroll, X.B.; Errulat, D.; Murugesu, M.; Jenkins, D.M. Late Lanthanide Macrocyclic Tetra-NHC Complexes. Inorg. Chem. 2022, 61, 1611–1619. [Google Scholar] [CrossRef]
- Okujima, T.; Inaba, H.; Mori, S.; Takase, M.; Uno, H. Synthesis of Azulitriphyrin(2.1.1). J. Porphyr. Phthalocyanines 2020, 24, 394–400. [Google Scholar]
- Larsen, S.; McCormick-McPherson, L.J.; Teat, S.J.; Ghosh, A. Azulicorrole. ACS Omega 2019, 4, 6737–6745. [Google Scholar] [CrossRef]
- Edwin, A.; Sulfikarali, T.; Kuppadakkath, G.; Gokulnath, S. Unprecedented Formation of Azulicorroles from the Scrambling of Azulitripyrrane. J. Porphyr. Phthalocyanines 2023. [Google Scholar] [CrossRef]
- Berlicka, A.; Sprutta, N.; Latos-Grażyński, L. Dithiaethyneazuliporphyrin—A Contracted Heterocarbaporphyrin. Chem. Commun. 2006, 42, 3346–3348. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.; Gryko, D.; Gryko, D.T. Synthesis of Corroles and Their Heteroanalogs. Chem. Rev. 2017, 117, 3102–3137, For further information of the synthesis and metalation of true corroles. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.E.; Alemayehu, A.B.; Conradie, J.; Beavers, C.M.; Ghosh, A. The Structural Chemistry of Metallocorroles: Combined X-ray Crystallography and Quantum Chemistry Studies Afford Unique Insights. Acc. Chem. Res. 2012, 45, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Aviv-Harel, I.; Gross, Z. Coordination Chemistry of Corroles with Focus on Main Group Elements. Coord. Chem. Rev. 2011, 255, 717–736. [Google Scholar] [CrossRef]
- Caulfield, K.P.; Conradie, J.; Arman, H.D.; Ghosh, A.; Tonzetich, Z.J. Iron(II) Corrole Anions. Inorg. Chem. 2019, 58, 15225–15235. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-C.; Fite, S.; Fridman, N.; Tumanskii, B.; Mahammed, A.; Gross, Z. Hydrogen Evolution Catalyzed by Corrole-Chelated Nickel Complexes, Characterized in all Catalysis Relevant Oxidation States. ACS Catal. 2022, 12, 4310–4317. [Google Scholar] [CrossRef]
- Fujino, K.; Hirata, Y.; Kawabe, Y.; Morimoto, T.; Srinivasan, A.; Toganoh, M.; Miseki, Y.; Kudo, A.; Furuta, H. Confusion and Neo-Confusion: Corrole Isomers with an NNNC Core. Angew. Chem. Int. Ed. 2011, 50, 6855–6859. [Google Scholar] [CrossRef] [PubMed]
- Toganoh, M.; Kawabe, Y.; Uno, H.; Furuta, H. Unique Interaction between Directly Linked Laminated π Planes in the Benzonorrole Dimer. Angew. Chem. Int. Ed. 2012, 51, 8753–8756. [Google Scholar] [CrossRef] [PubMed]
- Maurya, Y.K.; Ishikawa, T.; Kawabe, Y.; Ishida, M.; Toganoh, M.; Mori, S.; Yasutake, Y.; Fukatsu, S.; Furuta, H. Near-Infrared Phosphorescent Iridium(III) Benzonorrole Complexes Possessing Pyridine-based Axial Ligands. Inorg. Chem. 2016, 55, 6223–6230. [Google Scholar] [CrossRef] [PubMed]
- Skonieczny, J.; Latos-Grażyński, L.; Szterenberg, L. Reactivityof Silole within a Core-Modified Porphyrin Environment: Synthesis of 21-Silaphlorin and its Conversion to Carbacorrole. Chem. Eur. J. 2008, 14, 4861–4874. [Google Scholar] [CrossRef]
- Basumatary, B.; Hashiguchi, I.; Mori, S.; Shimizu, S.; Ishida, M.; Furuta, H. Copper 1,19-Diaza-21,24-dicarbacorrole: A Corrole Analogue with an N−N Linkage Stabilizes a Ground-State Singlet Organocopper Species. Angew. Chem. Int. Ed. 2020, 59, 15897–15901. [Google Scholar] [CrossRef] [PubMed]
- Adinarayana, B.; Thomas, A.P.; Suresh, C.H.; Srinivasan, A. A 6,11,16-Triarylbiphenylcorrole with an adj-CCNN Core: Stabilization of an Organocopper(III) Complex. Angew. Chem. Int. Ed. 2015, 54, 10478–10482. [Google Scholar] [CrossRef] [PubMed]
- Chitranshi, S.; Adinarayana, B.; Das, M.; Anila, S.; Suresh, C.H.; Srinivasan, A. Rh(I) and Organo-Rh(III) Complexes of meso-Triarylbiphenylcorrole. Inorg. Chem. 2023, 62, 336–341. [Google Scholar] [CrossRef]
- Szyszko, B. Phenanthrene-Embedded Carbaporphyrinoids and Related Systems: From Ligands to Cages and Molecular Switches. Eur. J. Org. Chem. 2022, 2022, e202200714. [Google Scholar] [CrossRef]
- Szyszko, B.; Bialonska, A.; Szterenberg, L.; Latos-Grażyński, L. Phenanthriporphyrin: An Antiaromatic Aceneporphyrinoid as a Ligand for a Hypervalent Organophosphorus(V) Moiety. Angew. Chem. Int. Ed. 2015, 54, 4932–4936. [Google Scholar] [CrossRef]
- Kupietz, K.; Białek, M.J.; Hassa, K.; Białońska, A.; Latos-Grażyński, L. Oxygenation of Phenanthriporphyrin and Copper(III) Phenanthriporphyrin: An Efficient Route to Phenanthribilinones. Inorg. Chem. 2019, 58, 12446–12456. [Google Scholar] [CrossRef]
- Kupietz, K.; Białek, M.J.; Białońska, A.; Szyszko, B.; Latos-Grażyński, L. Aromaticity Control via Modifications of a Macrocyclic Frame: 5,6-Dimethoxyphenanthriporphyrin and 5,6-Dioxophenanthriporphyrin. Org. Chem. Front. 2018, 5, 3068–3076. [Google Scholar] [CrossRef]
- Kupietz, K.; Białek, M.J.; Szyszko, B.; Sarwa, A.; Latos-Grażyński, L. Phenanthrene Cyclocarbonylation—Core Post-synthetic Modification of Phenanthriporphyrin. Org. Chem. Front. 2022, 9, 2968–2976. [Google Scholar] [CrossRef]
- Ke, X.-S.; Hong, Y.; Tu, P.; He, Q.; Lynch, V.M.; Kim, D.; Sessler, J.L. Hetero Cu(III)–Pd(II) Complex of a Dibenzo[g,p]chrysene-Fused Bis-dicarbacorrole with Stable Organic Radical Character. J. Am. Chem. Soc. 2017, 139, 15232–15238. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.-S.; Hong, Y.; Lynch, V.M.; Kim, D.; Sessler, J.L. Metal-Stabilized Quinoidal Dibenzo[g,p]chrysene-Fused Bis-dicarbacorrole System. J. Am. Chem. Soc. 2018, 140, 7579–7586. [Google Scholar] [CrossRef]
- Berlicka, A.; Latos-Grażyński, L.; Lis, T. Dithiaethyneporphyrin: An Atypical [18]Triphyrin(4.1.1) Frame for Contracted Porphyrins. Angew. Chem. Int. Ed. 2005, 44, 5288–5291. [Google Scholar] [CrossRef] [PubMed]
- Nojman, E.; Berlicka, A.; Szterenberg, L.; Latos-Grażyński, L. Nickel(II) and Palladium(II) Thiaethyneporphyrins. Intramolecular Metal(II)-η2-CC Interaction inside a Porphyrinoid Frame. Inorg. Chem. 2012, 51, 3247–3260. [Google Scholar] [CrossRef]
- Sinha, A.; Kumar, A.; Ravikanth, M. Synthesis, Structure, Spectral, and Anion Sensing Studies of an Aromatic meso-Fused Boron(III) Benzitriphyrin(2.1.1) Complex. Inorg. Chem. 2021, 60, 18094–18102. [Google Scholar] [CrossRef]
- Pacholska-Dudziak, E.; Ulatowski, F.; Ciunik, Z.; Latos-Grażyński, L. N-Fusion Approach in Construction of Contracted Carbaporphyrinoids: Formation of N-Fused Telluraporphyrin. Chem. Eur. J. 2009, 15, 10924–10929. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D.; Richter, D.T. Synthesis of the First Expanded Carbaporphyrinoid by the “4 + 1” Approach. J. Am. Chem. Soc. 1998, 120, 9965–9966. [Google Scholar] [CrossRef]
- Richter, D.T.; Lash, T.D. Oxidation with Dilute Aqueous Ferric Chloride Solutions Greatly Improves Yields in the “4 + 1” Synthesis of Sapphyrins. Tetrahedron Lett. 1999, 40, 6735–6738. [Google Scholar] [CrossRef]
- Richter, D.T.; Lash, D.T. Synthesis of Sapphyrins, Heterosapphyrins and Carbasapphyrins by a “4 + 1” Approach. J. Org. Chem. 2004, 69, 8842–8850. [Google Scholar] [CrossRef] [PubMed]
- Sessler, J.L.; Davis, J.M. Sapphyrins: Versatile Anion Binding Agents. Acc. Chem. Res. 2001, 34, 989–997, For further information on the synthesis and metalation of true sapphyrins and hetero-analogues. [Google Scholar] [CrossRef]
- Chatterjee, T.; Srinivasan, A.; Ravikanth, M.; Chandrasekar, T.K. Smaragdyrins and Sapphyrins Analogues. Chem. Rev. 2017, 117, 3329–3376. [Google Scholar] [CrossRef]
- Chen, Q.-C.; Soll, M.; Mizrahi, A.; Saltsman, I.; Fridman, N.; Saphier, M.; Gross, Z. One-Pot Synthesis of Contracted and Expanded Porphyrins with meso-CF3 Groups. Angew. Chem. Int. Ed. 2018, 57, 1006–1010. [Google Scholar] [CrossRef]
- Chen, Q.-C.; Fridman, N.; Diskin-Posner, Y.; Gross, Z. Palladium Complexes of Corroles and Sapphyrins. Chem. Eur. J. 2020, 26, 9481–9485. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Bolin, G.; Moreau, J.J.E.; Vernhet, C. A Facile Synthesis of New Tetrapyrrole Macrocyclic Derivatives. Formation of Bimetallic Transition Metal Complexes. J. Chem. Soc., Chem. Commun. 1991, 27, 211–213. [Google Scholar] [CrossRef]
- Carré, F.H.; Corriu, R.J.P.; Bolin, G.; Moreau, J.J.E.; Vernhet, C. Aminosilanes in Organic Synthesis. Preparation of New Expanded Porphyrin Ligands and Bimetallic Transition-Metal Complexes. Crystal Structure of a Tetrapyrrole Macrocycle Dirhodium Complex. Organometallics 1993, 12, 2478–2486. [Google Scholar] [CrossRef]
- Peterson, G.R.; Bampos, N. One-Pot Synthesis of Indene-Expanded Porphyrins. Angew. Chem. Int. Ed. 2010, 49, 3930–3933. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Osuka, A. Aromatic and Antiaromatic Gold(III) Hexaphyrins with Multiple Gold−Carbon Bonds. J. Am. Chem. Soc. 2005, 127, 8030–8031. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Kim, K.S.; Yoon, Z.S.; Noh, S.B.; Kim, D.; Osuka, A. Peripheral Fabrications of a Bis-Gold(III) Complex of [26]-Hexaphyrin(1.1.1.1.1.1) and Aromatic versus Antiaromatic Effect on Two-Photon Absorption Cross Section. J. Am. Chem. Soc. 2007, 129, 11344–11345. [Google Scholar] [PubMed]
- Muranaka, A.; Matsushita, O.; Yoshida, K.; Mori, S.; Suzuki, M.; Furuyama, T.; Uchiyama, M.; Osuka, A.; Kobayashi, N. Application of the Perimeter Model to the Assignment of the Electronic Absorption Spectra of Gold(III) Hexaphyrins with [4n + 2] and [4n] π-Electron Systems. Chem. Eur. J. 2009, 15, 3744–3751. [Google Scholar] [CrossRef]
- Mori, S.; Osuka, A. Heterobismetal Complexes of [26]-Hexaphyrin(1.1.1.1.1.1). Inorg. Chem. 2008, 47, 3937–3939. [Google Scholar] [CrossRef]
- Naoda, K.; Mori, H.; Osuka, A. GoldIII-IridiumIII Hybrid Complexes of Hexaphyrin(1.1.1.1.1.1). Chem. Asian J. 2013, 8, 1395–1398. [Google Scholar] [CrossRef]
- Mori, S.; Shimizu, S.; Taniguchi, R.; Osuka, A. Group 10 Metal Complexes of meso-Aryl-Substituted [26]Hexaphyrins with a Metal- Carbon Bond. Inorg. Chem. 2005, 44, 4127–4129. [Google Scholar] [CrossRef]
- Tanaka, T.; Sugita, T.; Tokuji, S.; Saito, S.; Osuka, A. Metal Complexes of Chiral Mobius Aromatic [28]Hexaphyrin(1.1.1.1.1.1): Enantiomeric Separation, Absolute Stereochemistry, and Asymmetric Synthesis. Angew. Chem. Int. Ed. 2010, 49, 6619–6621. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Osuka, A. Redox-Induced Palladium Migrations that Allow Reversible Topological Changes between Palladium(II) Complexes of Mobius Aromatic [28]Hexaphyrin and Huckel Aromatic [26]Hexaphyrin. Angew. Chem. Int. Ed. 2010, 49, 9488–9491. [Google Scholar] [CrossRef]
- Yoneda, T.; Osuka, A. Synthesis of a [26]Hexaphyrin Bis-PdII Complex with a Characteristic Aromatic Circuit. Chem. Eur. J. 2013, 19, 7314–7318. [Google Scholar] [CrossRef]
- Gokulnath, S.; Yamaguchi, K.; Toganoh, M.; Mori, S.; Uno, H.; Furuta, H. Singly N-Confused [26]Hexaphyrin: A Binucleating Porphyrinoid Ligand for Mixed Metals in Different Oxidation States. Angew. Chem. Int. Ed. 2011, 50, 2302–2306. [Google Scholar] [CrossRef]
- Liu, L.; Hu, Z.; Zhang, F.; Liu, Y.; Xu, L.; Zhou, M.; Tanaka, T.; Osuka, A. Benzene- and Pyridine-incorporated Octaphyrins with different Coordination Modes toward two PdII Centers. Nat. Commun. 2020, 11, 6202. [Google Scholar] [CrossRef]
- Szyszko, B.; Sprutta, N.; Chwalisz, P.; Stepień, M.; Latos-Grażyński, L. Huckel and Mobius Expanded para-Benziporphyrins: Synthesis and Aromaticity Switching. Chem. Eur. J. 2014, 20, 1985–1997. [Google Scholar] [CrossRef]
- Tanaka, Y.; Saito, S.; Mori, S.; Aratani, N.; Shinokubo, H.; Shibata, N.; Higuchi, Y.; Yoon, Z.S.; Kim, K.S.; Noh, S.B.; et al. Metalation of Expanded Porphyrins: A Chemical Trigger used to Produce Molecular Twisting and Mobius Aromaticity. Angew. Chem. Int. Ed. 2008, 47, 681–684. [Google Scholar] [CrossRef]
- Simkowa, I.; Latos-Grażyński, L.; Stępień, M. π Conjugation Transmitted Across a d-Electron Metallocene in Ferrocenothiaporphyrin Macrocycles. Angew. Chem. Int. Ed. 2010, 49, 7665–7669. [Google Scholar] [CrossRef]
- Grocka, I.; Latos-Grażyński, L.; Stępień, M. Ruthenocenoporphyrinoids: Conformation Determines Macrocyclic π Conjugation Transmitted Across a d-Electron Metallocene. Angew. Chem. Int. Ed. 2013, 52, 1044–1048. [Google Scholar] [CrossRef]
- Pacholska-Dudziak, E.; Szczepaniak, M.; Ksiazek, A.; Latos-Grażyński, L. A Porphyrin Skeleton Containing a Palladacyclopentadiene. Angew. Chem. Int. Ed. 2013, 52, 8898–8903. [Google Scholar] [CrossRef]
- Pacholska-Dudziak, E.; Vetter, G.; Goratowska, A.; Bialonska, A.; Latos-Grażyński, L. Chemistry Inside a Porphyrin Skeleton: Platinacyclopentadiene from Tellurophene. Chem. Eur. J. 2020, 26, 16011–16018. [Google Scholar] [CrossRef]
- Vetter, G.; Białońska, A.; Krzyszowska, P.; Koniarz, S.; Pacholska-Dudziak, E. Two Rhodium(III) Ions Confined in a [18]Porphyrin Frame: 5,10,15,20-Tetraaryl-21,23-Dirhodaporphyrin. Chem. Eur. J. 2022, 28, e202201513. [Google Scholar] [CrossRef]
- Rao, Y.; Kim, T.; Park, K.H.; Peng, F.; Liu, L.; Liu, Y.; Wen, B.; Liu, S.; Kirk, S.R.; Wu, L.; et al. π-Extended “Earring” Porphyrins with Multiple Cavities and Near-Infrared Absorption. Angew. Chem. Int. Ed. 2016, 55, 6438–6442. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, F.; Rao, Y.; Wen, B.; Xu, L.; Zhou, M.; Tanaka, T.; Osuka, A.; Song, J. Synthesis, Structures and Near-IR Absorption of Heterole-Fused Earring Porphyrins. Angew. Chem. Int. Ed. 2019, 58, 8124–8128. [Google Scholar] [CrossRef]
- Song, J.; Aratani, N.; Heo, J.H.; Kim, D.; Shinokubo, H.; Osuka, A. Directly Pd(II)-Bridged Porphyrin Belts with Remarkable Curvatures. J. Am. Chem. Soc. 2010, 132, 11868–11869. [Google Scholar] [CrossRef]
- Yin, B.; Deng, F.; Zhou, W.; Rao, Y.; Xu, L.; Zhou, M.; Osuka, A.; Song, J. PdII and NiII Complexes of Earring Porphyrins with Distorted Coordination Geometries. Asian J. Org. Chem. 2022, 2000, e202200556. [Google Scholar]
- Fields, K.B.; Engle, J.T.; Sripothongnak, S.; Kim, C.; Zhang, X.P.; Ziegler, C.J. Cobalt Carbaporphyrin-catalyzed Cyclopropanation. Chem. Commun. 2011, 47, 749–751. [Google Scholar] [CrossRef]
- Jay-ar, B.; Hung, C.H. Ni and Zn N-confused Porphyrin Complexes as Recyclable Catalysts for High Efficiency Solvent-free CO2 fixation into Cyclic Carbonates. Catal. Sci. Technol. 2021, 11, 2144–2154. [Google Scholar]
- Babu, B.; Mack, J.; Nyokong, T. Sn(IV) N-confused Porphyrins as Photosensitizer Dyes for Photodynamic Therapy in the Near IR Region. Dalton Trans. 2020, 49, 15180–15183. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Engelmann, F.M.; Mayer, I.; Toma, H.E.; Baptista, M.S.; Maeda, H.; Osuka, A.; Furuta, H. Doubly N-Confused Porphyrins as Efficient Sensitizers for Singlet Oxygen Generation. Chem. Lett. 2003, 32, 244–245. [Google Scholar] [CrossRef]






































































































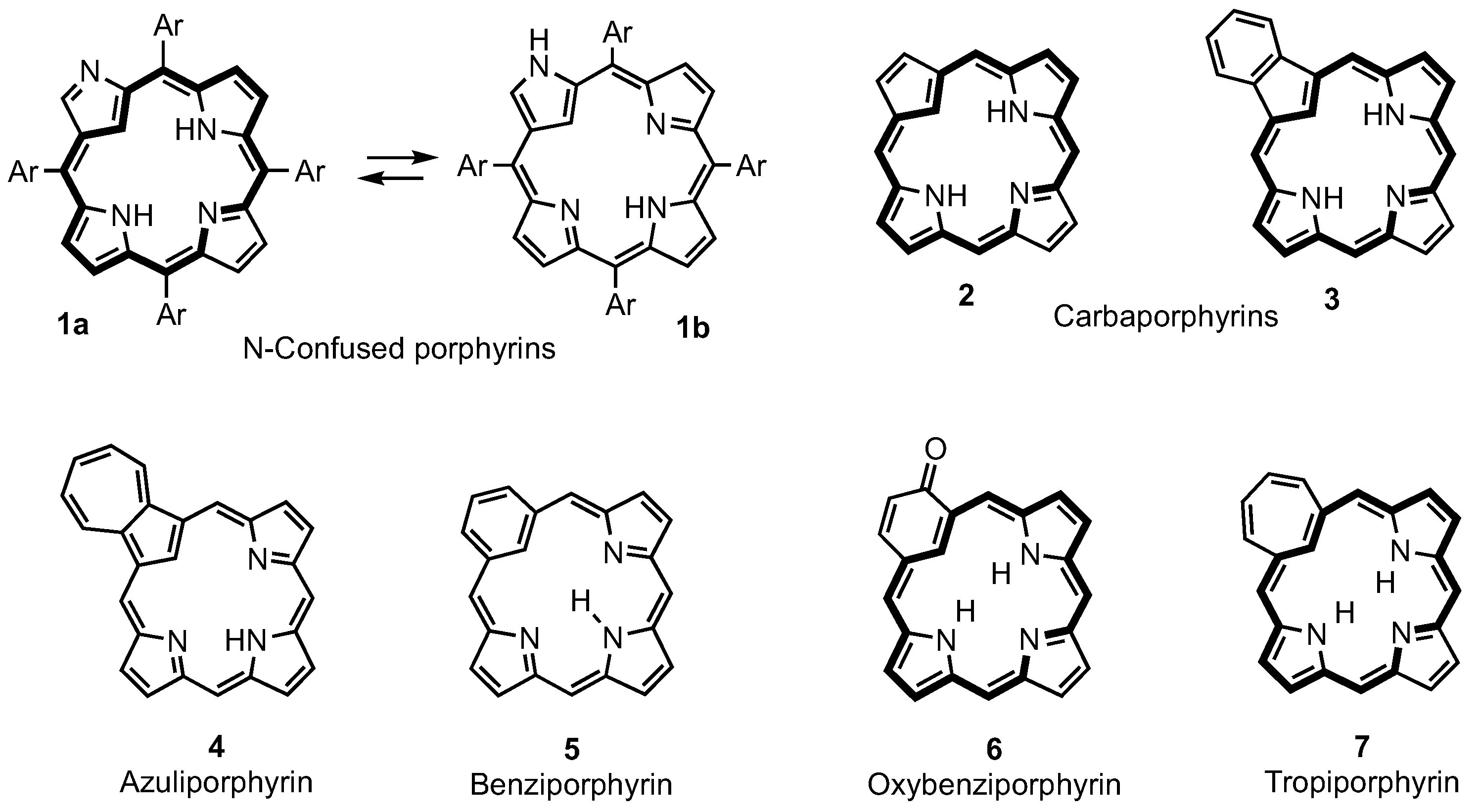{kind=link}
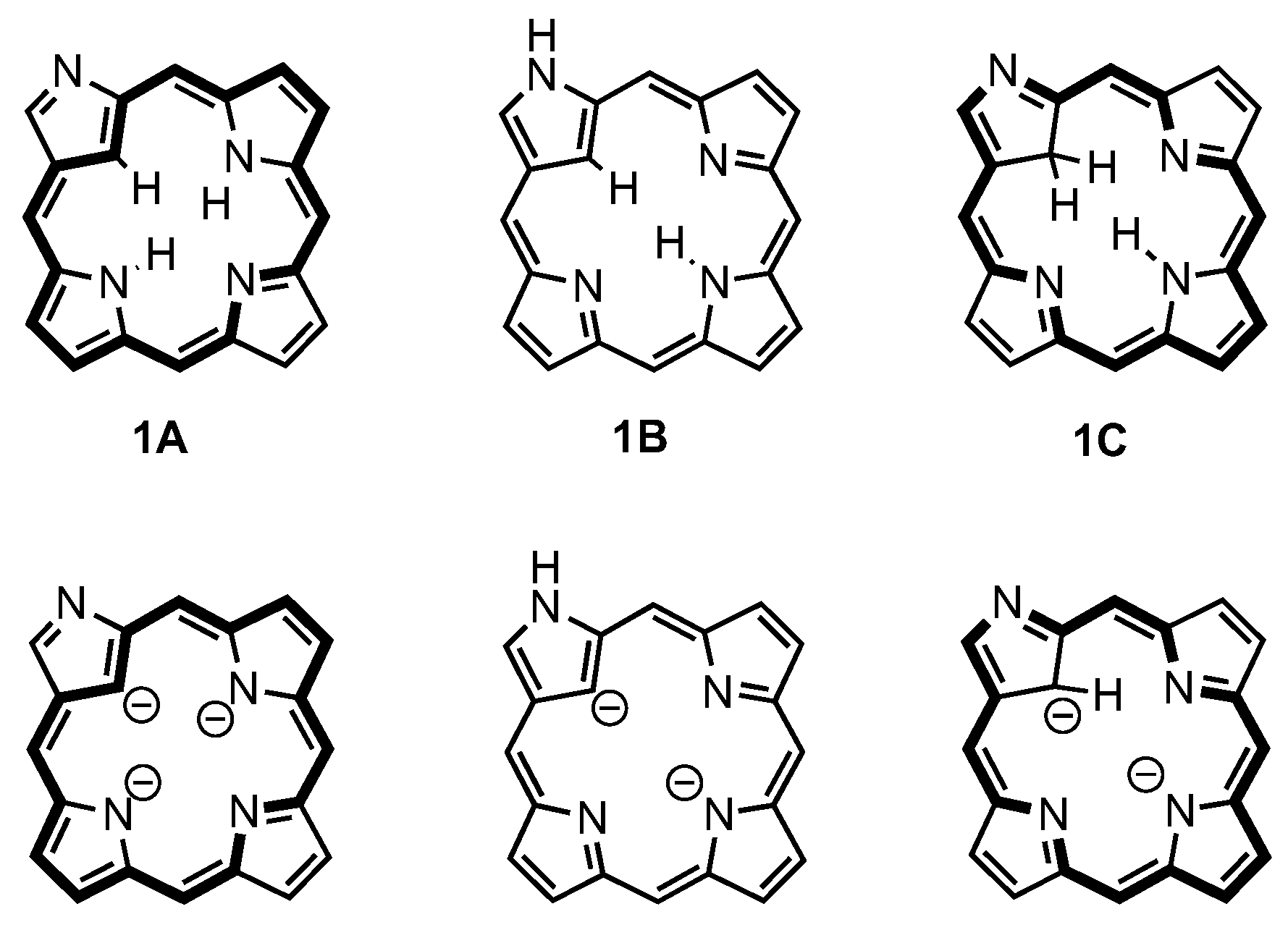{kind=link}
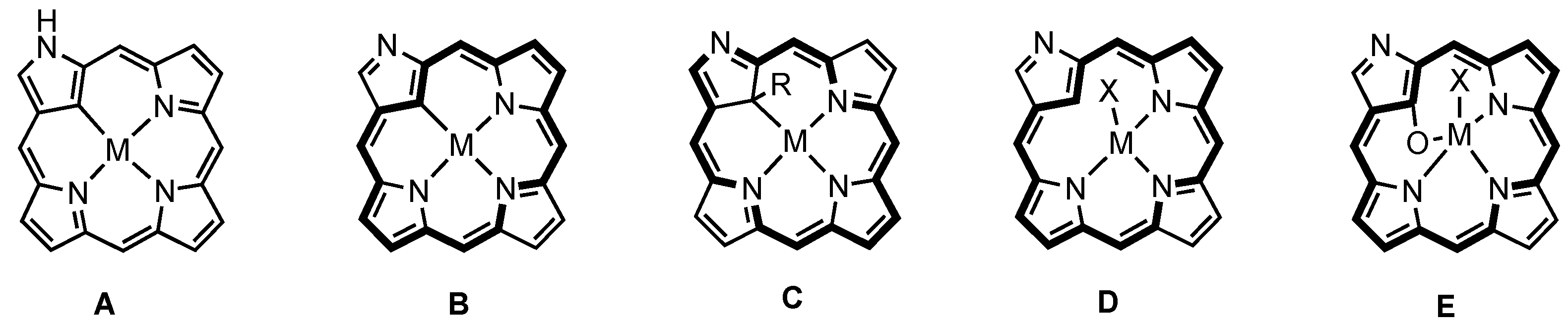{kind=link}
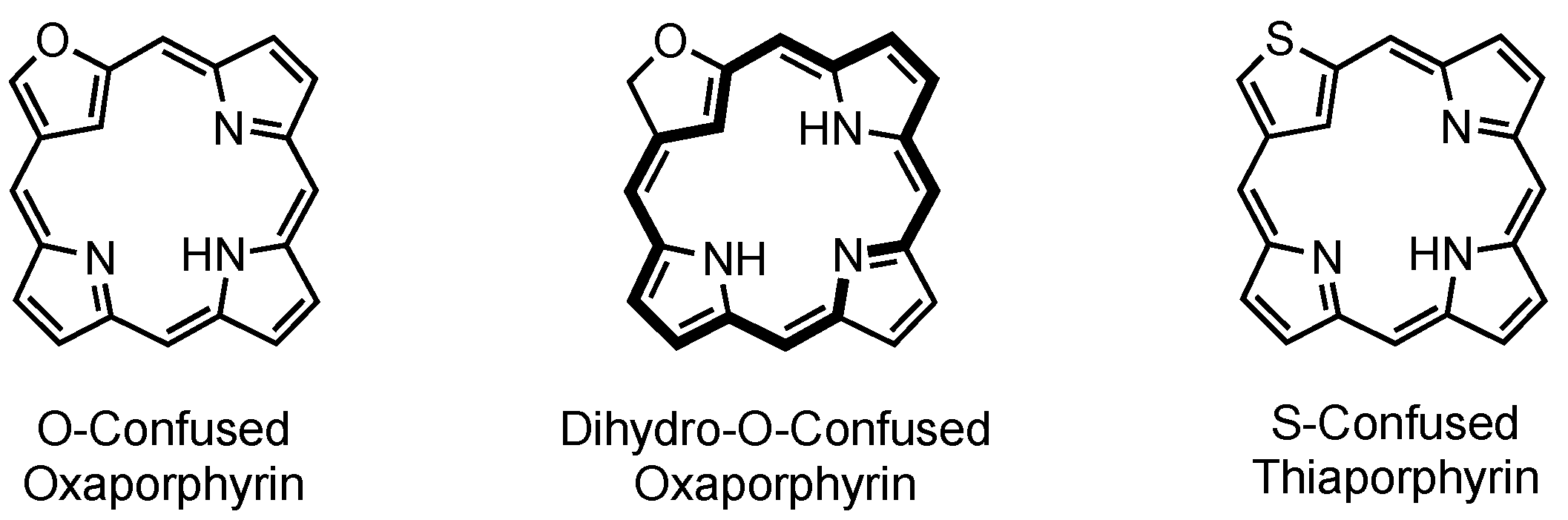{kind=link}
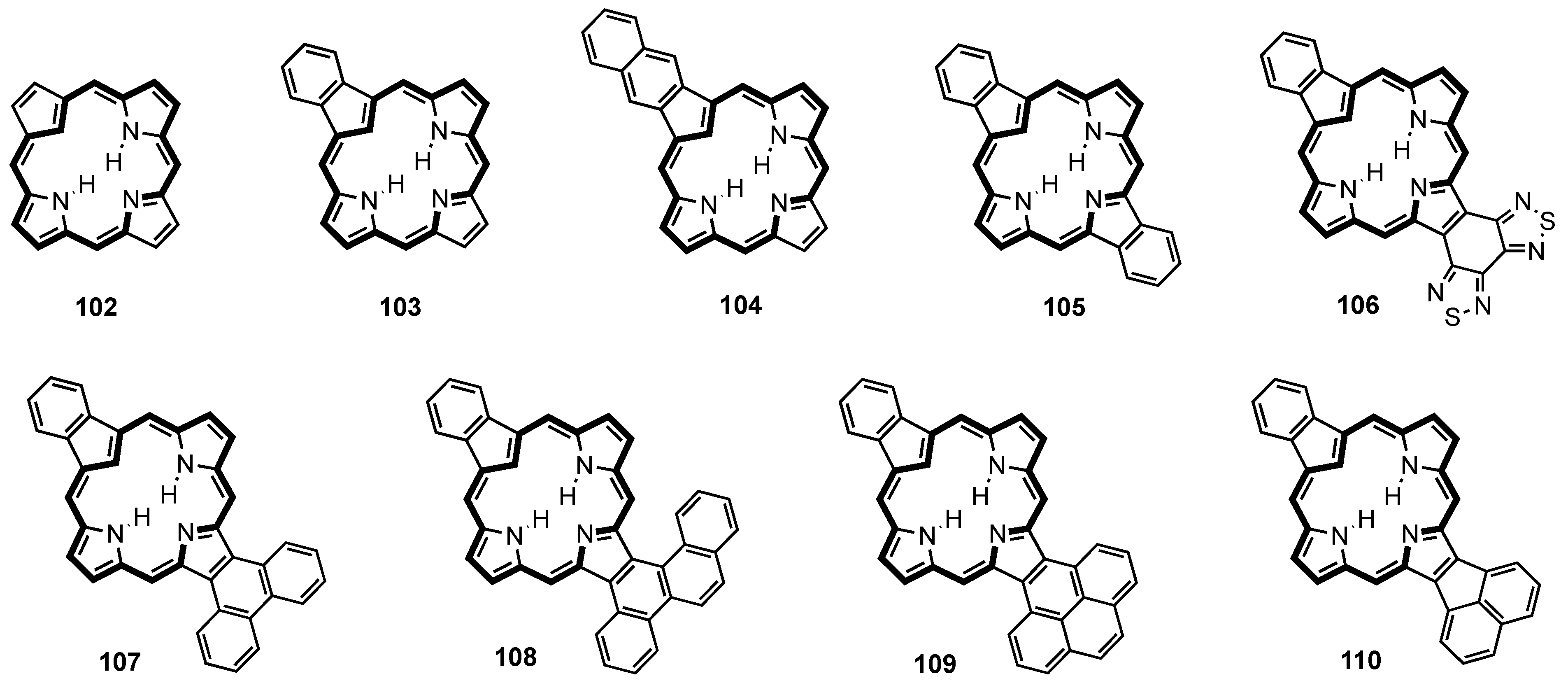{kind=link}
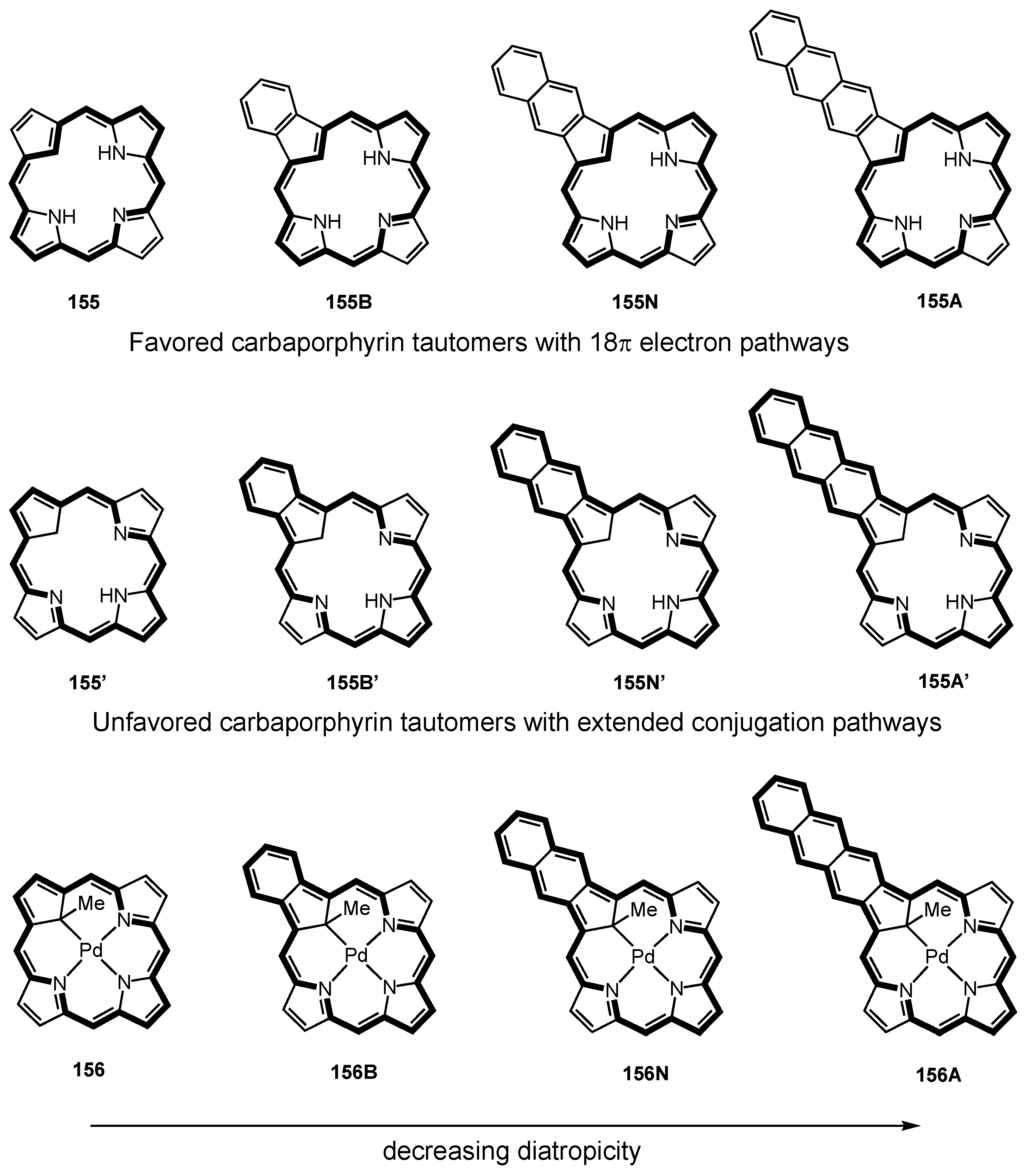{kind=link}
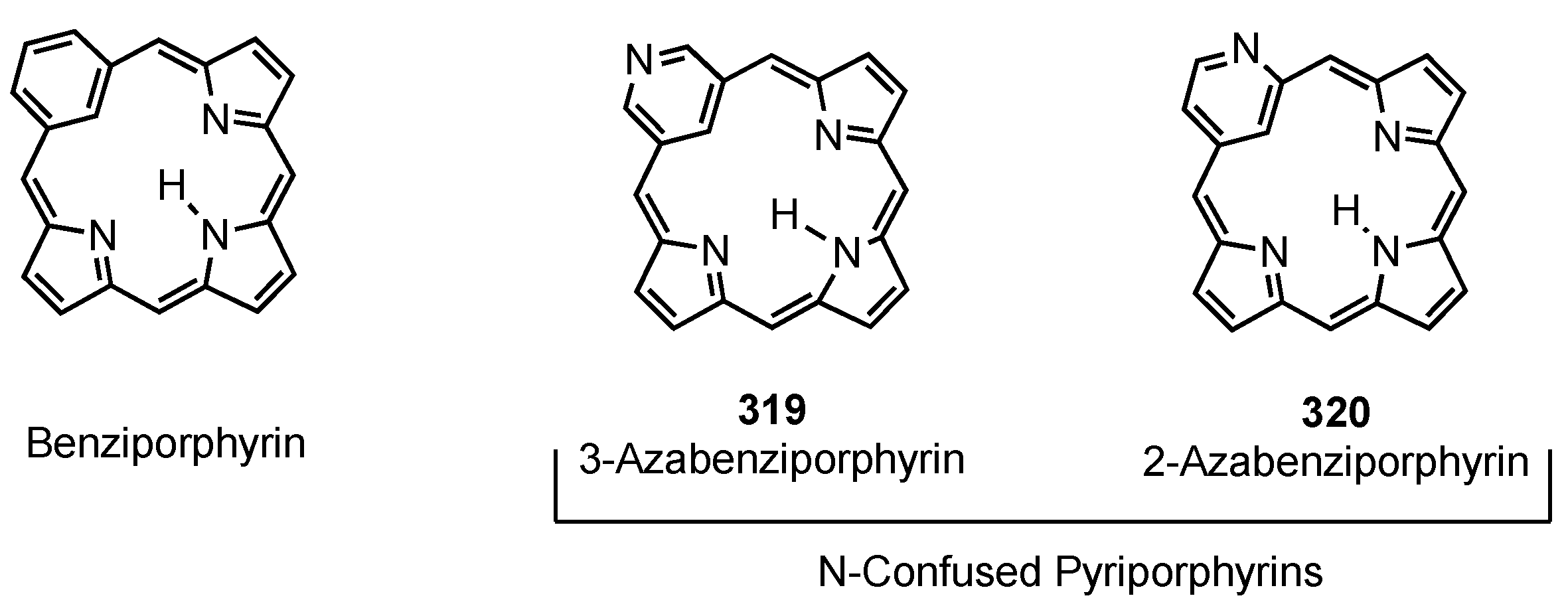{kind=link}
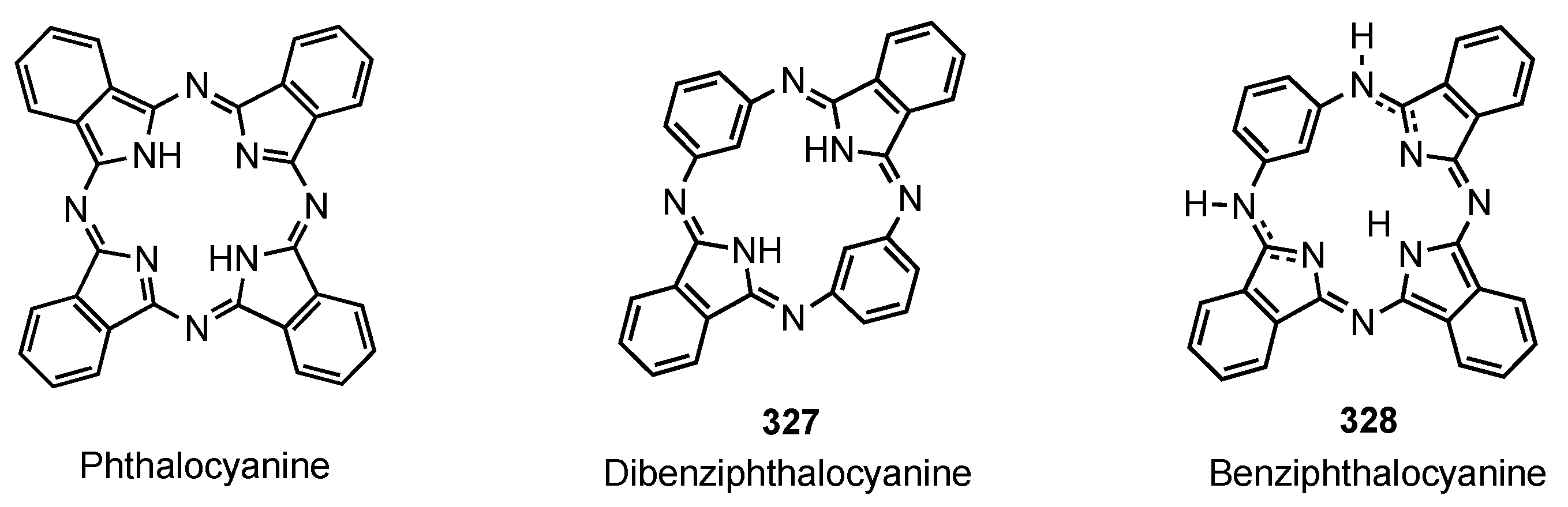{kind=link}
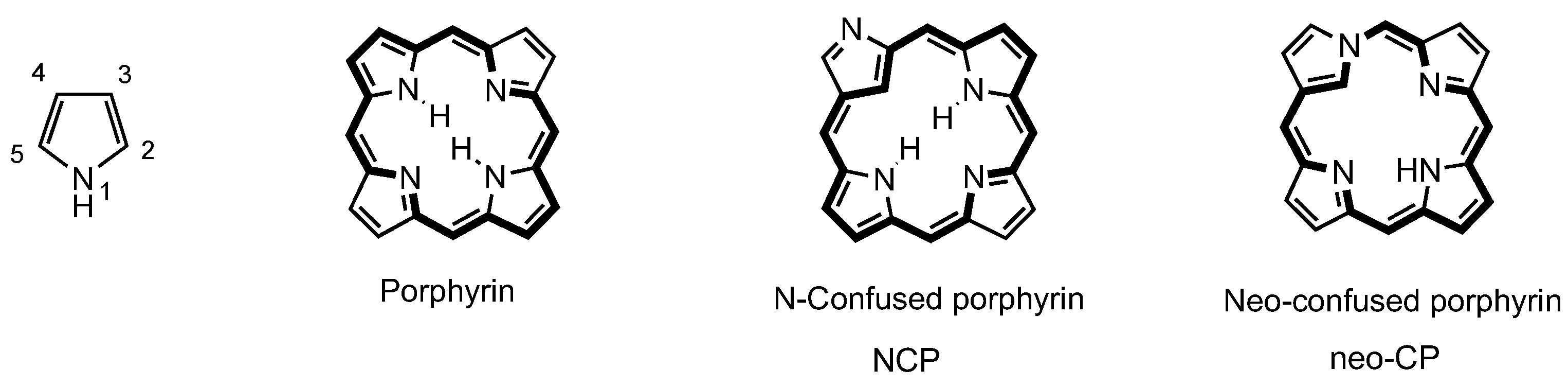{kind=link}
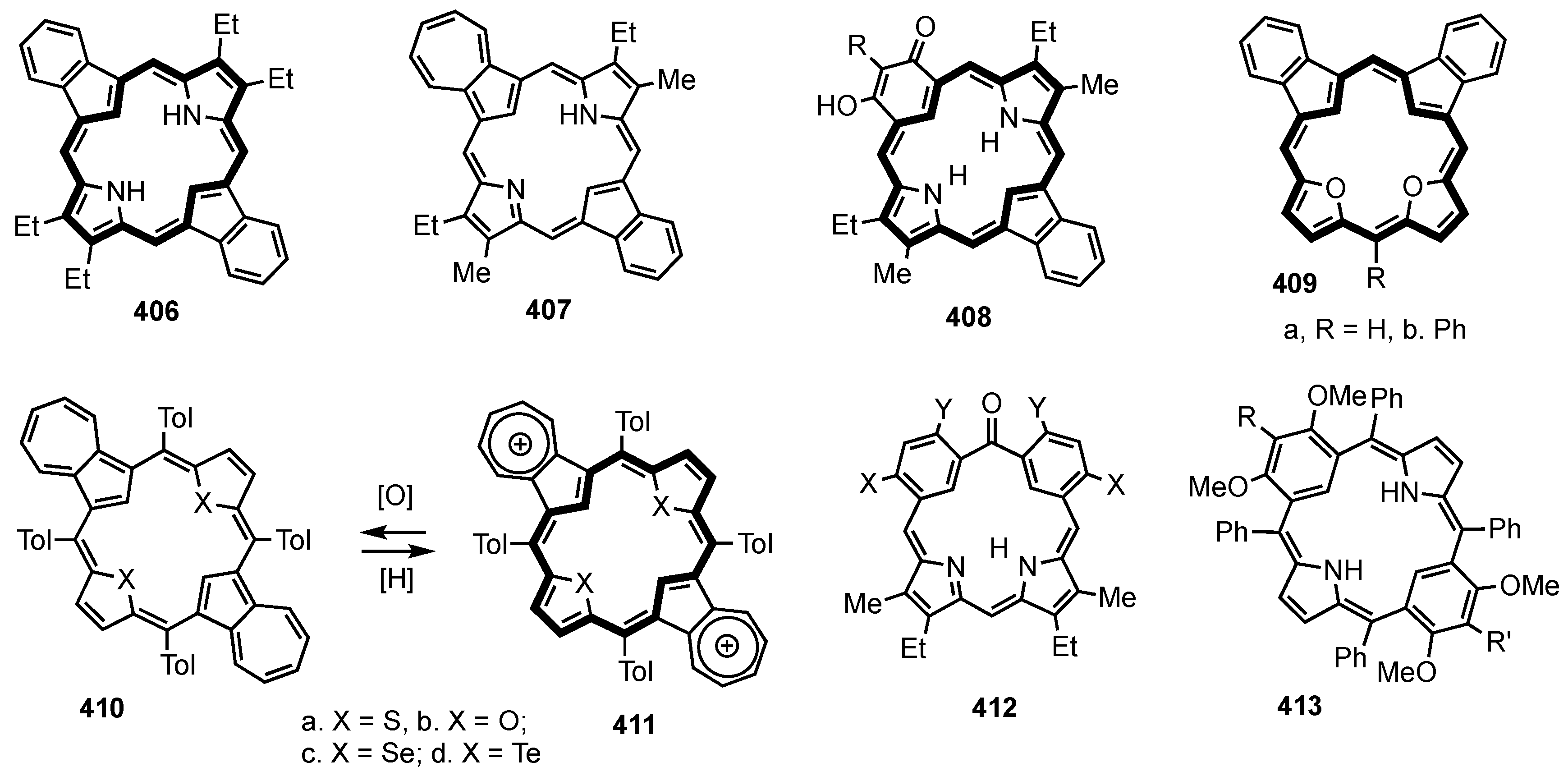{kind=link}
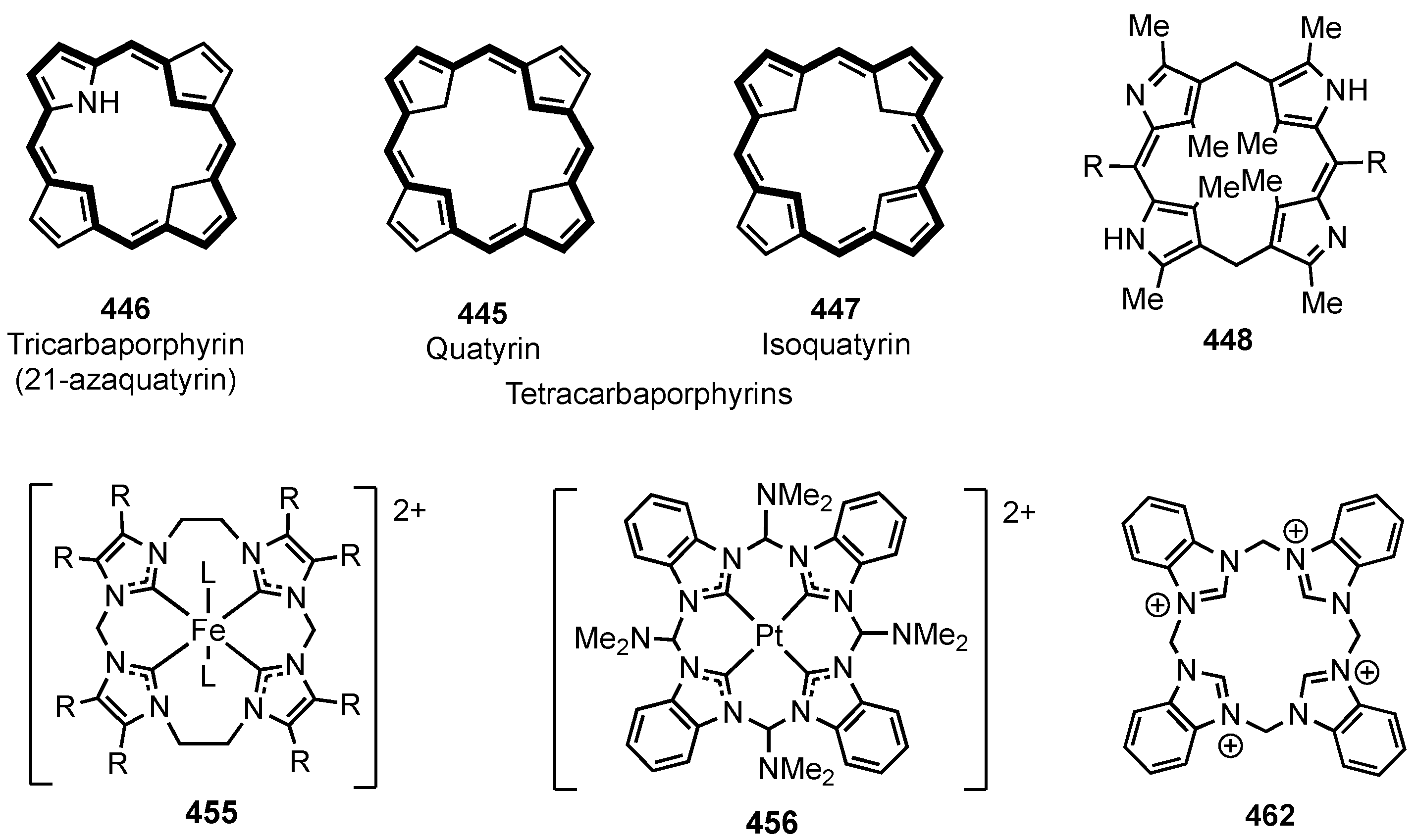{kind=link}
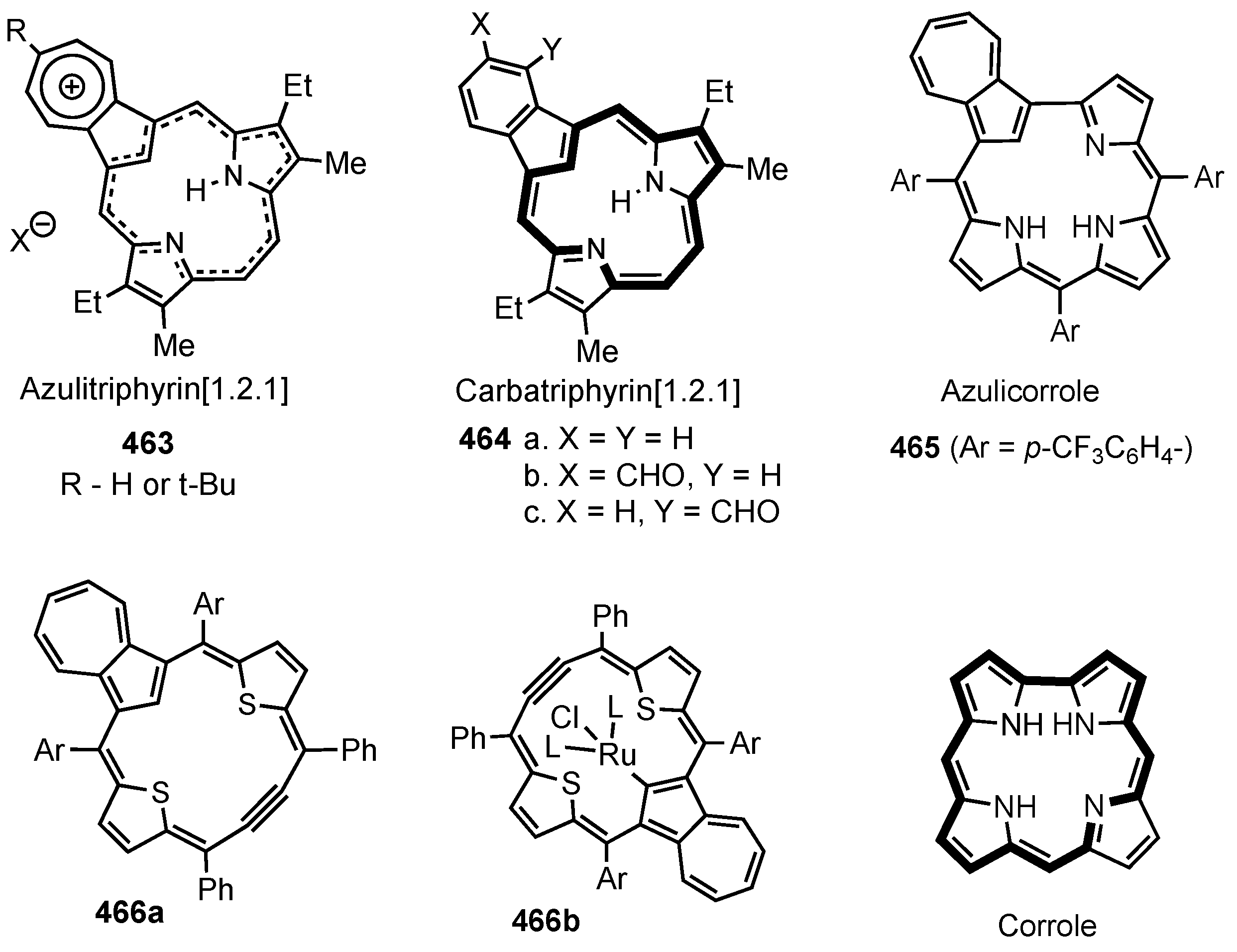{kind=link}
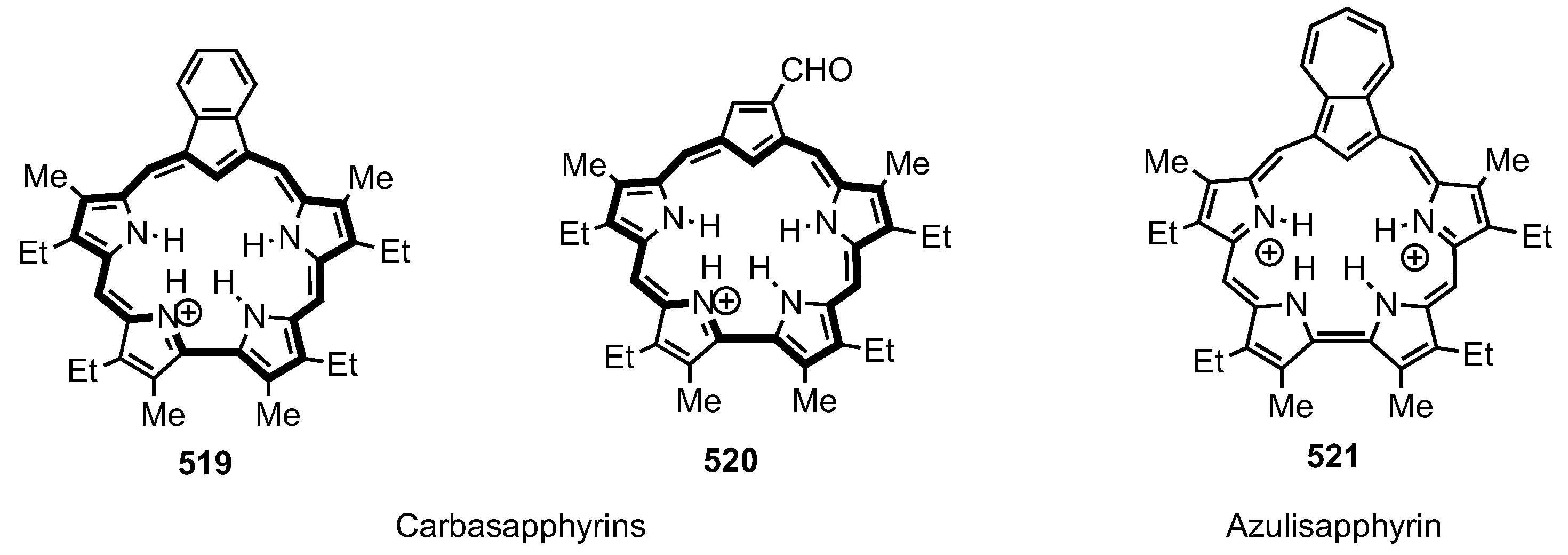{kind=link}
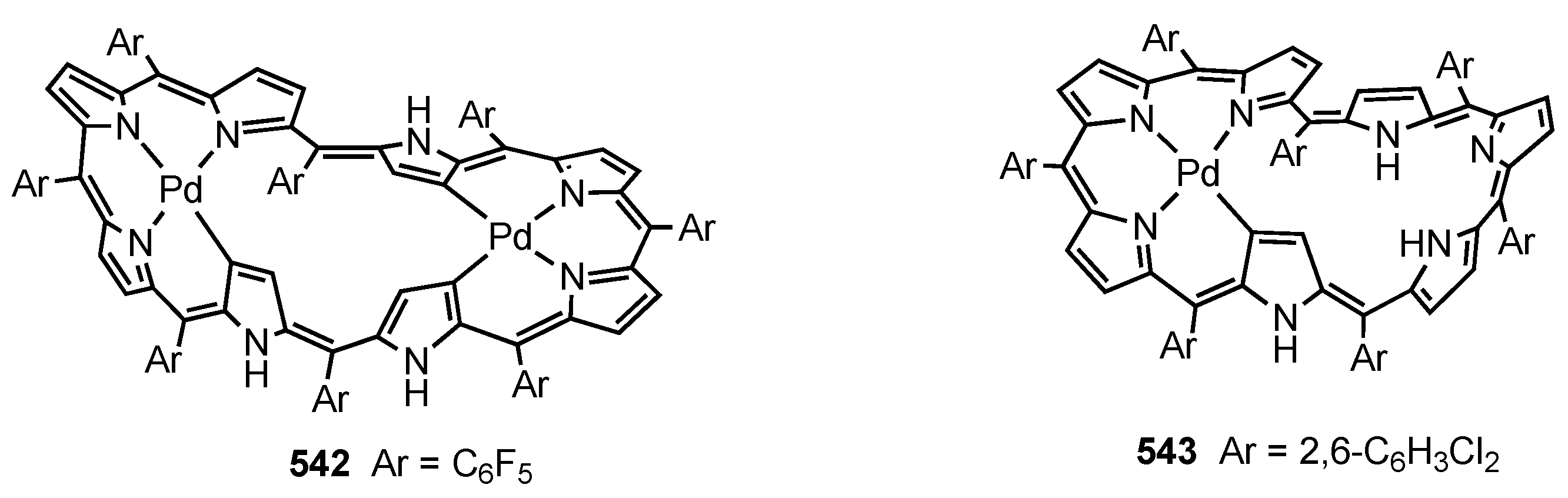{kind=link}
{kind=link}
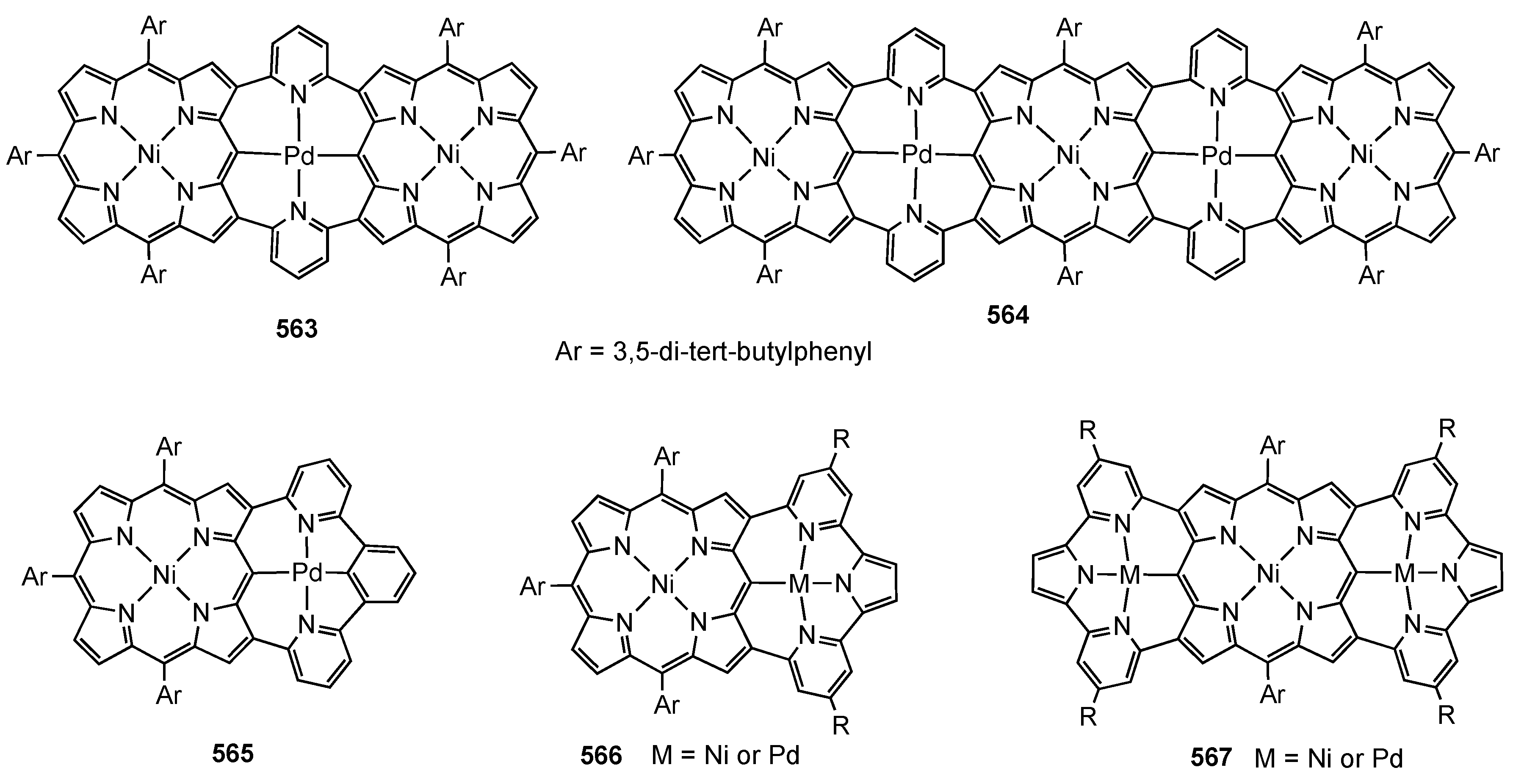{kind=link}
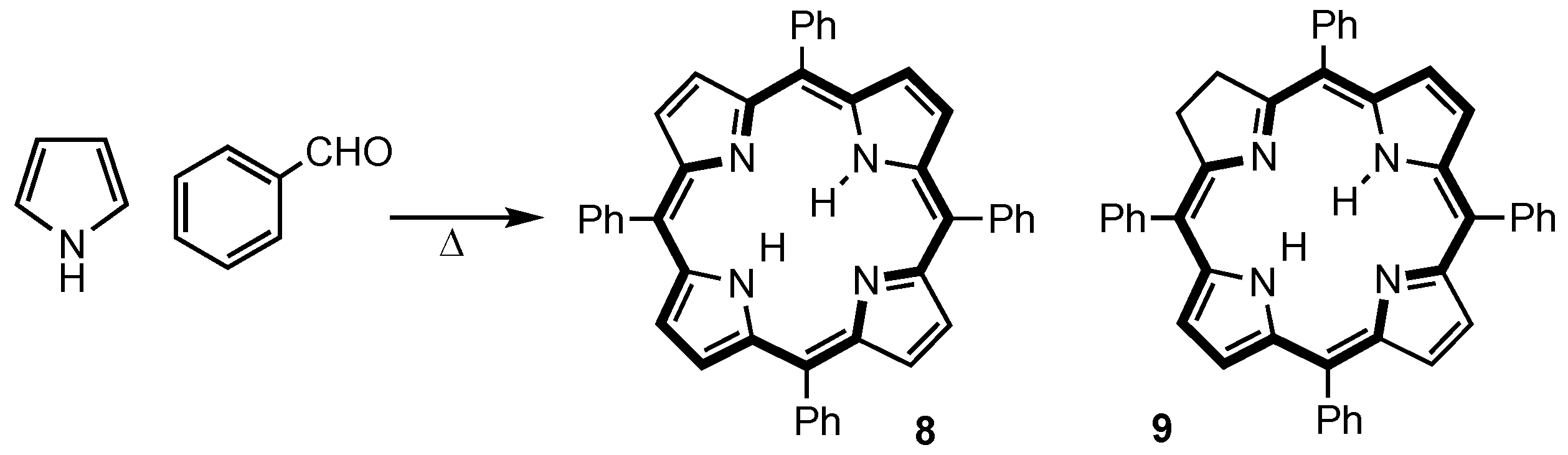{kind=link}
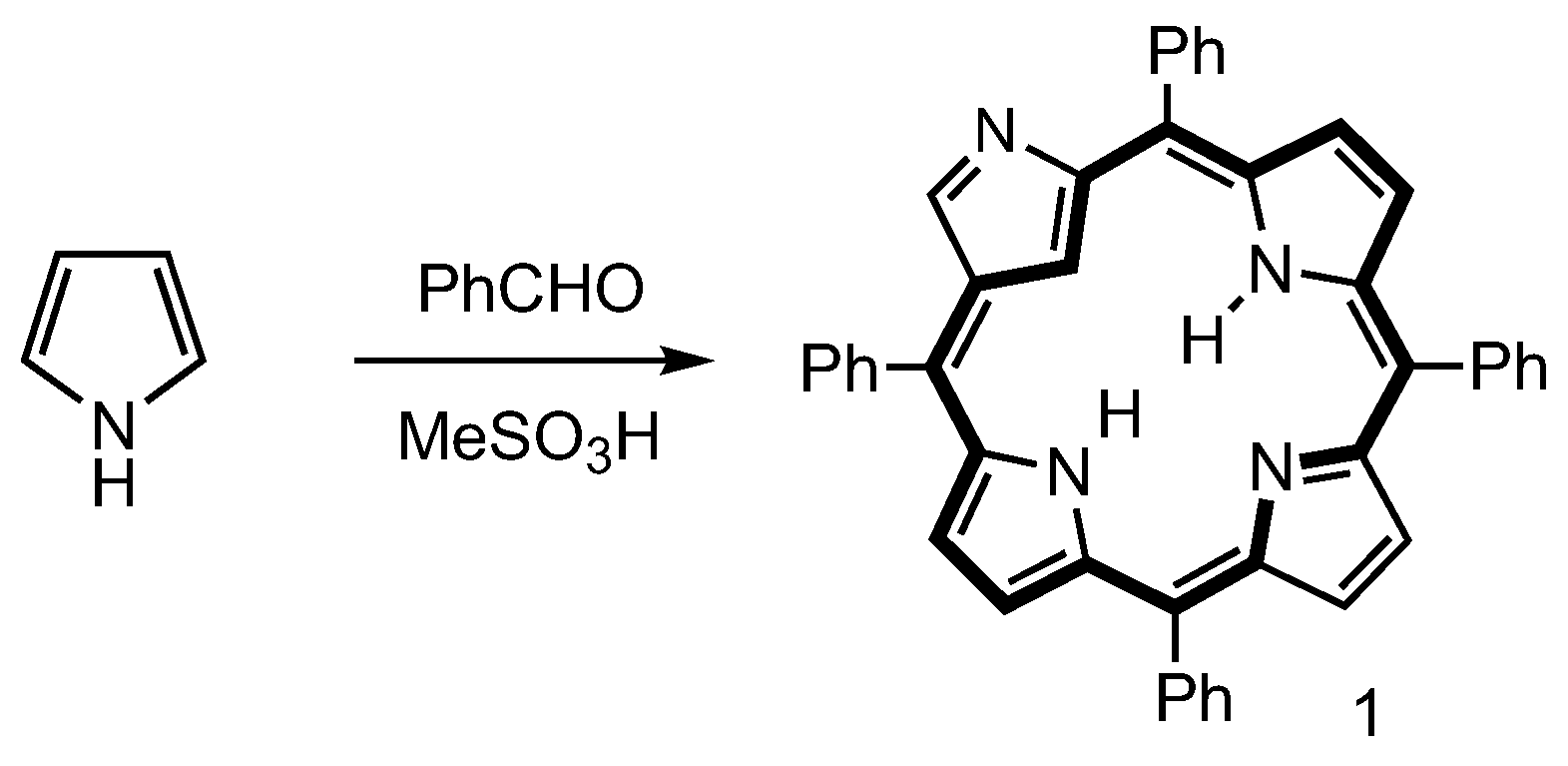{kind=link}
{kind=link}
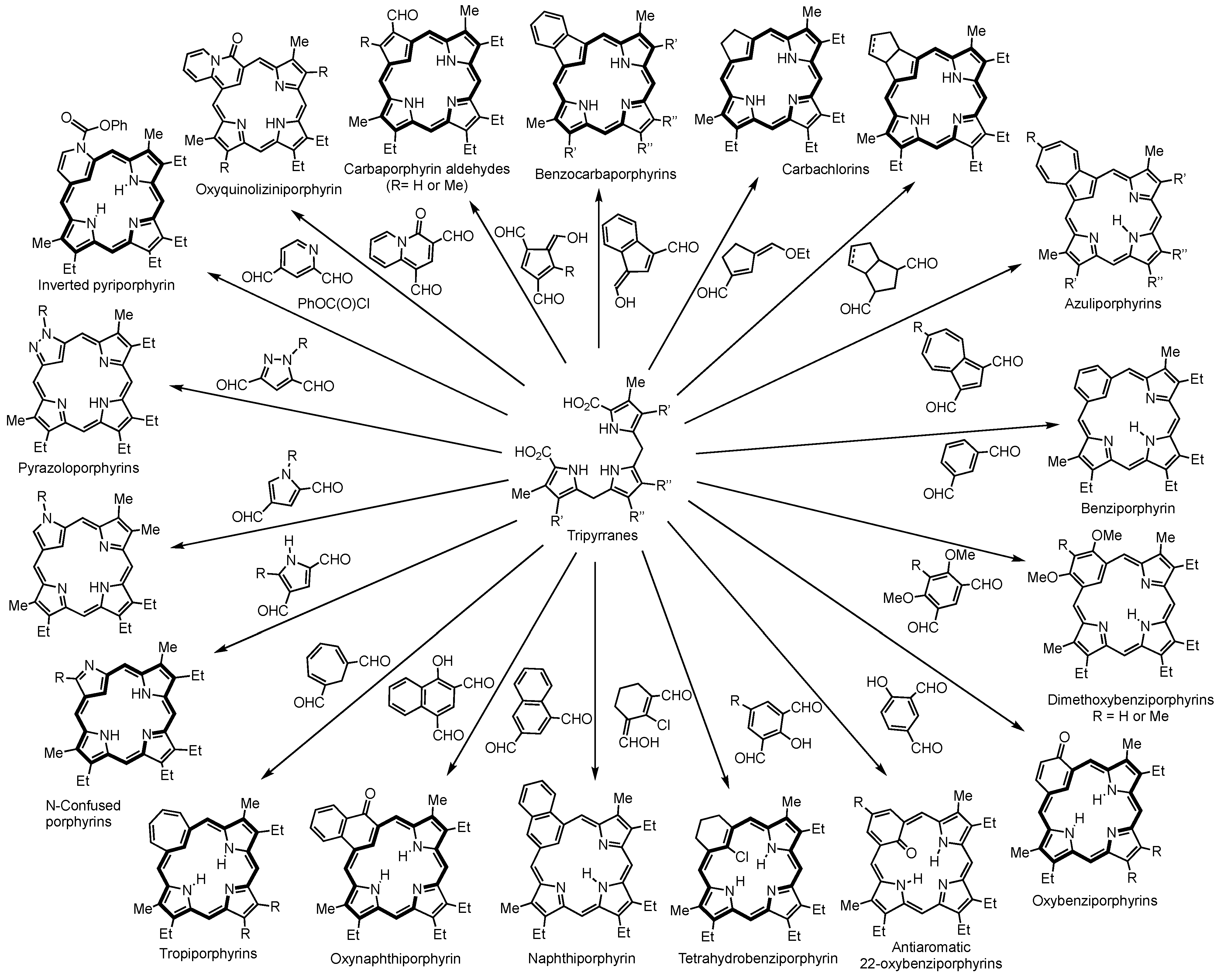{kind=link}
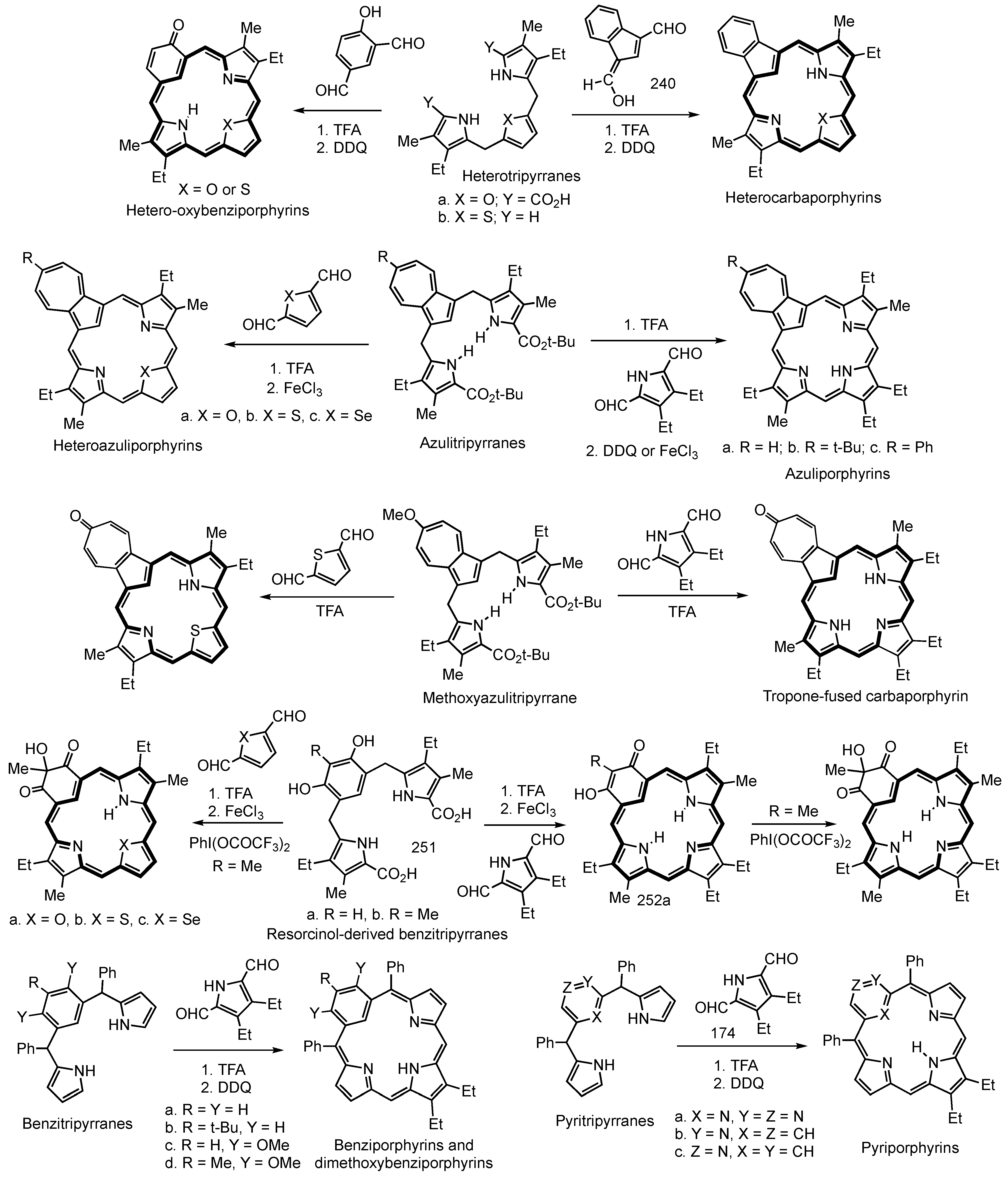{kind=link}
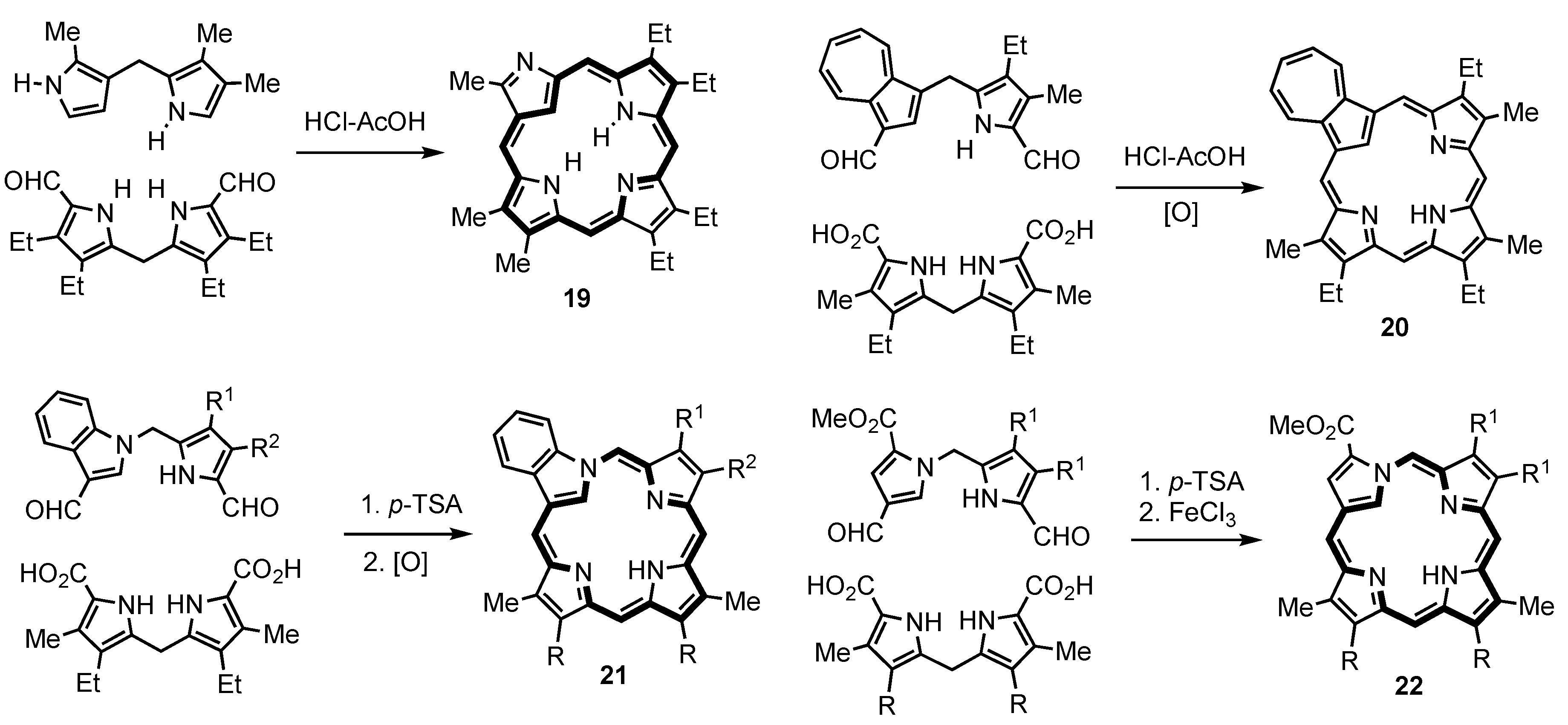{kind=link}
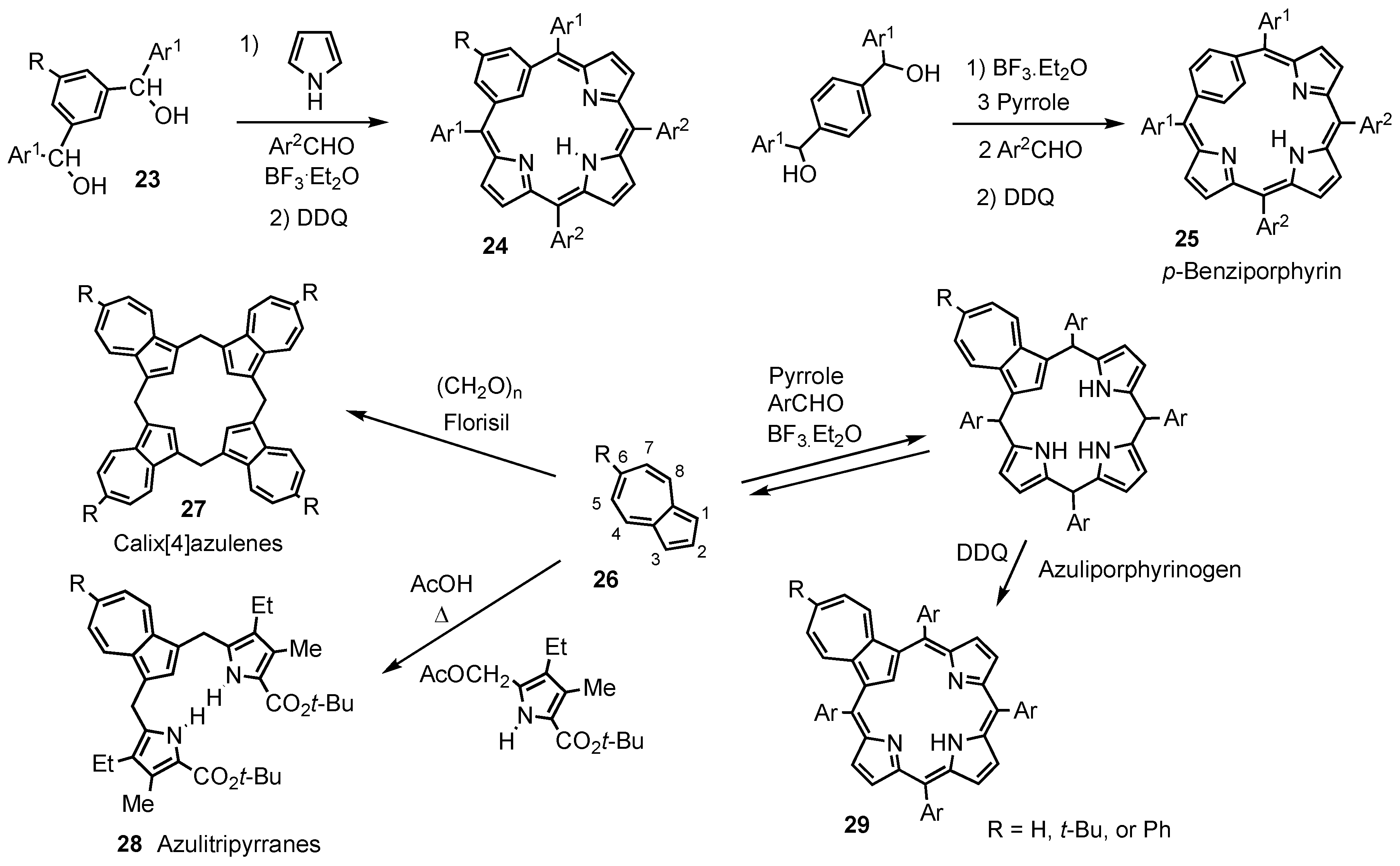{kind=link}
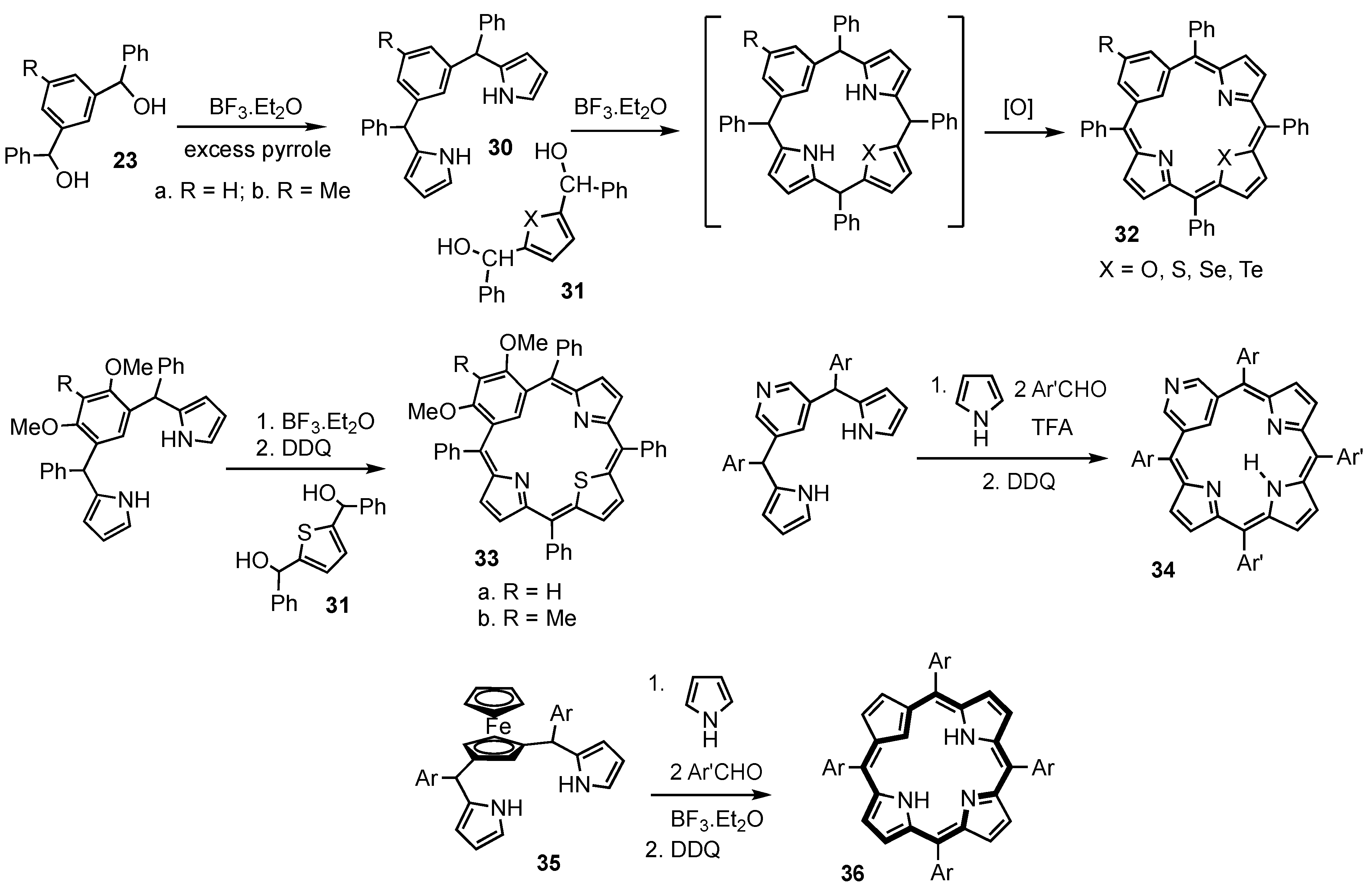{kind=link}
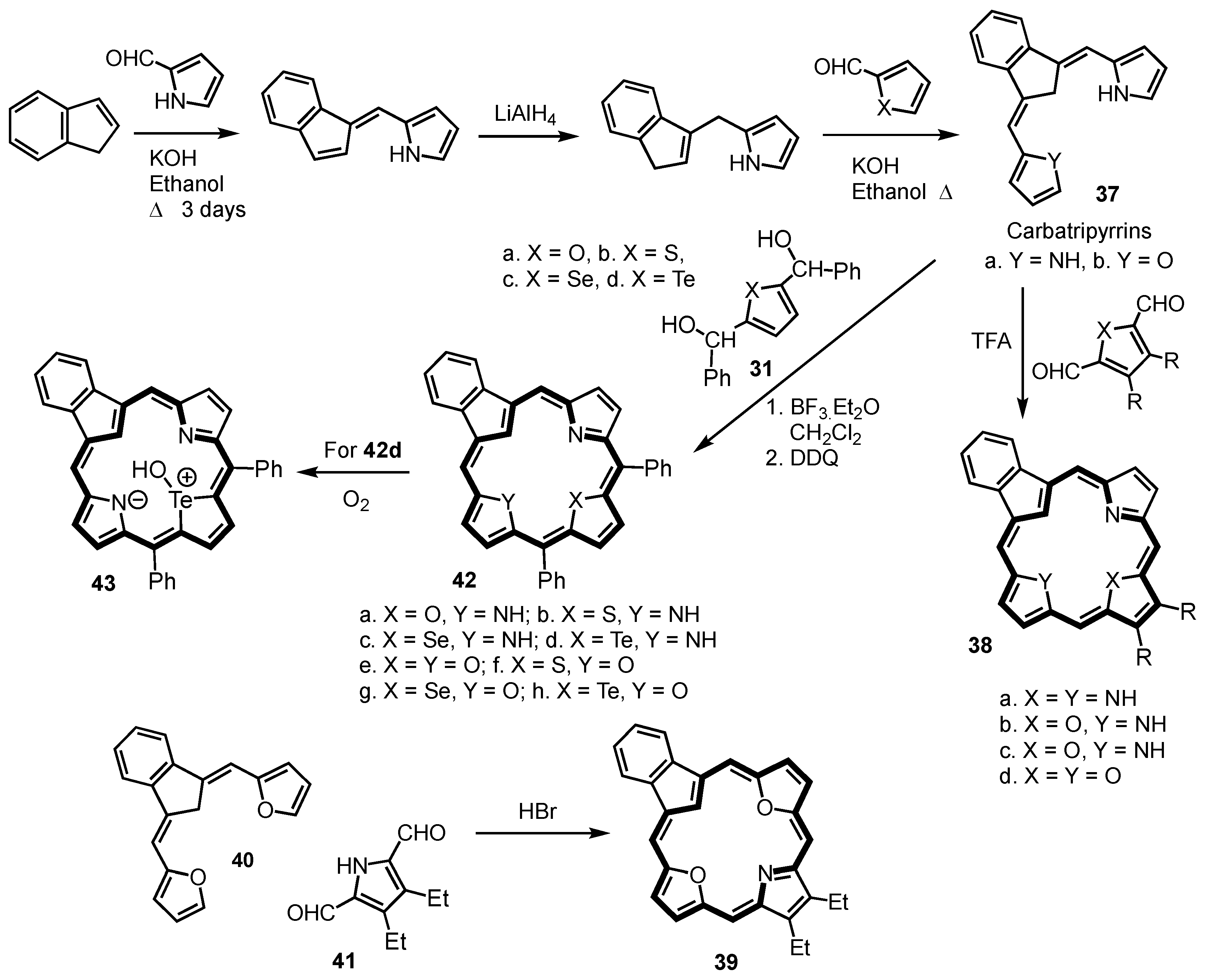{kind=link}
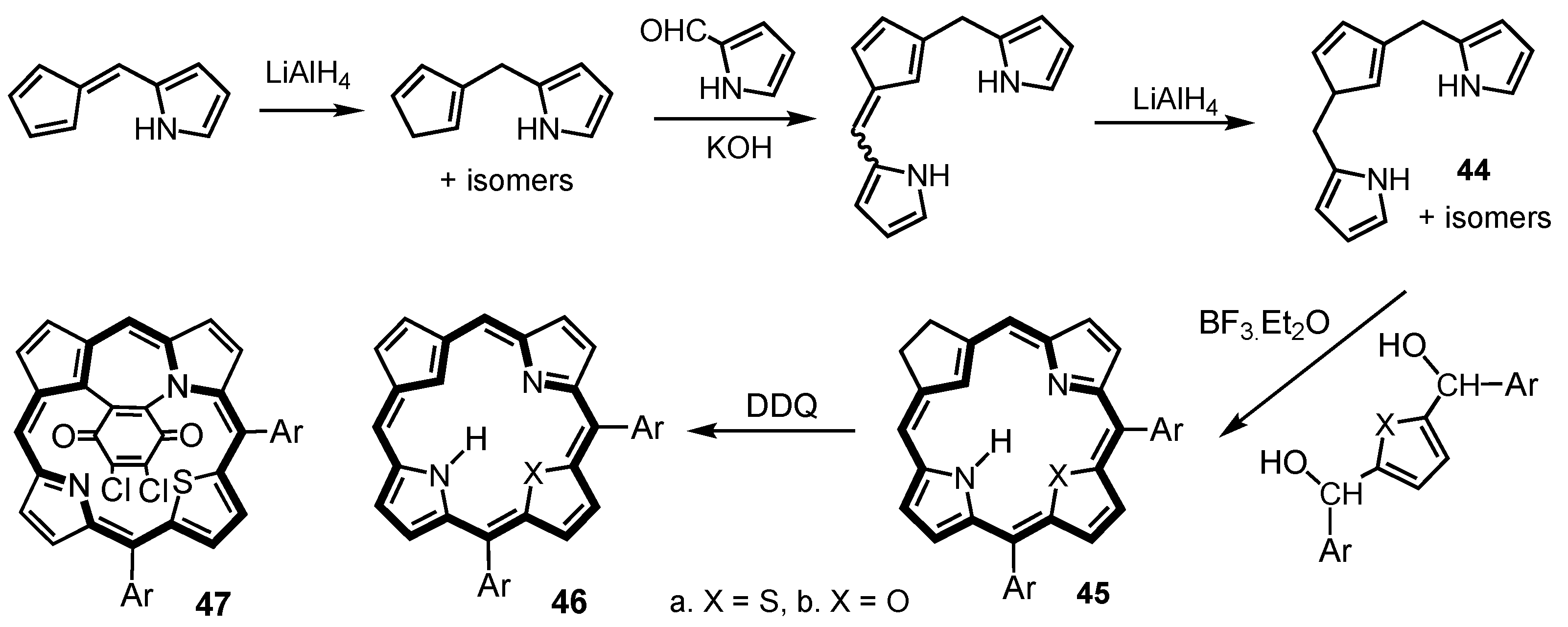{kind=link}
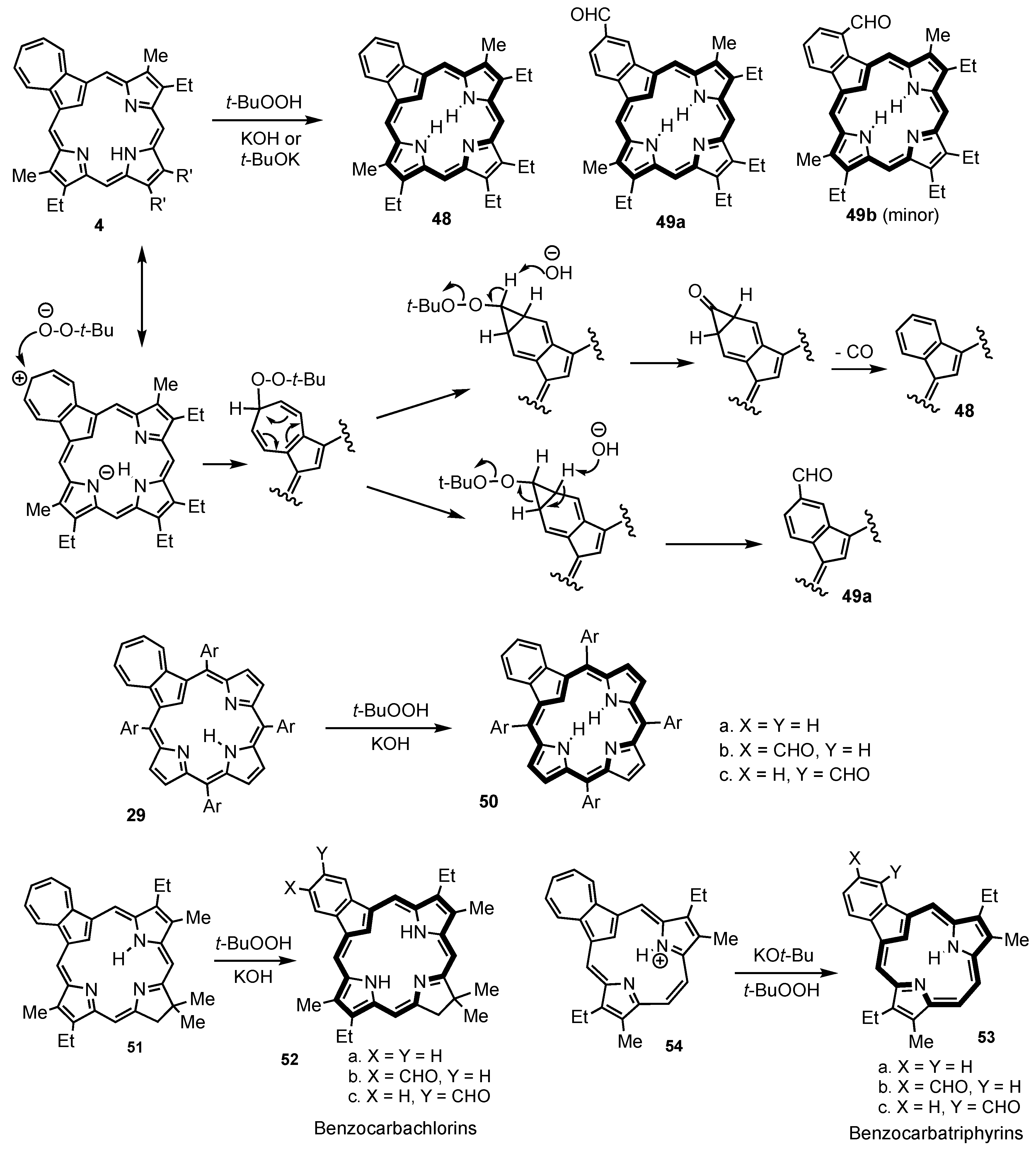{kind=link}
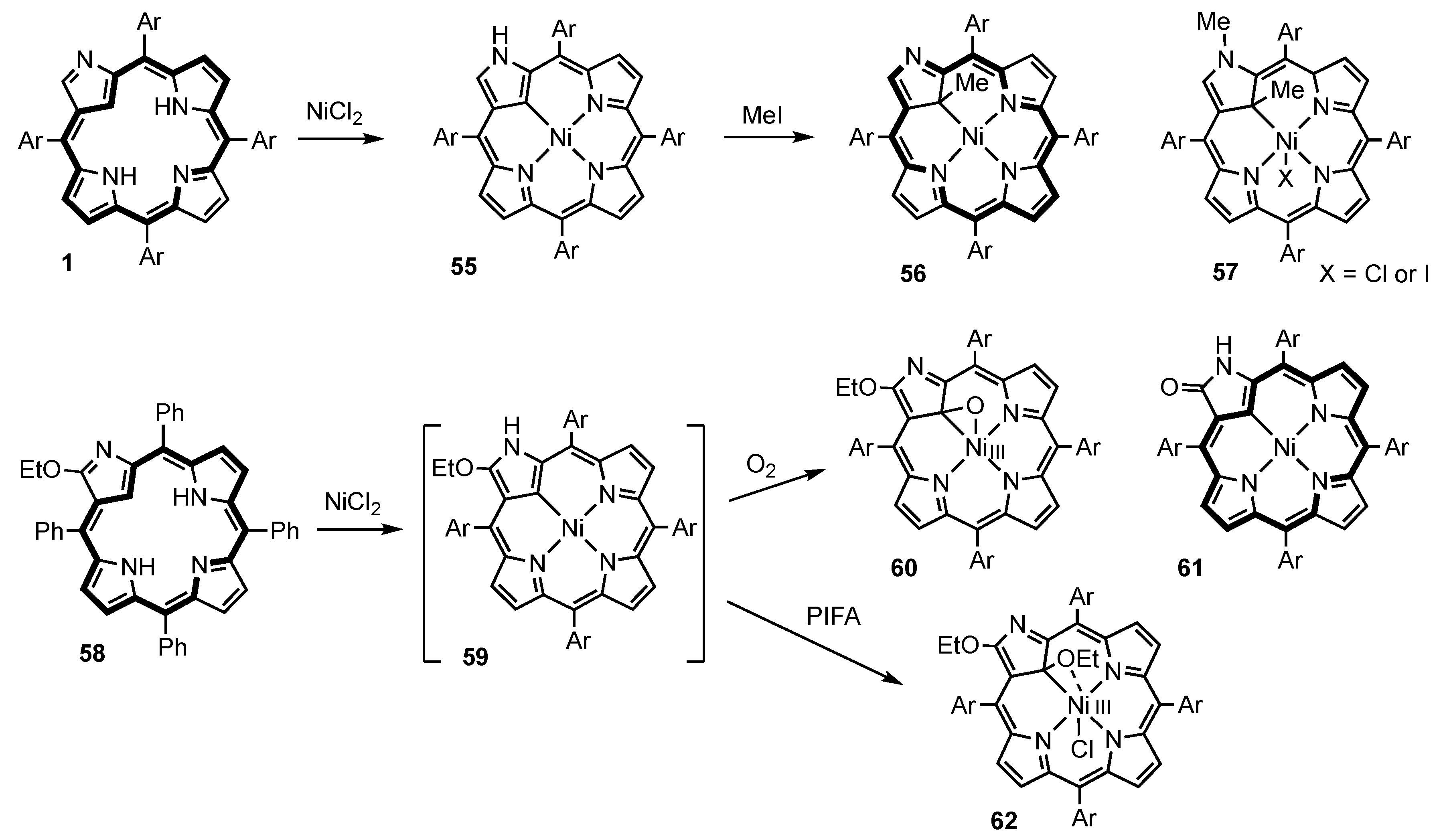{kind=link}
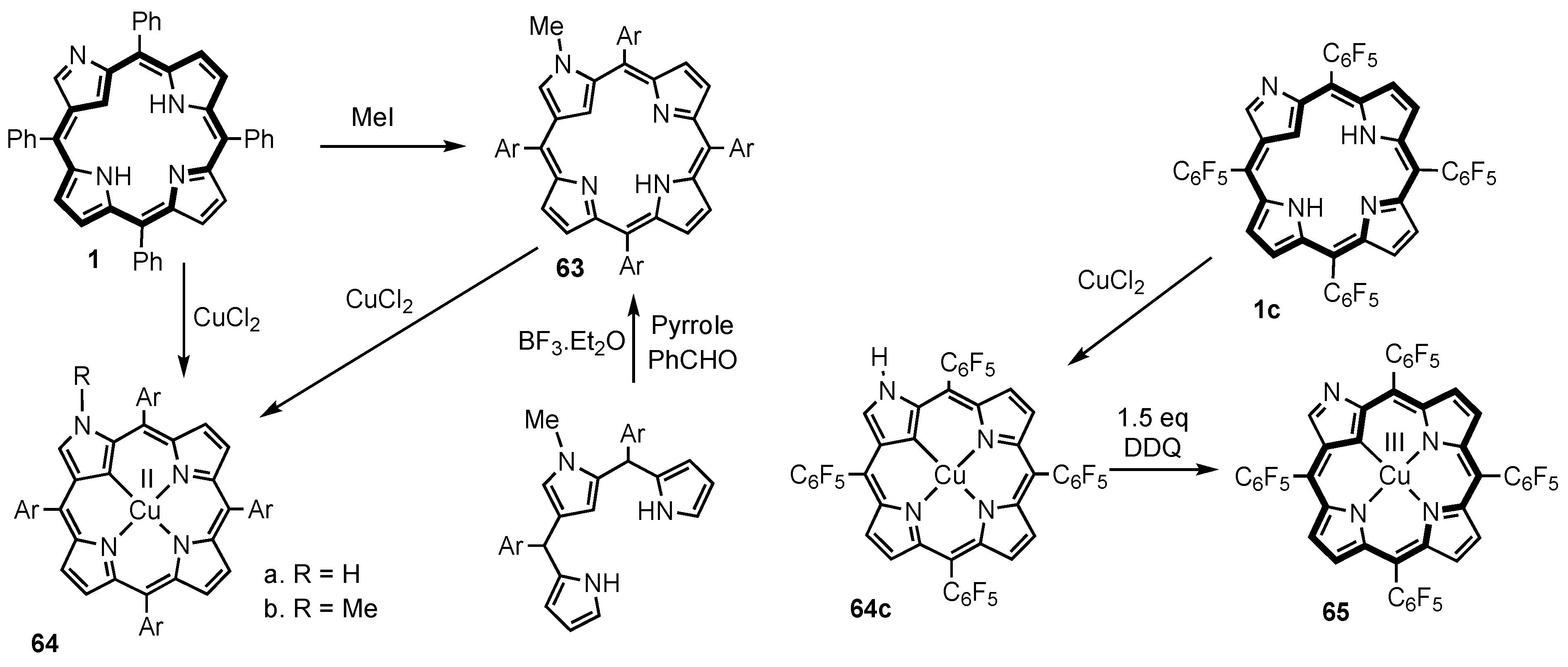{kind=link}
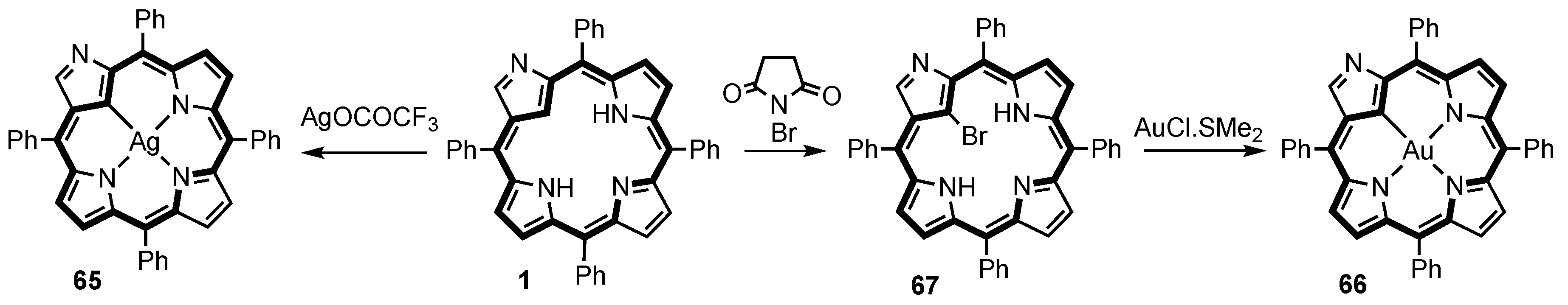{kind=link}
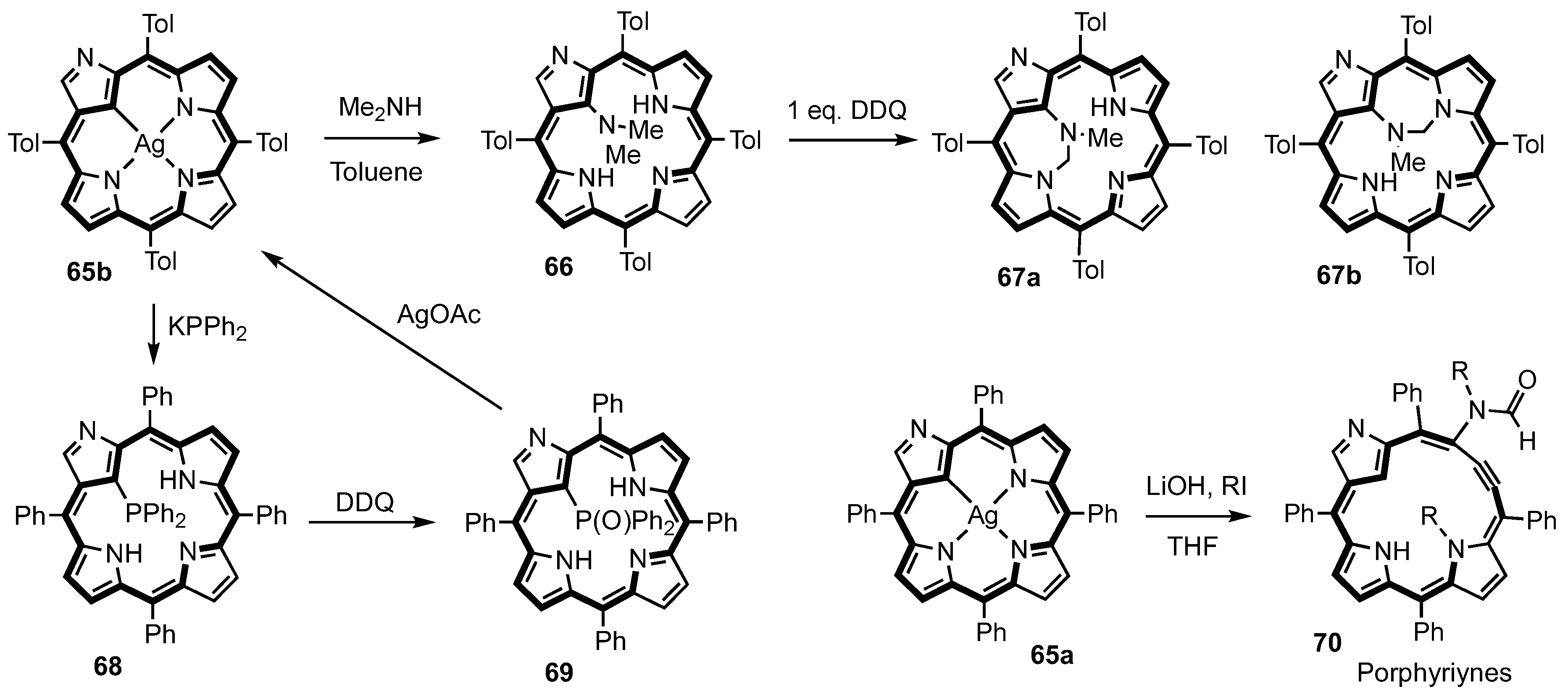{kind=link}
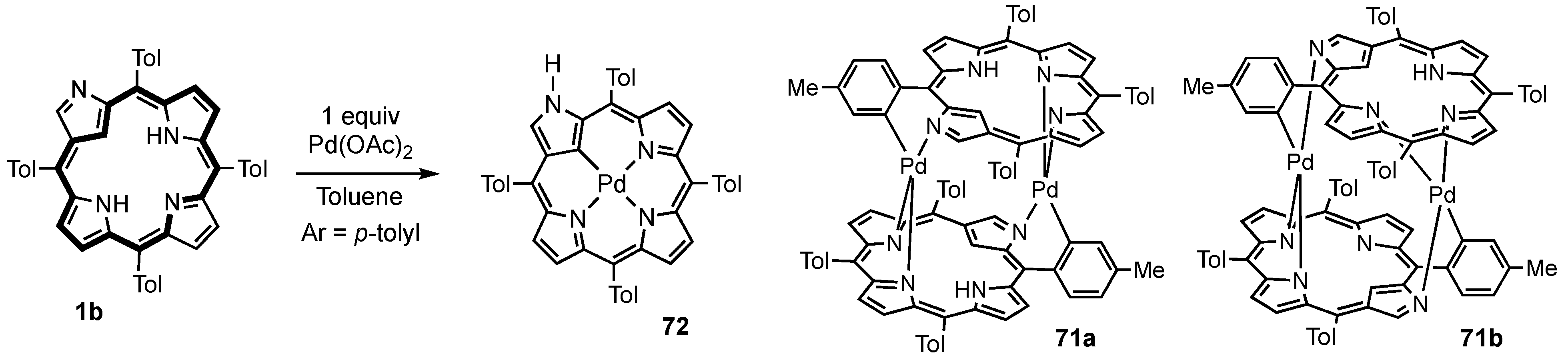{kind=link}
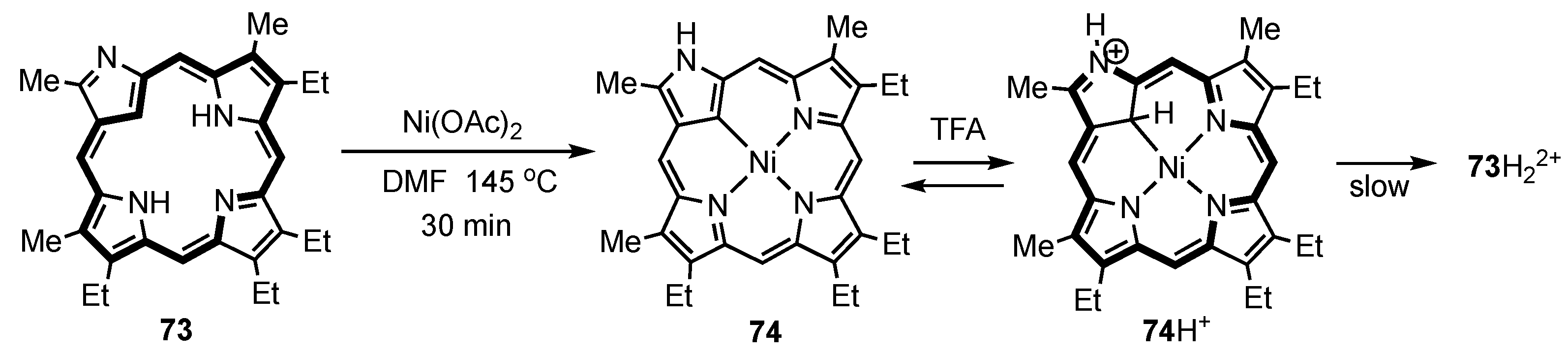{kind=link}
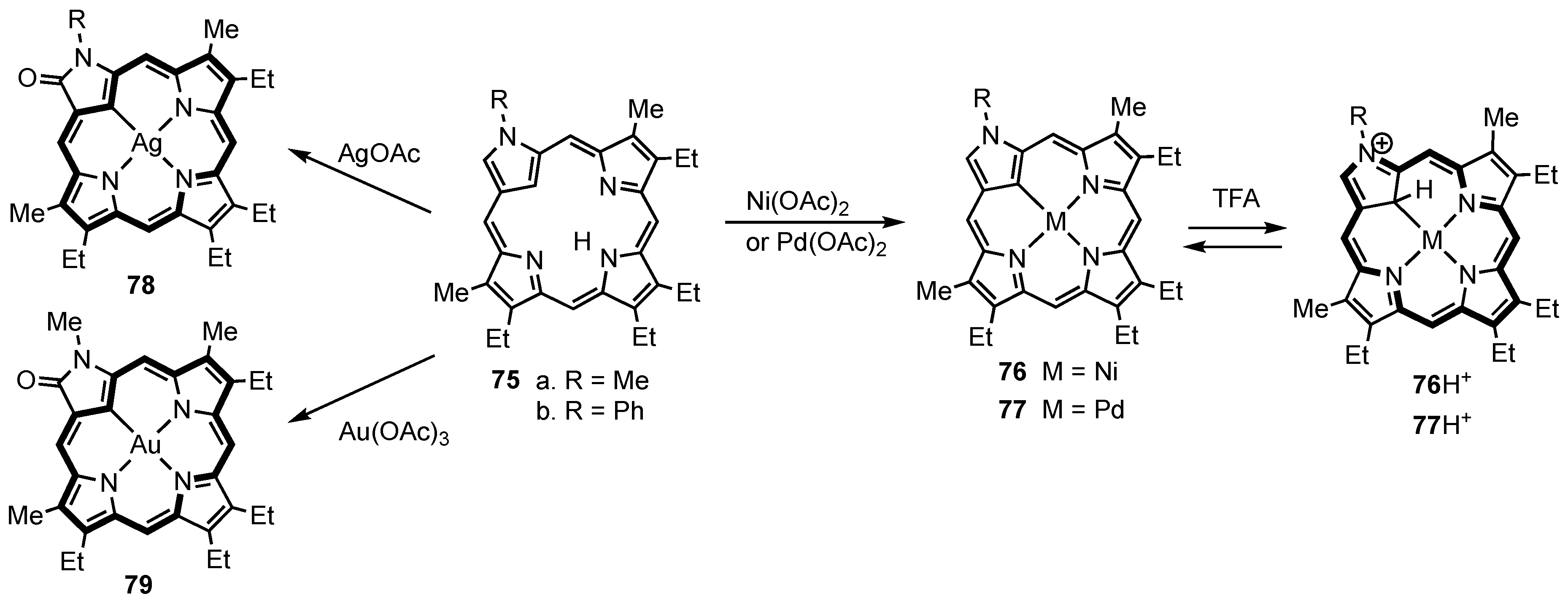{kind=link}
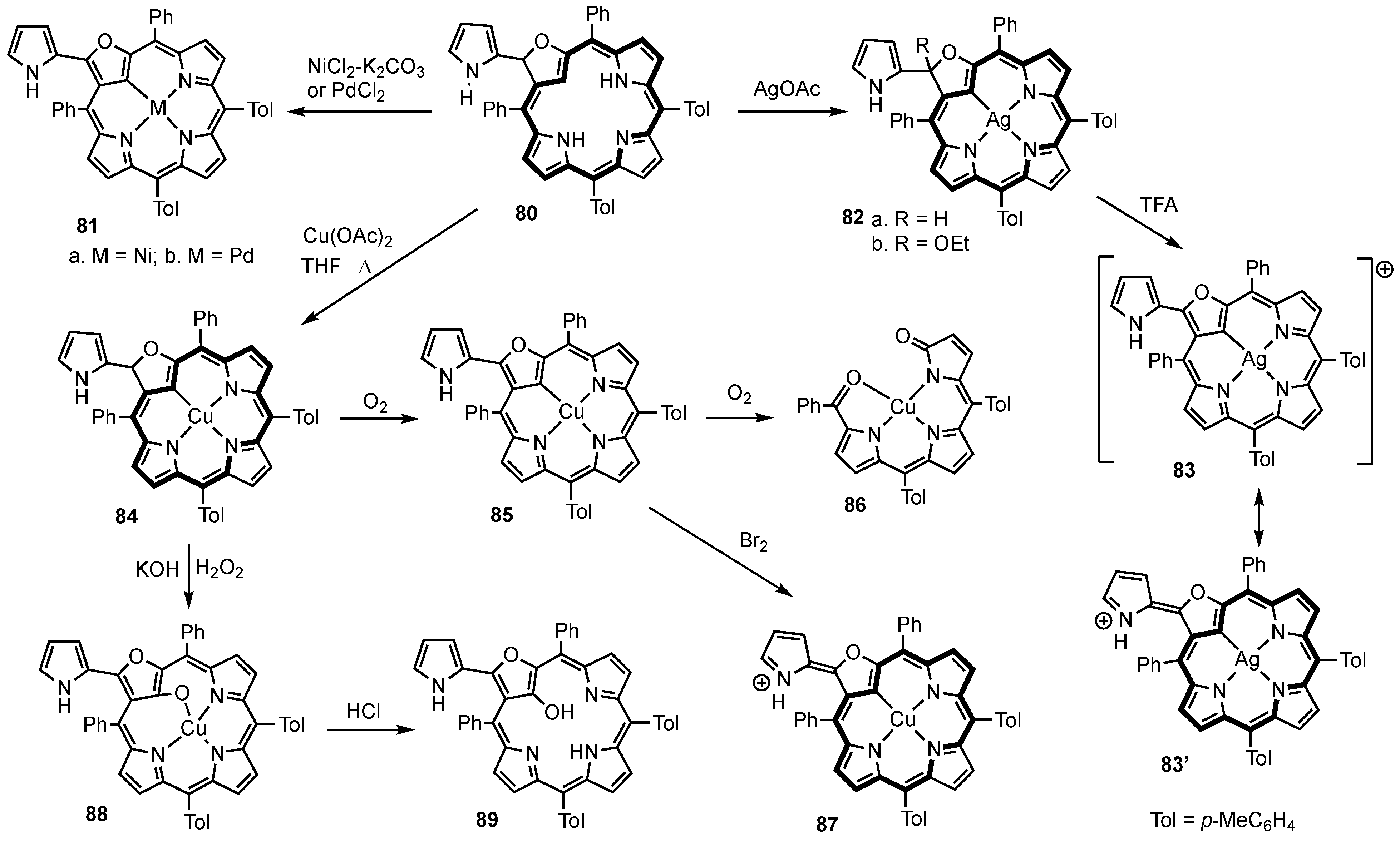{kind=link}
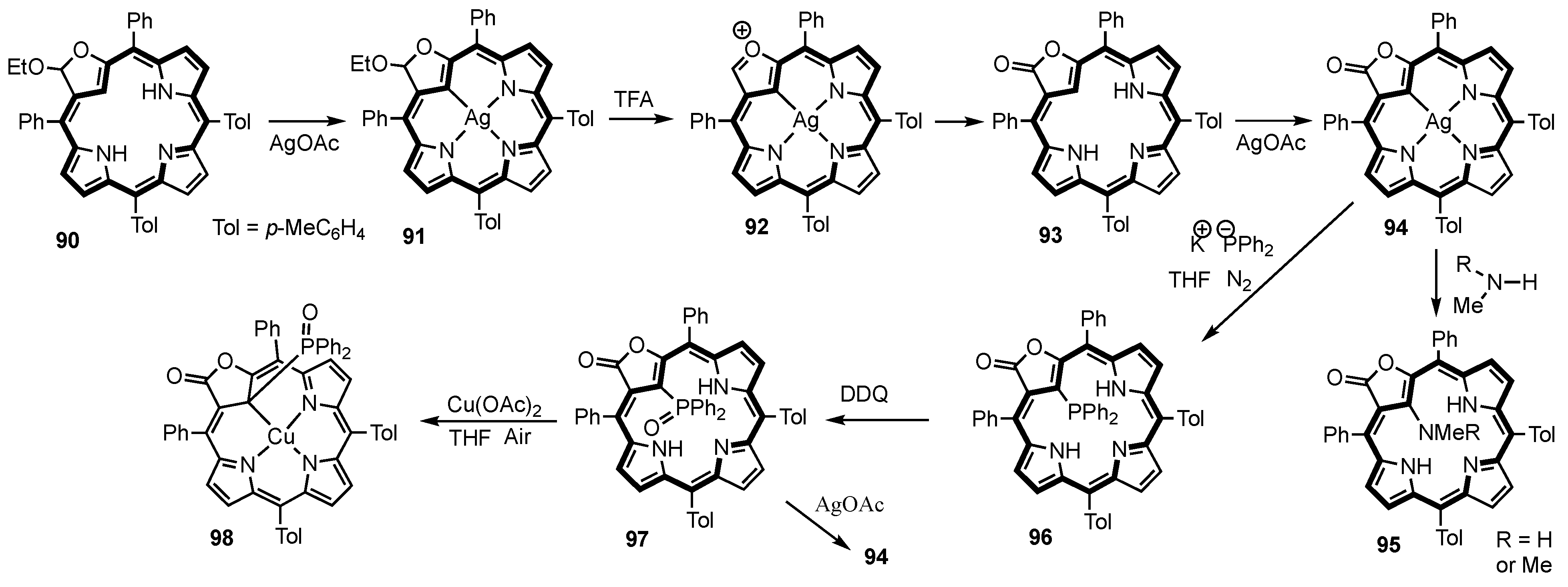{kind=link}
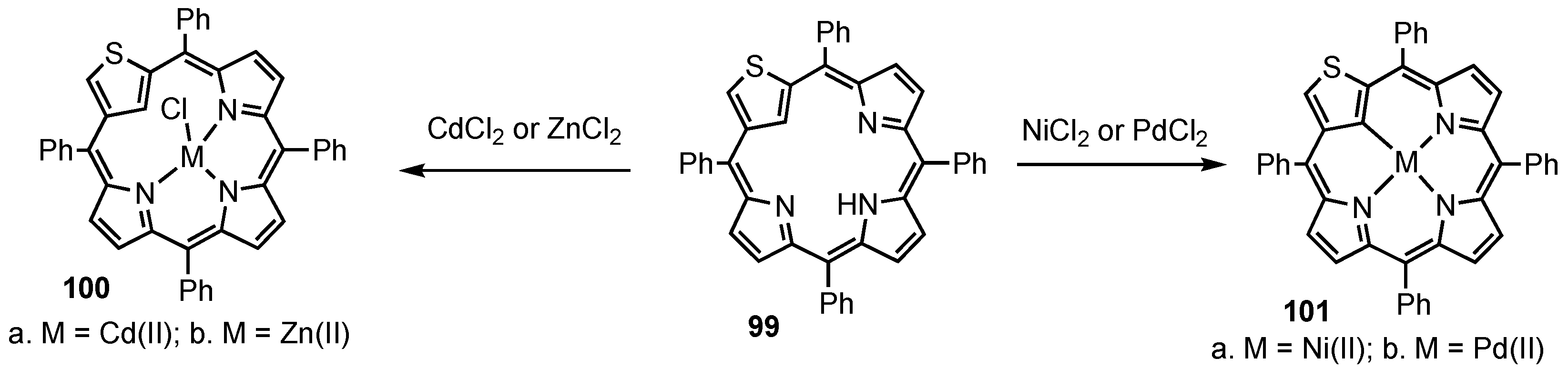{kind=link}
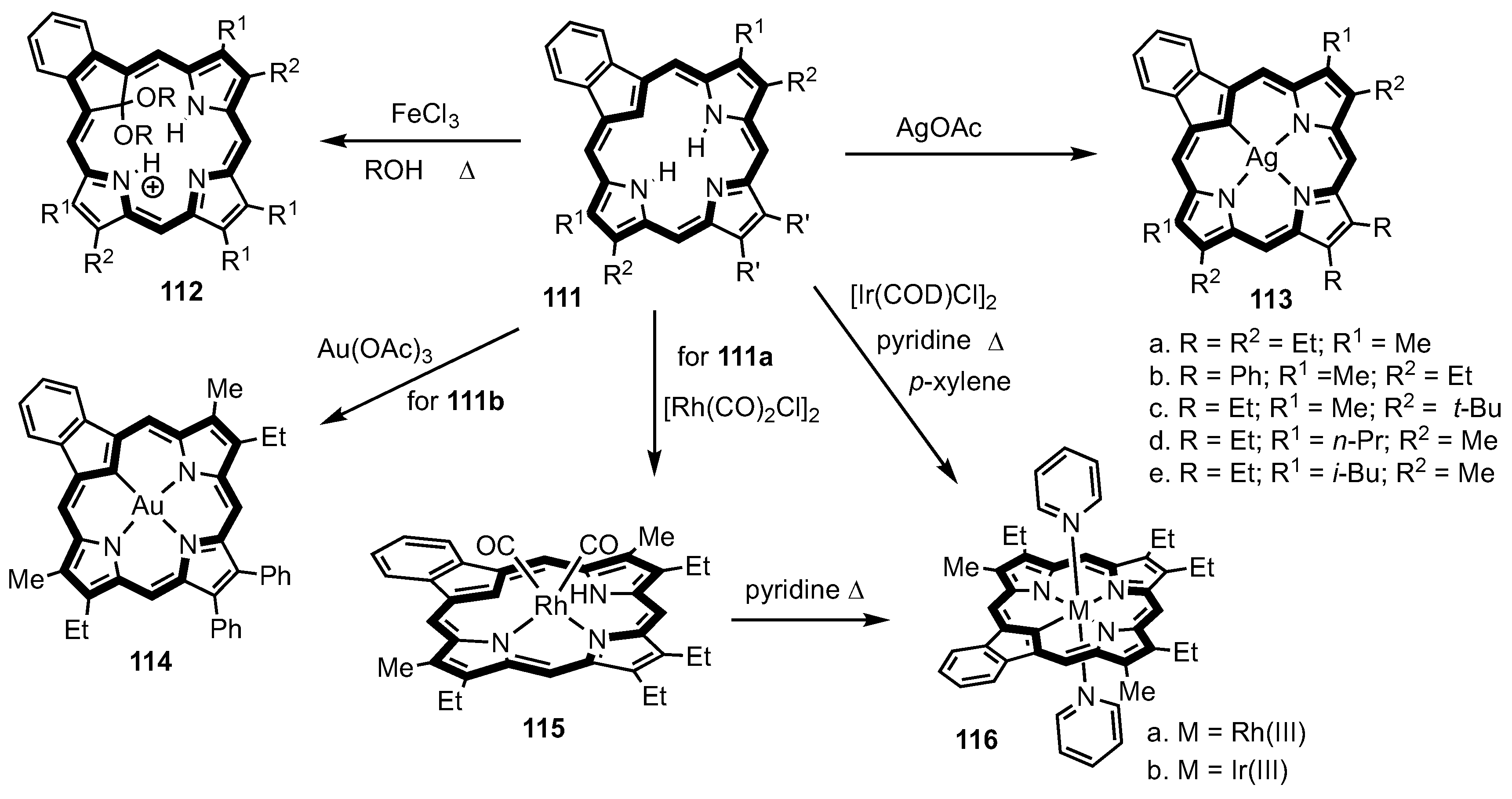{kind=link}
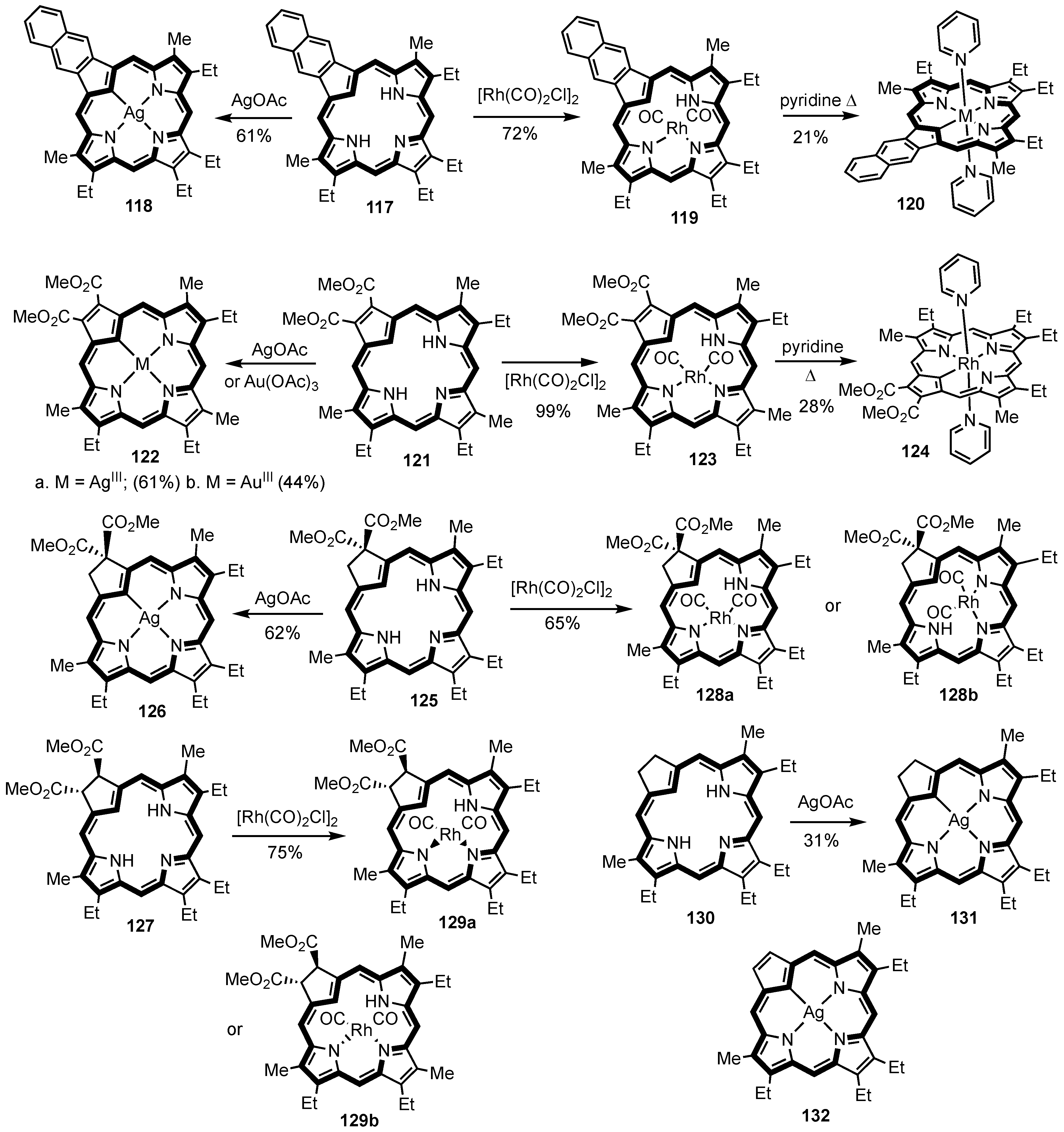{kind=link}
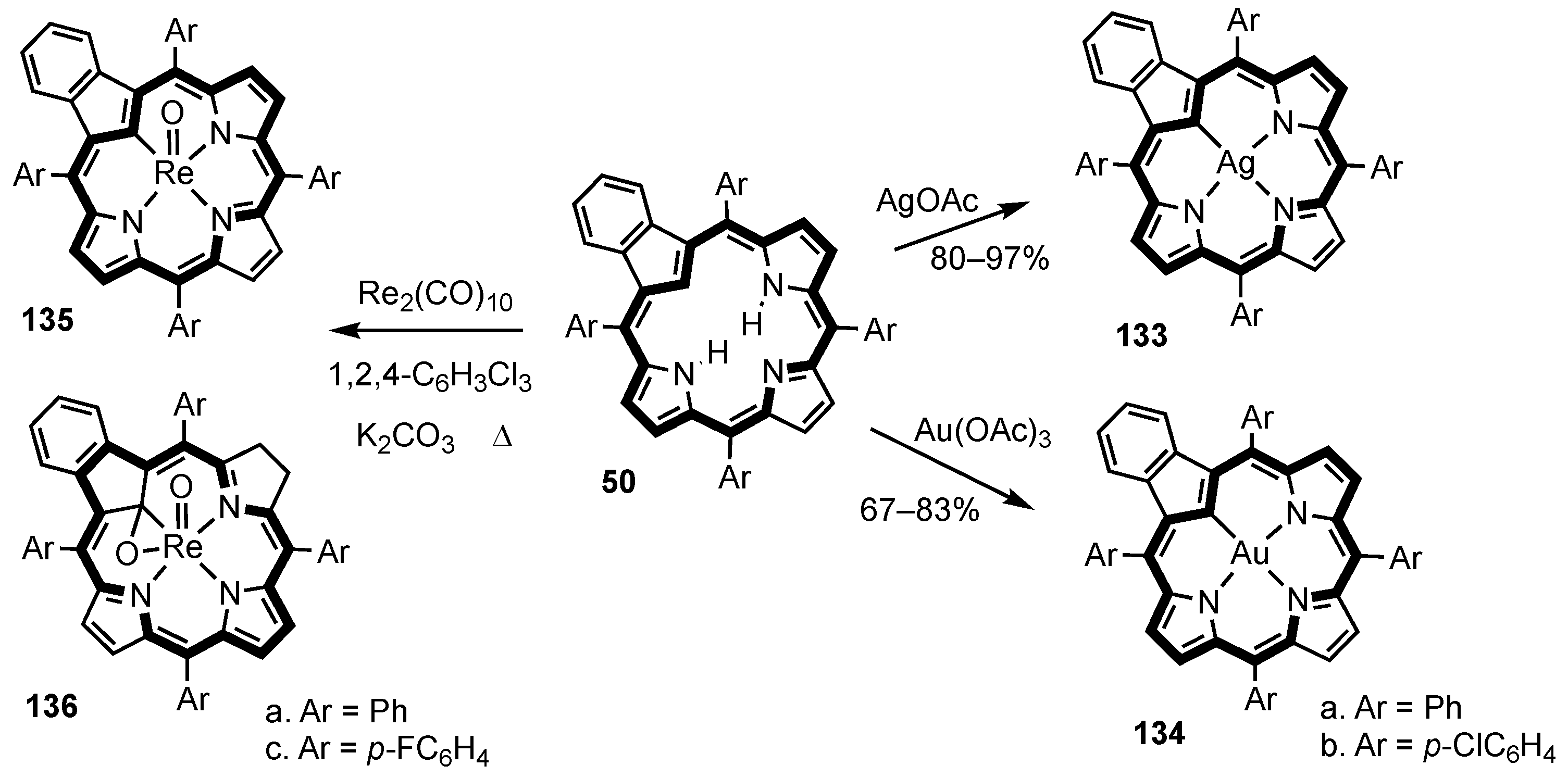{kind=link}
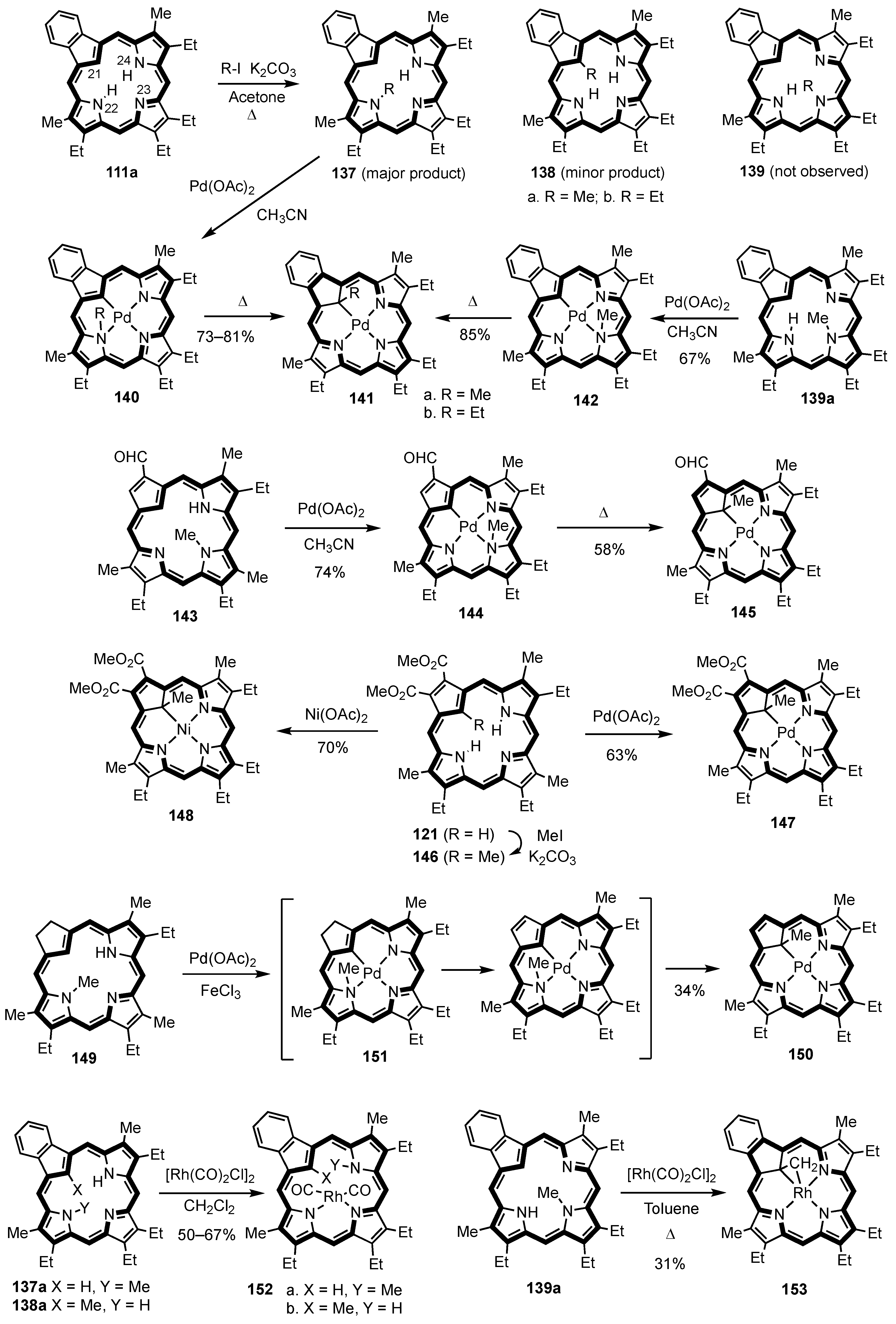{kind=link}
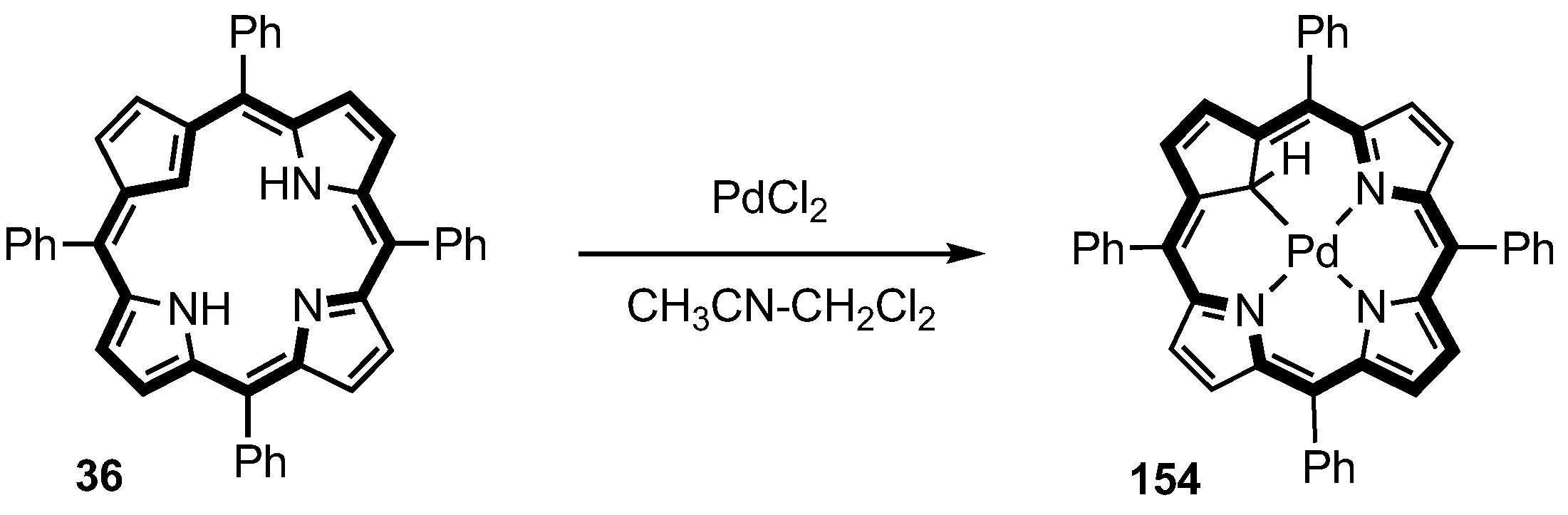{kind=link}
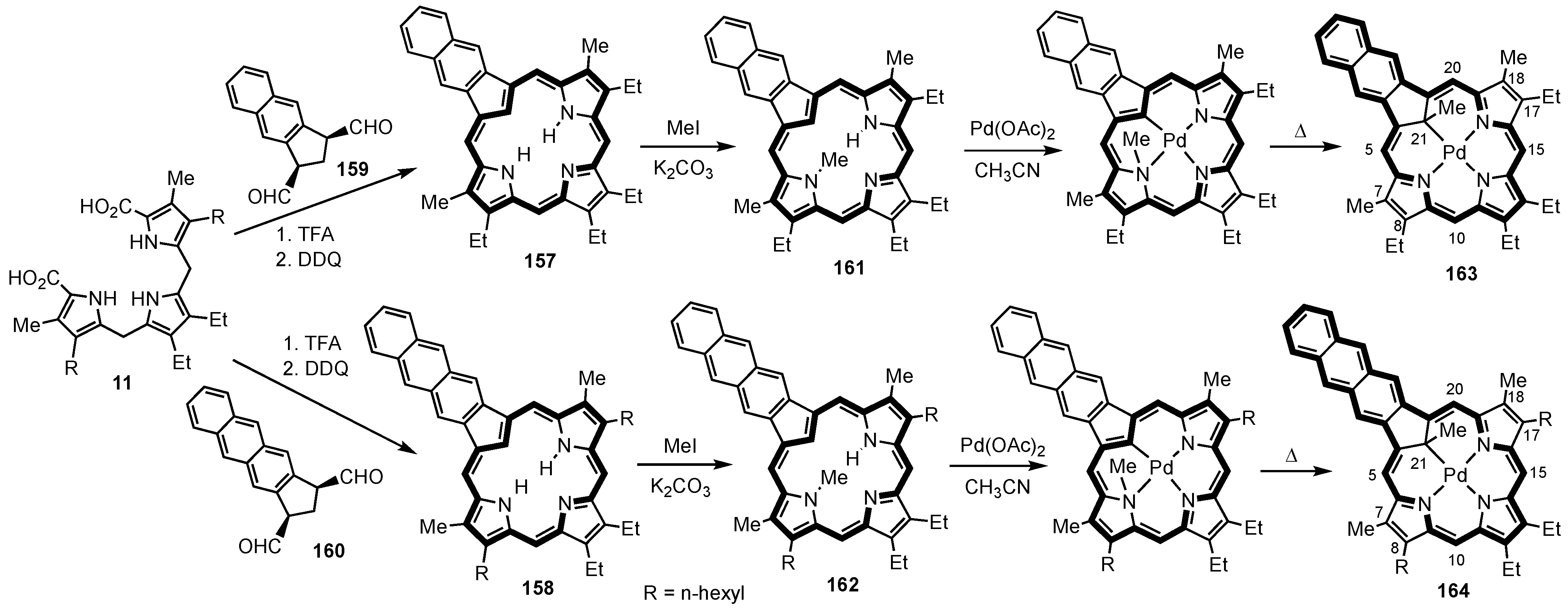{kind=link}
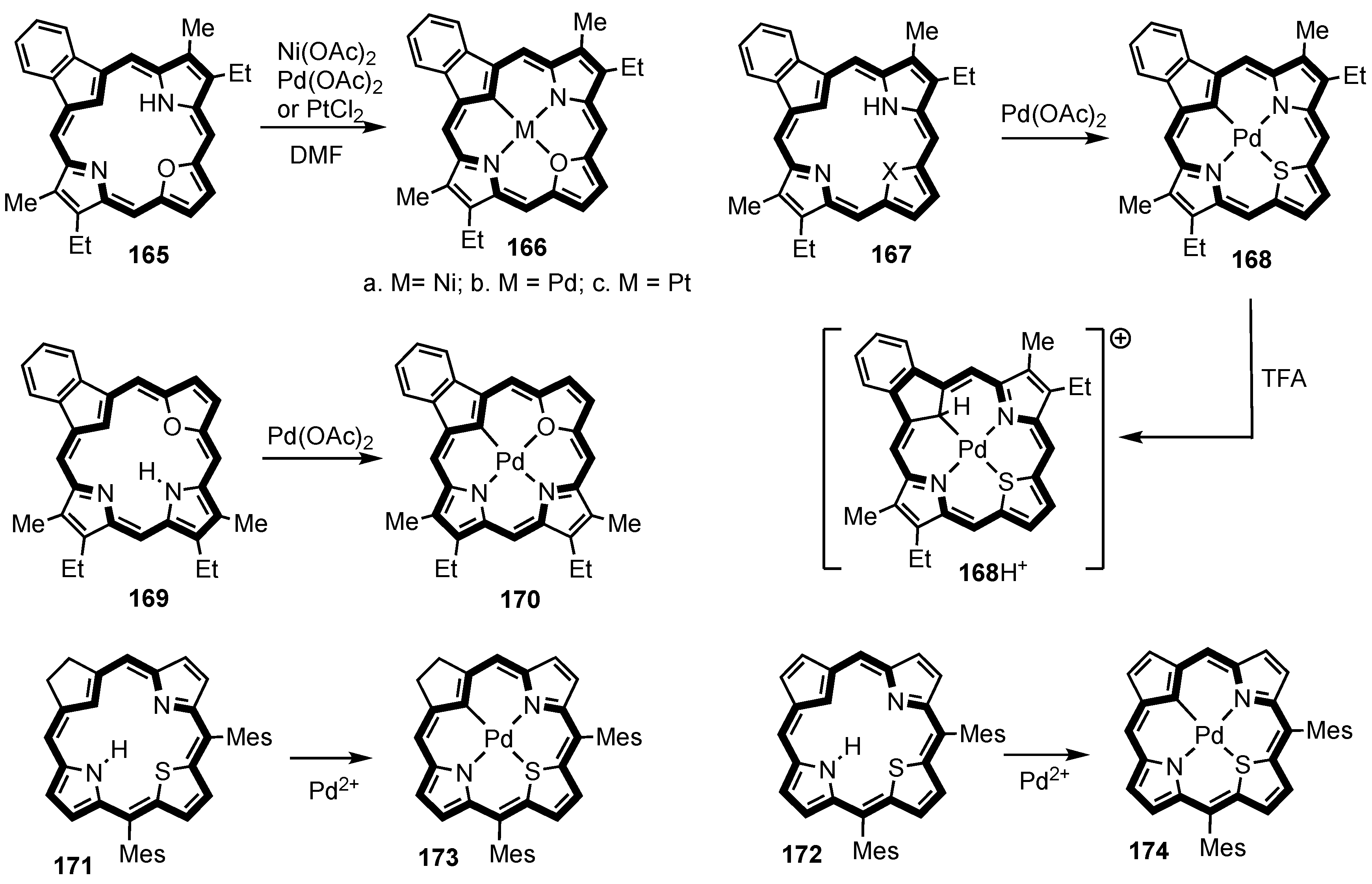{kind=link}
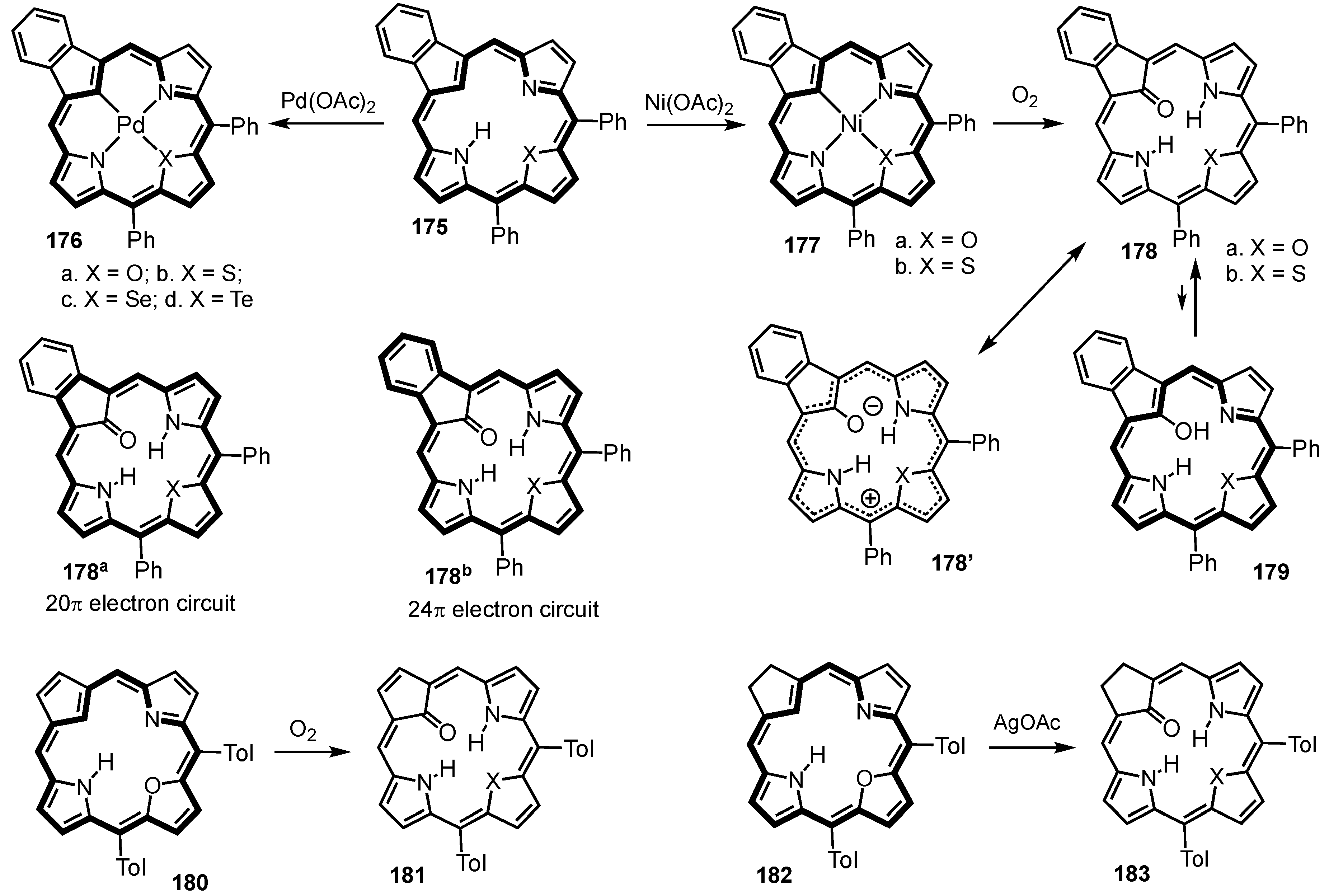{kind=link}
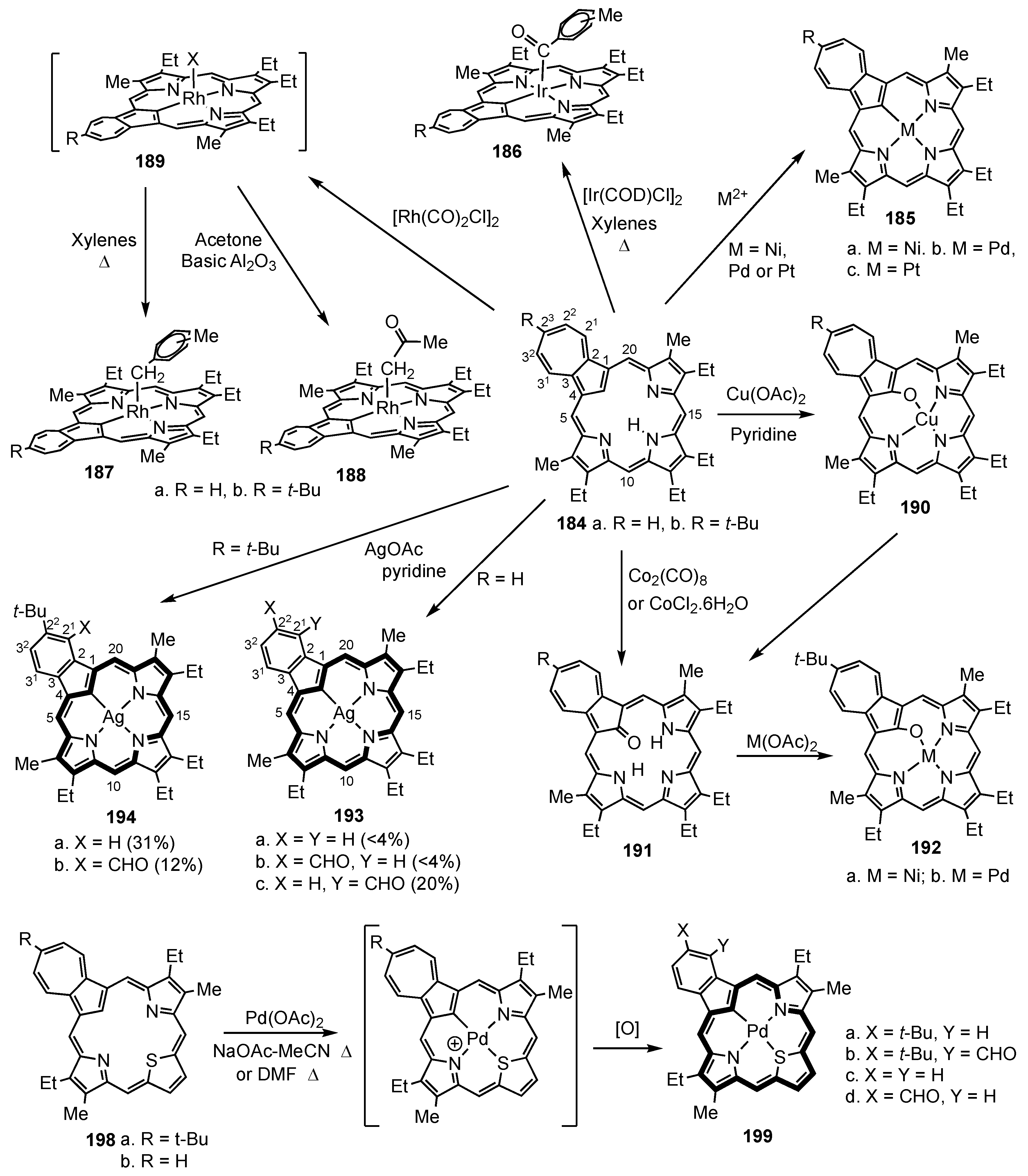{kind=link}
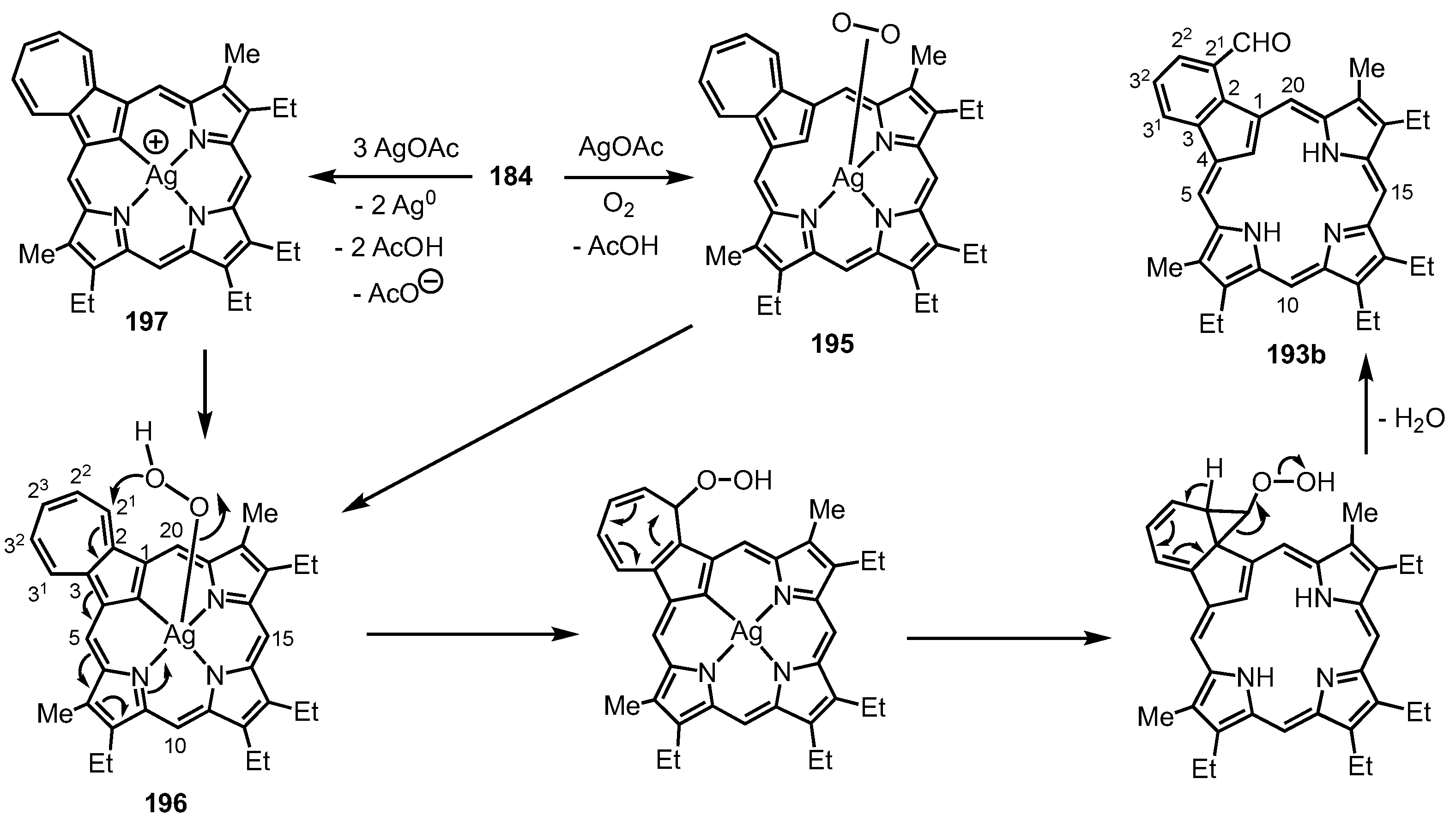{kind=link}
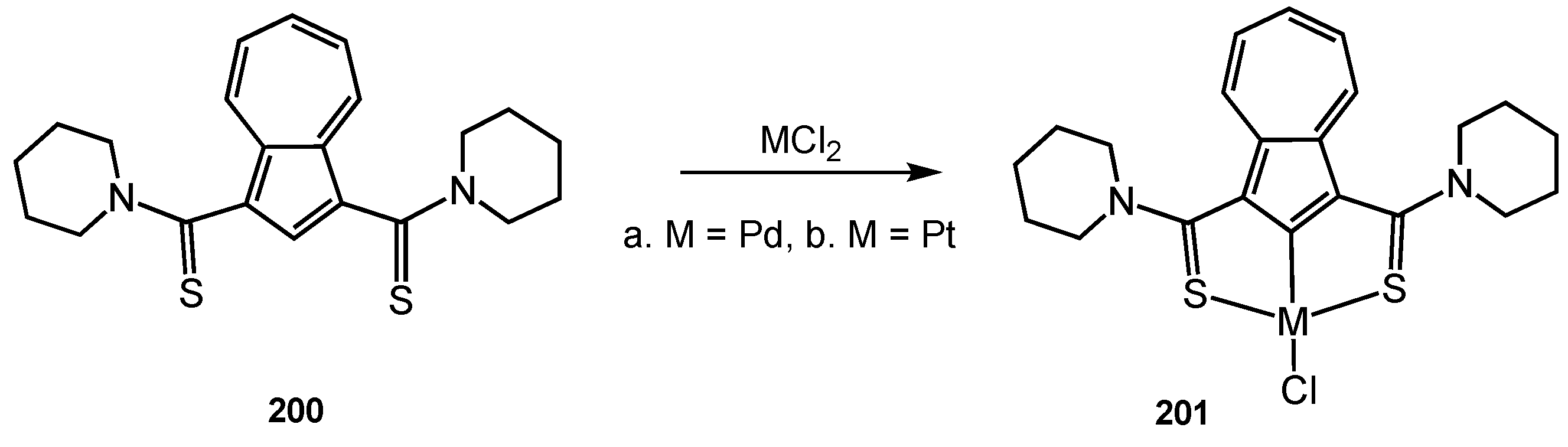{kind=link}
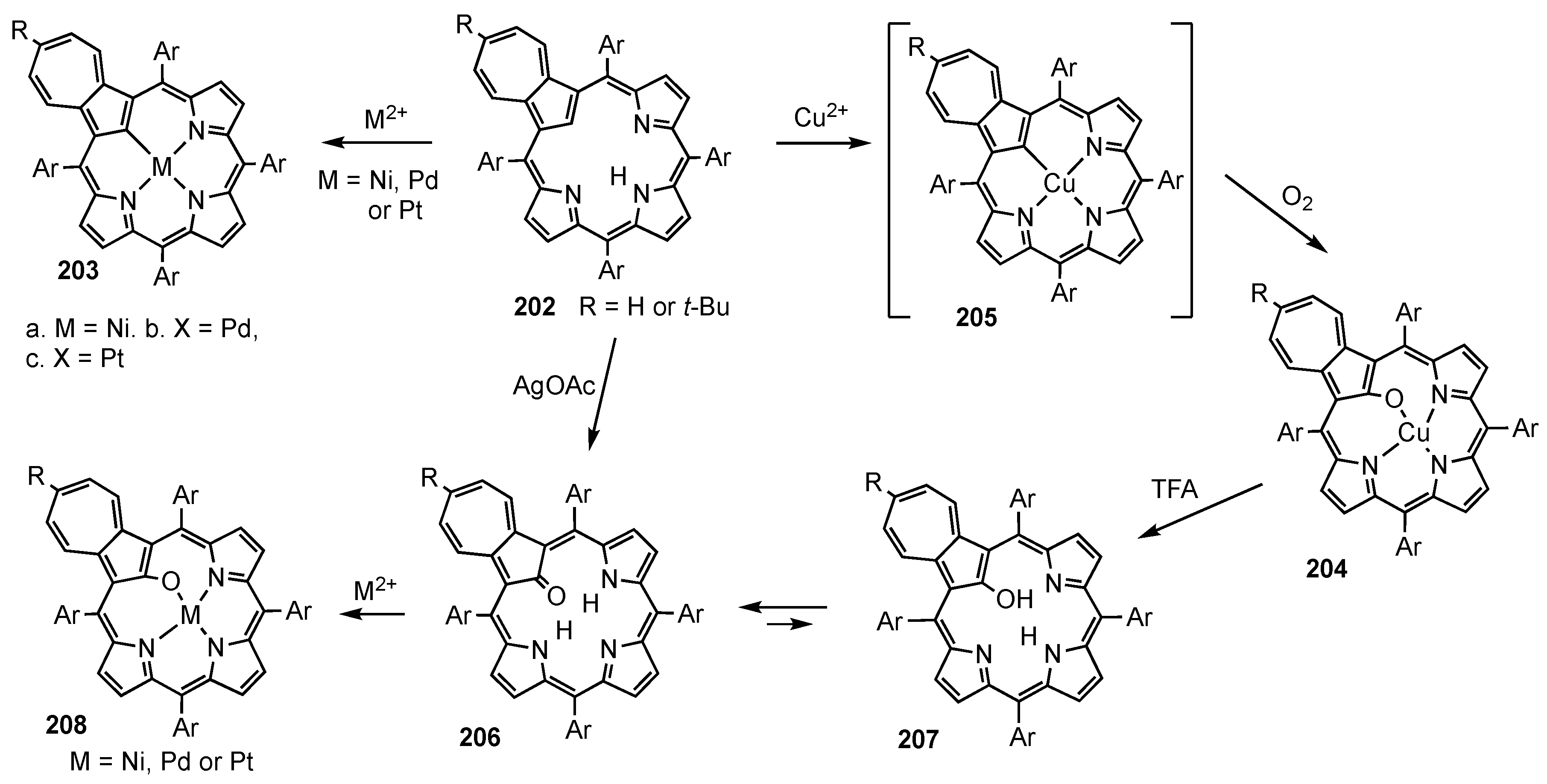{kind=link}
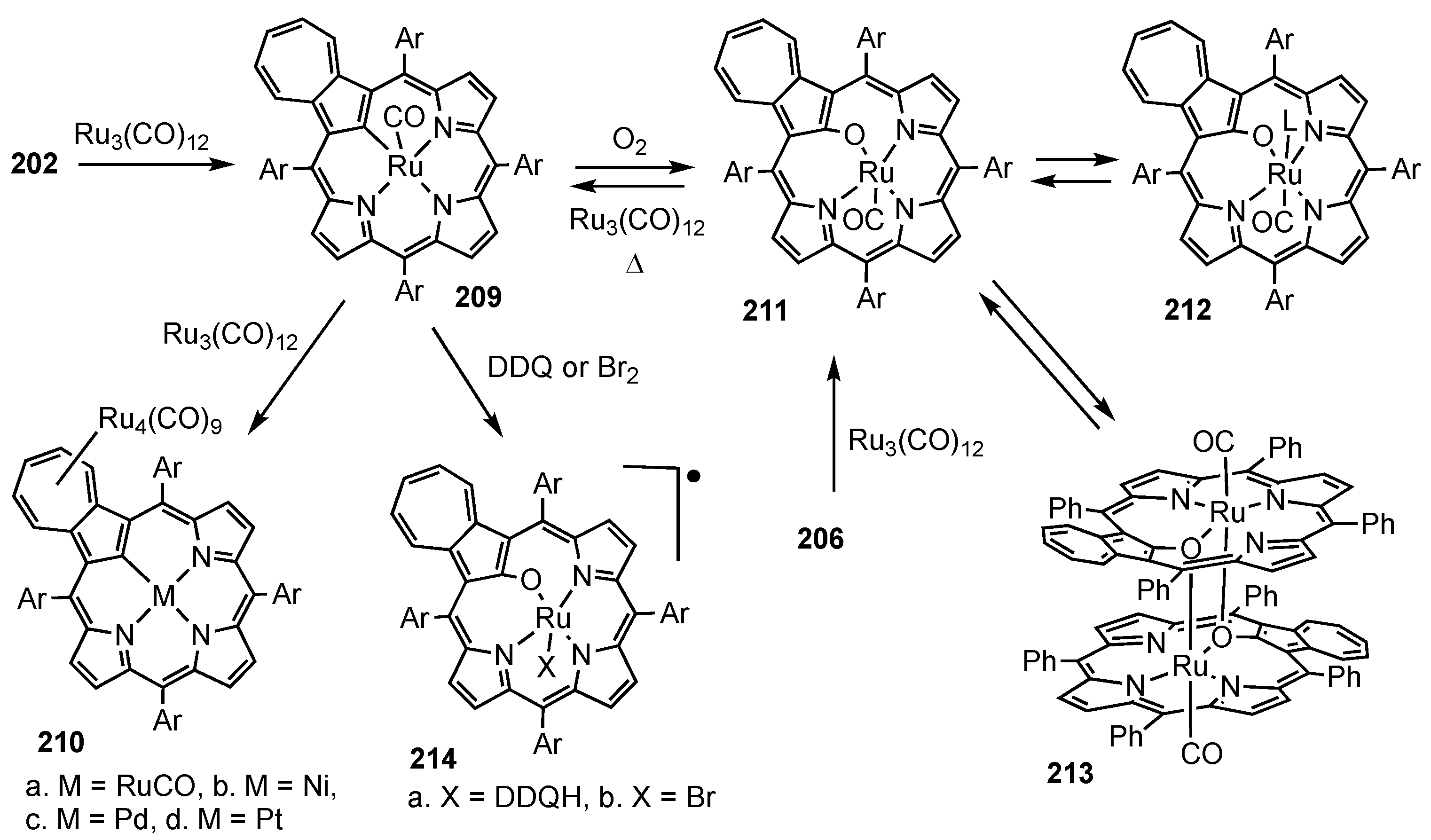{kind=link}
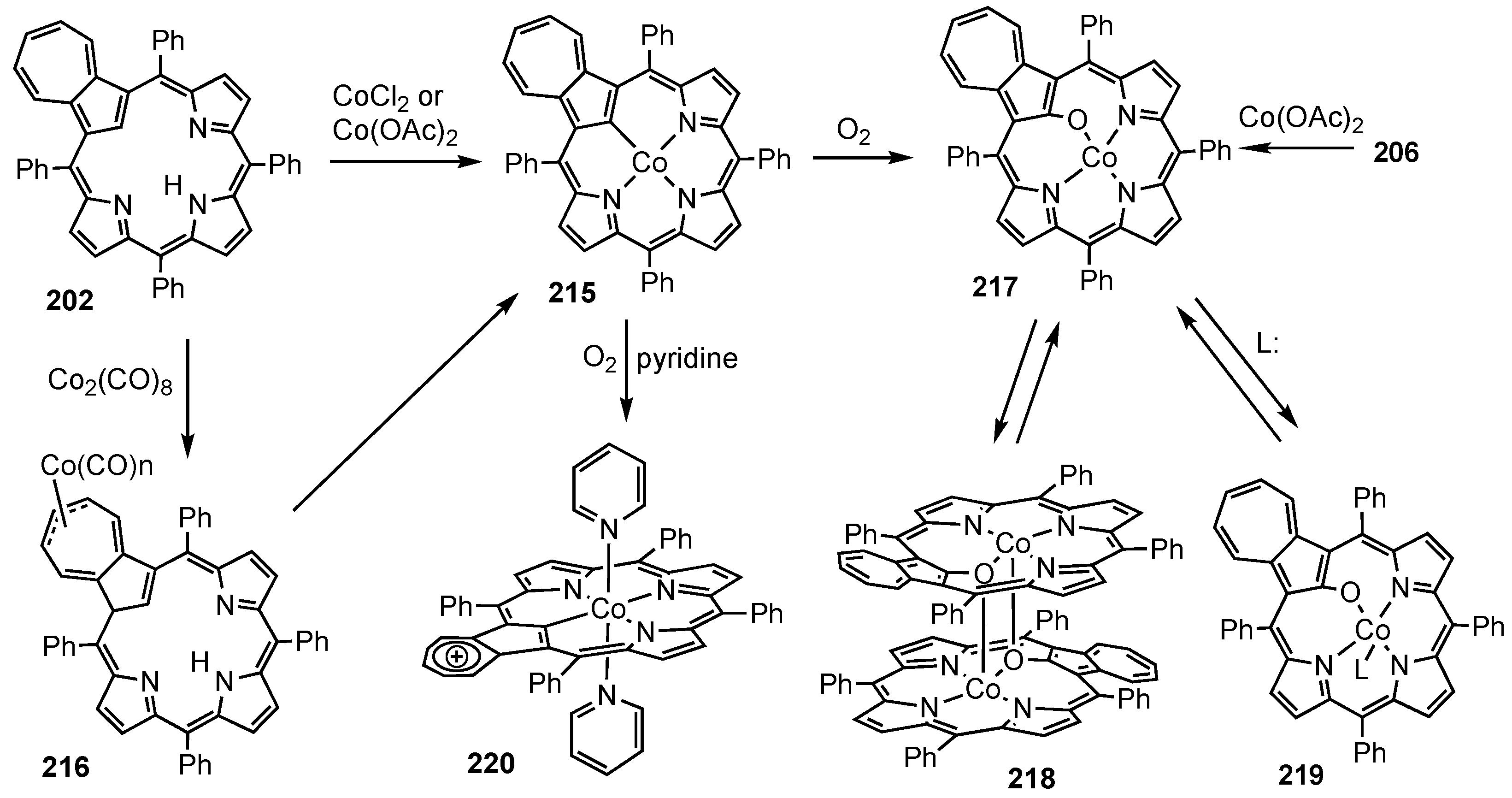{kind=link}
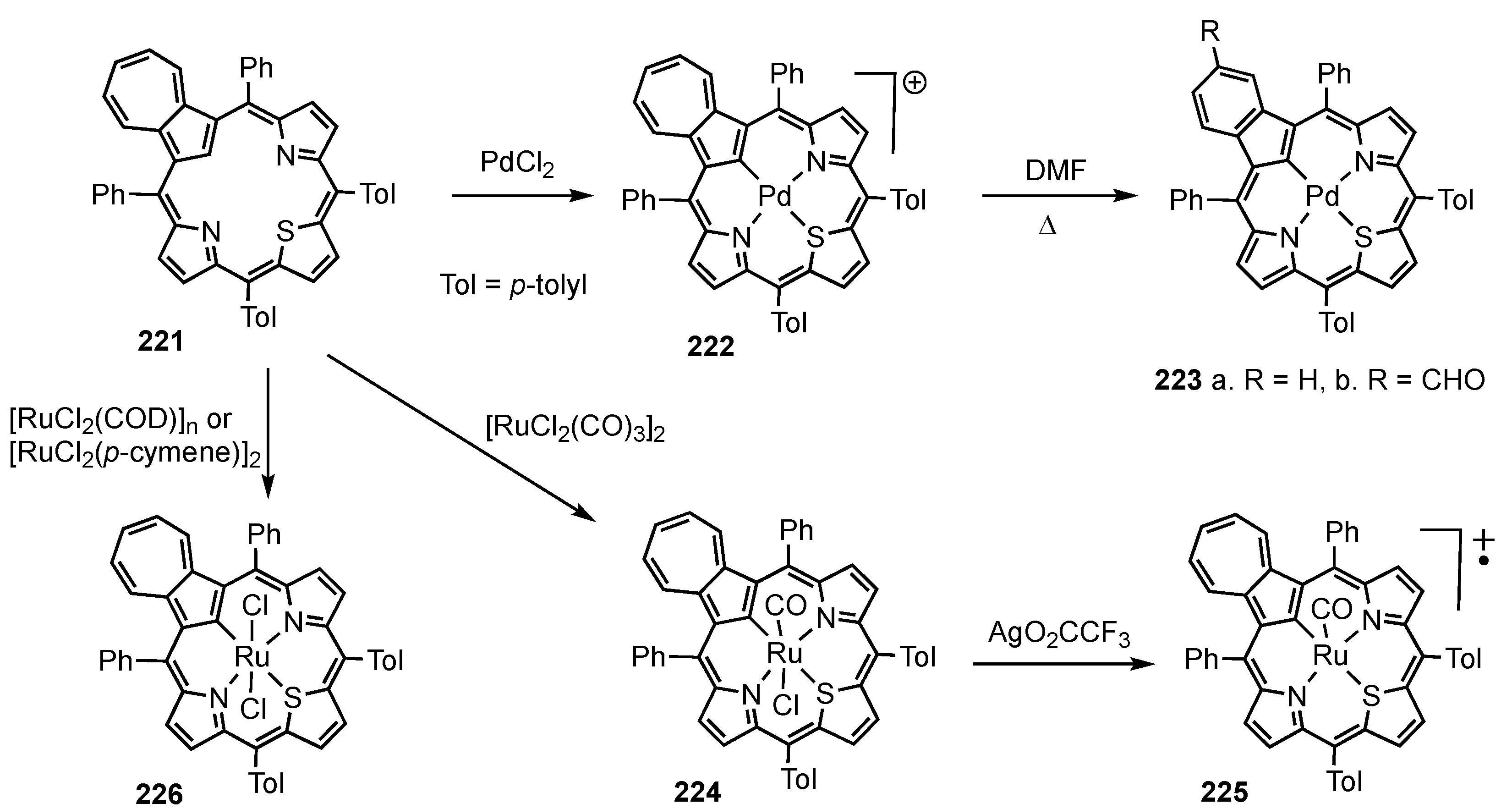{kind=link}
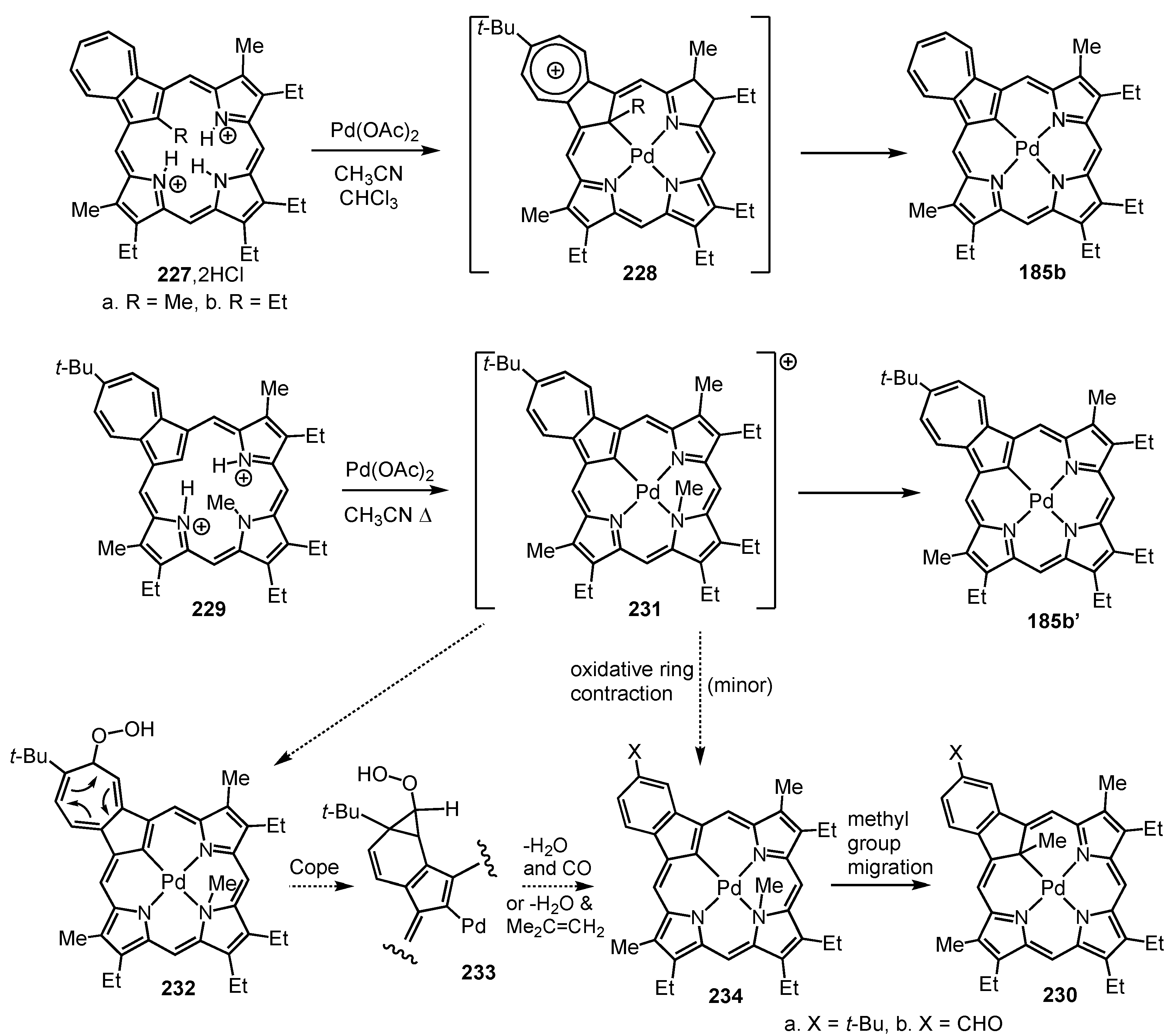{kind=link}
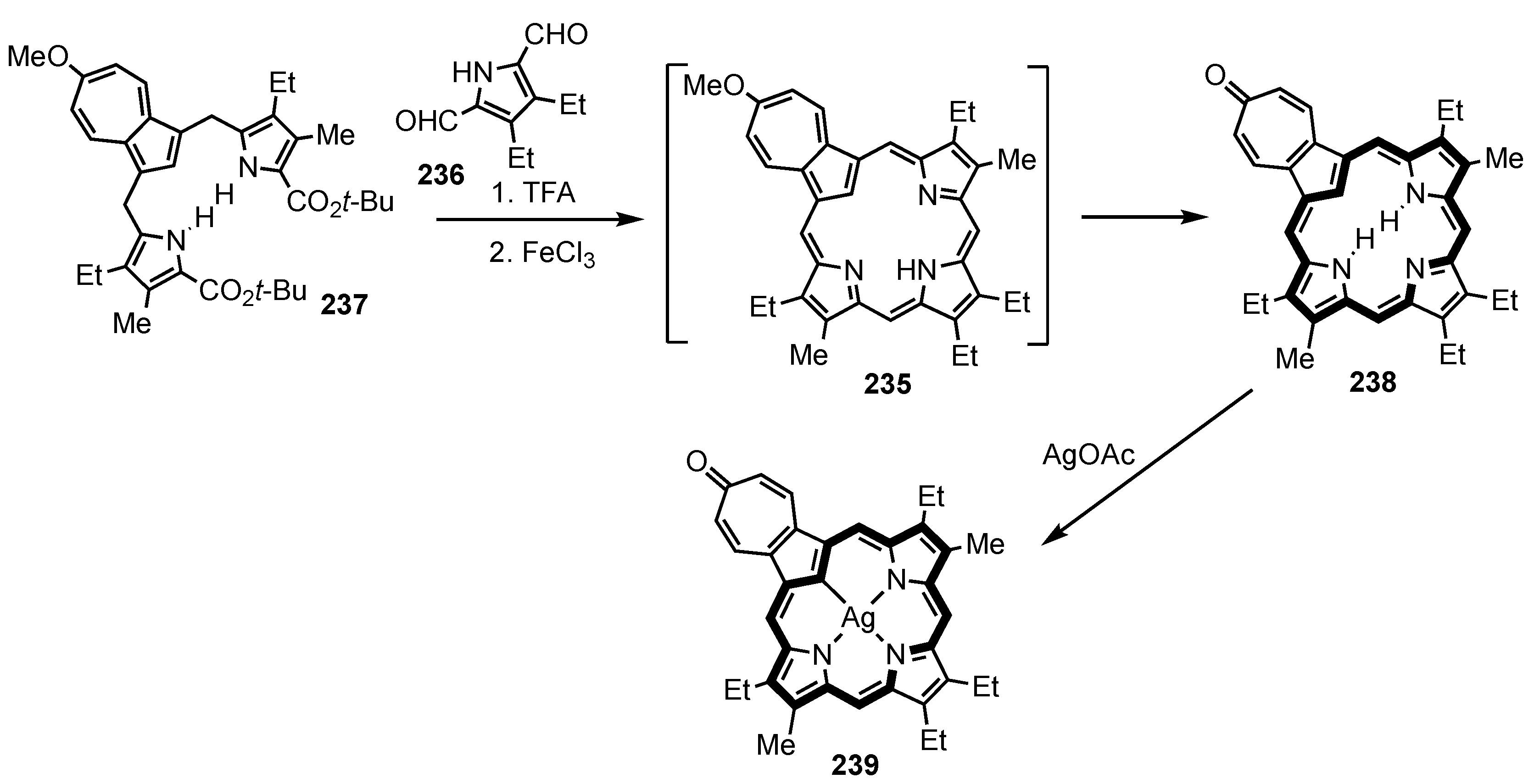{kind=link}
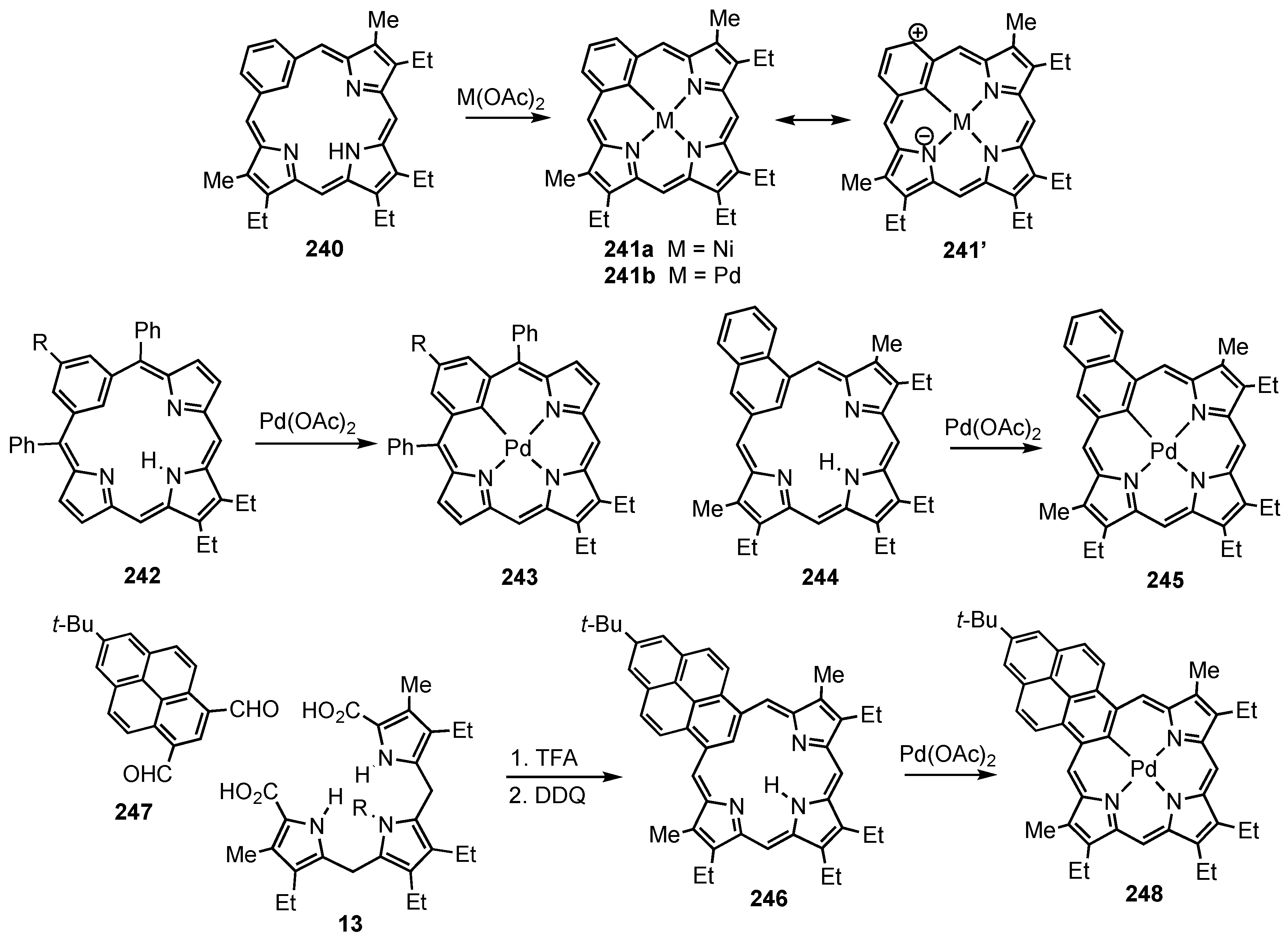{kind=link}
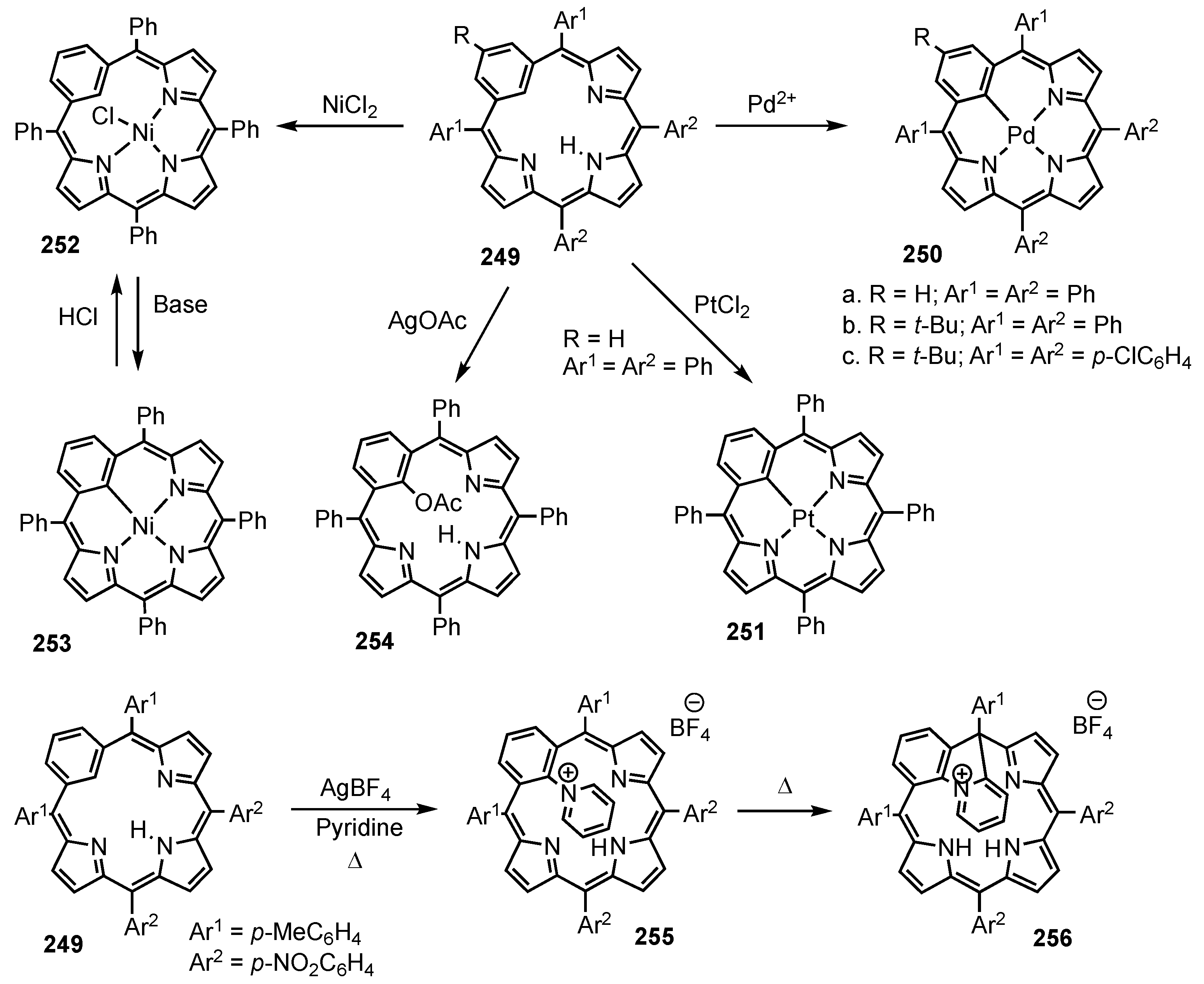{kind=link}
{kind=link}
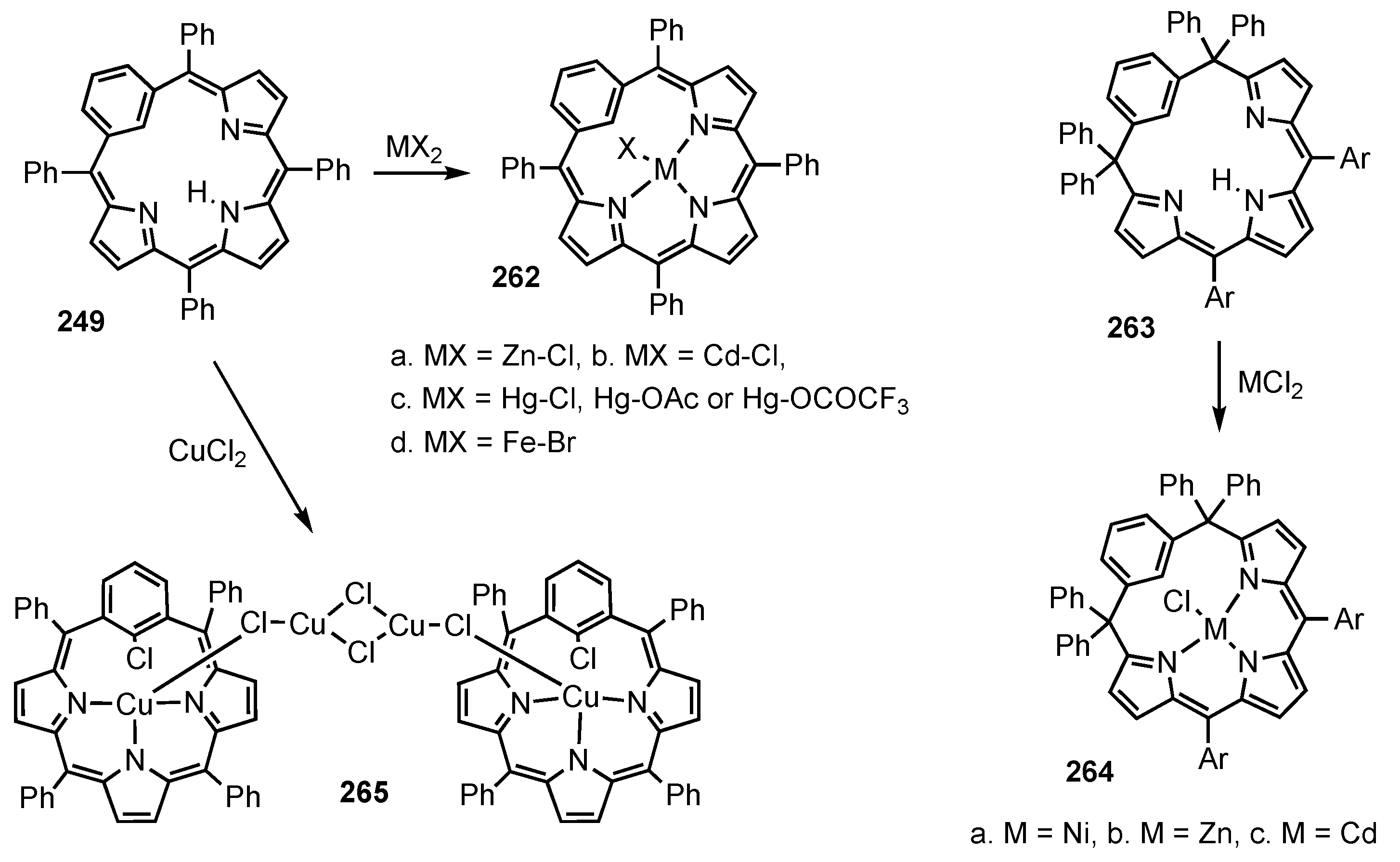{kind=link}
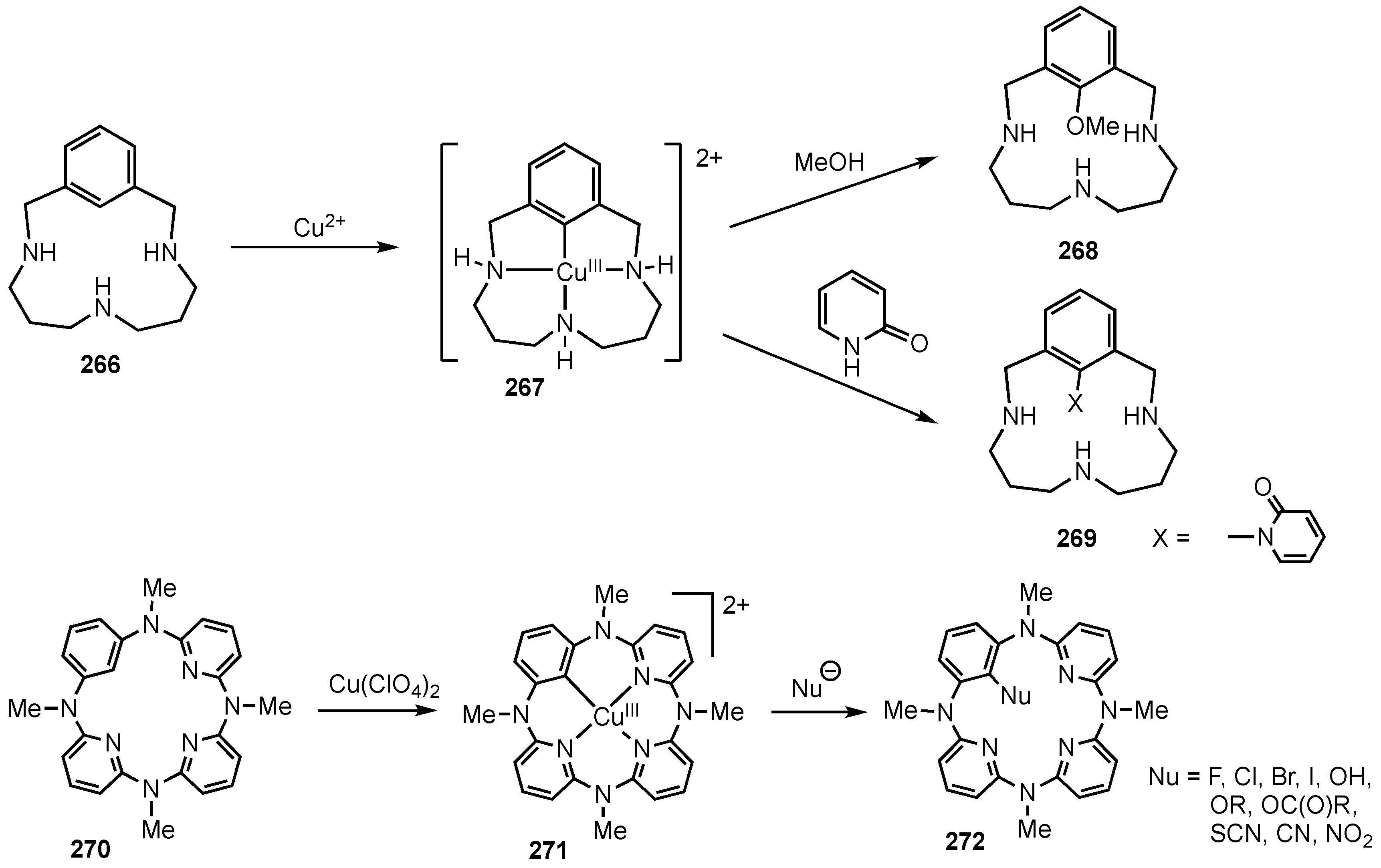{kind=link}
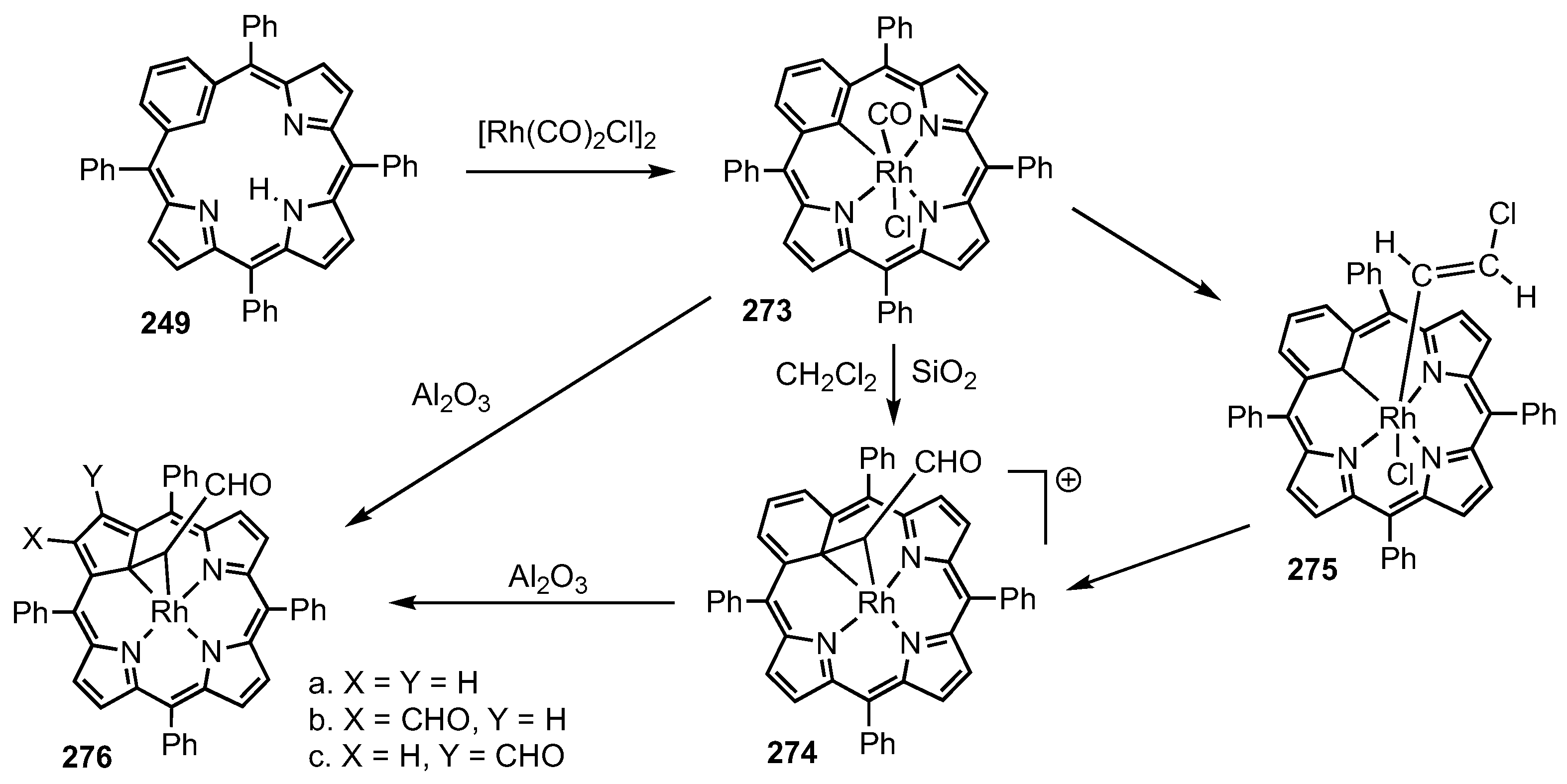{kind=link}
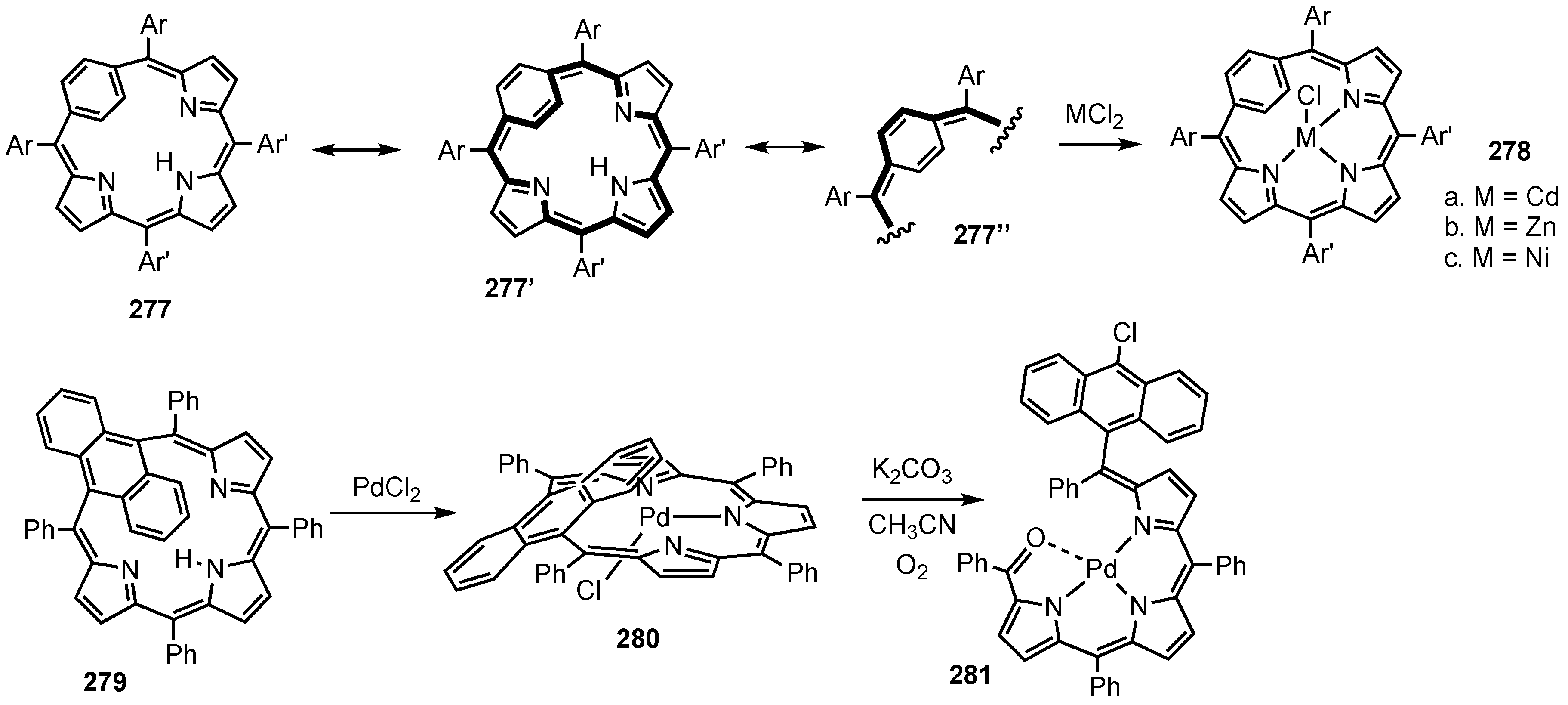{kind=link}
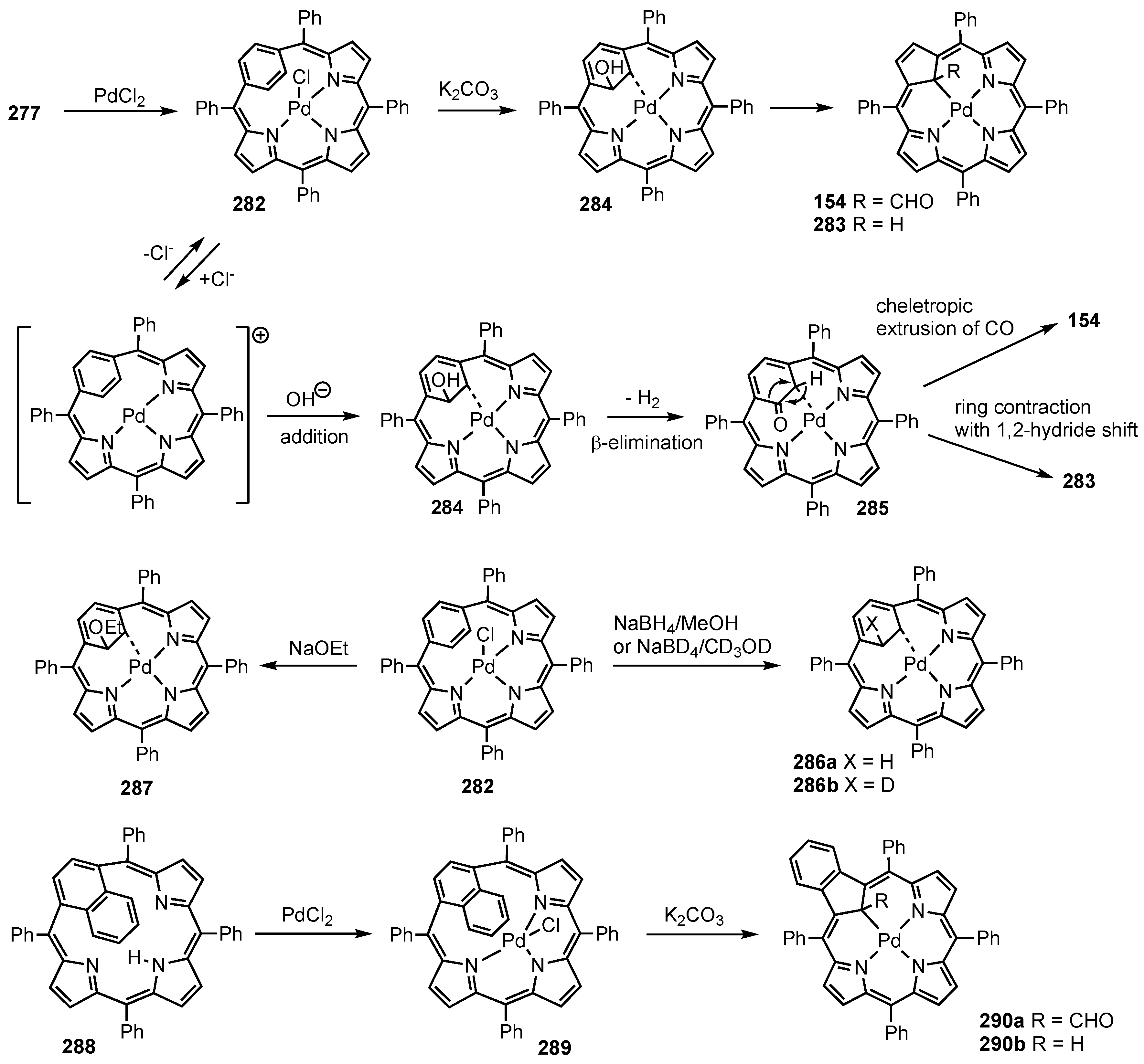{kind=link}
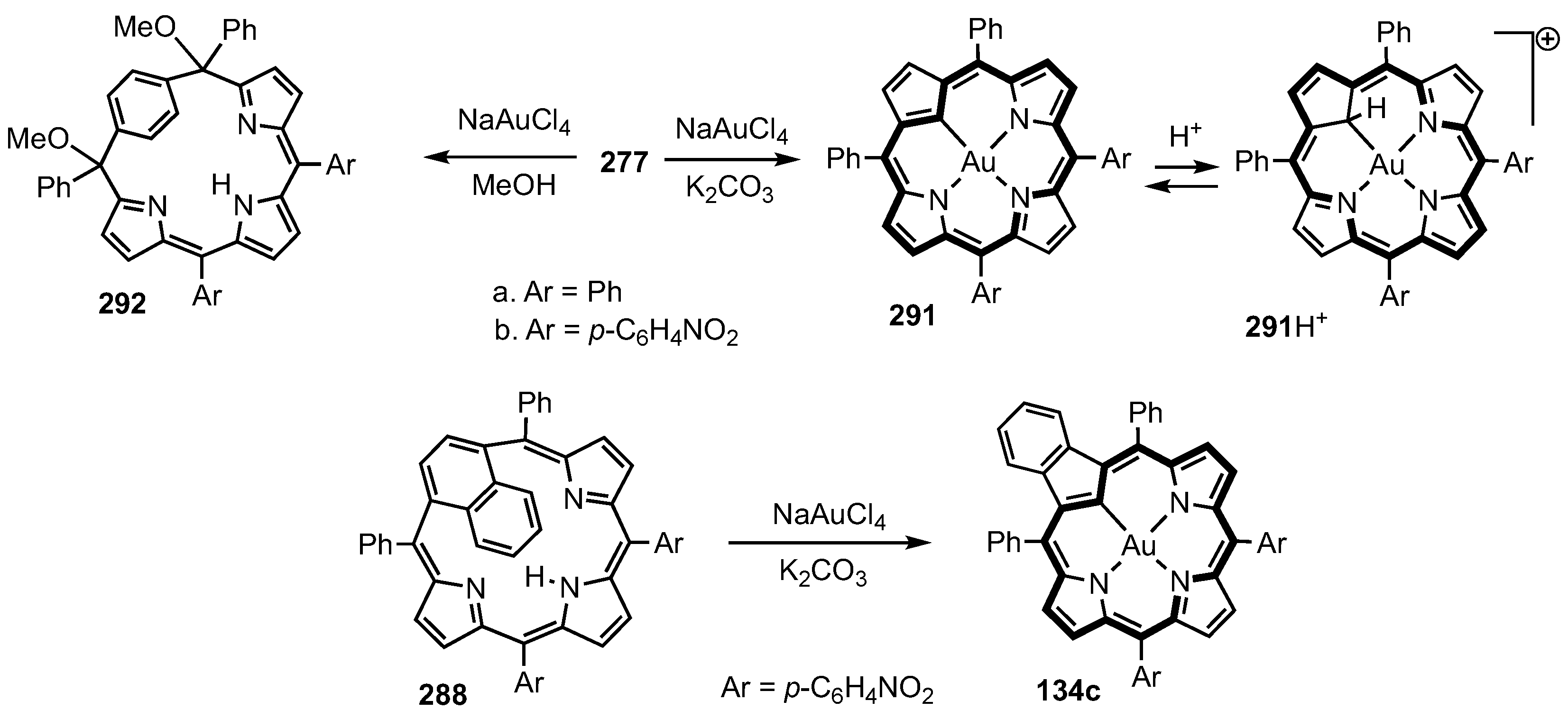{kind=link}
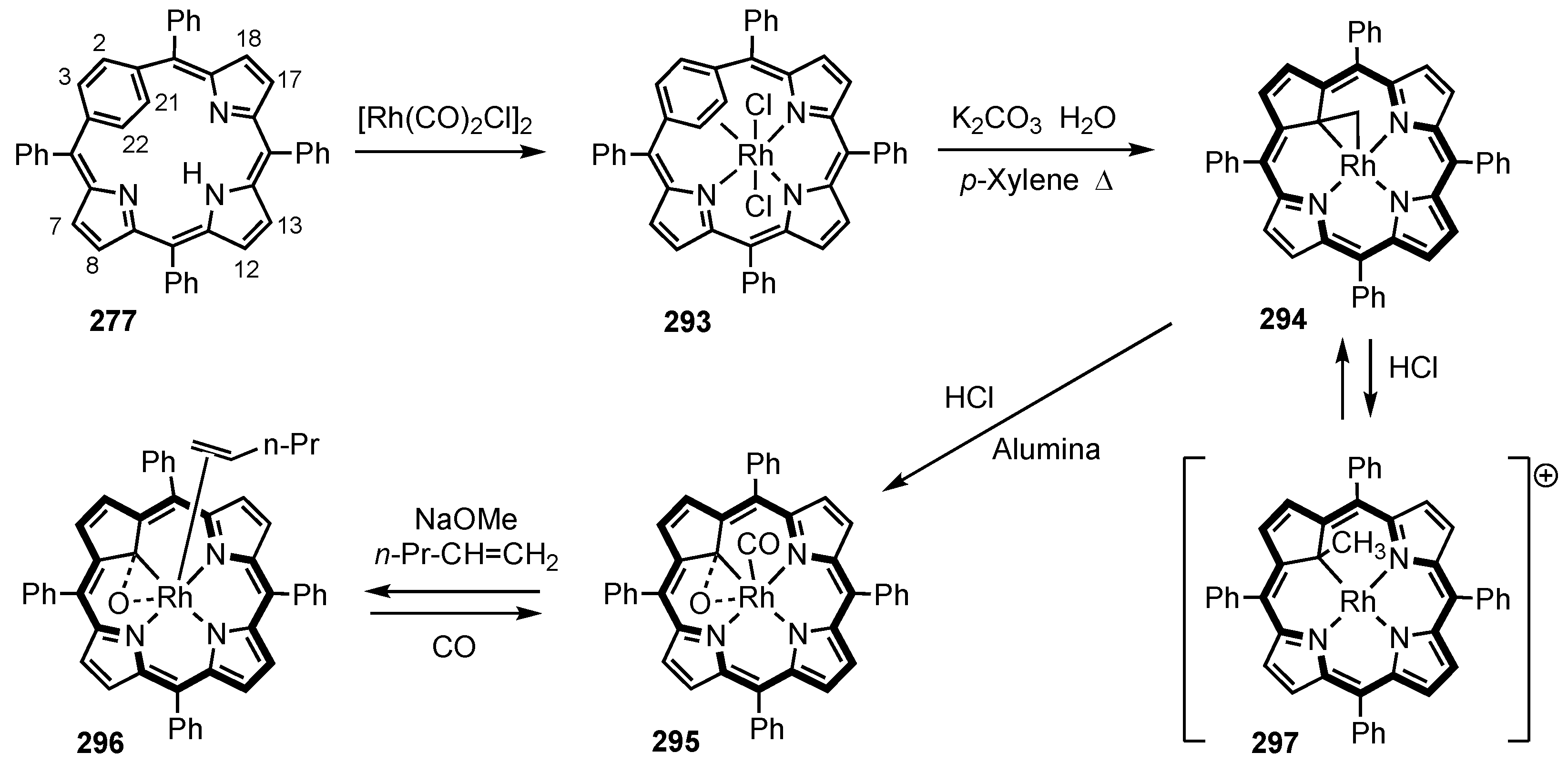{kind=link}
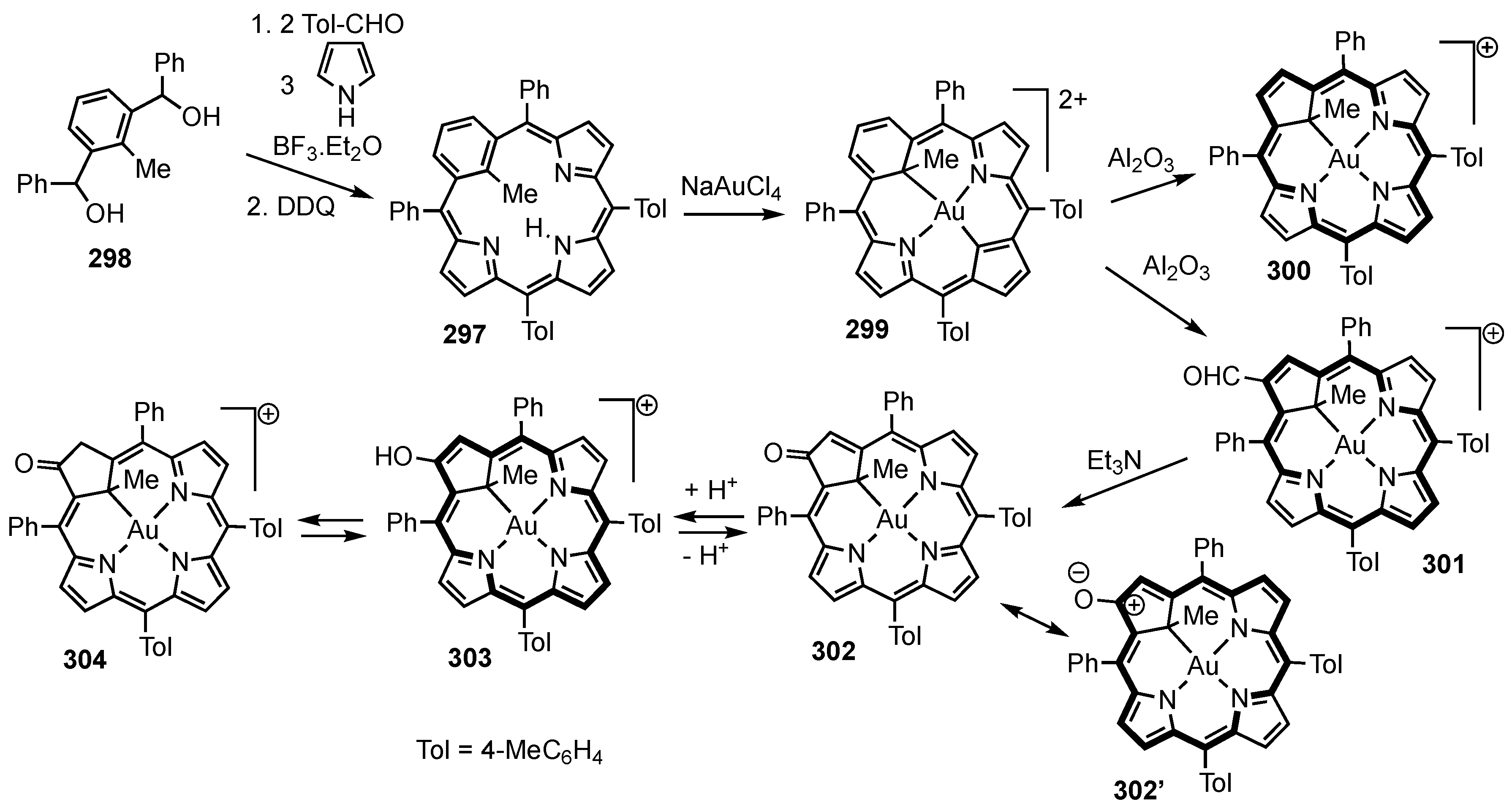{kind=link}
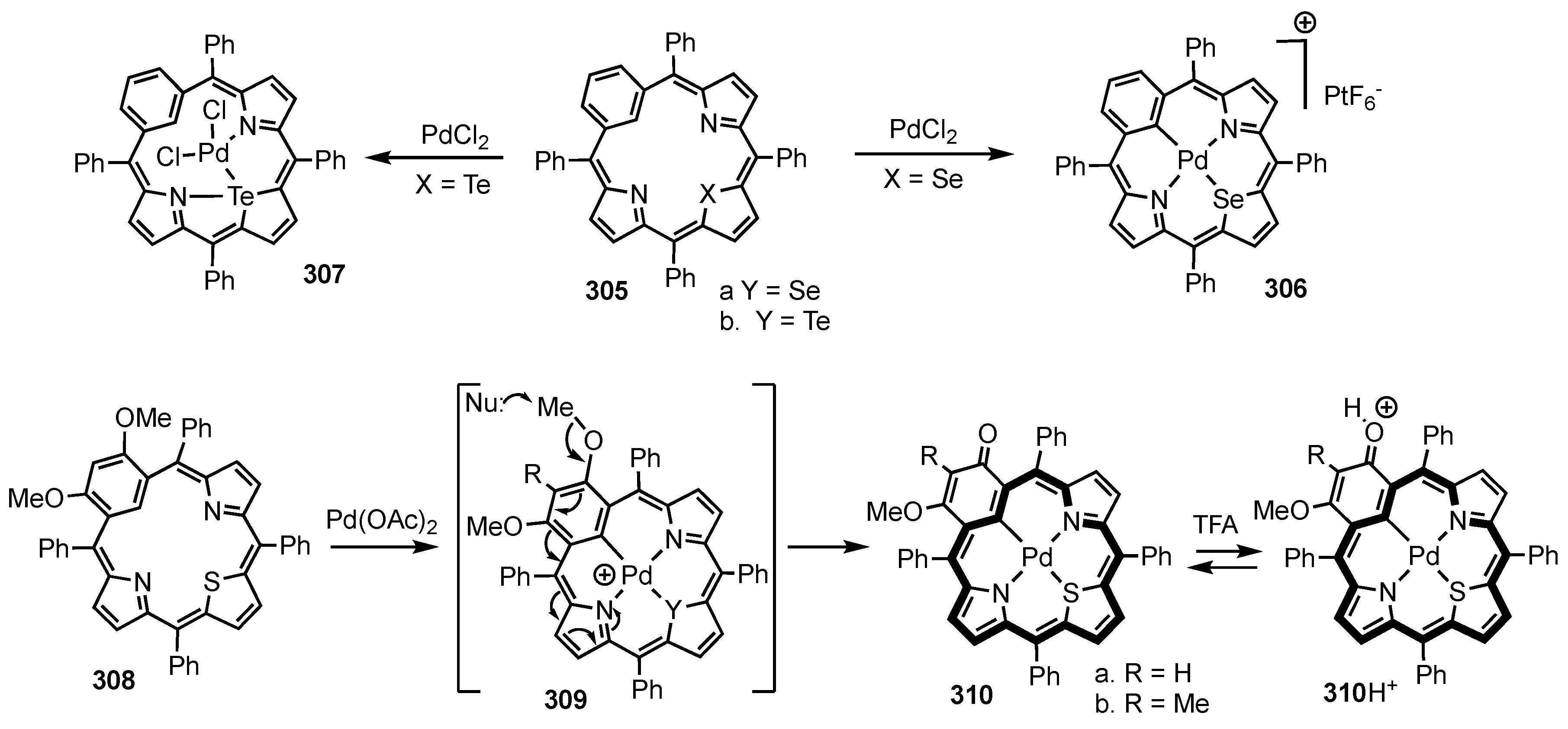{kind=link}
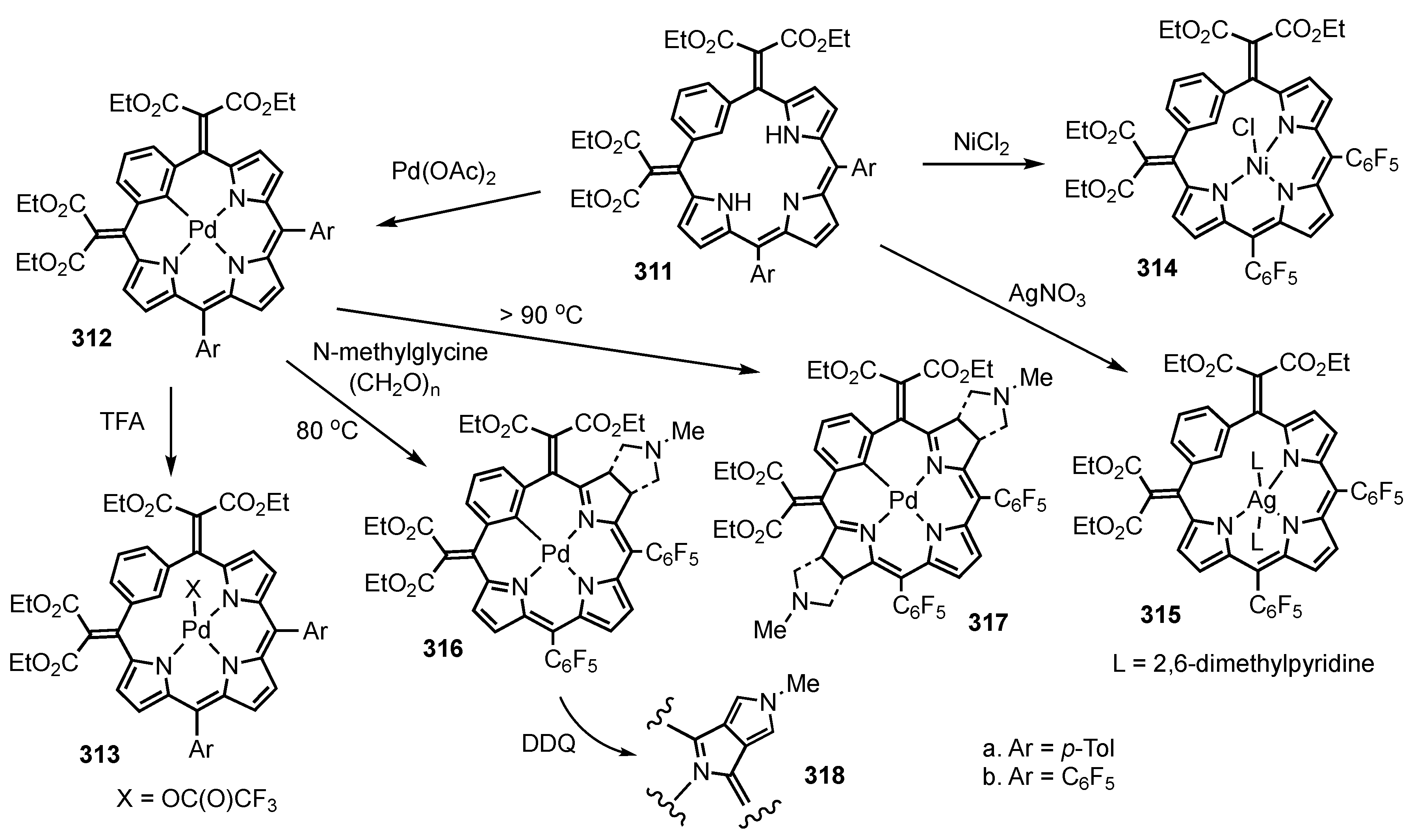{kind=link}
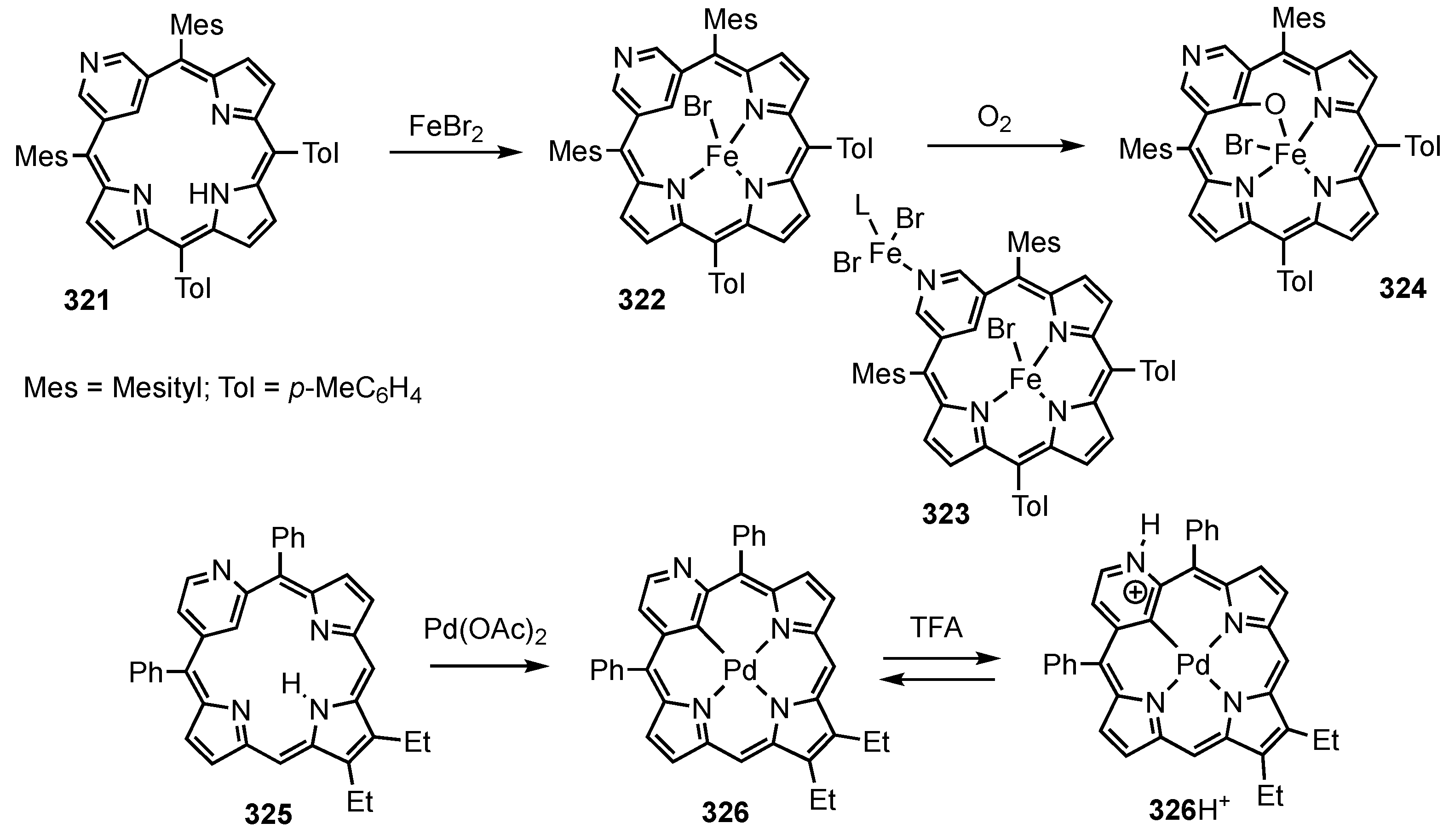{kind=link}
{kind=link}
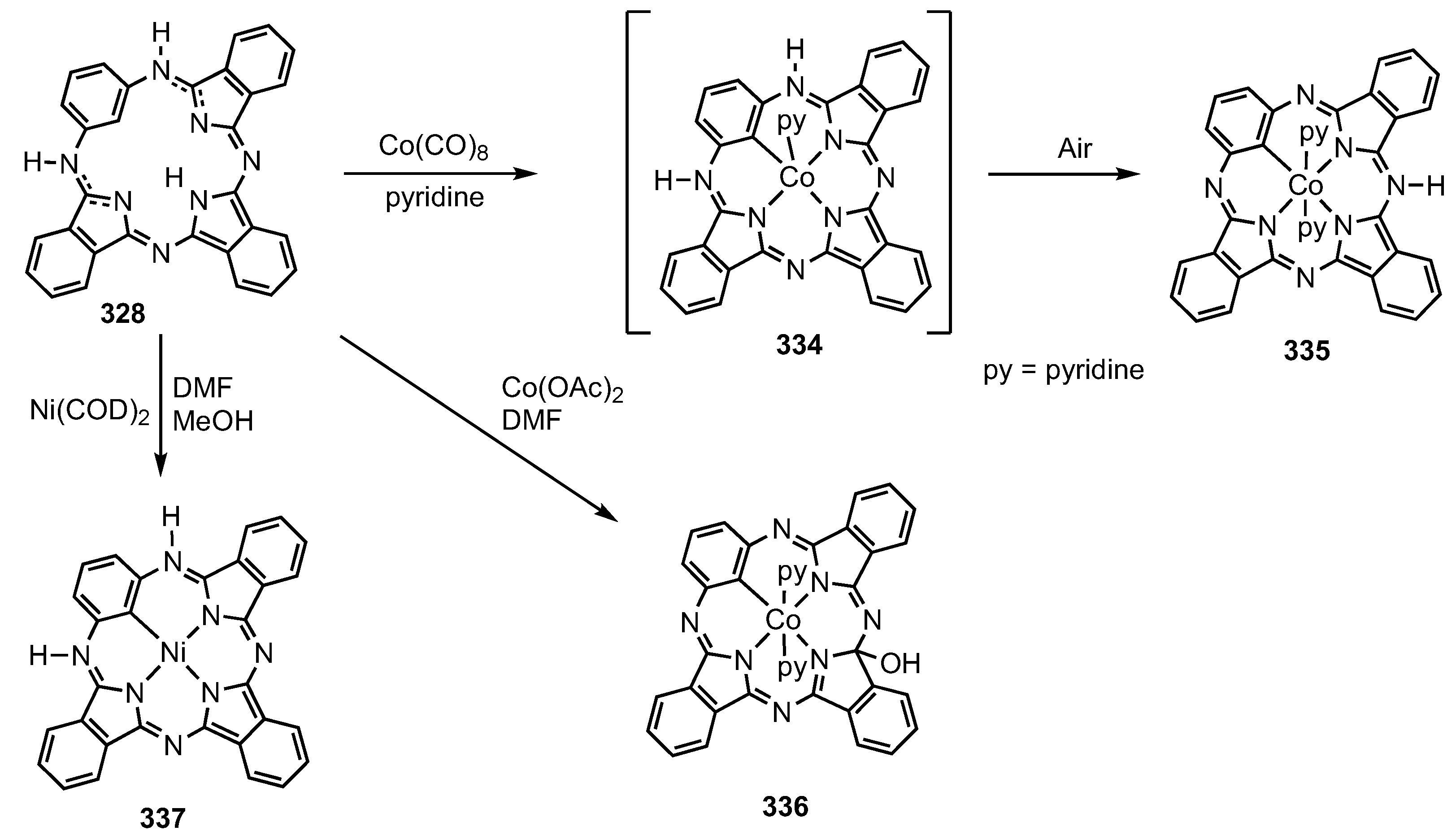{kind=link}
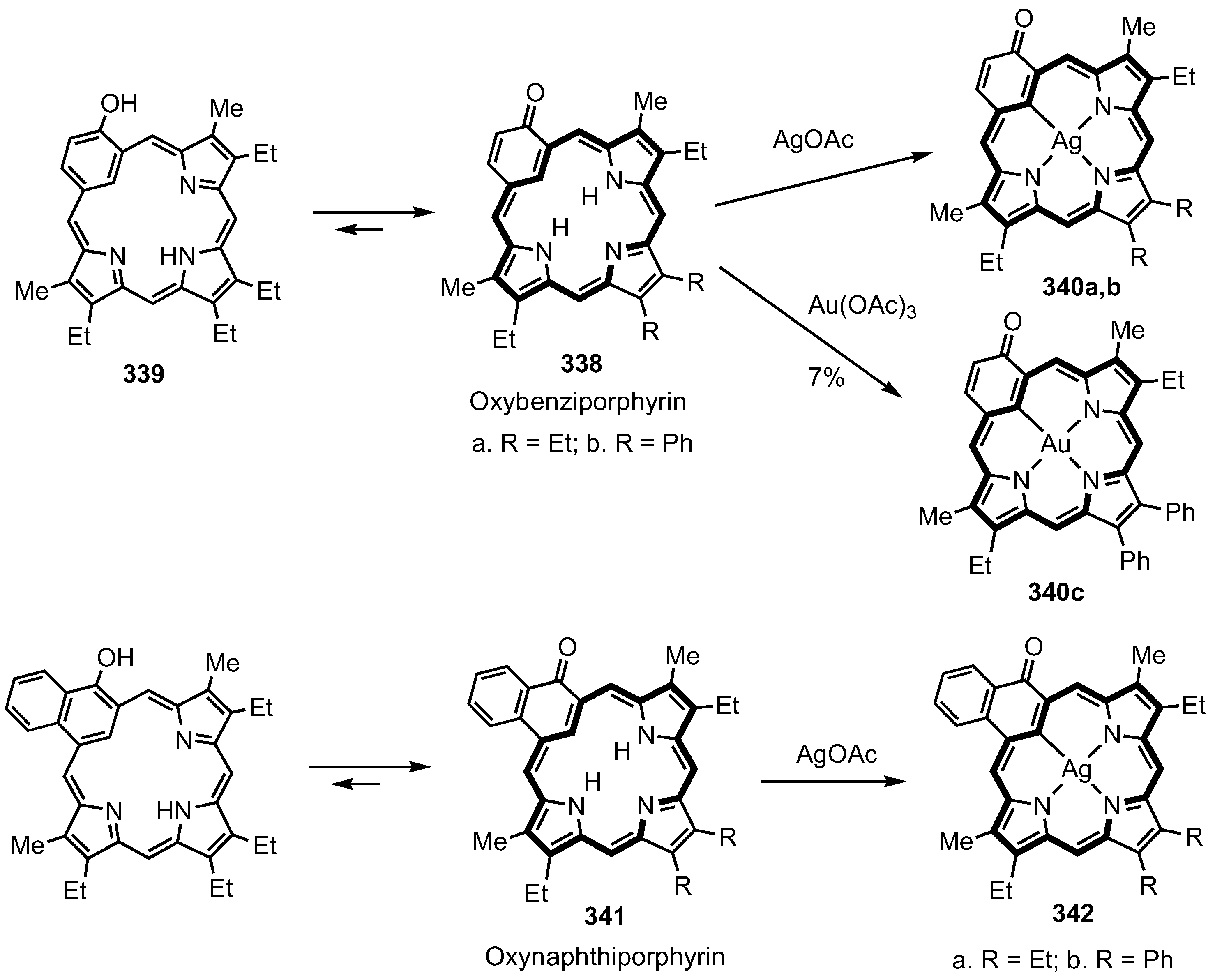{kind=link}
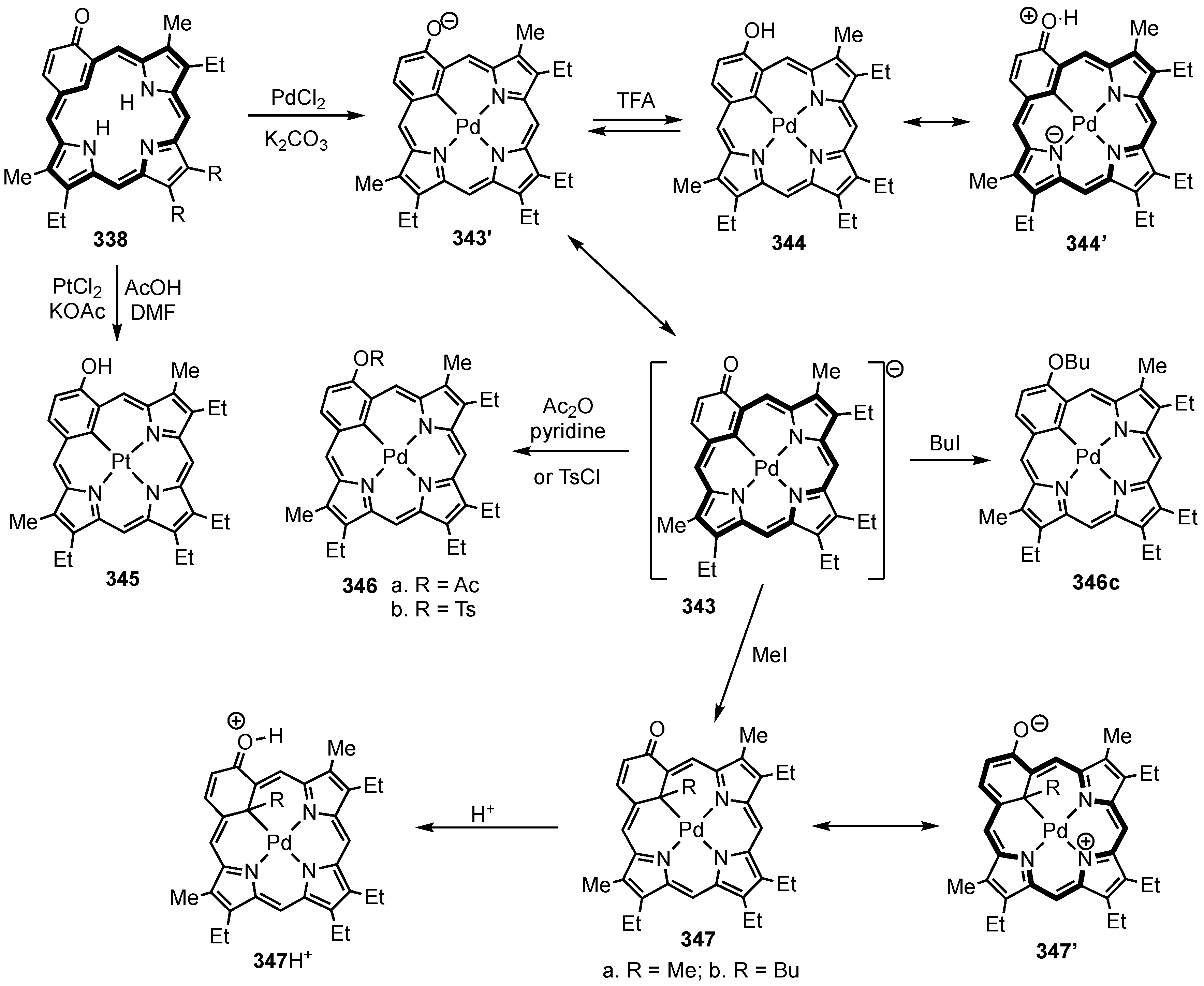{kind=link}
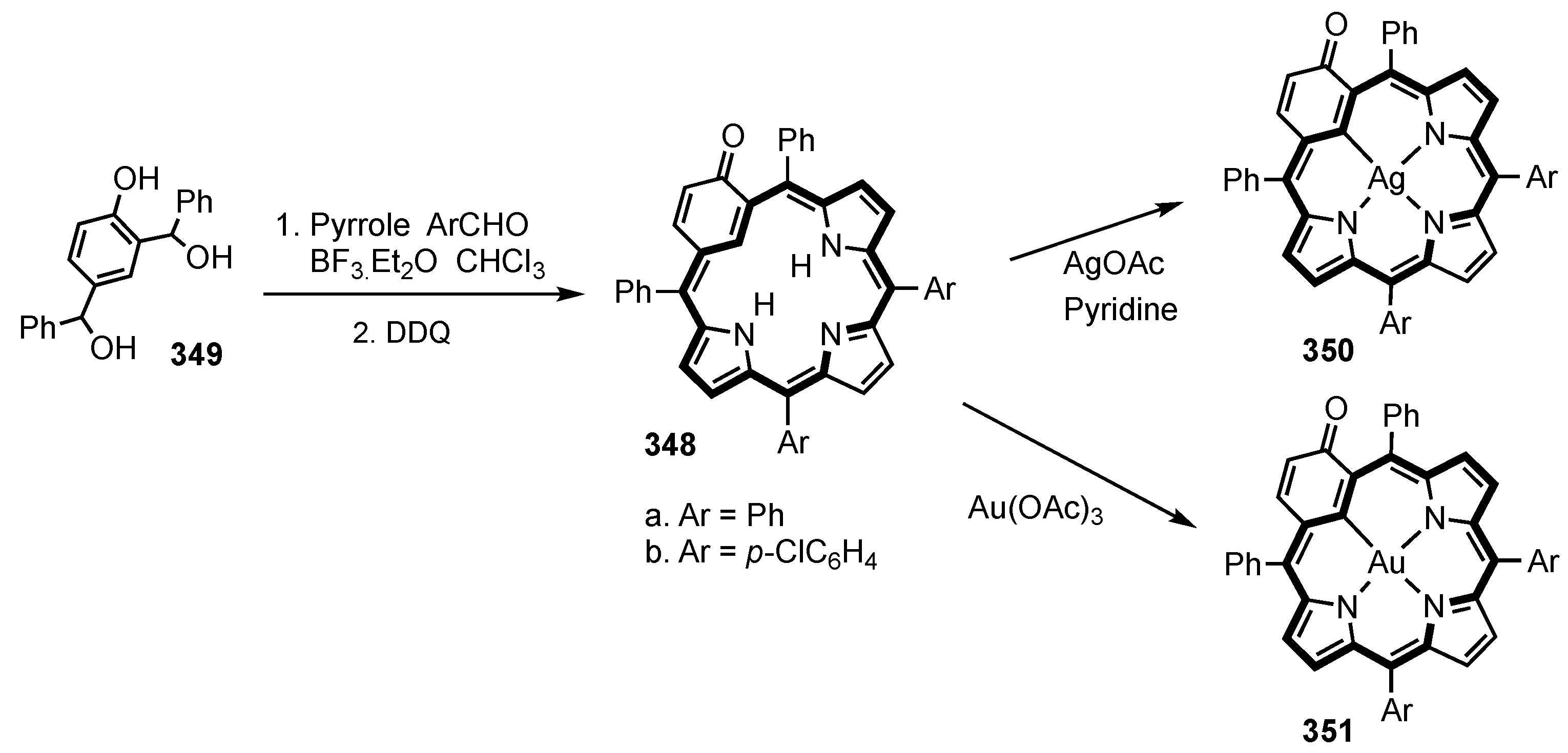{kind=link}
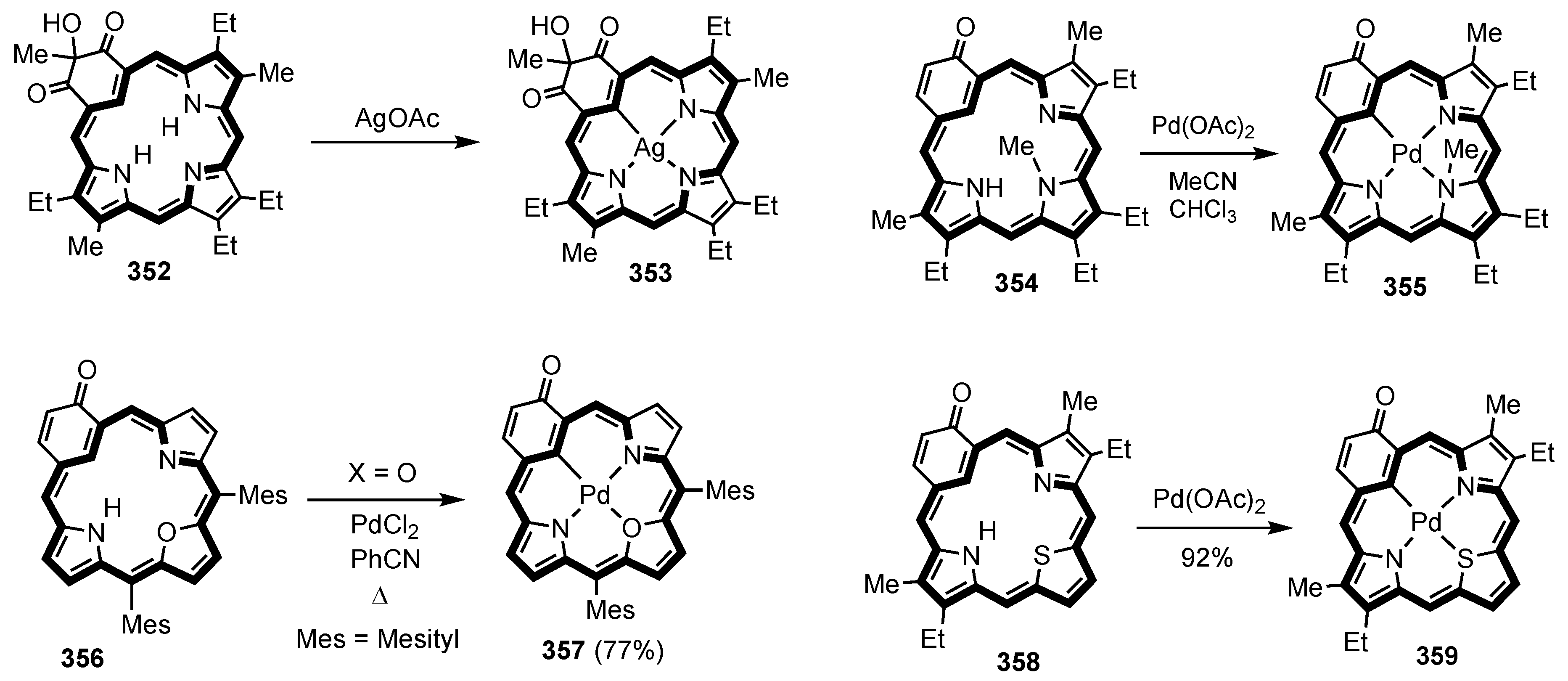{kind=link}
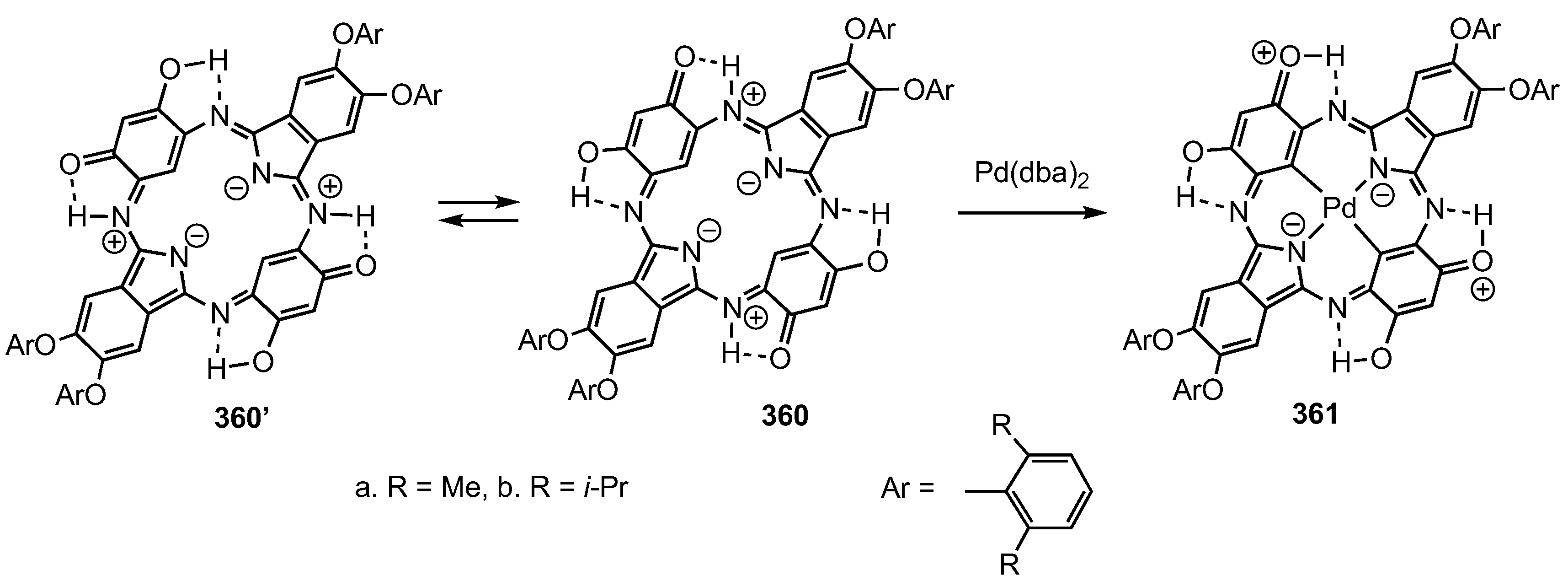{kind=link}
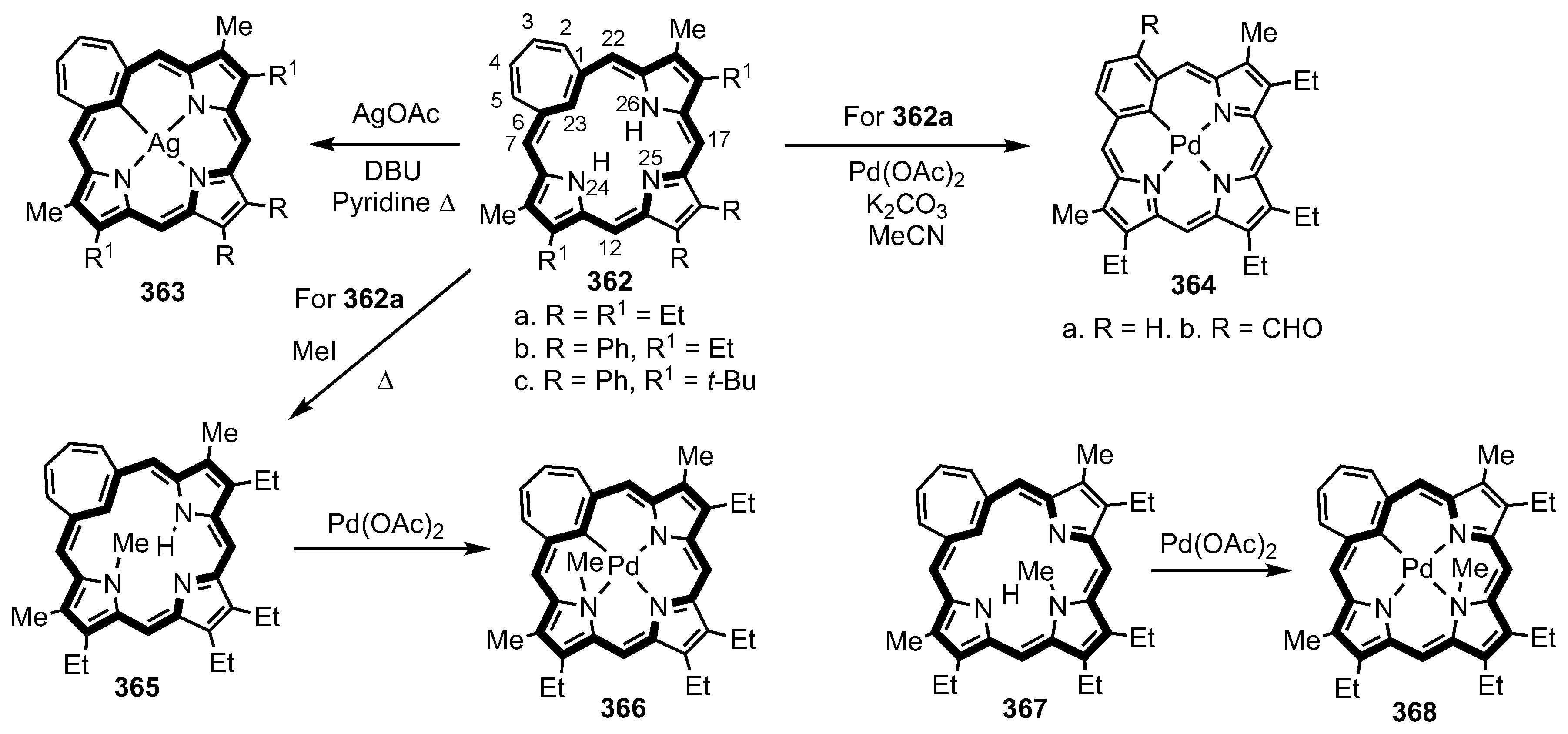{kind=link}
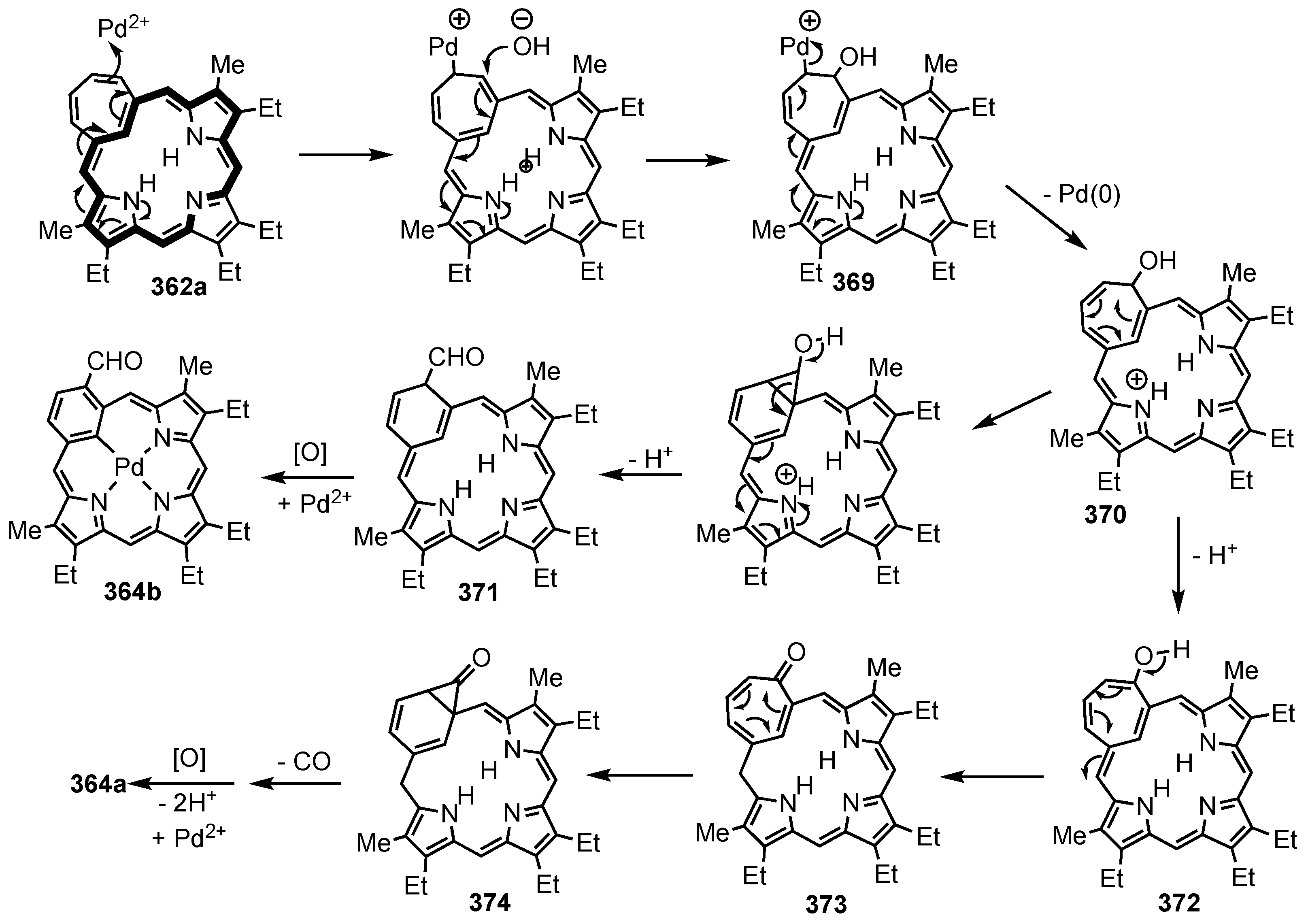{kind=link}
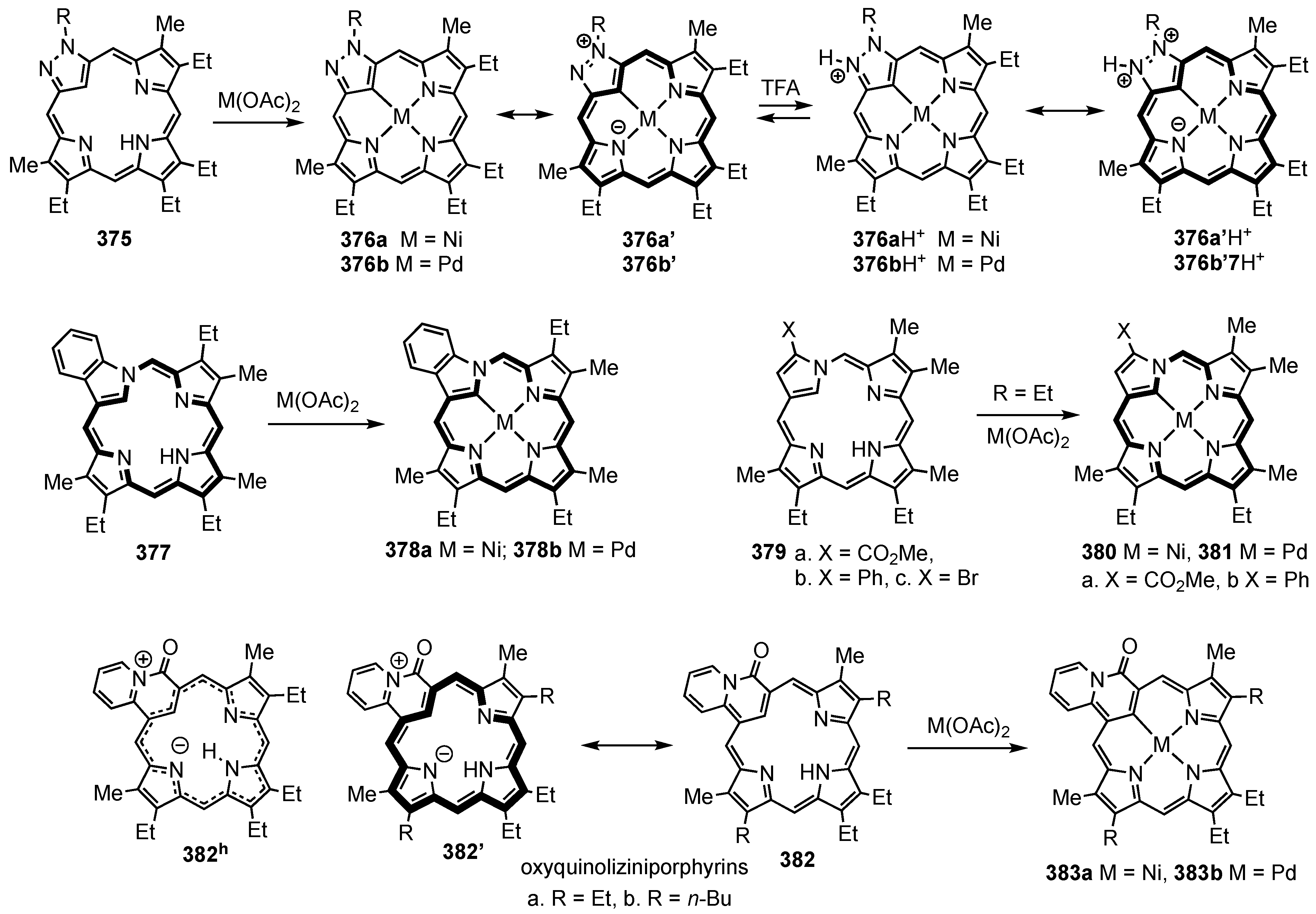{kind=link}
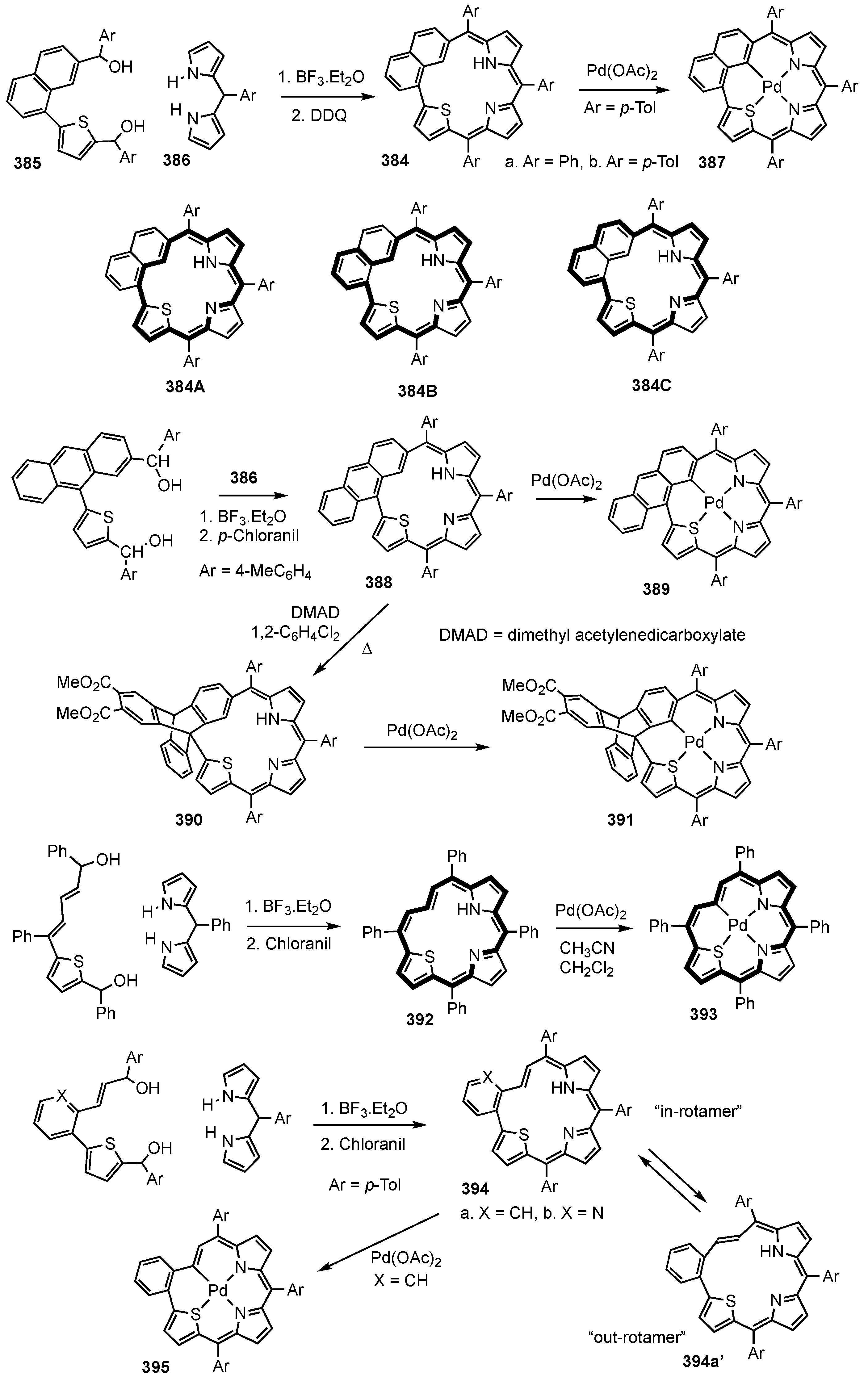{kind=link}
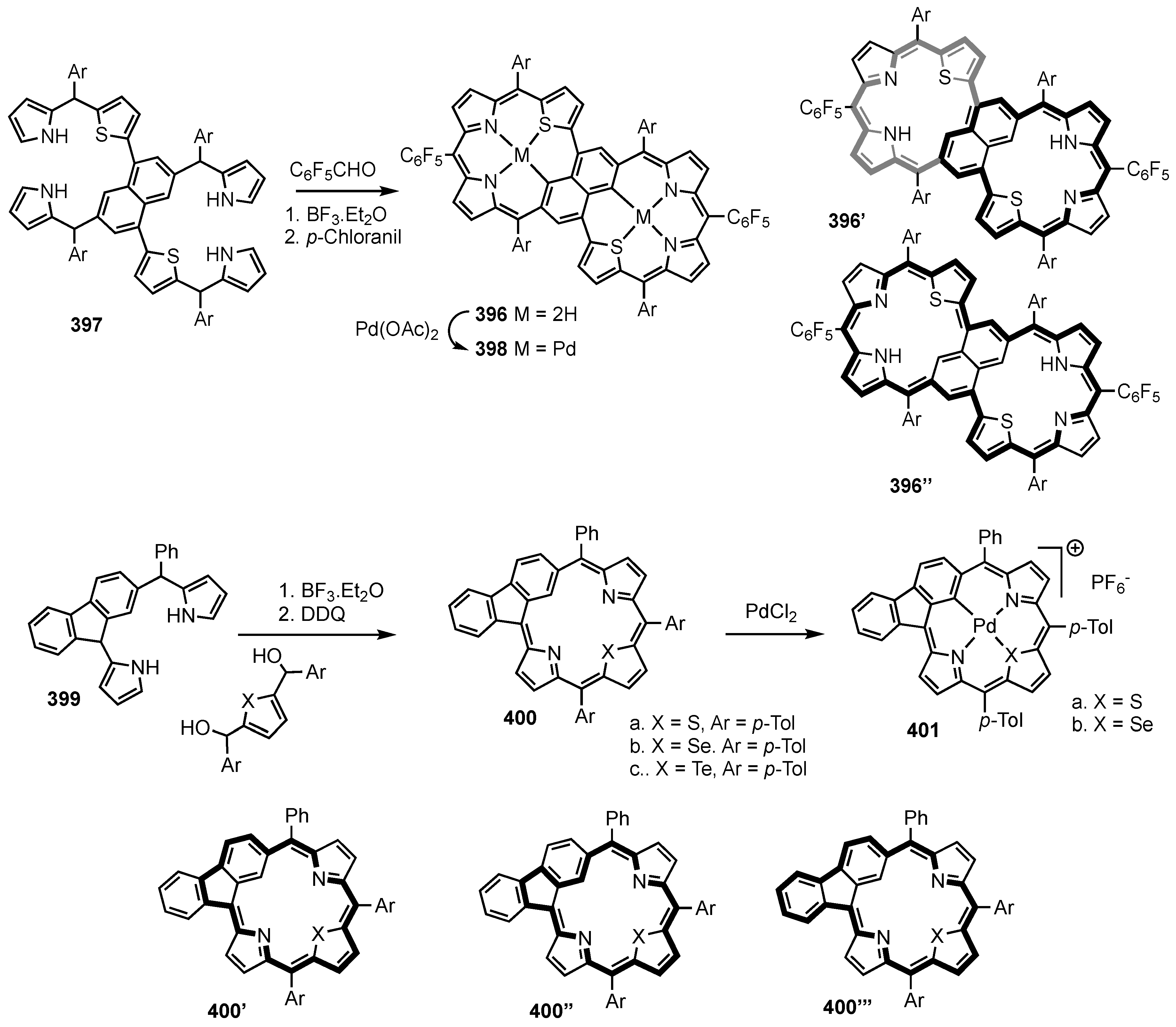{kind=link}
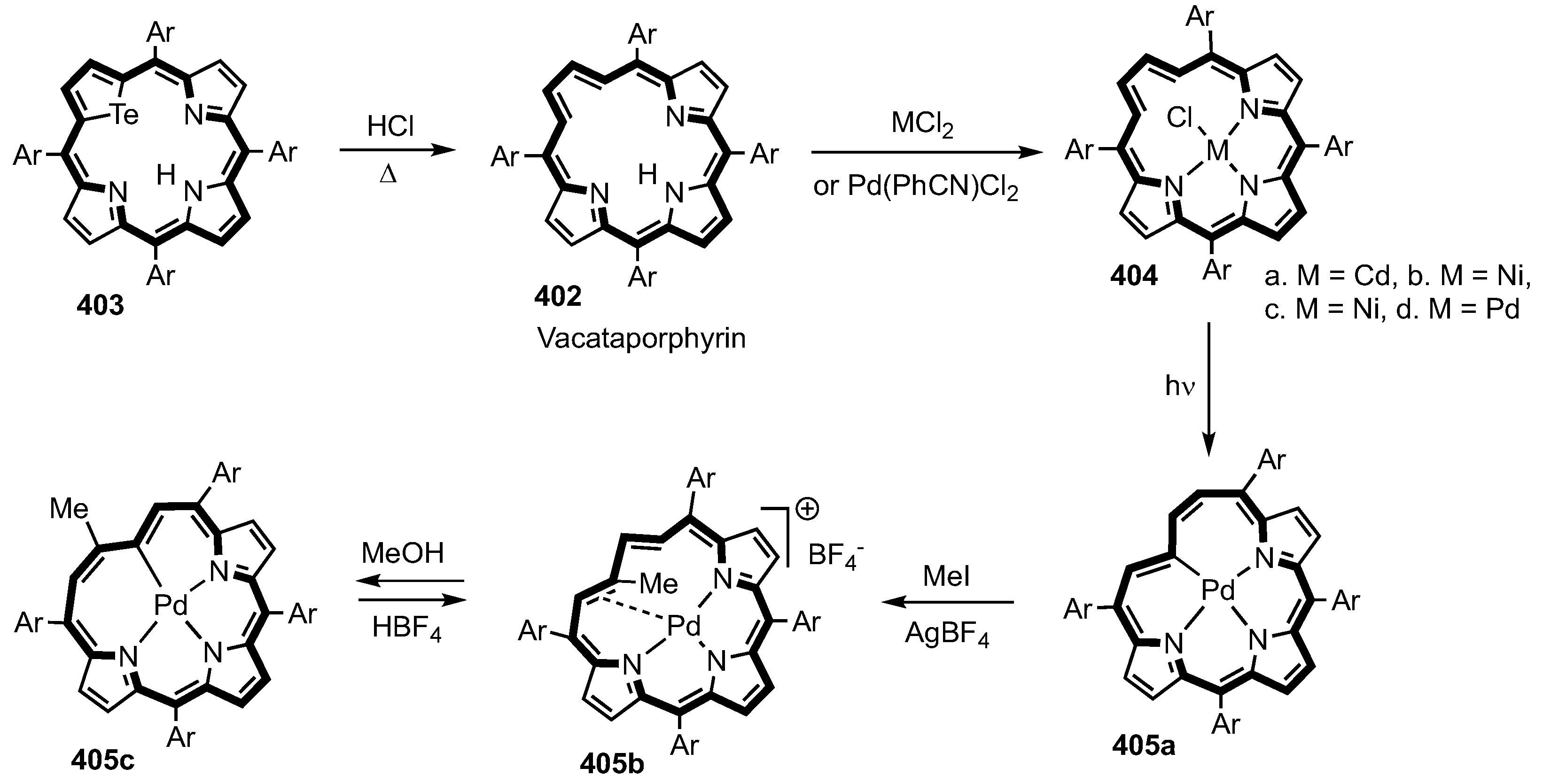{kind=link}
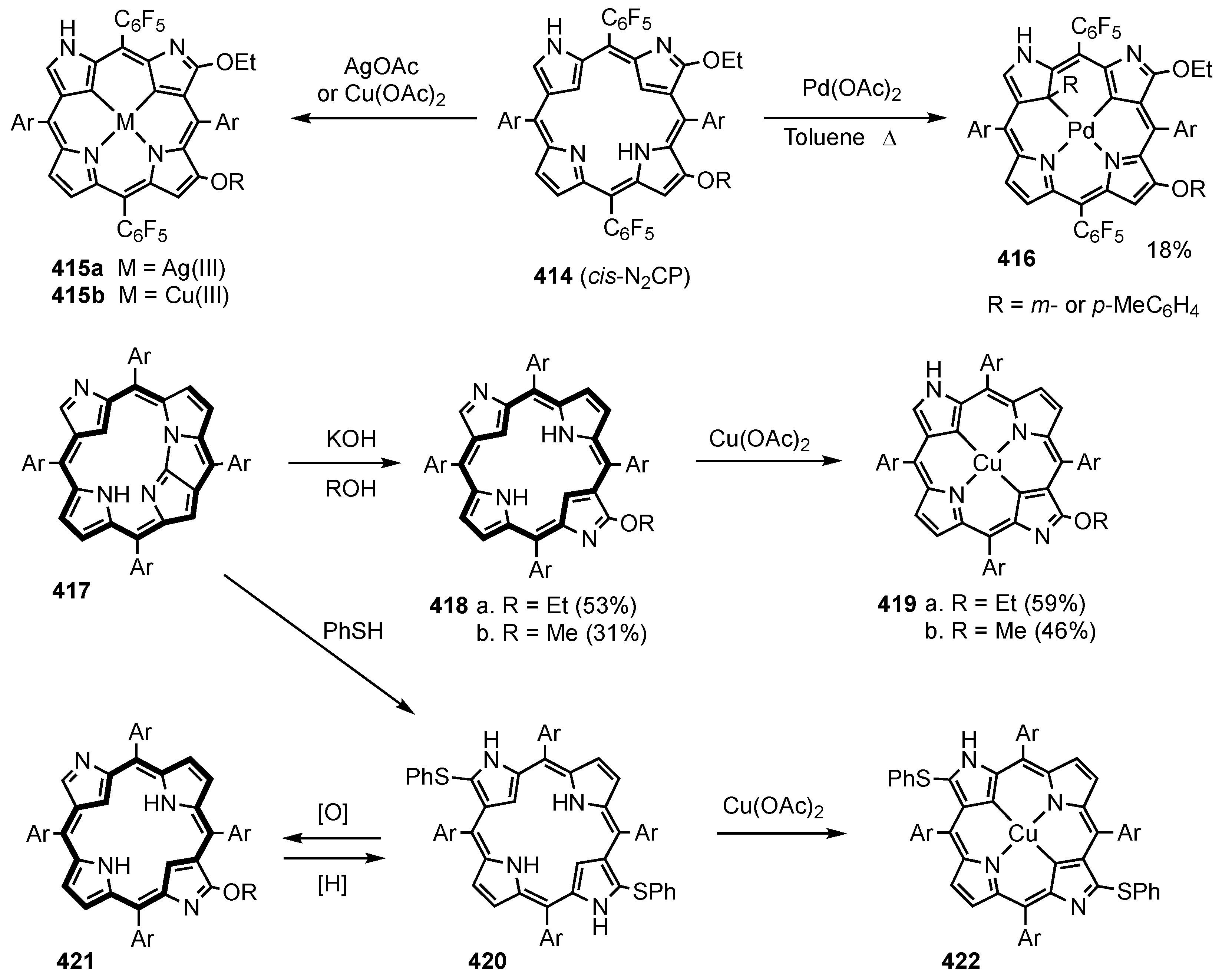{kind=link}
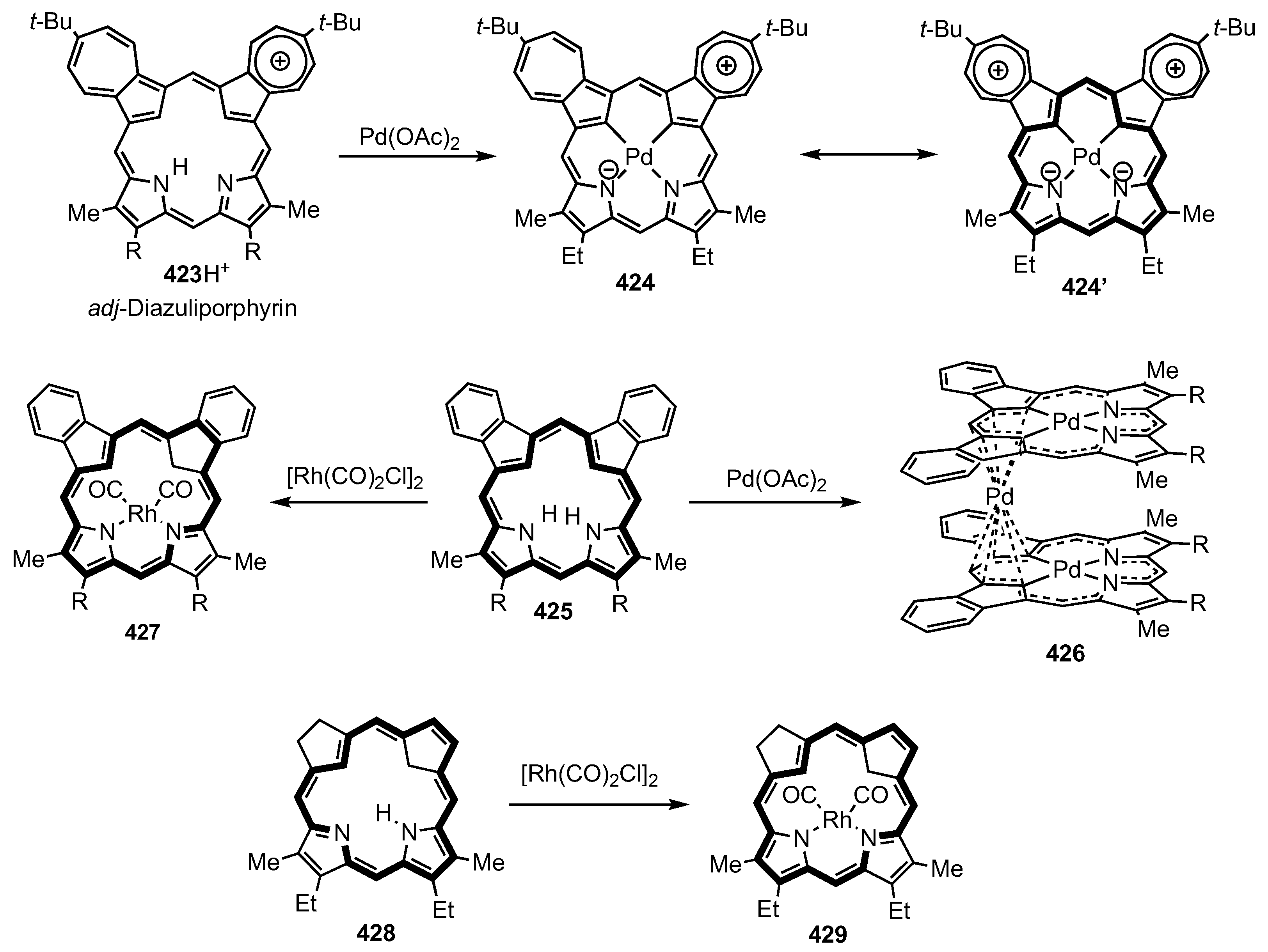{kind=link}
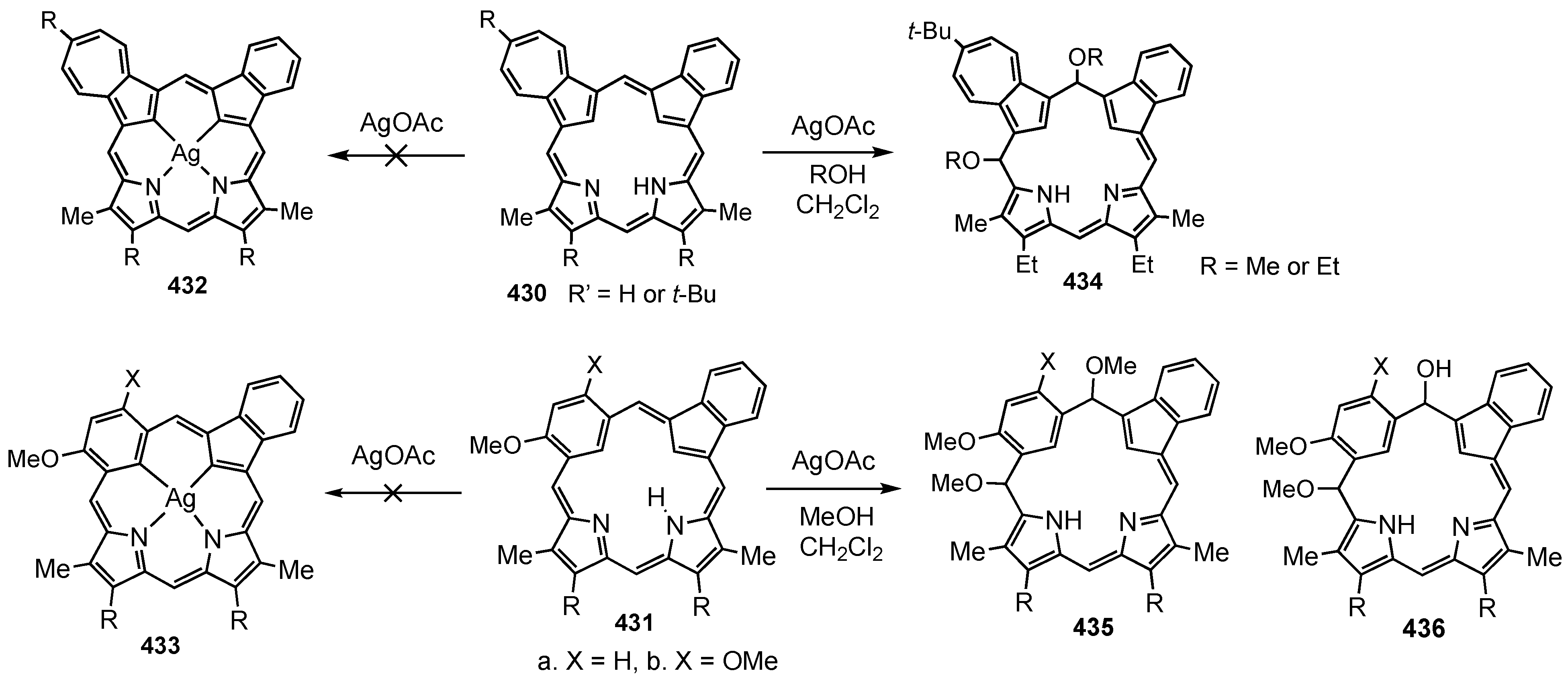{kind=link}
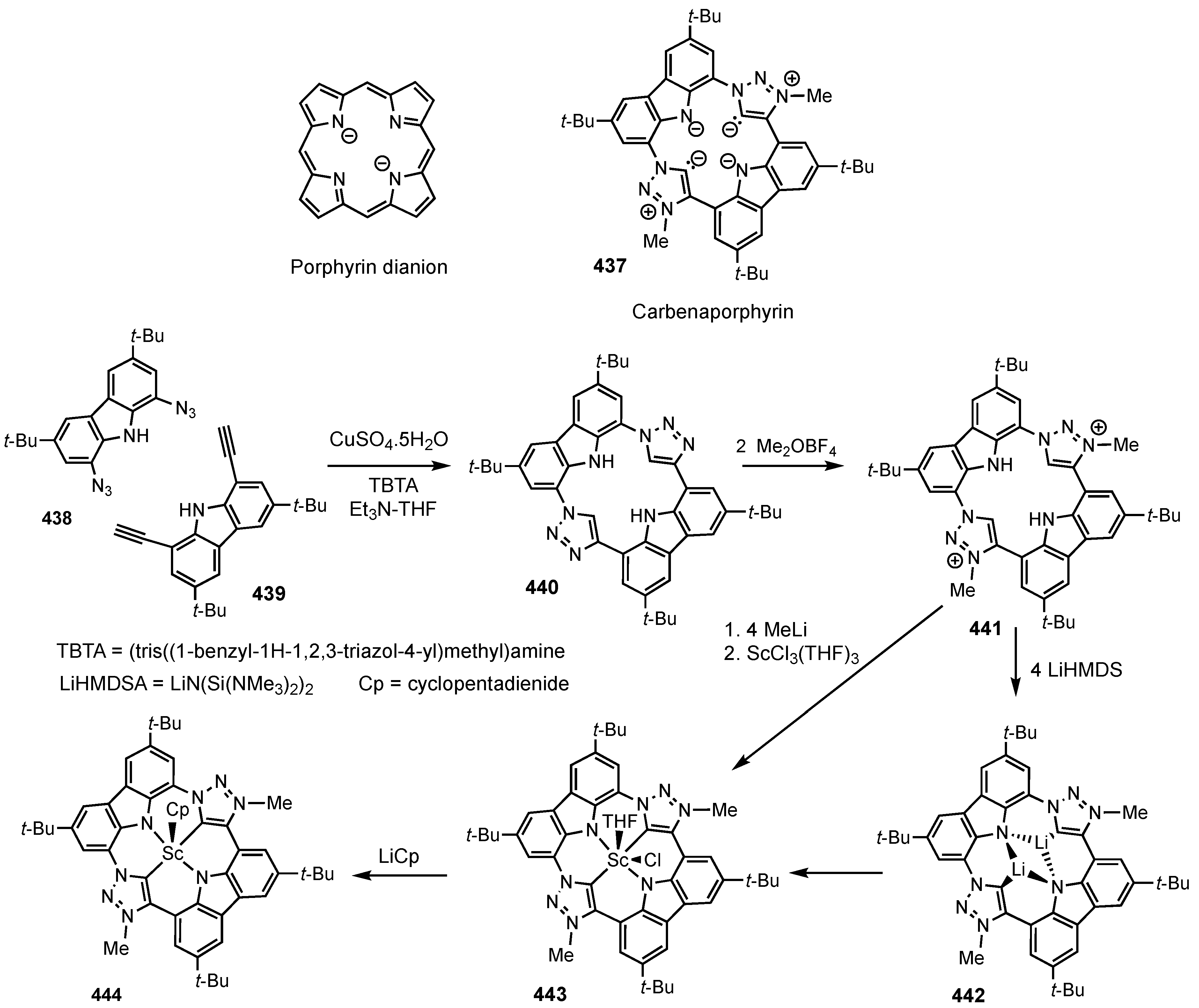{kind=link}
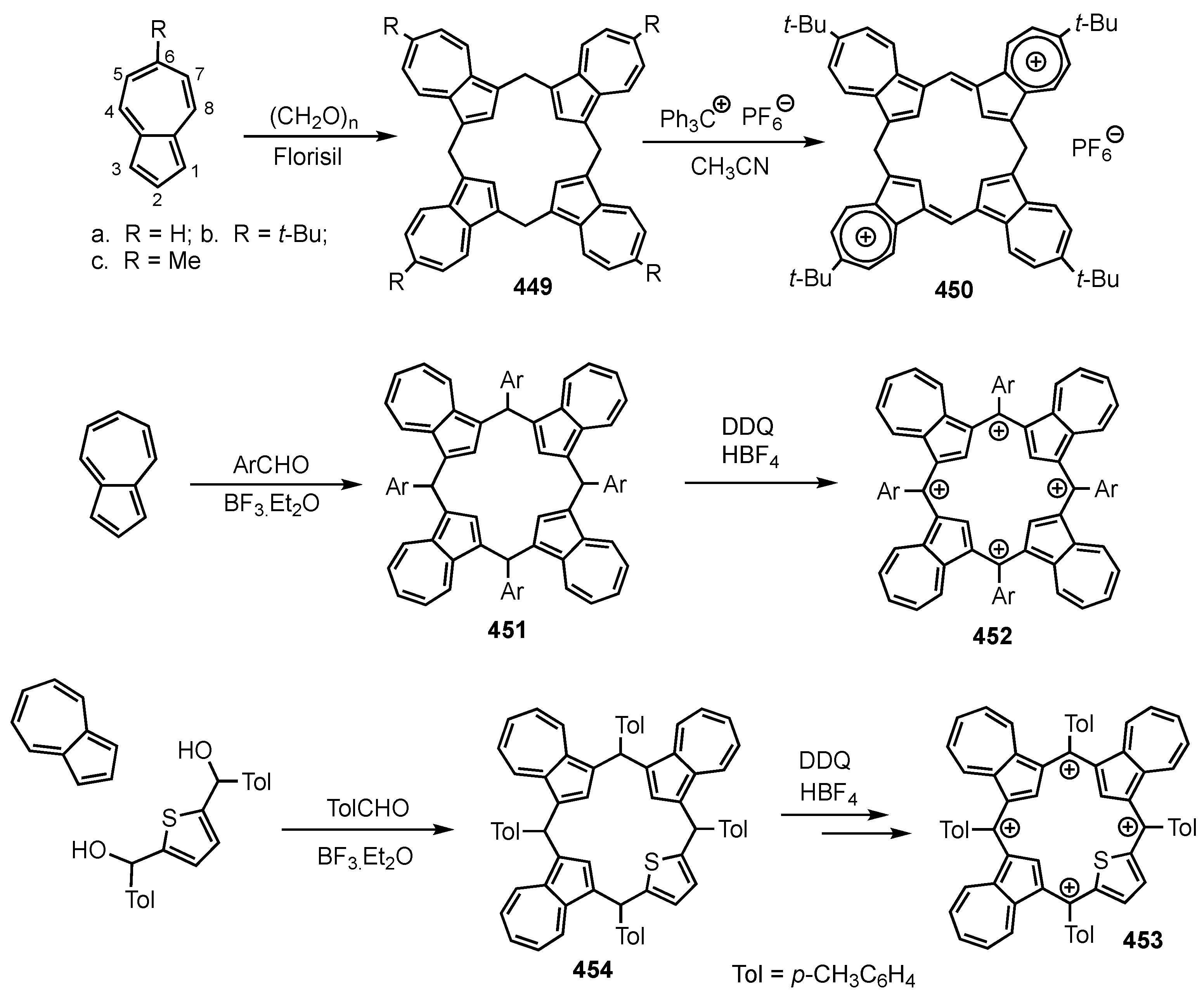{kind=link}
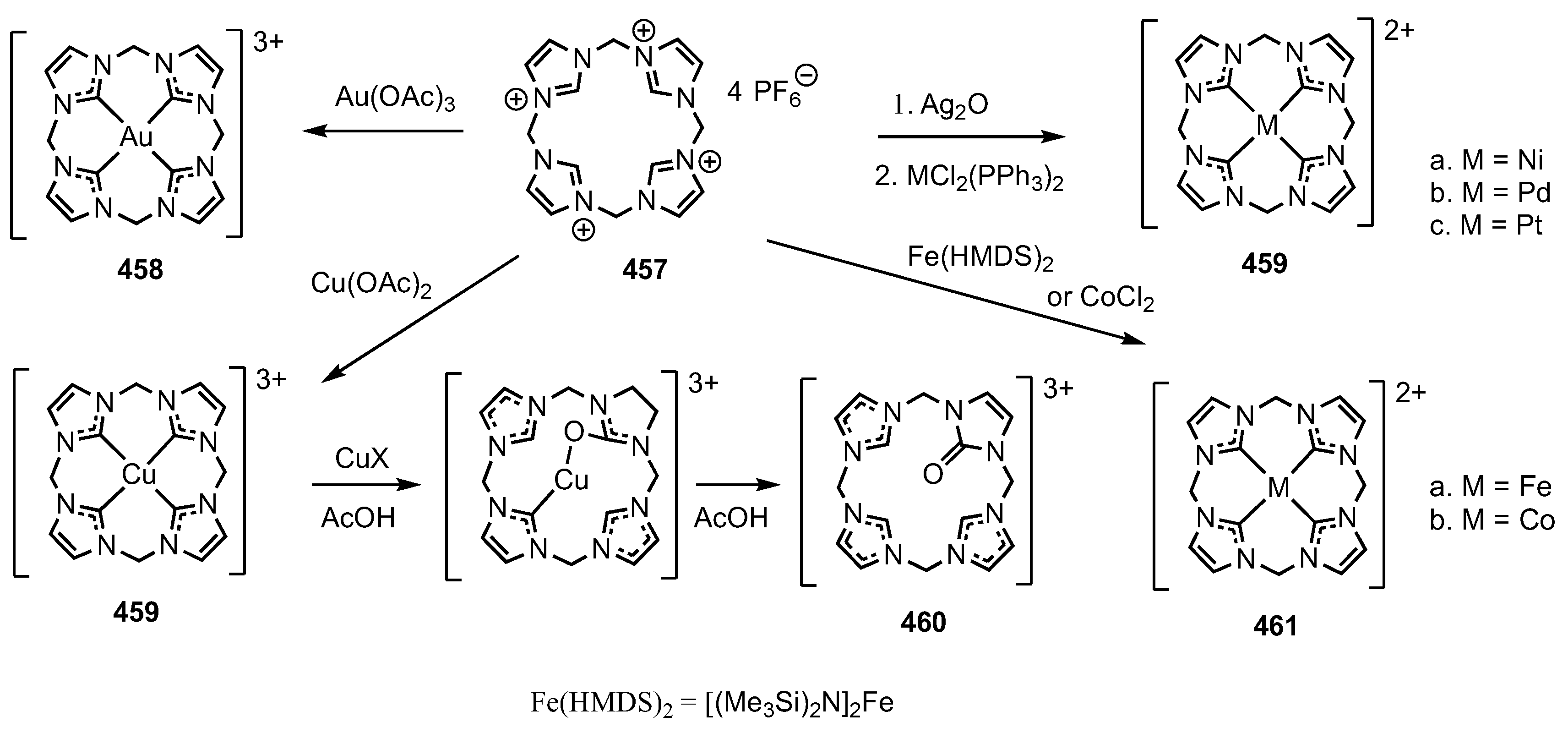{kind=link}
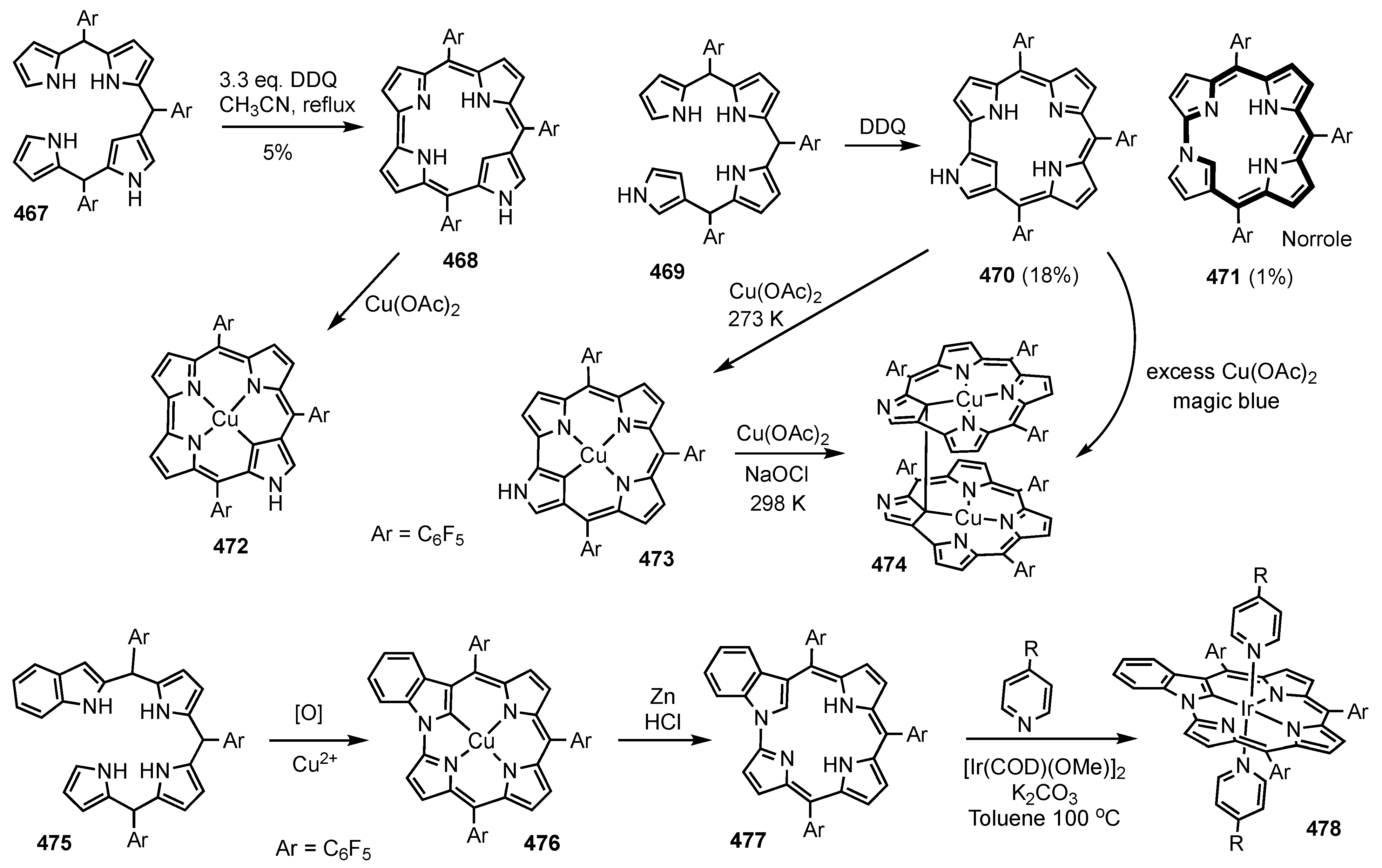{kind=link}
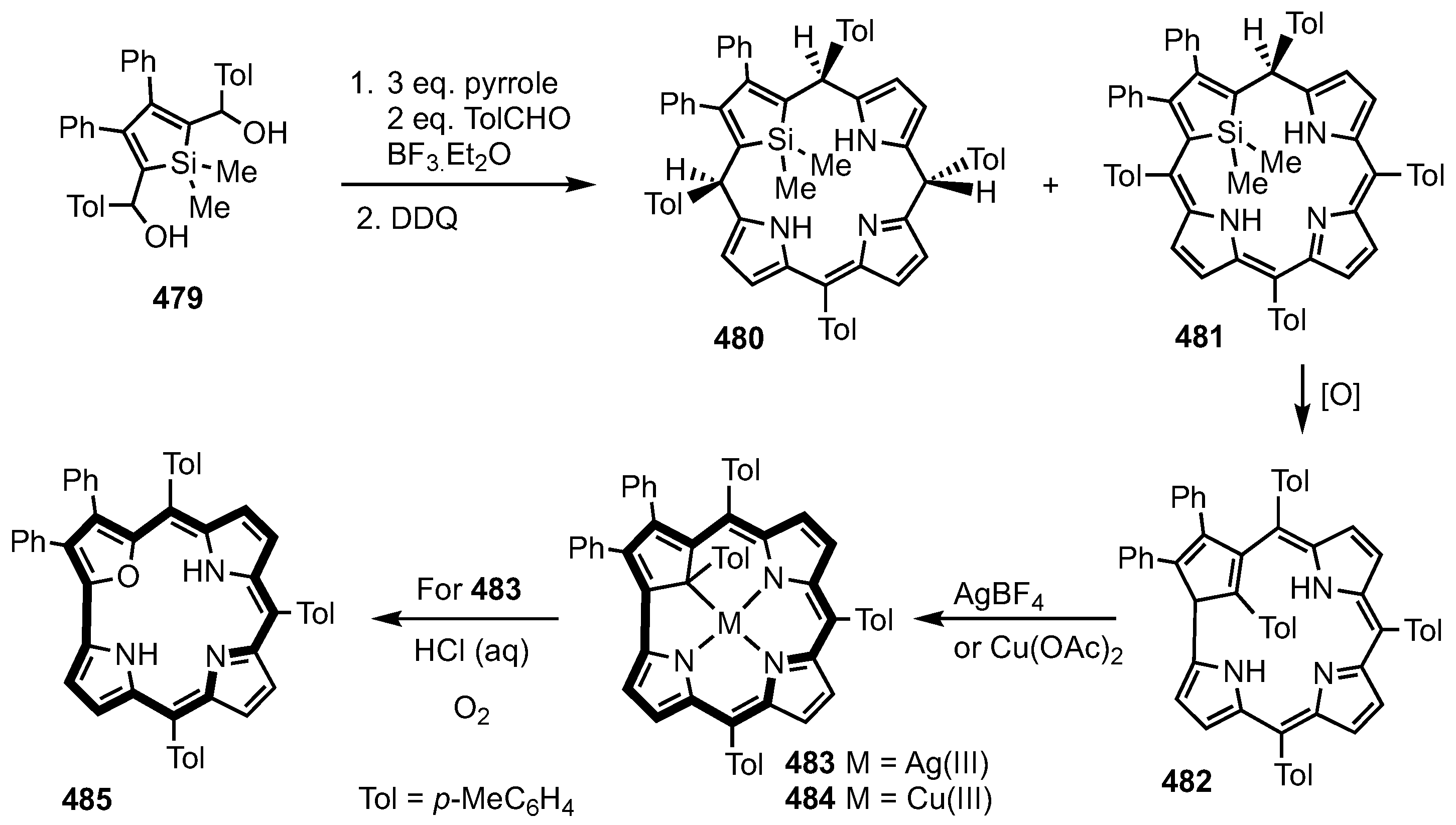{kind=link}
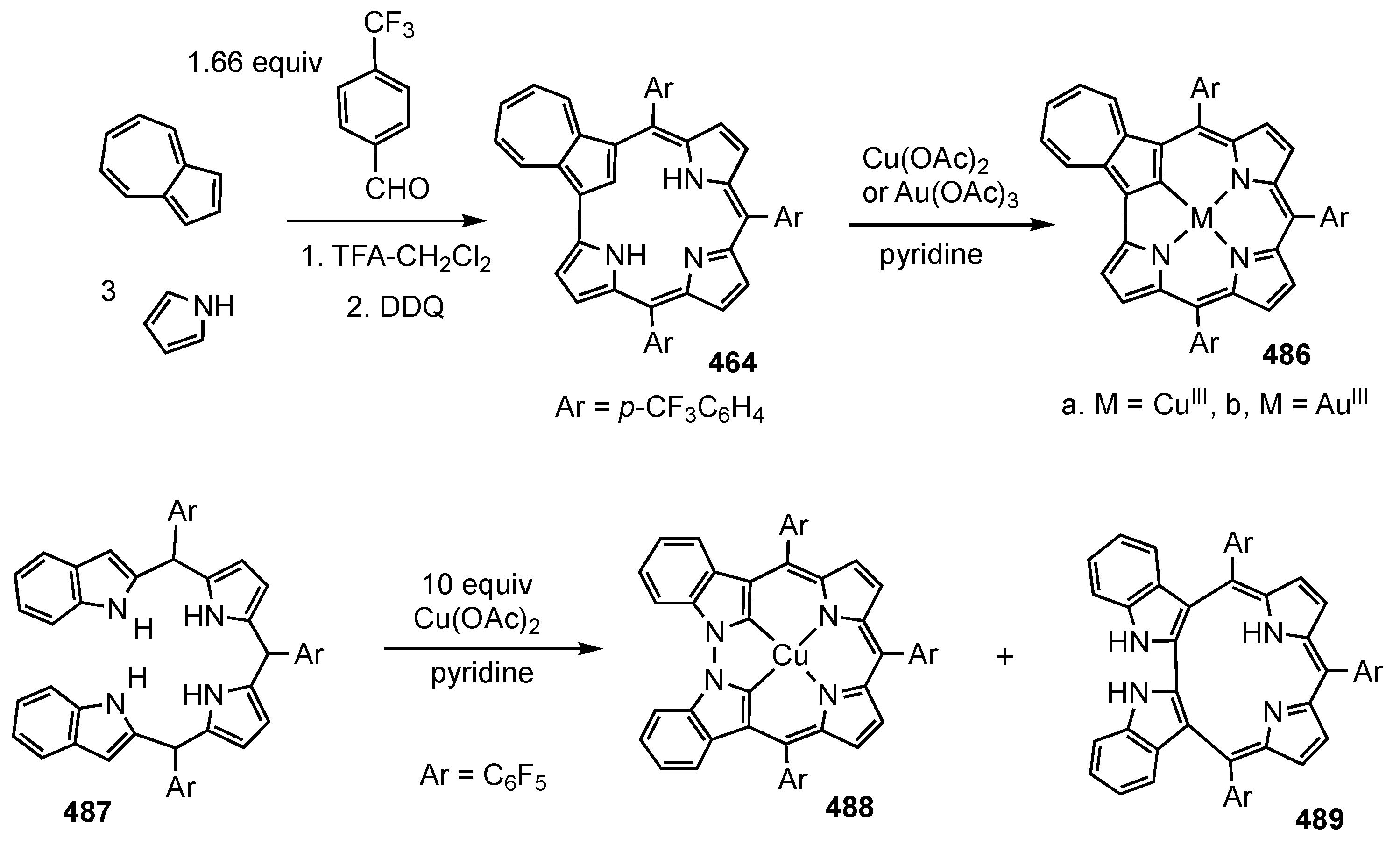{kind=link}
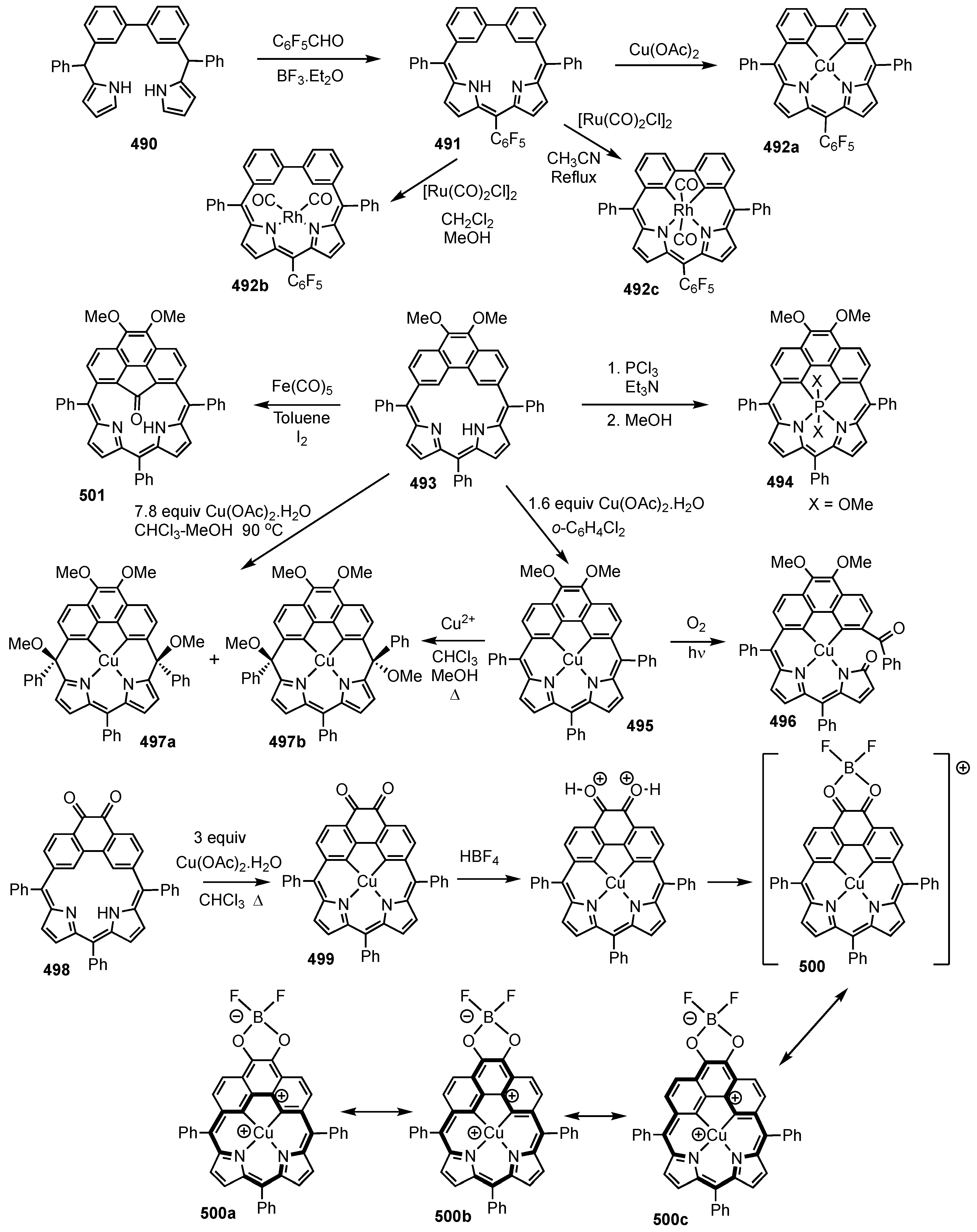{kind=link}
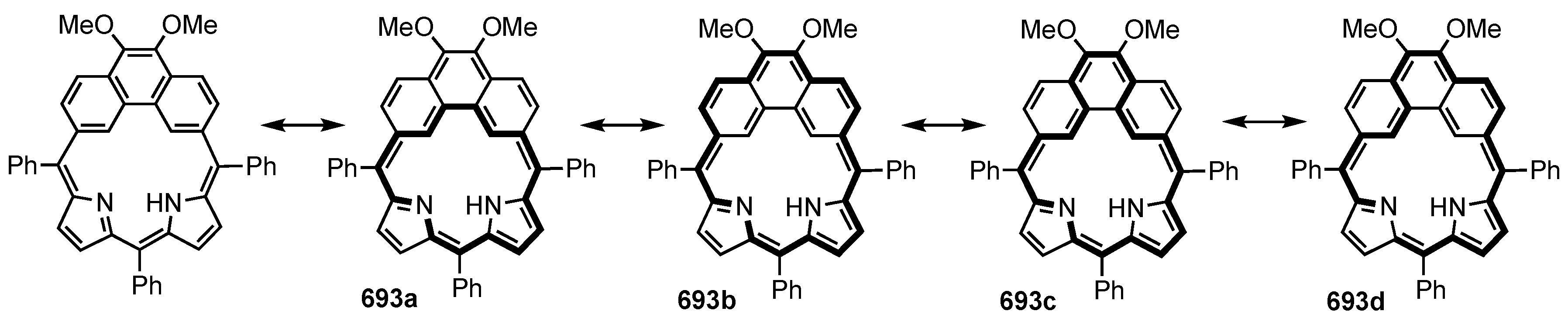{kind=link}
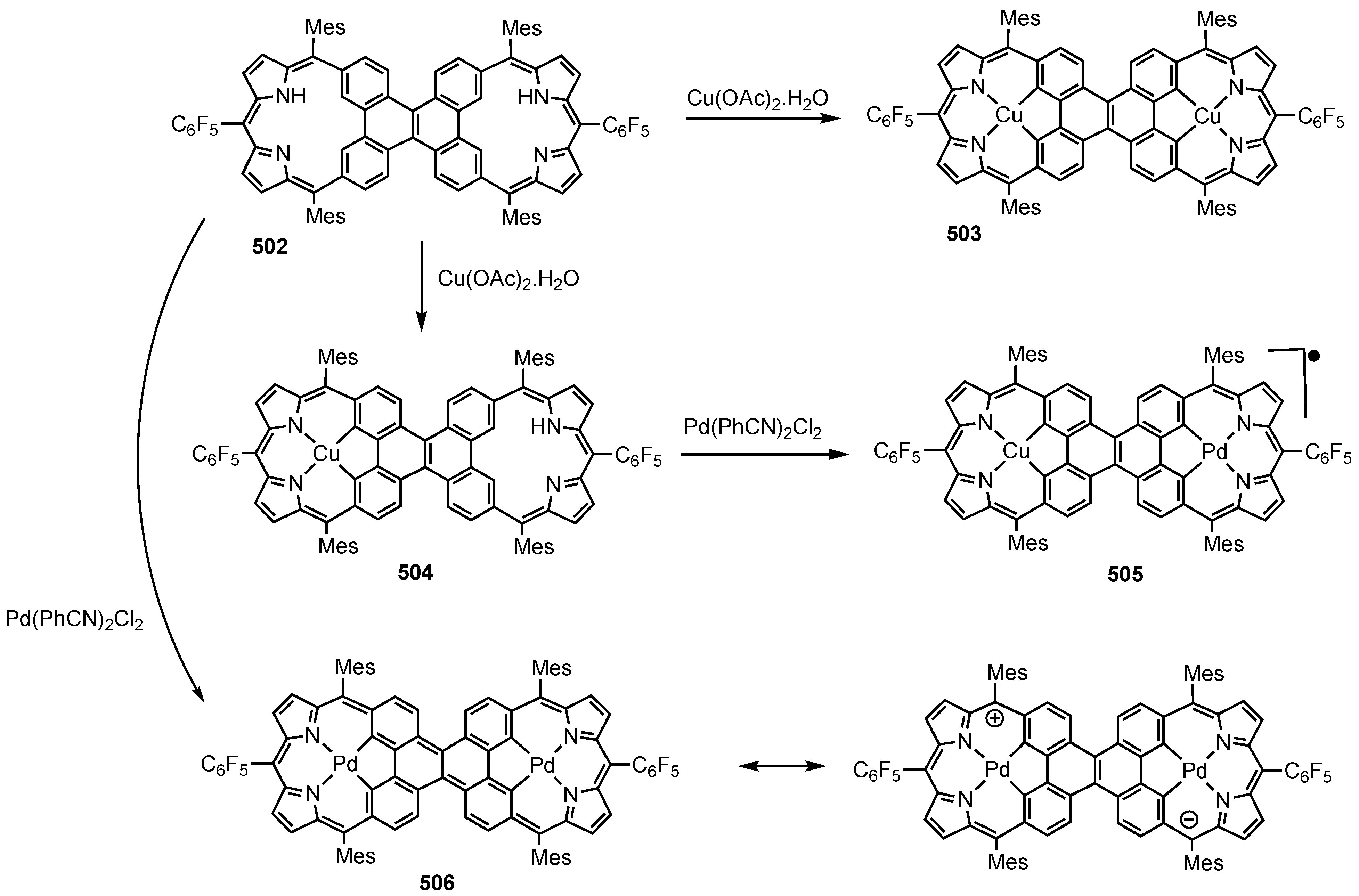{kind=link}
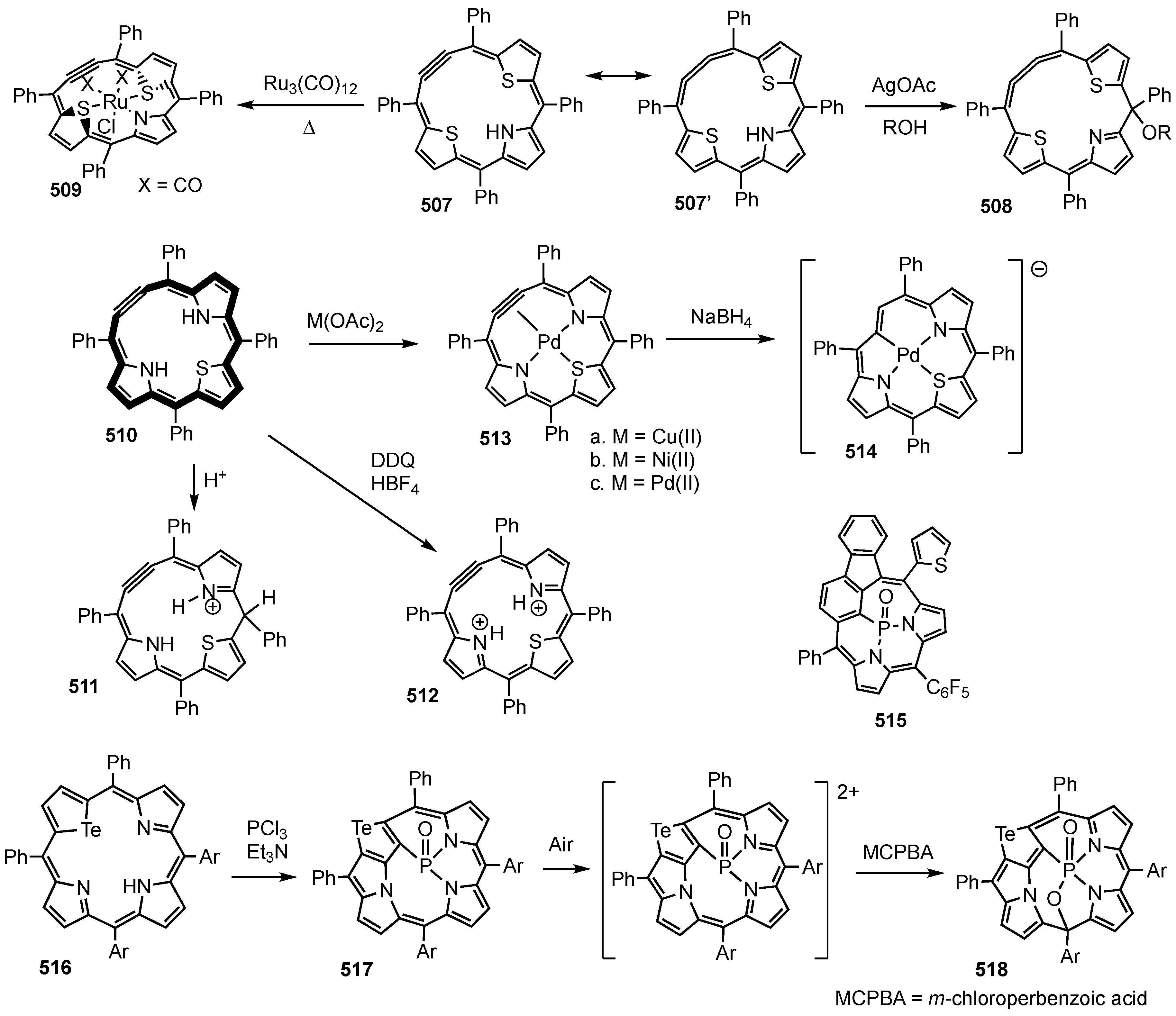{kind=link}
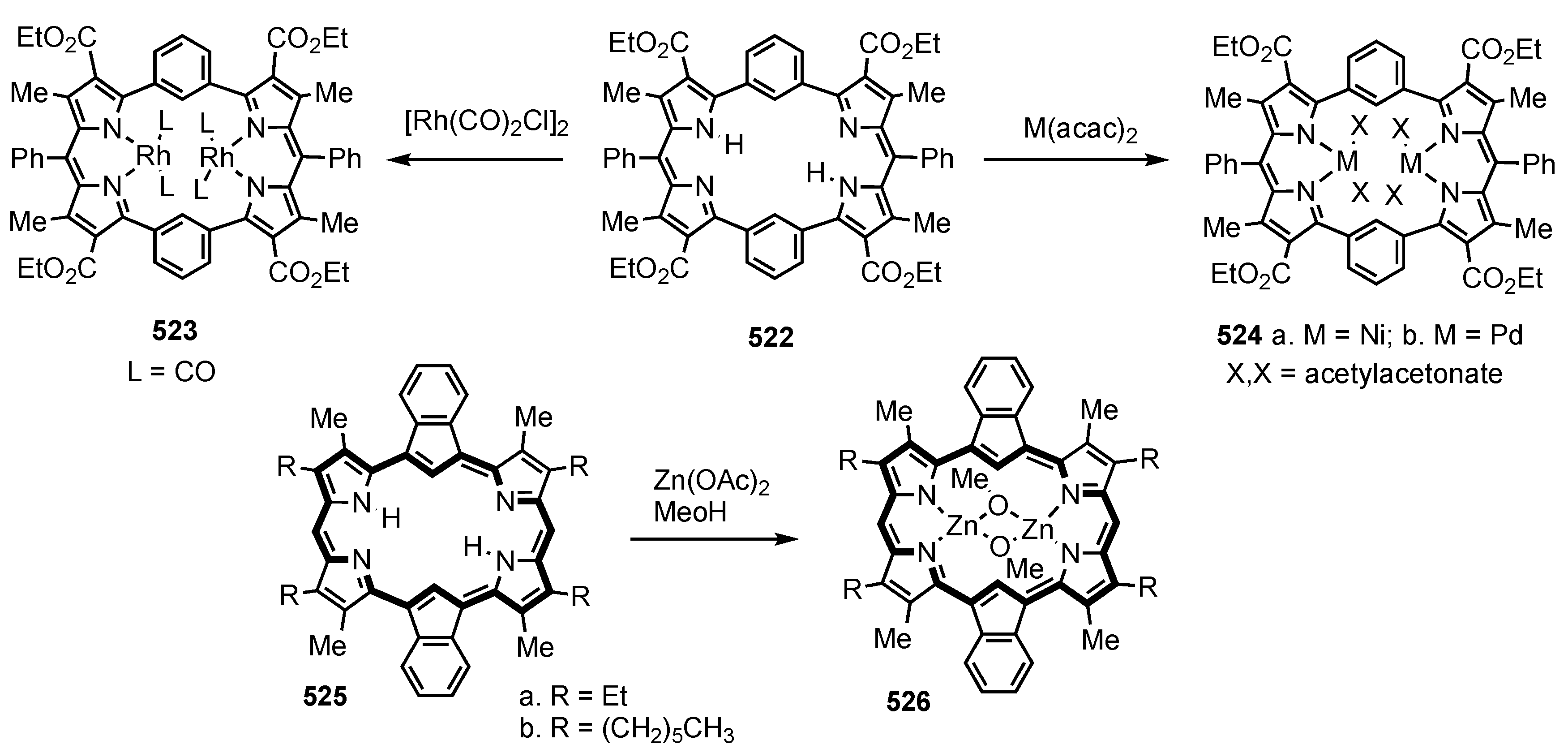{kind=link}
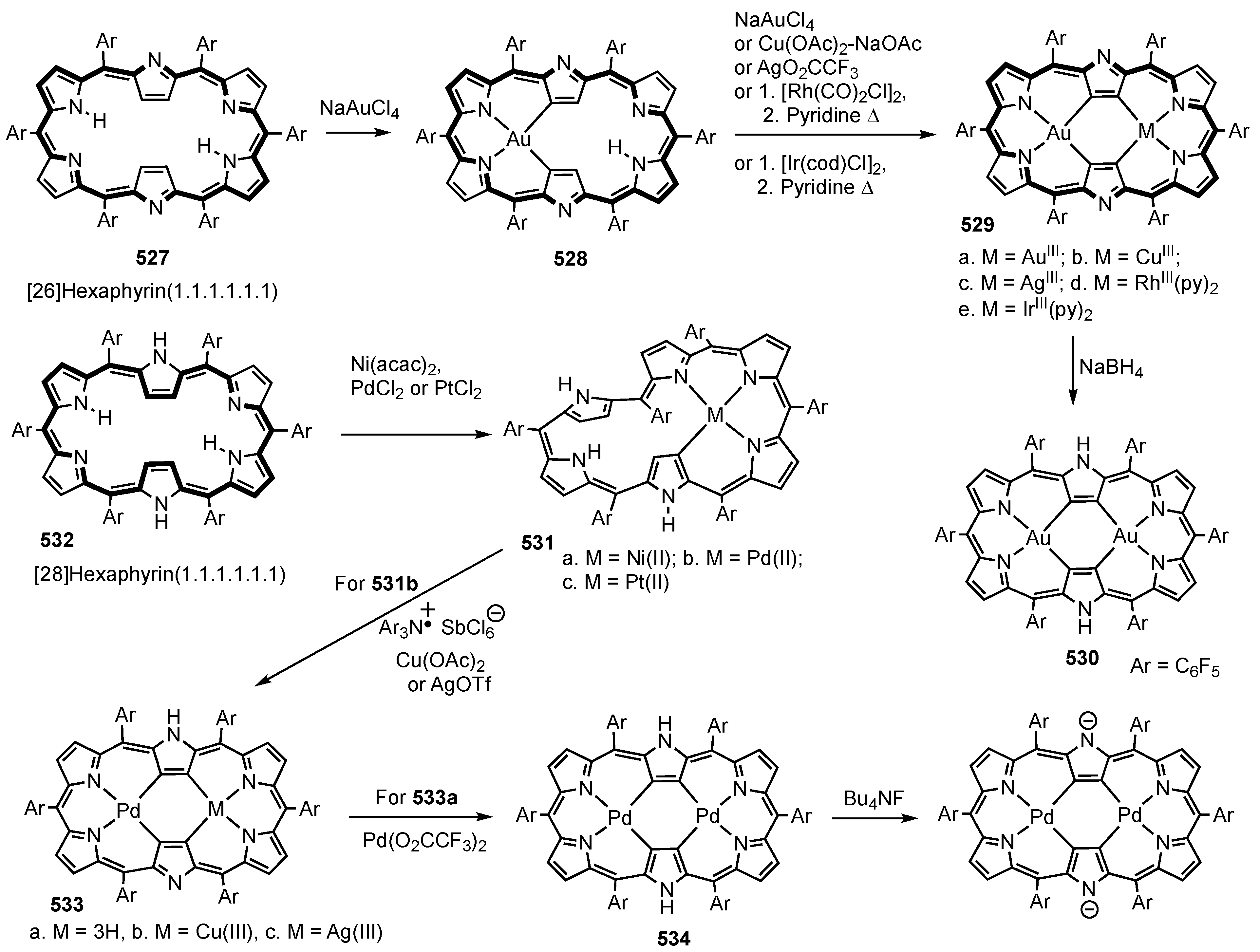{kind=link}
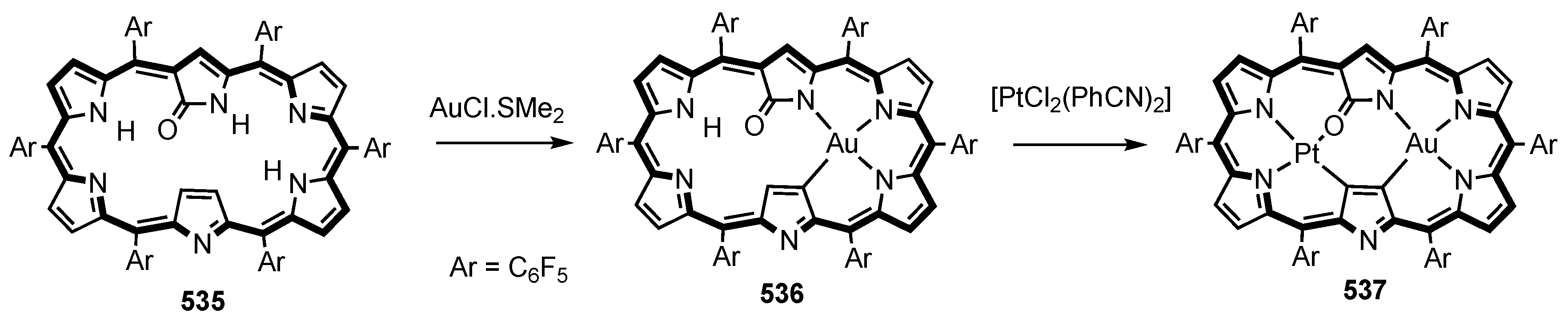{kind=link}
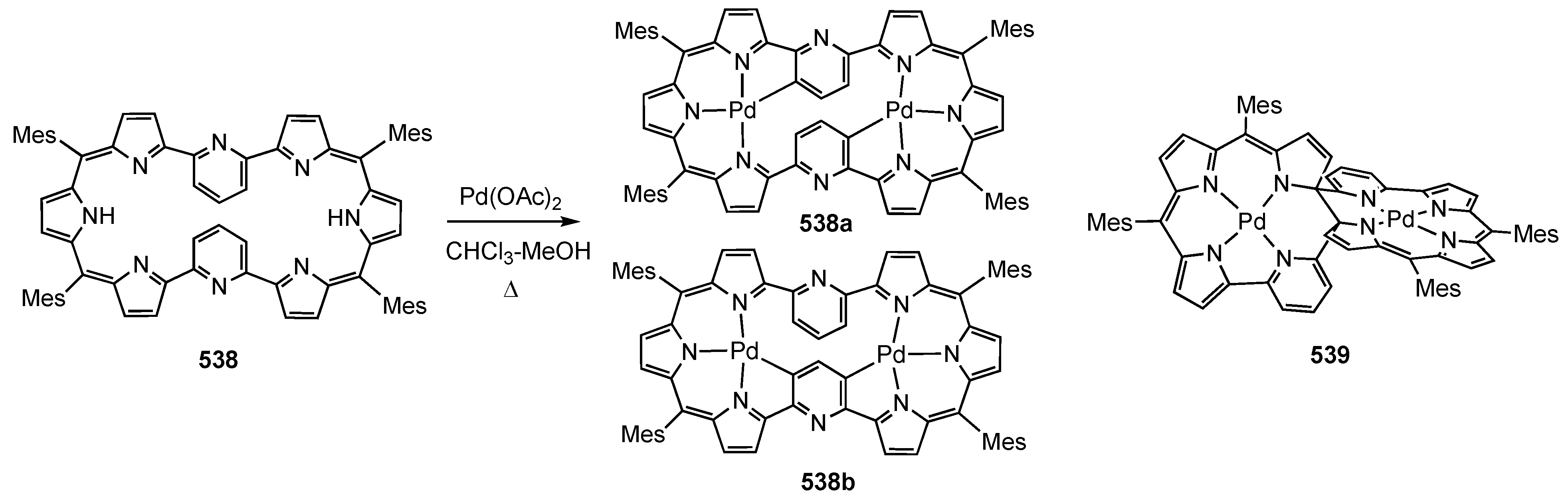{kind=link}
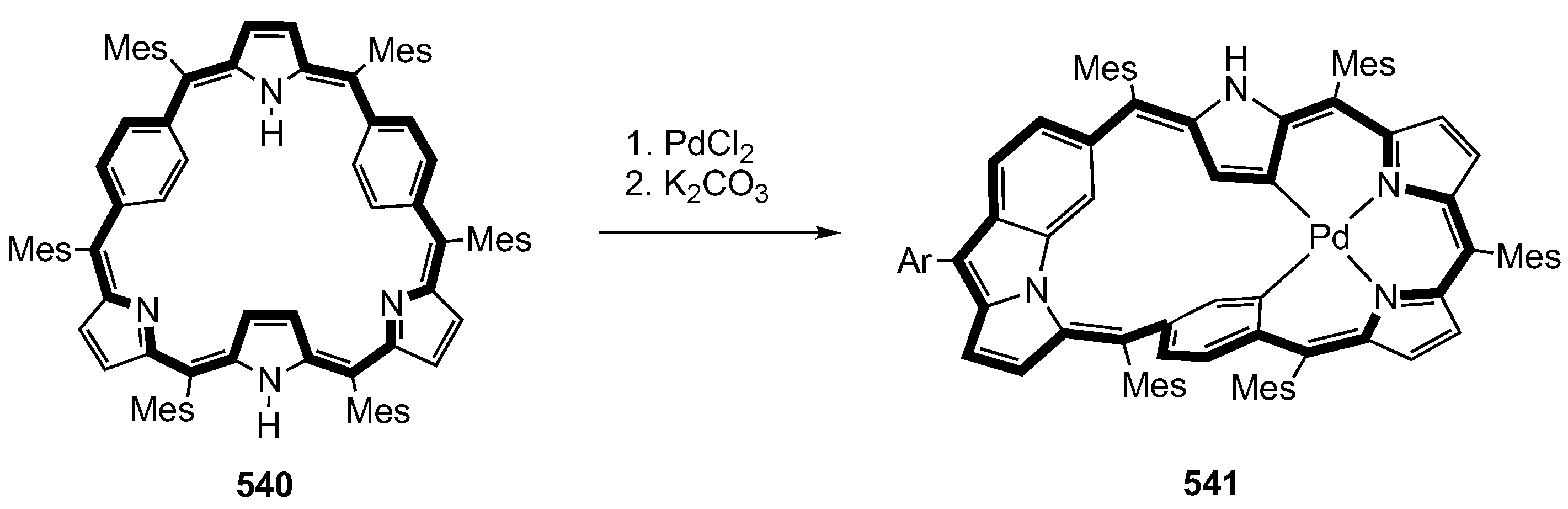{kind=link}
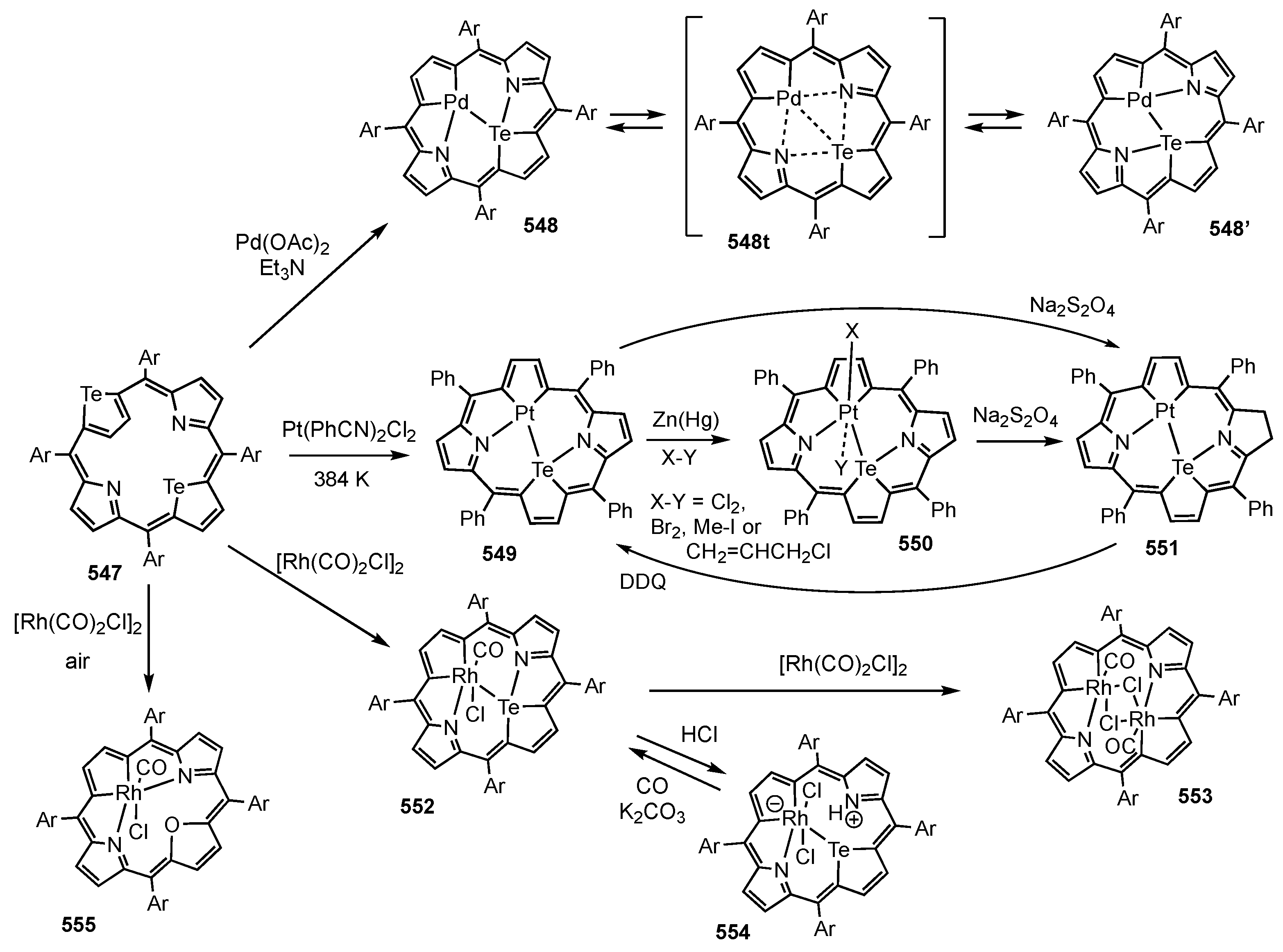{kind=link}
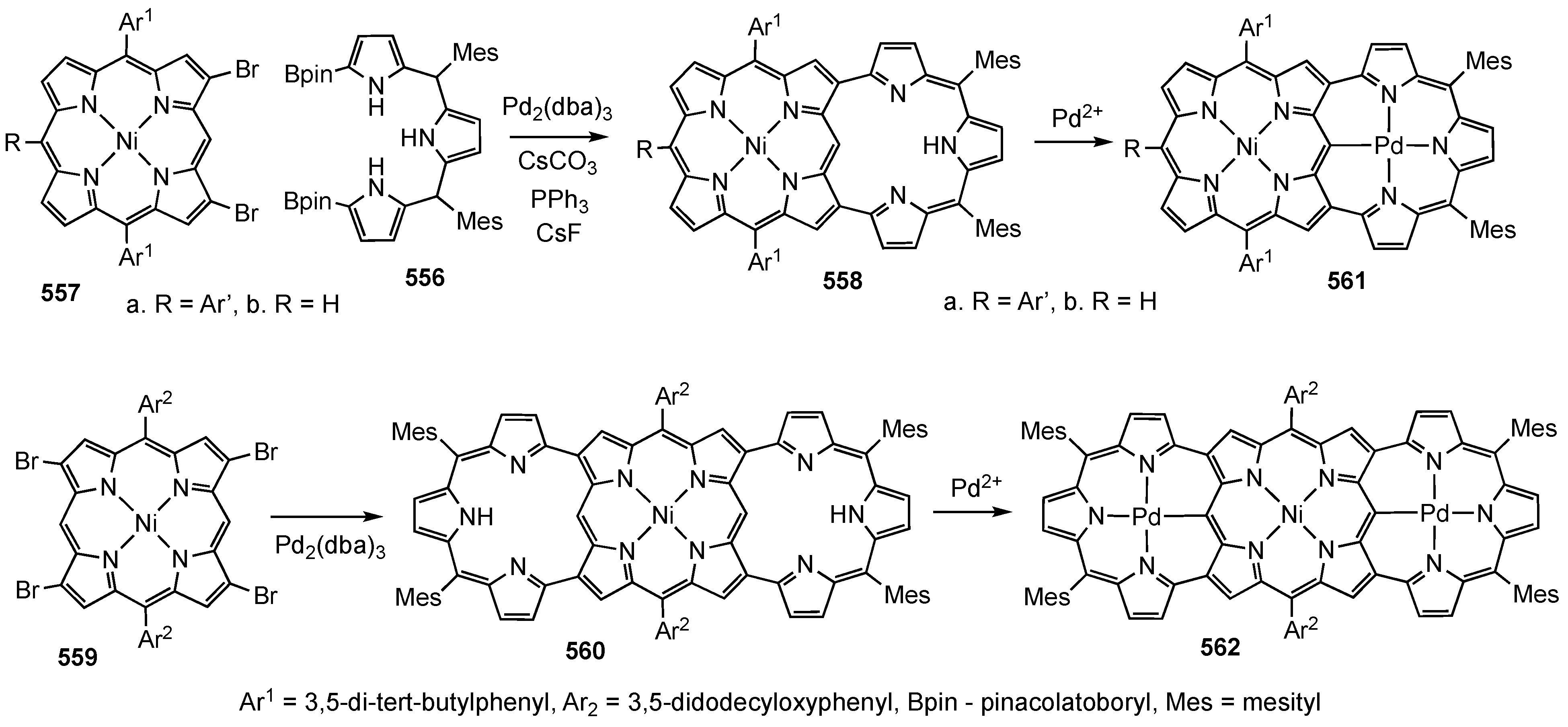{kind=link}
| 150 | 141 | 163 | 164 | |
|---|---|---|---|---|
| 5,20-H | 10.42 | 10.27 | 9.85 | 9.54 |
| 10,15-H | 10.00 | 9.56 | 9.13 | 8.84 |
| 7,18-Me | 3.49 | 3.33 | 3.16 | 3.05 |
| 21-Me | −4.46 | −3.21 | −2.18 | −1.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lash, T.D. Organometallic Chemistry within the Structured Environment Provided by the Macrocyclic Cores of Carbaporphyrins and Related Systems. Molecules 2023, 28, 1496. https://doi.org/10.3390/molecules28031496
Lash TD. Organometallic Chemistry within the Structured Environment Provided by the Macrocyclic Cores of Carbaporphyrins and Related Systems. Molecules. 2023; 28(3):1496. https://doi.org/10.3390/molecules28031496
Chicago/Turabian StyleLash, Timothy D. 2023. "Organometallic Chemistry within the Structured Environment Provided by the Macrocyclic Cores of Carbaporphyrins and Related Systems" Molecules 28, no. 3: 1496. https://doi.org/10.3390/molecules28031496
APA StyleLash, T. D. (2023). Organometallic Chemistry within the Structured Environment Provided by the Macrocyclic Cores of Carbaporphyrins and Related Systems. Molecules, 28(3), 1496. https://doi.org/10.3390/molecules28031496