


A Knowledge-Graph-Based Multimodal Deep Learning Framework for Identifying Drug–Drug Interactions



Abstract
1. Introduction
2. Results and Discussion
2.1. Effect of Threshold on Model Performance
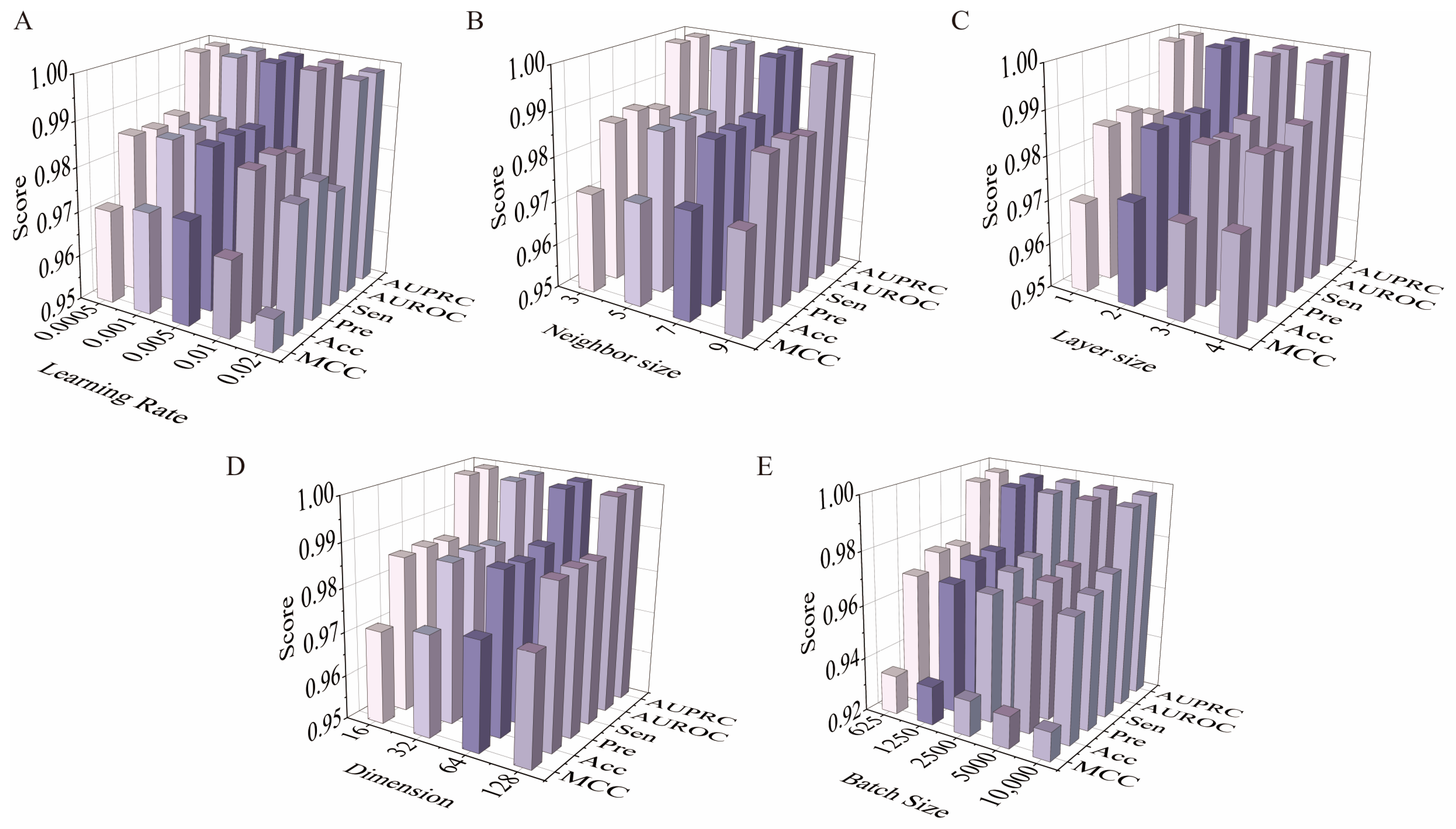
2.2. Optimization of Model Parameter
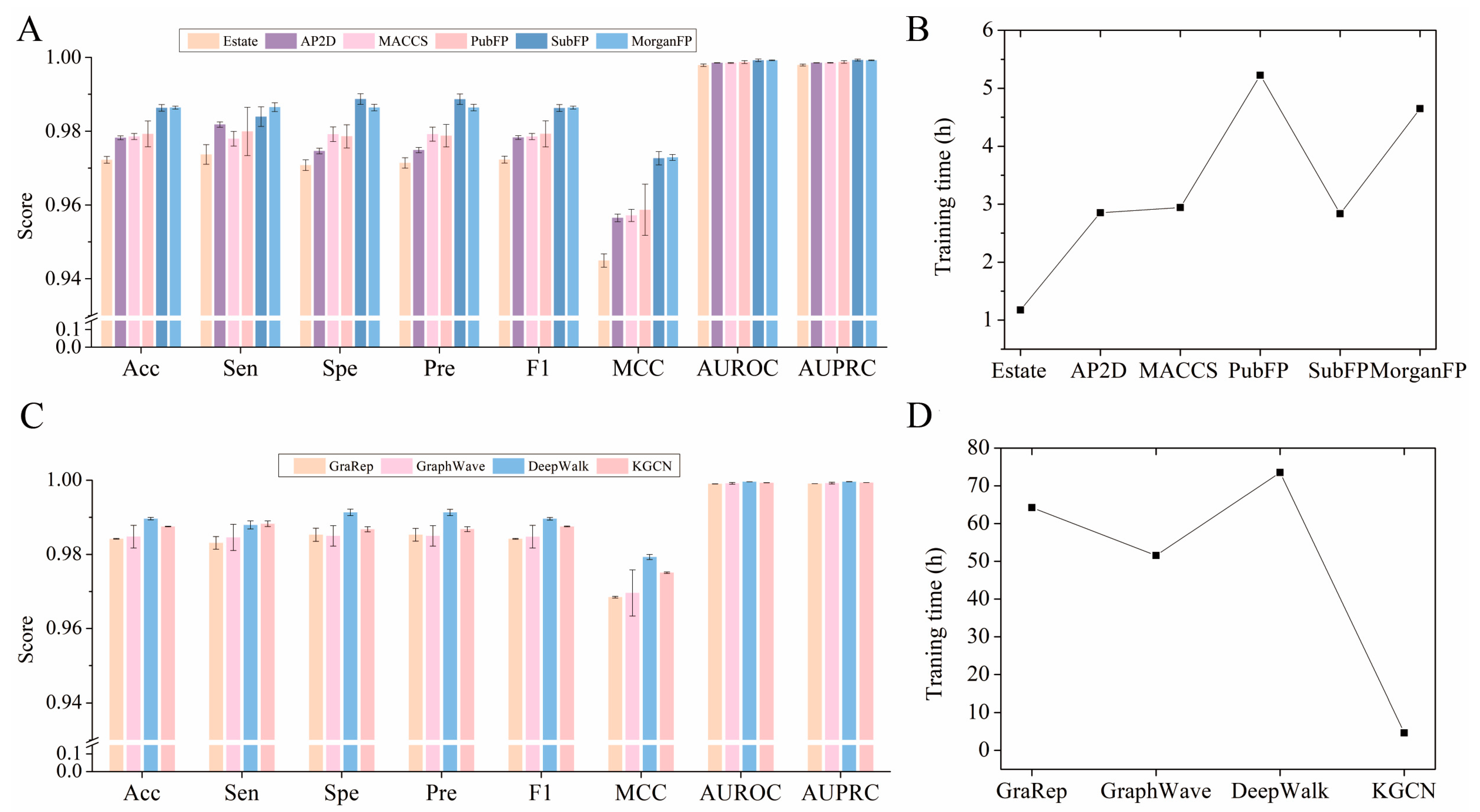
2.3. Comparison of Various Embedding and Fingerprint Features
2.4. Comparison with Other Methods
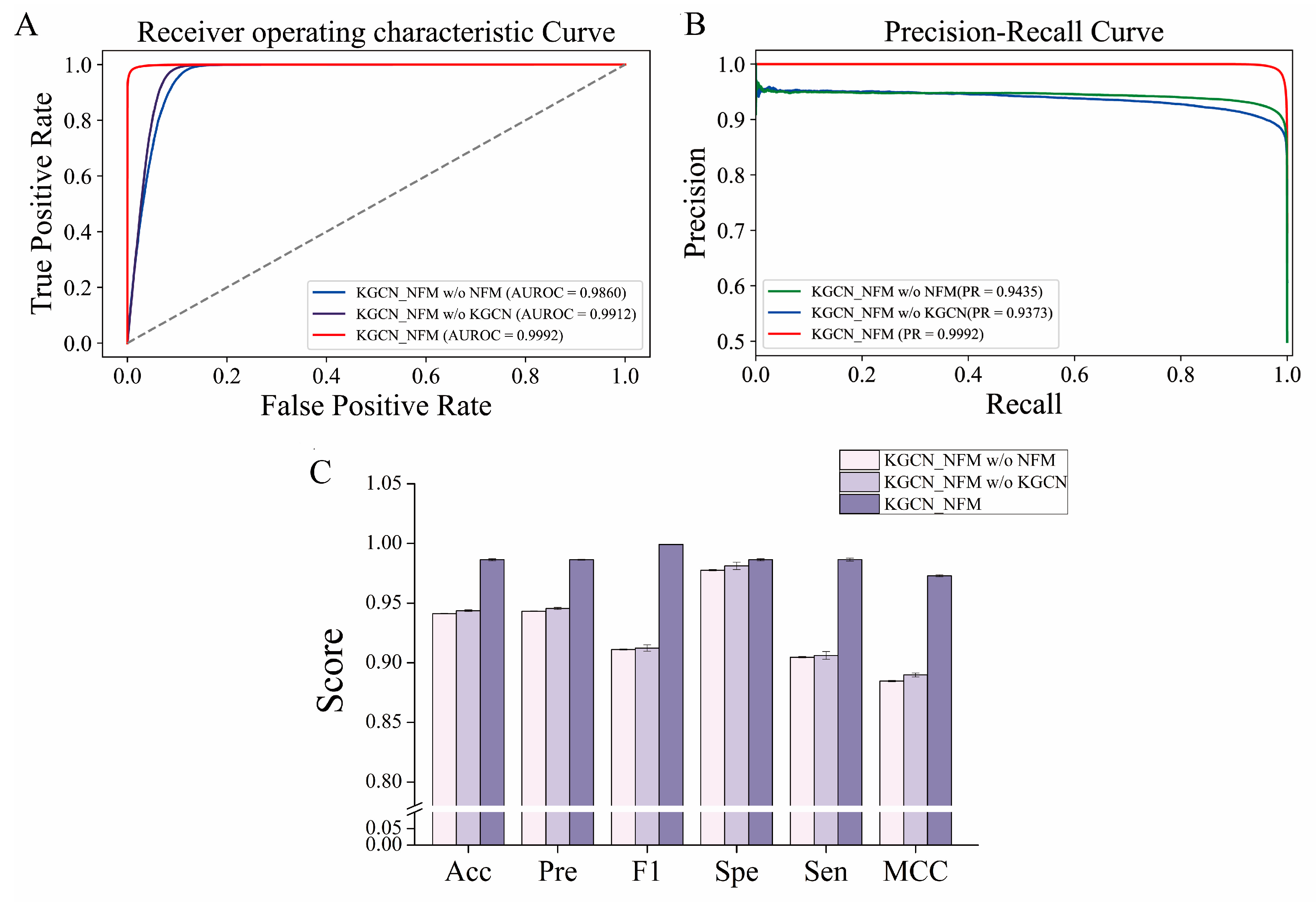
2.5. Ablation Study
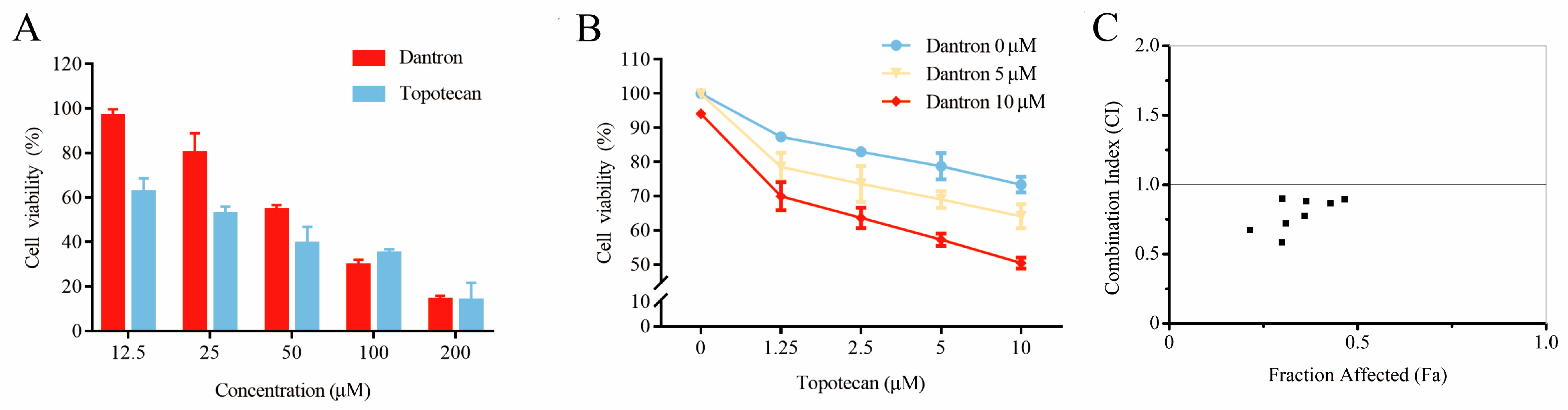
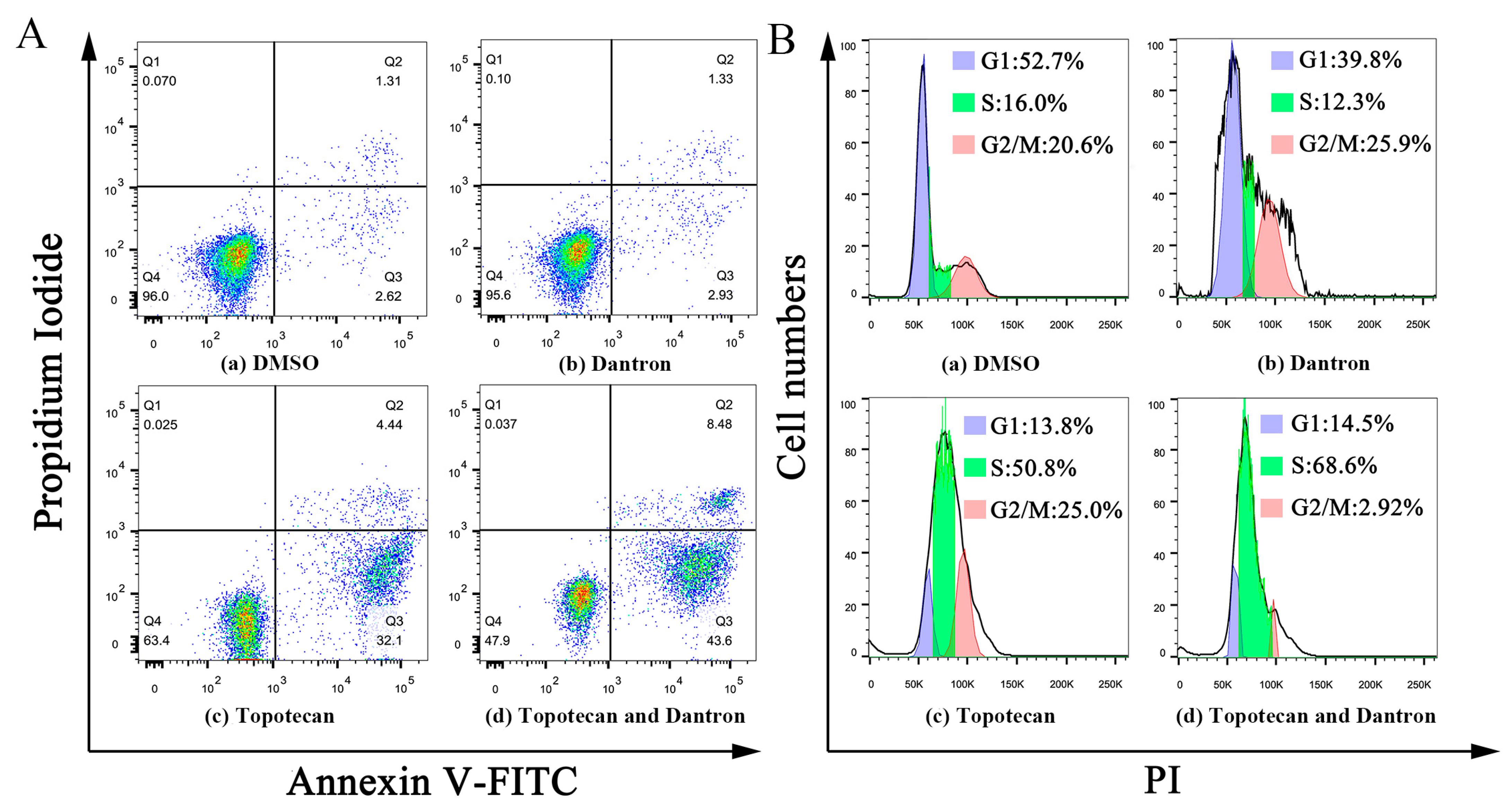
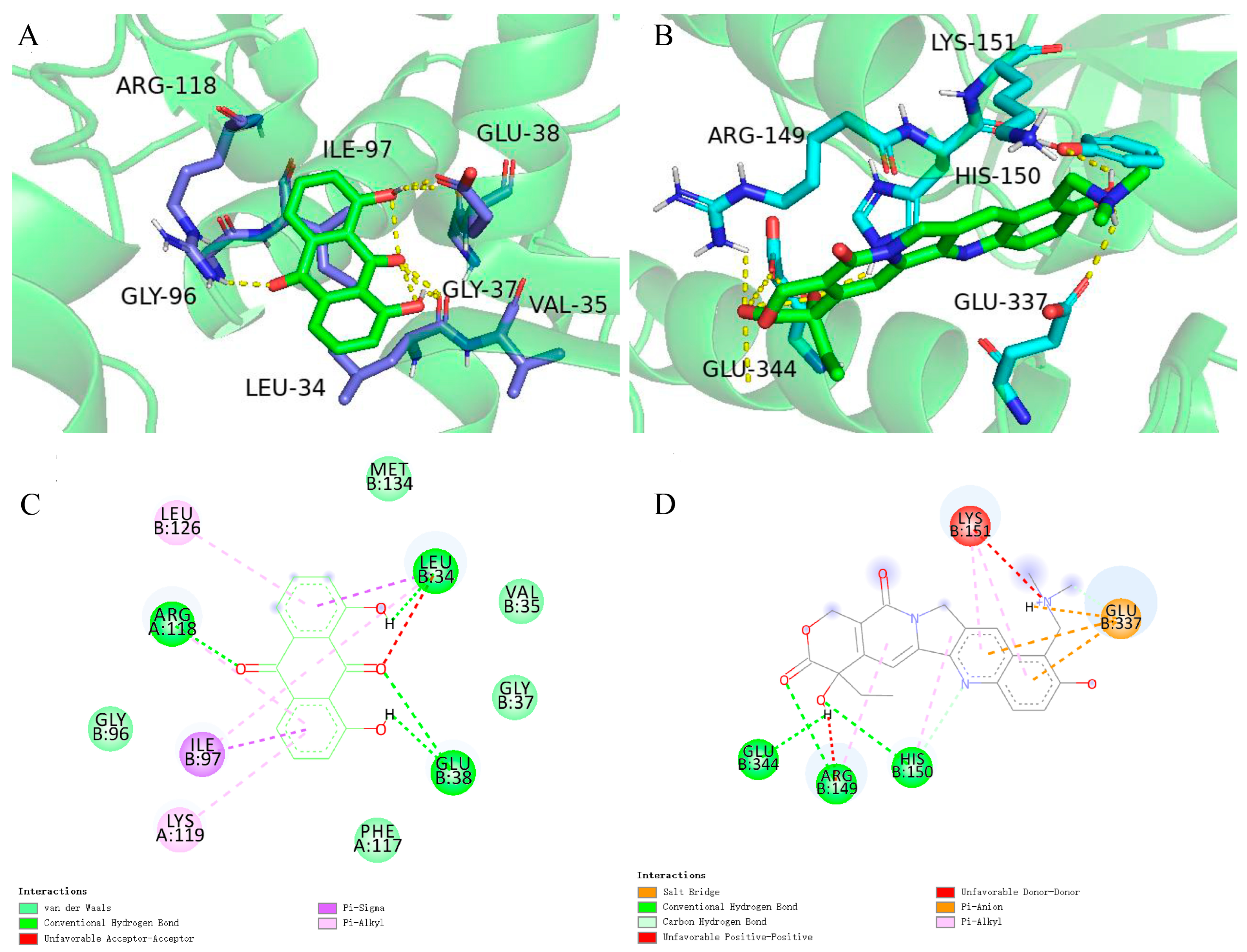
2.6. Case Study
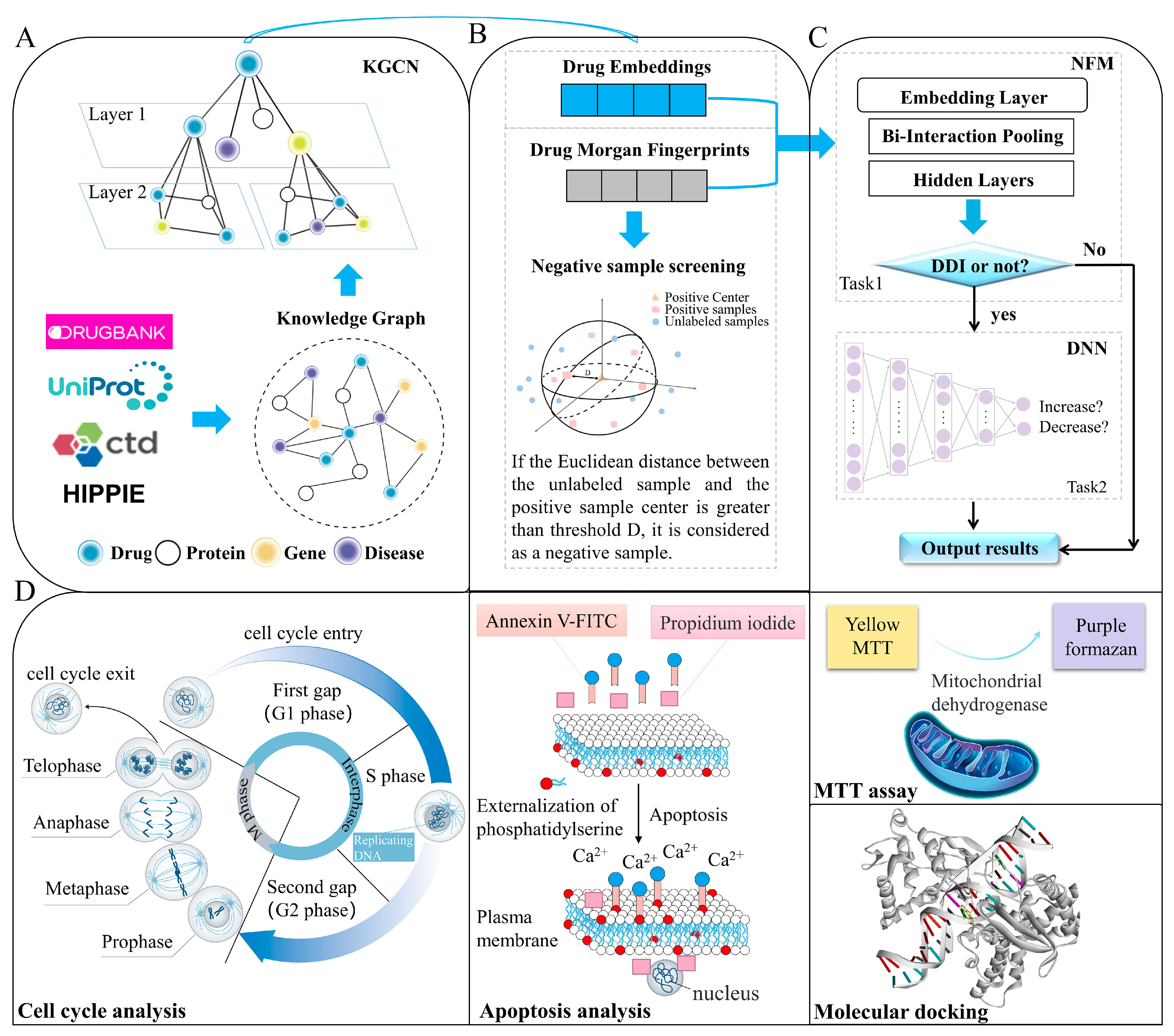
3. Materials and Methods
3.1. Construction of Knowledge Graph and Dataset
3.2. Characterization of Drug–Drug Interactions
3.3. Selection of Reliable Negative Sample
3.4. Construction of Identification Model
3.4.1. KGCN Layer
3.4.2. NFMs and DNN layer
3.5. Baselines
3.6. Performance Evaluation
3.7. Cytotoxicity Assays and Synergistic Effect
3.8. Apoptosis and Cell Cycle Analysis
3.9. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nagai, N. Drug interaction studies on new drug applications: Current situations and regulatory views in Japan. Drug Metab. Pharmacokinet. 2010, 25, 3–15. [Google Scholar] [CrossRef]
- Strandell, J.; Bate, A.; Lindquist, M.; Edwards, I.R. Drug-drug interactions—A preventable patient safety issue? Br. J. Clin. Pharmacol. 2008, 65, 144–146. [Google Scholar] [CrossRef]
- Tatonetti, N.P.; Ye, P.P.; Daneshjou, R.R.; Altman, B. Data-driven prediction of drug effects and interactions. Sci. Transl. Med. 2012, 4, 125ra31. [Google Scholar] [CrossRef]
- Percha, B.; Altman, R.B. Informatics confronts drug-drug interactions. Trends Pharmacol. Sci. 2013, 34, 178–184. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Deep learning improves prediction of drug-drug and drug-food interactions. Proc. Natl. Acad. Sci. USA 2018, 115, E4304–E4311. [Google Scholar] [CrossRef]
- Whitebread, S.; Hamon, J.; Bojanic, D.; Urban, L. Keynote review: In Vitro safety pharmacology profiling: An essential tool for successful drug development. Drug Discov. Today 2005, 10, 1421–1433. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, J.; Guo, F. Identification of drug-side effect association via multiple information integration with centered kernel alignment. Neurocomputing 2019, 325, 211–224. [Google Scholar] [CrossRef]
- Vilar, S.; Uriarte, E.; Santana, L.; Lorberbaum, T.; Hripcsak, G.; Friedman, C.; Tatonetti, N.P. Similarity-based modeling in large-scale prediction of drug-drug interactions. Nat. Protoc. 2014, 9, 2147–2163. [Google Scholar] [CrossRef]
- Rohani, N.; Eslahchi, C. Drug-drug interaction predicting by neural network using integrated similarity. Sci. Rep. 2019, 9, 13645. [Google Scholar] [CrossRef]
- Sridhar, D.; Fakhraei, S.; Getoor, L. A probabilistic approach for collective similarity-based drug-drug interaction prediction. Bioinformatics 2016, 32, 3175–3182. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, Y.; Cui, Y.; Liua, S.; Zhang, W. Predicting drug-drug interactions using multi-modal deep auto-encoders based network embedding and positive-unlabeled learning. Methods 2020, 179, 37–46. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinform. 2017, 18, 18–29. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Y.; Li, D.; Yue, X. Manifold regularized matrix factorization for drug-drug interaction prediction. J. Biomed. Inform. 2018, 88, 90–97. [Google Scholar] [CrossRef]
- Gottlieb, A.; Stein, G.Y.; Oron, Y.; Ruppin, E.; Sharan, R. INDI: A computational framework for inferring drug interactions and their associated recommendations. Mol. Syst. Biol. 2012, 8, 592. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.; Yan, G. Drug-target interaction prediction by random walk on the heterogeneous network. Mol. BioSystems 2012, 8, 1970. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef]
- Perozzi, B.; Al-Rfou, R.; Skiena, S. DeepWalk: Online learning of social representations. In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 24–27 August 2014; pp. 701–710. [Google Scholar] [CrossRef]
- Donnat, C.; Zitnik, M.; Hallac, D.; Leskovec, J. Learning structural node embeddings via diffusion wavelets. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, England, 19–23 August 2018; pp. 1320–1329. [Google Scholar] [CrossRef]
- Cao, S.; Lu, W.; Xu, Q. GraRep: Learning graph representations with Global Structural Information. In Proceedings of the 24th ACM International Conference on Knowledge Discovery & Knowledge Management, New York, NY, USA, 19–23 October 2015; pp. 891–900. [Google Scholar] [CrossRef]
- Nickel, M.; Murphy, K.; Tresp, V.; Gabrilovich, E. A review of relational machine learning for knowledge graphs. Proc. IEEE 2016, 104, 11–33. [Google Scholar] [CrossRef]
- Wang, Q.; Mao, Z.; Wang, B.; Guo, L. Knowledge graph embedding: A survey of approaches and applications. IEEE Trans. Knowl. Data Eng. 2017, 29, 2724–2743. [Google Scholar] [CrossRef]
- Bordes, A.; Usunier, N.; Garcia-Duran, A. Translating embeddings for modeling multi-relational data. In Proceedings of the Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 5–10 December 2013; pp. 2787–2795. [Google Scholar]
- Wang, Z.; Zhang, J.; Feng, J.; Chen, Z. Knowledge graph embedding by translating on hyperplanes. In Proceedings of the 28th AAAI Conference on Artificial Intelligence, Quebec City, QC, Canada, 27–31 July 2014; pp. 1112–1119. [Google Scholar]
- Lin, Y.; Liu, Z.; Sun, M.; Liu, Y.; Zhu, X. Learning entity and relation embeddings for knowledge graph completion. In Proceedings of the 29th AAAI Conference on Artificial Intelligence, Austin, TX, USA, 25–30 January 2015; pp. 2181–2187. [Google Scholar]
- Abdelaziz, I.; Fokoue, A.; Hassanzadeh, O.; Zhang, P.; Sadoghi, M. Large-scale structural and textual similarity-based mining of knowledge graph to predict drug-drug interactions. J. Web Semant. 2017, 44, 104–117. [Google Scholar] [CrossRef]
- Nickel, M.; Rosasco, L.; Poggio, T. Holographic embeddings of knowledge graphs. In Proceedings of the 30th Association-for-the-Advancement-of-Artificial-Intelligence (AAAI) Conference on Artificial Intelligence, Phoenix, AZ, USA, 12–17 February 2016; pp. 1955–1961. [Google Scholar]
- Zitnik, M.; Zupan, B. Collective pairwise classification for multi-way analysis of disease and drug data. In Proceedings of the 21st Pacific Symposium on Biocomputing (PSB), Fairmont Orchid, HI, USA, 4–8 January 2016; pp. 81–92. [Google Scholar]
- Nickel, M.; Tresp, V.; Kriegel, H. A three-way model for collective learning on multi-relational data. In Proceedings of the 28th International Conference on Machine Learning, Bellevue, WA, USA, 28 June–2 July 2011; pp. 809–816. [Google Scholar]
- Wu, Z.; Pan, S.; Chen, F.; Liu, S. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4–24. [Google Scholar] [CrossRef]
- Feng, Y.H.; Zhang, S.W.; Shi, J.Y. DPDDI: A deep predictor for drug-drug interactions. BMC Bioinform. 2020, 21, 419. [Google Scholar] [CrossRef]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, M.; Xie, X.; Li, W.; Guo, M. Knowledge graph convolutional networks for recommender systems. In Proceedings of the World Wide Web Conference (WWW 2019), San Francisco, CA, USA, 13–17 May 2019; pp. 3307–3313. [Google Scholar]
- Alaimo, S.; Giugno, R.; Pulvirenti, A. Recommendation techniques for drug-target interaction prediction and drug repositioning. In Data Mining Techniques for the Life Sciences; Springer: Berlin/Heidelberg, Germany, 2016; pp. 441–462. [Google Scholar] [CrossRef]
- Ye, Q.; Hsieh, C.; Yang, Z.; Kang, J.; Chen, D.; Cao, S. A unified drug-target interaction prediction framework based on knowledge graph and recommendation system. Nat. Commun. 2021, 12, 6775. [Google Scholar] [CrossRef]
- He, X.; Chua, T. Neural factorization machines for sparse predictive analytics. In Proceedings of the 40th International ACM SIGIR Conference on Research and Development in Information Retrieval, Shinjuku, Japan, 7–11 August 2017; pp. 355–364. [Google Scholar]
- Lin, X.; Quan, Z.; Wang, Z.; Ma, T.; Zeng, X. KGNN: Knowledge graph neural network for drug-drug interaction prediction. In Proceedings of the Twenty-Ninth International Joint Conference on Artificial Intelligence, Yokohama, Japan, 7–15 January 2020; pp. 2739–2745. [Google Scholar]
- Rozemberczki, B.; Kiss, O.; Sarkar, R. Karate Club: An api oriented open-source python framework for unsupervised learning on graphs. In Proceedings of the ACM International on Conference on Information and Knowledge Management, Galway, Ireland, 19–23 October 2020; pp. 3125–3132. [Google Scholar]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Guo, J.; Wang, Q.; Zhang, Y.; Sun, W.; Zhang, S.; Li, Y.; Wang, J.; Bao, Y. Functional daidzein enhances the anticancer effect of topotecan and reverses BCRP-mediated drug resistance in breast cancer. Pharmacol. Res. 2019, 147, 104387. [Google Scholar] [CrossRef]
- Li, D.D.; Sun, T.; Wu, X.Q.; Chen, S.P.; Deng, R.; Jiang, S.; Feng, G.K.; Pan, J.; Zhang, X.S.; Zeng, Y.X.; et al. The inhibition of autophagy sensitises colon cancer cells with wild-type p53 but not mutant p53 to topotecan treatment. PLoS ONE 2012, 7, e45058. [Google Scholar] [CrossRef]
- Zhou, R.; Wang, L.; Xu, X.; Chen, J.; Hu, L.H.; Chen, L.; Shen, X. Danthron activates AMP-activated protein kinase and regulates lipid and glucose metabolism in vitro. Acta Pharmacol. Sin. 2013, 34, 1061–1069. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The comparative toxicogenomics database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Alanis-Lobato, G.; Andrade-Navarro, M.A.; Schaefer, M.H. HIPPIE v2.0: Enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res. 2017, 45, D408–D414. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 29–34. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org/ (accessed on 1 January 2023).
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Huang, Q.; Wang, Y.; Xie, Y.; Dai, Z.; Zou, X.; Li, Z. Seq-SymRF: A random forest model predicts potential miRNA-disease associations based on information of sequences and clinical symptoms. Sci. Rep. 2020, 10, 17901. [Google Scholar] [CrossRef]
- Hamilton, W.L.; Ying, R.; Leskovec, J. Inductive representation learning on large graphs. In Proceedings of the 31st Annual Conference on Neural Information Processing Systems (NIPS), Long Beach, CA, USA, 4–9 December 2017; pp. 1024–1034. [Google Scholar]
- Zhang, Y.; Pan, C.; Sun, J.; Tang, C. Multiple sclerosis identification by convolutional neural network with dropout and parametric ReLU. J. Comput. Sci. 2018, 28, 1–10. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Karim, M.R.; Cochez, M.; Jares, J.B.; Uddin, M.; Beyan, O.; Decker, S. Drug-drug interaction prediction based on knowledge graph embeddings and convolutional-lstm network. In Proceedings of the 10th ACM International Conference on Bioinformatics, Niagara Falls, NY, USA, 7–10 September 2019; pp. 113–123. [Google Scholar]
- Su, X.; Hu, L.; You, Z.; Hu, P.; Zhao, B. Attention-based knowledge graph representation learning for predicting drug-drug interactions. Brief. Bioinform. 2022, 23, bbac140. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Park, Y.; Marcotte, E.M. Flaws in evaluation schemes for pair-input computational predictions. Nat. Methods 2012, 9, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Guney, E. Revisiting cross-validation of drug similarity based classifiers using paired data. Genom. Comput. Biol. 2018, 4, e100047. [Google Scholar] [CrossRef]
- Swift, M.L. GraphPad Prism, data analysis, and scientific graphing. J. Chem. Inf. Comput. Sci. 1997, 37, 411–412. [Google Scholar] [CrossRef]
- Chou, T. Preclinical versus clinical drug combination studies. Leuk. Lymphoma 2008, 49, 2059–2080. [Google Scholar] [CrossRef]
- Bijnsdorp, I.V.; Giovannetti, E.; Peters, G.J. Analysis of drug interactions. Methods Mol. Biol. 2011, 731, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Threshold | Acc (%) | Sen (%) | Spe (%) | Pre (%) | F1 | MCC | AUC | AUPR |
|---|---|---|---|---|---|---|---|---|
| UN | 96.20 ± 0.13 | 96.97 ± 0.23 | 95.42 ± 0.33 | 95.50 ± 0.30 | 0.9622 ± 0.13 | 0.9241 ± 0.27 | 0.9908 ± 0.06 | 0.9882 ± 0.08 |
| 0.6 AED | 96.40 ± 0.09 | 97.07 ± 0.29 | 95.74 ± 0.18 | 95.80 ± 0.16 | 0.9643 ± 0.09 | 0.9282 ± 0.19 | 0.9917 ± 0.02 | 0.9895 ± 0.03 |
| 0.8 AED | 96.46 ± 0.05 | 97.18 ± 0.22 | 95.75 ± 0.17 | 95.81 ± 0.15 | 0.9649 ± 0.05 | 0.9295 ± 0.11 | 0.9919 ± 0.01 | 0.9898 ± 0.02 |
| 1.0 AED | 97.06 ± 0.06 | 97.50 ± 0.16 | 96.62 ± 0.12 | 96.65 ± 0.11 | 0.9707 ± 0.06 | 0.9413 ± 0.11 | 0.9954 ± 0.01 | 0.9949 ± 0.01 |
| 1.2 AED | 98.64 ± 0.04 | 98.65 ± 0.12 | 98.64 ± 0.09 | 98.64 ± 0.09 | 0.9864 ± 0.04 | 0.9729 ± 0.08 | 0.9992 ± 0.01 | 0.9992 ± 0.01 |
| Datasets | Methods | Acc (%) | Sen (%) | Spe (%) | Pre (%) | F1 | MCC | AUC | AUPR |
|---|---|---|---|---|---|---|---|---|---|
| benchmark dataset | RF | 89.50 ± 0.04 | 93.63 ± 0.01 | 85.37 ± 0.07 | 86.48 ± 0.06 | 0.8992 ± 0.04 | 0.7927 ± 0.08 | 0.9393 ± 0.02 | 0.8914 ± 0.04 |
| KGNN | 97.09 ± 0.58 | 98.56 ± 0.20 | 95.63 ± 1.03 | 95.79 ± 0.93 | 0.9714 ± 0.55 | 0.9424 ± 1.15 | 0.9924 ± 0.38 | 0.9911 ± 0.34 | |
| CNN-LSTM | 97.55 ± 0.96 | 97.95 ± 0.66 | 97.14 ± 2.06 | 97.21 ± 1.97 | 0.9756 ± 0.93 | 0.9512 ± 1.94 | 0.9958 ± 0.31 | 0.9950 ± 0.38 | |
| KGE_NFM | 96.76 ± 0.05 | 97.83 ± 0.33 | 95.70 ± 0.26 | 95.79 ± 0.23 | 0.9679 ± 0.06 | 0.9356 ± 0.11 | 0.9929 ± 0.02 | 0.9912 ± 0.02 | |
| DDKG | 87.94 ± 0.82 | 96.96 ± 1.99 | 78.52 ± 1.35 | 82.46 ± 0.91 | 0.8911 ± 1.05 | 0.7707 ± 1.25 | 0.9217 ± 1.13 | 0.8920 ± 2.26 | |
| Our (Task 1) | 98.75 ± 0.01 | 98.83 ± 0.08 | 98.68 ± 0.07 | 98.68 ± 0.07 | 0.9875 ± 0.01 | 0.9751 ± 0.02 | 0.9993 ± 0.01 | 0.9994 ± 0.01 | |
| Our (Task 2) | 96.84 ± 0.01 | 97.42 ± 0.04 | 96.00 ± 0.07 | 97.32 ± 0.04 | 0.9737 ± 0.01 | 0.9344 ± 0.01 | 0.9955 ± 0.01 | 0.9970 ± 0.01 | |
| KEGG-DDI | RF | 90.51 ± 0.11 | 93.19 ± 0.14 | 87.83 ± 0.18 | 88.45 ± 0.15 | 0.9075 ± 0.11 | 0.8113 ± 0.25 | 0.9676 ± 0.04 | 0.9664 ± 0.05 |
| KGNN | 87.45 ± 0.38 | 91.98 ± 1.27 | 82.91 ± 0.84 | 84.39 ± 0.44 | 0.8800 ± 0.48 | 0.7525 ± 0.98 | 0.9348 ± 0.33 | 0.9108 ± 0.37 | |
| CNN-LSTM | 98.58 ± 0.03 | 96.25 ± 0.47 | 94.93 ± 0.97 | 95.04 ± 0.89 | 0.9563 ± 0.61 | 0.9716 ± 0.05 | 0.9882 ± 0.26 | 0.9862 ± 0.27 | |
| KGE_NFM | 84.22 ± 0.62 | 86.82 ± 1.69 | 81.57 ± 1.21 | 82.91 ± 0.76 | 0.8468 ± 0.80 | 0.6878 ± 1.49 | 0.9203 ± 0.50 | 0.9090 ± 0.50 | |
| DDKG | 83.07 ± 0.61 | 84.48 ± 3.49 | 81.66 ± 2.65 | 82.13 ± 1.47 | 0.8324 ± 1.09 | 0.6624 ± 1.81 | 0.9191 ± 0.61 | 0.9202 ± 0.65 | |
| Our | 98.85 ± 0.03 | 98.36 ± 0.05 | 98.80 ± 0.04 | 98.79 ± 0.04 | 0.9857 ± 0.03 | 0.9716 ± 0.05 | 0.9987 ± 0.01 | 0.9988 ± 0.01 |
| Methods | Traditional CV | PW-CV | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acc (%) | Sen (%) | Spe (%) | Pre (%) | F1 | AUC | AUPR | Acc (%) | Sen (%) | Spe (%) | Pre (%) | F1 | AUC | AUPR | |
| RF | 89.50 | 90.63 | 85.37 | 86.48 | 0.8992 | 0.9393 | 0.8914 | 67.10 | 45.72 | 89.09 | 81.16 | 0.5840 | 0.8006 | 0.7851 |
| KGNN | 97.09 | 98.56 | 95.63 | 95.79 | 0.9714 | 0.9924 | 0.9911 | 38.96 | 55.02 | 22.41 | 42.08 | 0.4760 | 0.4704 | 0.5861 |
| CNN-LSTM | 97.55 | 97.95 | 97.14 | 97.21 | 0.9756 | 0.9958 | 0.9950 | 51.39 | 74.63 | 27.66 | 52.08 | 0.5247 | 0.6115 | 0.6227 |
| KGE_NFM | 96.76 | 97.83 | 95.70 | 95.79 | 0.9679 | 0.9929 | 0.9912 | 72.56 | 56.69 | 88.43 | 84.32 | 0.6727 | 0.7256 | 0.8148 |
| DDKG | 87.94 | 96.96 | 78.52 | 82.46 | 0.8911 | 0.9217 | 0.8920 | 86.74 | 97.13 | 76.03 | 80.73 | 0.8815 | 0.8925 | 0.8333 |
| Our | 98.75 | 98.83 | 98.68 | 98.68 | 0.9875 | 0.9993 | 0.9914 | 83.18 | 82.45 | 83.95 | 84.39 | 0.8324 | 0.9083 | 0.8775 |
| DrugBank ID | Name | Structure | Evidence |
|---|---|---|---|
| DB14783 | Diroximel fumarate |  | Drugs.com |
| DB11793 | Niraparib |  | Drugs.com |
| DB09269 | Phenylacetic acid |  | PMID: 33218116 |
| DB15091 | Upadacitinib |  | Drugs.com |
| DB11942 | Selinexor |  | Drugs.com |
| DB04816 | Dantron |  | unconfirmed |
| DB03419 | Uracil |  | PMID: 22543158 |
| DB01208 | Sparfloxacin |  | Drugs.com |
| DB01059 | Norfloxacin |  | Drugs.com |
| DB02690 | NU1025 |  | PMID:10914735 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Chen, M.; Liu, J.; Peng, D.; Dai, Z.; Zou, X.; Li, Z. A Knowledge-Graph-Based Multimodal Deep Learning Framework for Identifying Drug–Drug Interactions. Molecules 2023, 28, 1490. https://doi.org/10.3390/molecules28031490
Zhang J, Chen M, Liu J, Peng D, Dai Z, Zou X, Li Z. A Knowledge-Graph-Based Multimodal Deep Learning Framework for Identifying Drug–Drug Interactions. Molecules. 2023; 28(3):1490. https://doi.org/10.3390/molecules28031490
Chicago/Turabian StyleZhang, Jing, Meng Chen, Jie Liu, Dongdong Peng, Zong Dai, Xiaoyong Zou, and Zhanchao Li. 2023. "A Knowledge-Graph-Based Multimodal Deep Learning Framework for Identifying Drug–Drug Interactions" Molecules 28, no. 3: 1490. https://doi.org/10.3390/molecules28031490
APA StyleZhang, J., Chen, M., Liu, J., Peng, D., Dai, Z., Zou, X., & Li, Z. (2023). A Knowledge-Graph-Based Multimodal Deep Learning Framework for Identifying Drug–Drug Interactions. Molecules, 28(3), 1490. https://doi.org/10.3390/molecules28031490