

Synthesis of Novel 1-Oxo-2,3,4-trisubstituted Tetrahydroisoquinoline Derivatives, Bearing Other Heterocyclic Moieties and Comparative Preliminary Study of Anti-Coronavirus Activity of Selected Compounds

 , , , , ,
, , , , ,
Abstract
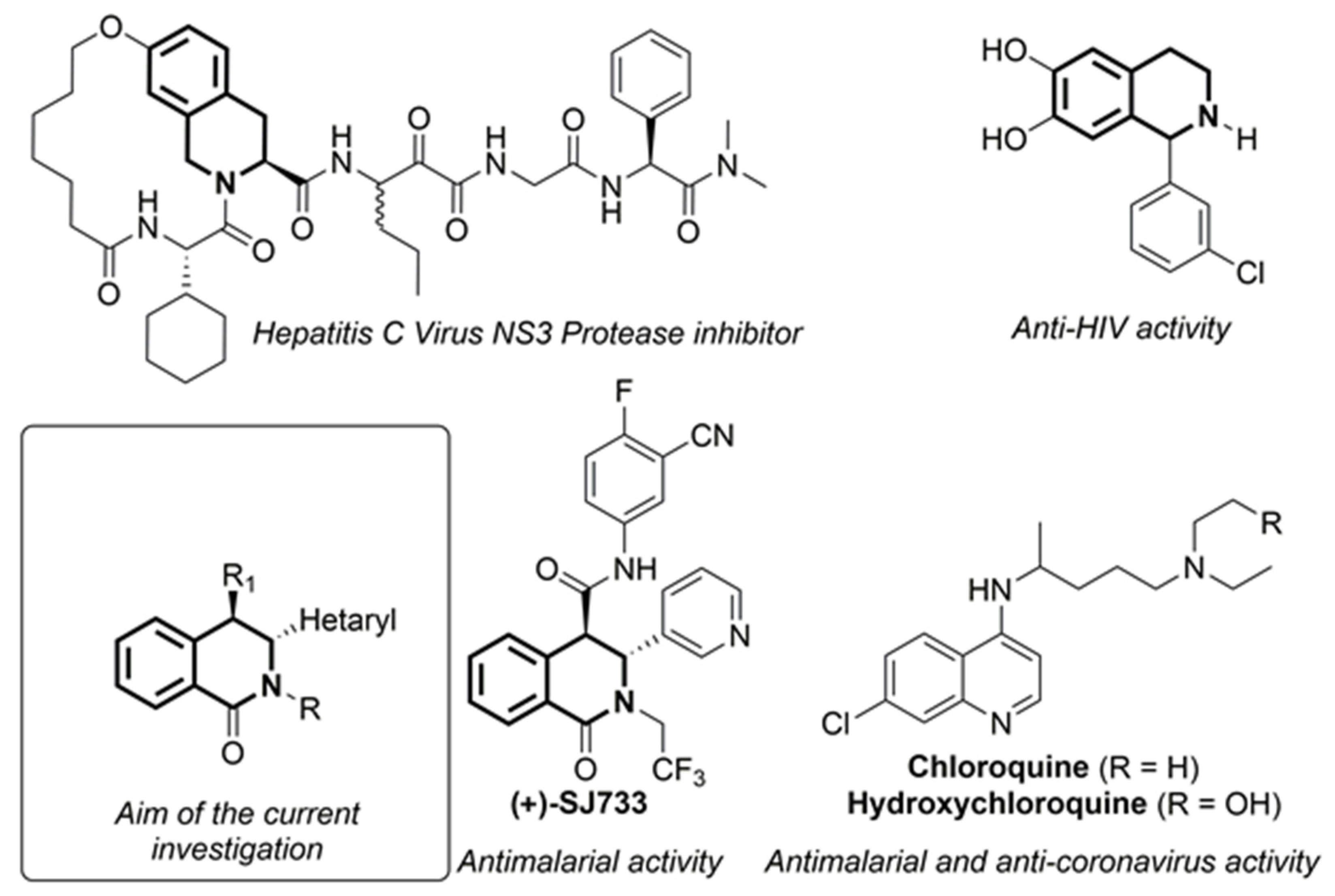
1. Introduction
2. Results and Discussion
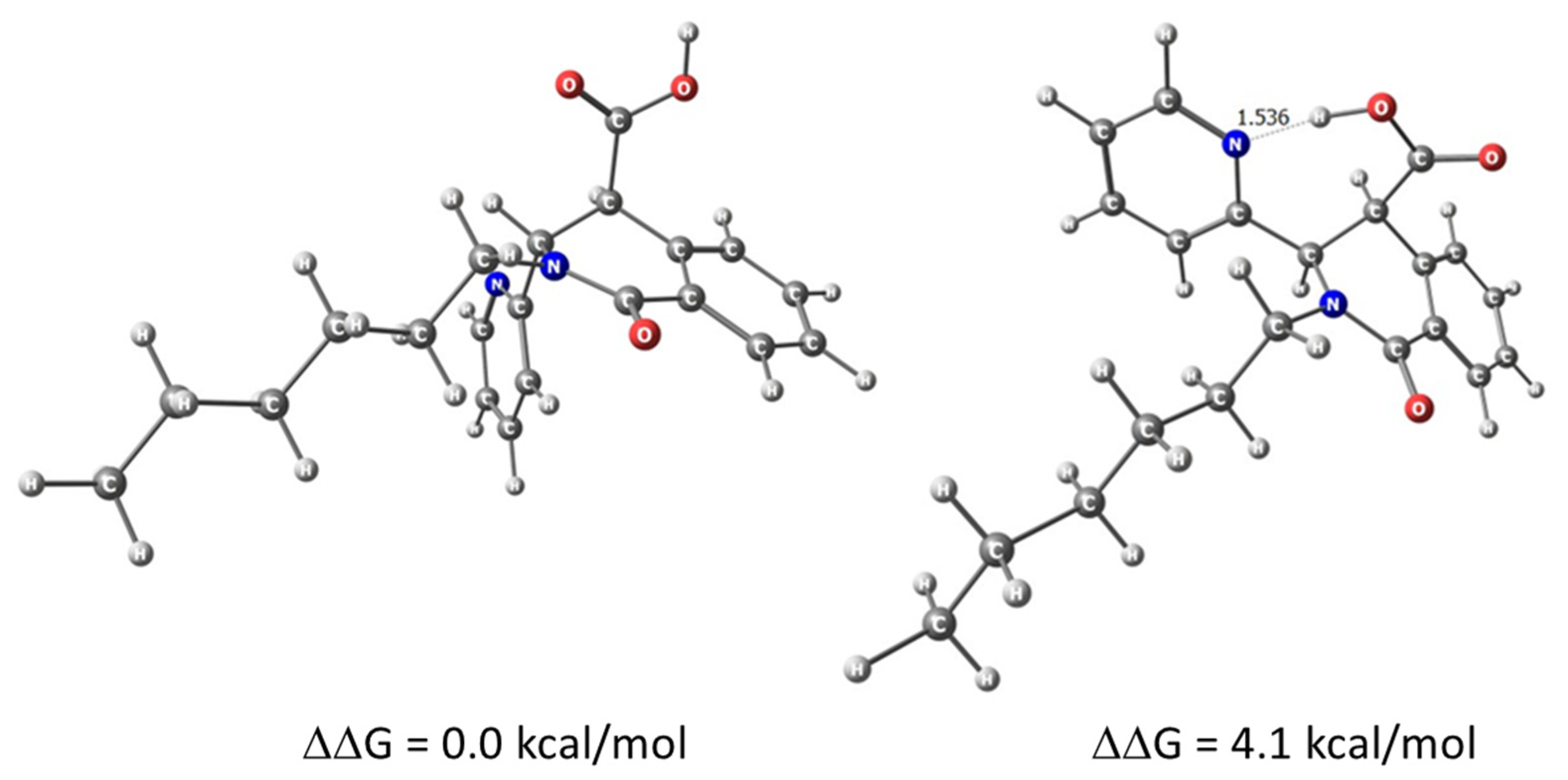
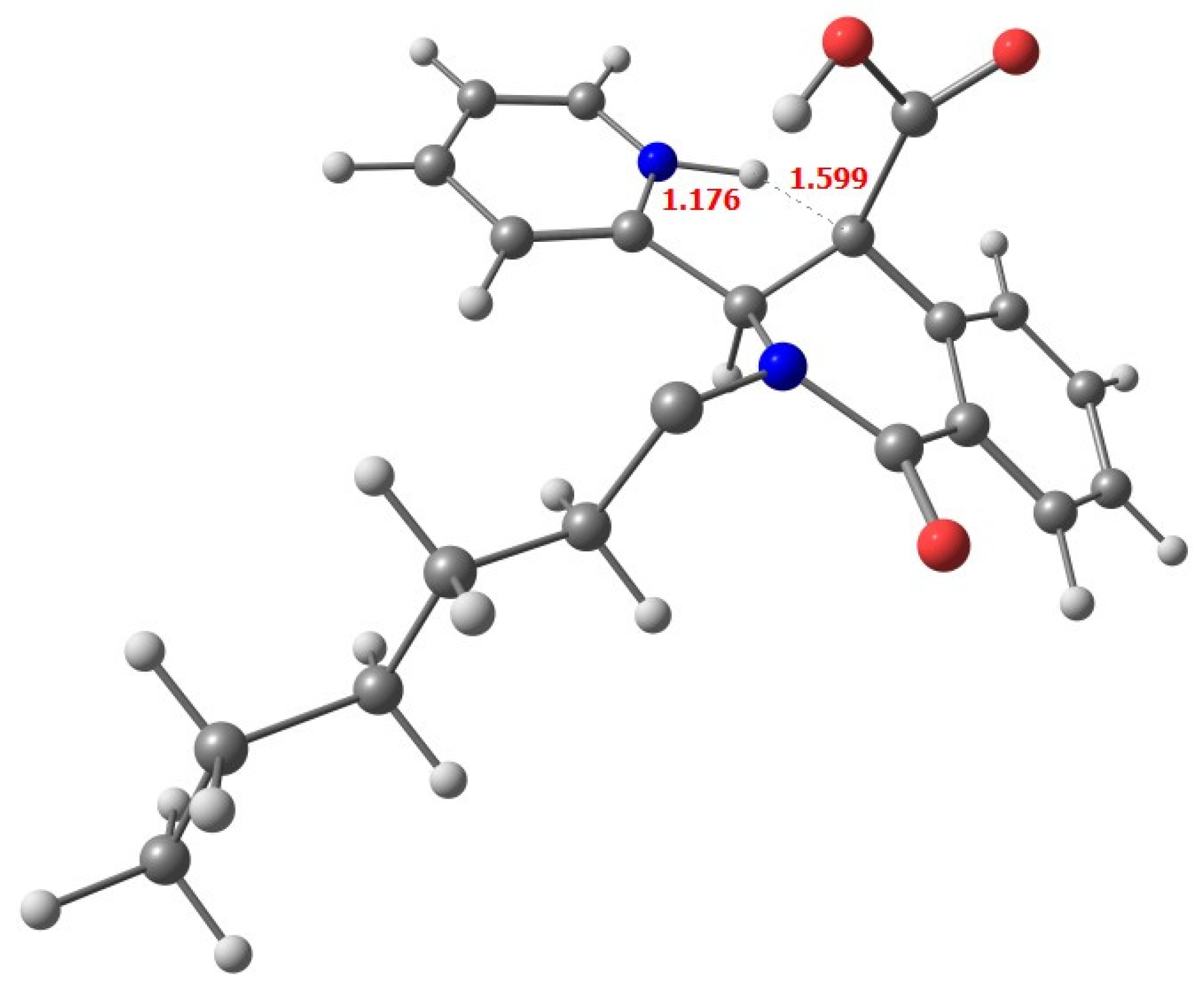
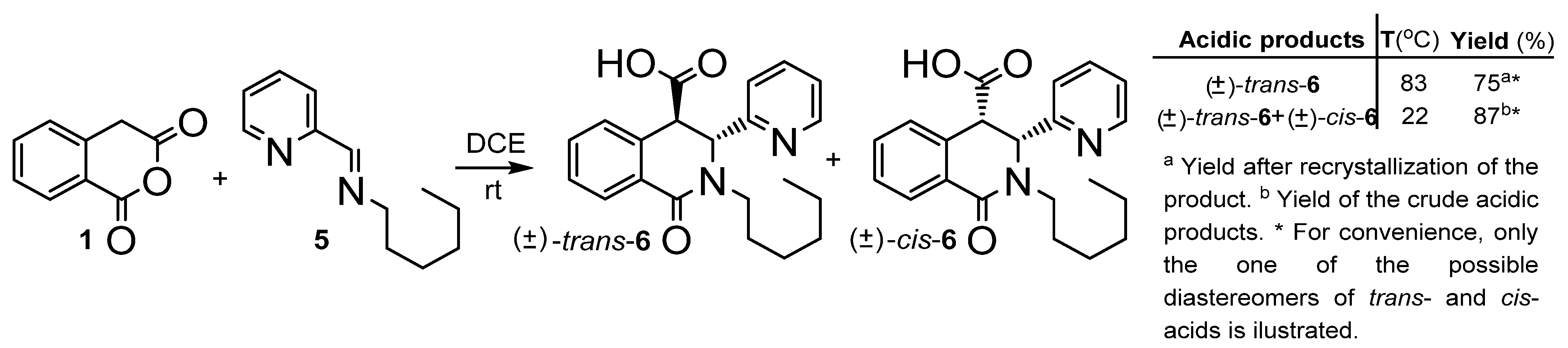
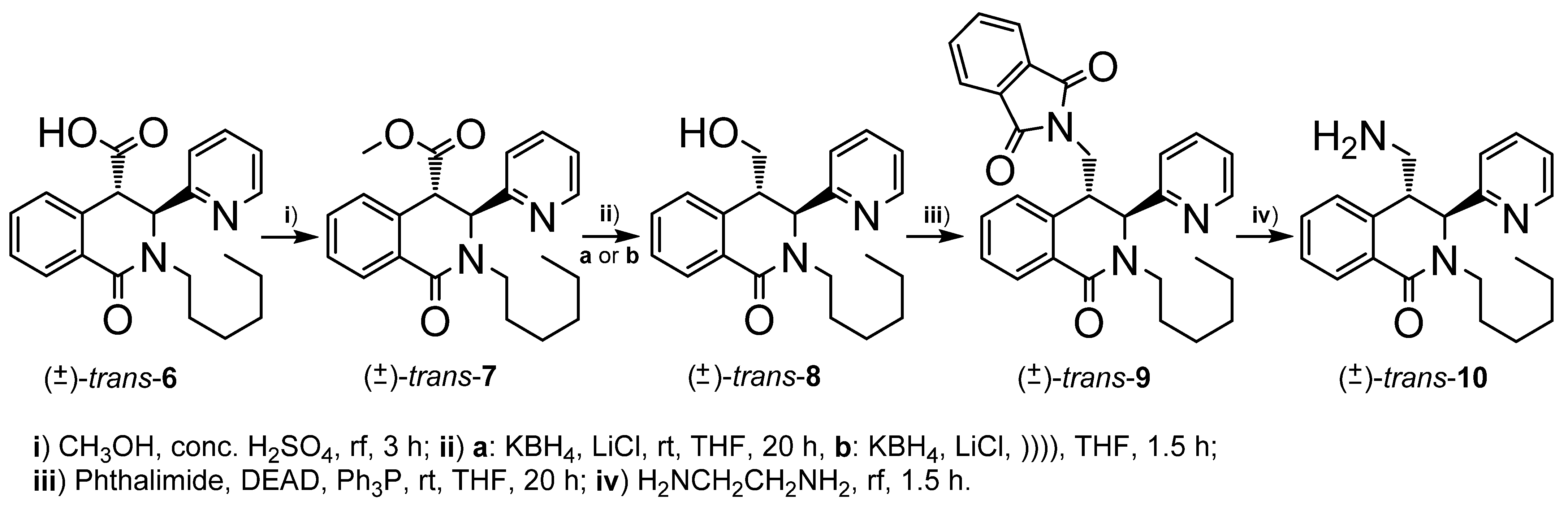
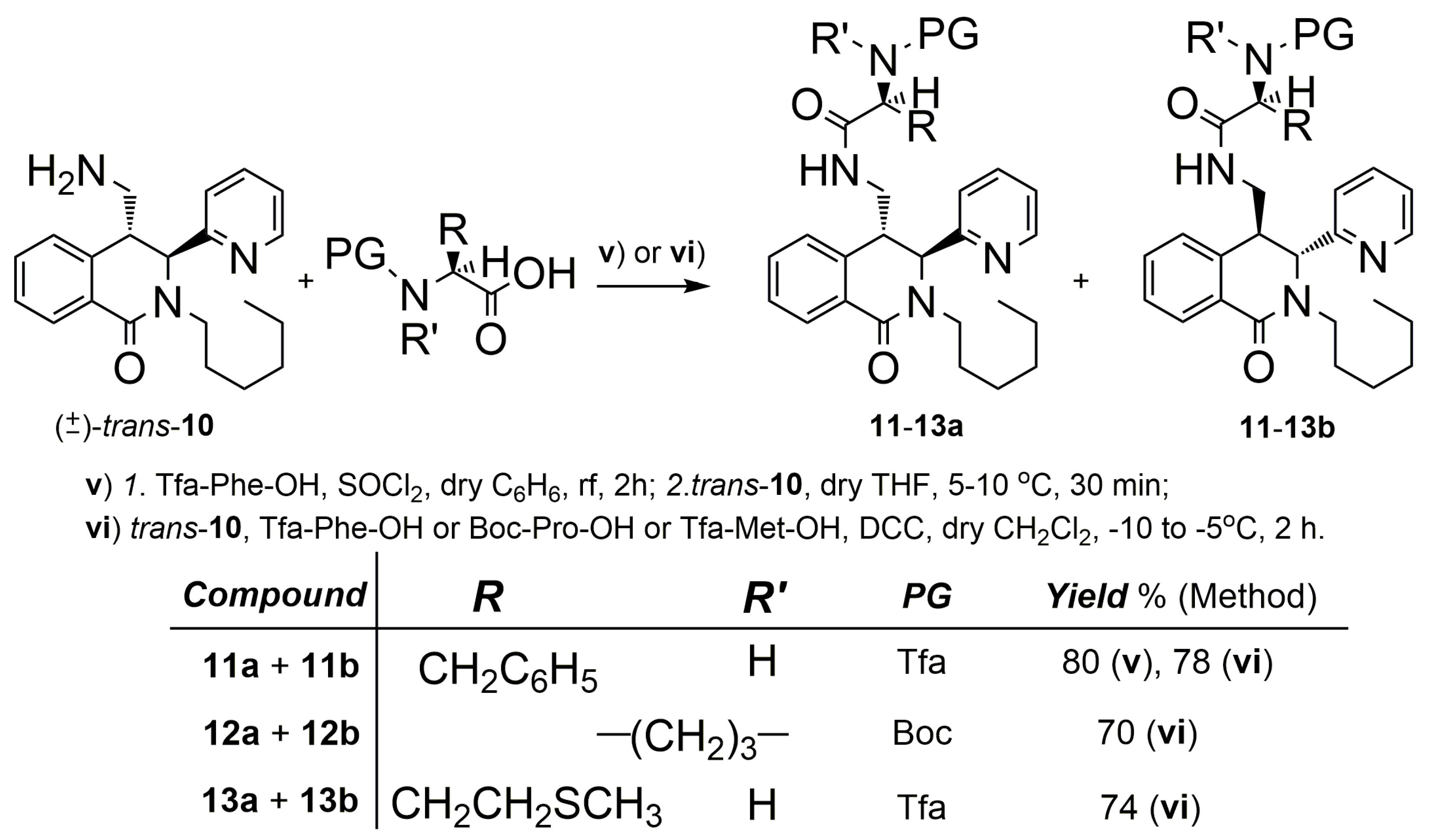
2.1. Synthesis
2.2. Virology
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Preparation of rel-(3R,4R)-3-(1H-indol-3-yl)-2-(2-methoxyethyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (3)
3.2.2. Synthesis of Amides with DIC as Coupling Agent (General Procedure)
rel-(3R,4R)-3-(1H-indol-3-yl)-N-isopropyl-N-(isopropylcarbamoyl)-2-(2-methoxyethyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxamide (4a)
rel-(3R,4R)-N-isopropyl-N-(isopropylcarbamoyl)-2-(2-methoxyethyl)-1-oxo-3-(1-(piperidin-1-ylmethyl)-1H-Indol-3-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxamide (4b)
rel-(3R,4R)-3-(1H-indol-3-yl)-2-(2-methoxyethyl)-4-(morpholine-4-carbonyl)-3,4-dihydroisoquinolin-1(2H)-one (4c)
3.2.3. Synthesis of Amides with TBTU as Coupling Agent (General Procedure)
rel-(3R,4R)-4-(1H-imidazole-1-carbonyl)-3-(1H-indol-3-yl)-2-(2-methoxyethyl)-3,4-dihydroisoquinolin-1(2H)-one (4d)
rel-(3R,4R)-3-(1H-indol-3-yl)-2-(2-methoxyethyl)-4-(4-methylpiperazine-1-carbonyl)-3,4-dihydroisoquinolin-1(2H)-one (4e)
3.2.4. rel-(3R,4R)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acids and rel-(3S,4R)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acids (trans-6 and cis-6)
3.2.5. rel-(3R,4R)-methyl 2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylate (trans-7)
3.2.6. rel-(3R,4R)-2-hexyl-4-(hydroxymethyl)-3-(pyridin-2-yl)-3,4-dihydroisoquinolin-1(2H)-one (trans-8)
3.2.7. rel-2-(((3S,4R)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)isoindoline-1,3-dione (trans-9)
3.2.8. rel-(3R,4S)-4-(aminomethyl)-2-hexyl-3-(pyridin-2-yl)-3,4-dihydroisoquinolin-1(2H)-one (trans-10)
3.2.9. (S)-N-((3R,4S)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)-3-phenyl-2-(2,2,2-trifluoroacetamido)propanamide and (S)-N-((3S,4R)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)-3-phenyl-2-(2,2,2-trifluoroacetamido)propanamide (trans-11a + trans-11b)
3.2.10. Acylation of trans-10 through the Carbodiimide Method (General Procedure)
(S)-N-(((3R,4S)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)-3-phenyl-2-(2,2,2-trifluoroacetamido)propanamide and (S)-N-(((3S,4R)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)-3-phenyl-2-(2,2,2-trifluoroacetamido)propanamide (trans-11a + trans-11b)
tert-butyl (S)-2-(((3R,4S)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate and tert-butyl (S)-2-(((3S,4R)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate (trans-12a + trans-12b)
(S)-N-(((3R,4S)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)-4-(methylthio)-2-(2,2,2-trifluoroacetamido)butanamide and (S)-N-(((3S,4R)-2-hexyl-1-oxo-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinolin-4-yl)methyl)-4-(methylthio)-2-(2,2,2-trifluoroacetamido)butanamide (trans-13a + trans-13b)
3.3. Microbiology
3.3.1. Cytotoxicity Assay
3.3.2. Antiviral Activity Assay
3.3.3. Pre-Treatment of Healthy Cells
3.3.4. Effect on Viral Adsorption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Phillipson, J.D.; Roberts, M.F.; Zenk, M.H. (Eds.) The Chemistry and Biology of Isoquinoline Alkaloids, 1st ed.; Springer: Berlin/Heidelberg, Germany, 1985; pp. 47–63. [Google Scholar]
- Scott, J.D.; Williams, R.M. Chemistry and Biology of the Tetrahydroisoquinoline Antitumor Antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef]
- Rothweiler, U.; Czarna, A.; Krajewski, M.; Ciombor, J.; Kalinski, C.; Khazak, V.; Ross, G.; Skobeleva, N.; Weber, L.; Holak, T.A. Isoquinolin-1-one Inhibitors of the MDM2–p53 Interaction. Chem. Med. Chem. 2008, 3, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Gitto, R.; Francica, E.; De Sarro, G.; Scicchitano, F.; Chimirri, A. Solution-Phase Parallel Synthesis of Novel 1,2,3,4-Tetrahydroisoquinolin-1-ones as Anticonvulsant Agents. Chem. Pharm. Bull. 2008, 56, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Billamboz, M.; Bailly, F.; Lion, C.; Calmels, C.; Andréola, M.; Witvrouw, M.; Christ, F.; Debyser, Z.; De Luca, L.; Chimirri, A.; et al. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: Influence of the alkylation of position 4. Eur. J. Med. Chem. 2011, 46, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.X.; George Njoroge, F.; Pichardo, J.; Prongay, A.; Butkiewicz, N.; Yao, N.; Madison, V.; Girijavallabhan, V. Potent 7-Hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic Acid-Based Macrocyclic Inhibitors of Hepatitis C Virus NS3 Protease. J. Med. Chem. 2006, 49, 567–574. [Google Scholar] [CrossRef]
- Humphries, P.S.; Benbow, J.W.; Bonin, P.D.; Boyer, D.; Doran, S.D.; Frisbie, R.K.; Piotrowski, D.W.; Balan, G.; Bechle, B.M.; Conn, E.L.; et al. Synthesis and SAR of 1,2,3,4-tetrahydroisoquinolin-1-ones as novel G-protein-coupled receptor 40 (GPR40) antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 2400–2403. [Google Scholar] [CrossRef]
- Glushkov, V.A.; Arapov, K.A.; Minova, O.N.; Ismailova, N.G.; Syropyatov, B.Y.; Shklyaev, Y.V. Synthesis and anticoagulant activity of 1-aryl derivatives of tetrahydroisoquinolines. Pharm. Chem. J. 2006, 40, 363–366. [Google Scholar] [CrossRef]
- Son, H.J.; Han, S.H.; Lee, J.A.; Lee, C.S.; Seo, J.W.; Chi, D.Y.; Hwang, O. 2-Acetyl-7-hydroxy-6-methoxy-1-methyl-1,2,3,4-tetrahydroisoquinoline exhibits anti-inflammatory properties and protects the nigral dopaminergic neurons. Eur. J. Pharmacol. 2016, 771, 152–161. [Google Scholar] [CrossRef]
- Fang, Y.; Zhou, H.; Gu, Q.; Xu, J. Synthesis and evaluation of tetrahydroisoquinoline-benzimidazole hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 167, 133–145. [Google Scholar] [CrossRef]
- Guy, R.K.; Zhu, F.; Guiguemde, W.A.; Floyd, D.; Knapp, S.; Stein, P.; Castro, S. Substituted 2-Alkyl-1-Oxo-N-Phenyl-1,2,3,4-Tetrahydroisoquinoline-4-Carboxamides for Antimalarial Therapies. U.S. Patent 9416124B2, 16 August 2016. [Google Scholar]
- Floyd, D.M.; Stein, P.; Wang, Z.; Liu, J.; Castro, S.; Clark, J.A.; Connelly, M.; Zhu, F.; Holbrook, G.; Matheny, A.; et al. Hit-to-Lead Studies for the Antimalarial Tetrahydroisoquinolone Carboxanilides. J. Med. Chem. 2016, 59, 7950–7962. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef]
- Kozekov, I.D.; Koleva, R.I.; Palamareva, M.D. New trans/cis tetrahydroisoquinolines. 1. trans-2-Benzyl-3-(1-methyl-1H-pyrrol-2-yl)-4-substituted-1,2,3,4-tetrahydroisoquinolin-1-ones and corresponding tetrahydroisoquinolines. J. Het. Chem. 2002, 39, 229–236. [Google Scholar] [CrossRef]
- Stoyanova, M.P.; Kozekov, I.D.; Palamareva, M.D. New trans/cis tetrahydroisoquinolines. 2. trans- and cis-3-(1-methyl-1H-pyrrol-2-yl)-1-(2H)-oxo2-phenethyl-1,2,3,4-tetrahydroisoquinolin-4-carboxylic acids and subsequent transformation. J. Het. Chem. 2003, 40, 795–803. [Google Scholar] [CrossRef]
- Burdzhiev, N.T.; Baramov, T.I.; Stanoeva, E.R.; Yanev, S.G.; Stoyanova, T.D.; Dimitrova, D.H.; Kostadinova, K.A. Synthesis of novel trans-4-(phthalimidomethyl)- and 4-(imidazol-1-ylmethyl)-3-indolyl-tetrahydroisoquinolinones as possible aromatase inhibitors. Chem. Pap. 2019, 73, 1263–1277. [Google Scholar] [CrossRef]
- Burdzhiev, N.T.; Stanoeva, E.R. Reaction between glutaric anhydride and N-benzylidenebenzylamine, and further transformations to new substituted piperidin-2-ones. Tetrahedron 2006, 62, 8318–8326. [Google Scholar] [CrossRef]
- Kandinska, M.I.; Kozekov, I.D.; Palamareva, M.D. Synthesis of New trans-2-Benzyl-3-(furan-2-yl)-4-substituted-1,2,3,4-tetrahydroisoquinolinones. Molecules 2006, 11, 403–414. [Google Scholar] [CrossRef]
- Yang, Y. Side Reactions in Peptide Synthesis, 1st ed.; Tsinghua University Press Limited, Elsevier Inc.: Beijing, China, 2016; pp. 95–97. [Google Scholar]
- Rudine, A.; Walter, M.; Wamser, C. Reaction of Dichloromethane with Pyridine Derivatives under Ambient Conditions. J. Org. Chem. 2010, 75, 4292–4295. [Google Scholar] [CrossRef]
- Kandinska, M.I.; Todorov, I.S.; Shivachev, B.; Bogdanov, M.G. trans-rac-2-Hexyl-1-oxo-3-(2-pyrid yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid. Acta Cryst. 2007, E63, o2544–o2546. [Google Scholar] [CrossRef]
- Haimova, M.A.; Mollov, N.M.; Ivanova, S.C.; Dimitrova, A.I.; Ognyanov, V.I. A highly stereoselective synthesis of 3,4-dihydro-1(2H)-isoquinolinones and 8-oxoberbines from homophthalic anhydride and azomethines. Tetrahedron 1977, 33, 331–336. [Google Scholar] [CrossRef]
- Cushman, M.; Gentry, J.; Dekow, F.W. Condensation of imines with homophthalic anhydrides. A convergent synthesis of cis- and trans-13-methyltetrahydroprotoberberines. J. Org. Chem. 1977, 42, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* bases set for third-row atoms. J. Comp. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Yildirim, S.Ö.; Akkurt, M.; Kandinska, M.I.; Bogdanov, M.G.; Büyükgüngör, O. Methyl trans-rac-2-hexyl-1-oxo-3-(2-pyridyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylate. Acta Cryst. 2008, 64, o1932. [Google Scholar] [CrossRef]
- Bonnaud, B.; Carlessi, A.; Bigg, D.C.H. Synthesis of novel Isoquinoline derivatives as potential CNS-agents. J. Het. Chem. 1993, 30, 257–265. [Google Scholar] [CrossRef]
- Burdzhiev, N.T. Available online: http://hdl.handle.net/10506/1506 (accessed on 2 February 2023).
- Fujita, T.; Iwasa, J.; Hansch, C. A New Substituent Constant, π, Derived from Partition Coefficients. J. Am. Chem. Soc. 1964, 86, 5175–5180. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Borenfreund, E.; Puerner, J.A. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol. Lett. 1985, 24, 119–124. [Google Scholar] [CrossRef]









{kind=link}
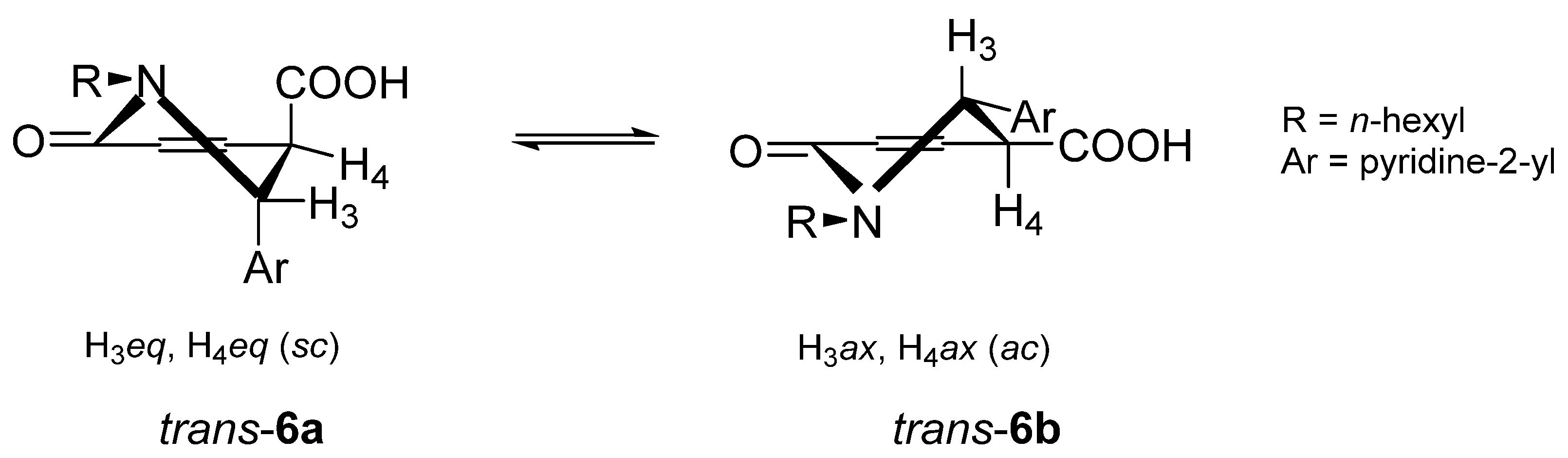{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G [kcal/mol] | μ | |
| trans-6a | 0.0 | 4.9475 |
| trans-6a’ * | 2.9 | 7.9465 |
| cis-6 | 4.1 | 13.5871 |
| trans-6b | 5.0 | 9.5114 |
 |  |  |  |
| Avir-1 [19] | Avir-2 [19] | Avir-3 [19] | Avir-4 [19] |
 |  |  |  |
| Avir-5 [30] | Avir-6 [17] | Avir-7 [17] | Avir-8 [18] |
| Compound Code | Cytotoxicity, MRC-5 | Antiviral Activity against 229E Strain | Cytotoxicity, HCT-8 | Antiviral Activity against OC-43 Strain | ||||
|---|---|---|---|---|---|---|---|---|
| CC50 (µM) | MTC (µM) | IC50 (µM) | SI | CC50 (µM) | MTC (µM) | IC50 (µM) | SI | |
| 4a | 670 ± 29 | 320 | - | - | 350 ± 15 | 150 | 47 ± 2 | 7.4 |
| 4c | 274 ± 12 | 100 | - | - | 645 ± 27 | 320 | 18 ± 1 | 35.8 |
| 4d | 299 ± 13 | 100 | - | - | 580 ± 25 | 320 | 100 ± 4 | 5.8 |
| 4e | 298 ± 13 | 100 | 60 ± 3 | 4.9 | 724 ± 31 | 320 | 80 ± 3 | 9 |
| Avir-1 | 321 ± 14 | 150 | - | 293 ± 12 | 100 | 105 ± 4 | 2.8 | |
| Avir-2 | 550 ± 23 | 200 | 249 ± 11 | 2.2 | 383 ± 16 | 100 | - | - |
| Avir-3 | 659 ± 28 | 320 | 70.3 ± 3 | 9.4 | 588 ± 25 | 150 | - | - |
| Avir-4 | 452 ± 19 | 150 | 20 ± 1 | 23 | 283 ± 12 | 100 | - | - |
| 11a + 11b | 490 ± 21 | 320 | 22 ± 1 | 22 | 471 ± 20 | 150 | - | - |
| Avir-5 | 670 ± 28 | 320 | 30.5 ± 2 | 22 | 522 ± 22 | 200 | 75 ± 3 | 7 |
| Avir-6 | 729 ± 31 | 320 | 110 ± 5 | 6.6 | 579 ± 24 | 200 | 38 ± 1 | 15.2 |
| Avir-7 | 280 ± 12 | 150 | 0.5 ± 0.03 | 560 | 280 ± 12 | 100 | 1 ± 0.06 | 280 |
| Avir-8 | 515 ± 22 | 200 | 1.4 ± 0.05 | 367 | 486 ± 21 | 150 | 0.5 ± 0.02 | 972 |
| Chloroquine | 60 ± 3 | 20 | 0.1 ± 0.01 | 600 | 65 ± 2 | 10 | 0.1 ± 0.01 | 650 |
| Hydroxychloroquine | 66 ± 3 | 20 | - | - | 130 ± 5 | 30 | 100 ± 4 | 1.3 |
| Compound Code | Decrease in Viral Titre (Δlg) | ||||
|---|---|---|---|---|---|
| 15 Min | 30 Min | 60 Min | 90 Min | 120 Min | |
| Avir-1 | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 |
| Avir-2 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 |
| Avir-3 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 |
| Avir-4 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| 11a + 11b | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Avir-5 | 0.75 | 1.0 | 1.0 | 1.0 | 1.0 |
| Avir-6 | 1.0 | 1.0 | 1.5 | 1.5 | 1.5 |
| Avir-7 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Avir-8 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 |
| Chloroquine | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 |
| Hydroxychloroquine | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 |
| Compound | Decrease in Viral Titre (Δlg) | ||||
|---|---|---|---|---|---|
| 15 Min | 30 Min | 60 Min | 90 Min | 120 Min | |
| Avir-1 | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 |
| Avir-2 | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 |
| Avir-3 | 2.0 | 1.75 | 1.75 | 1.0 | 1.0 |
| Avir-4 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| 11a + 11b | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 |
| Avir-5 | 0.75 | 1.0 | 1.0 | 1.0 | 1.0 |
| Avir-6 | 1.0 | 1.0 | 1.25 | 1.25 | 1.25 |
| Avir-7 | 0.5 | 0.5 | 0.75 | 0.75 | 0.75 |
| Avir-8 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Chloroquine | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 |
| Compound | Decrease in Viral Titre (Δlg) | ||||
|---|---|---|---|---|---|
| 15 Min | 30 Min | 60 Min | 90 Min | 120 Min | |
| Avir-1 | 0 | 1.0 | 1.75 | 2.0 | 2.0 |
| Avir-2 | 0 | 0.75 | 1.5 | 2.0 | 2.0 |
| Avir-3 | 0 | 1.0 | 1.25 | 1.5 | 1.5 |
| Avir-4 | 0 | 0.5 | 0.5 | 1.0 | 1.5 |
| 11a + 11b | 0.75 | 1.0 | 1.5 | 2.0 | 2.0 |
| Avir-5 | 0.75 | 1.0 | 1.25 | 1.5 | 2.0 |
| Avir-6 | 0.75 | 1.0 | 1.75 | 2.0 | 2.5 |
| Avir-7 | 0 | 0 | 1.0 | 1.75 | 2.0 |
| Avir-8 | 0 | 1.0 | 1.75 | 2.0 | 2.0 |
| Chloroquine | 0.5 | 2.0 | 2.0 | 2.0 | 2.5 |
| Hydroxychloroquine | 0 | 0 | 0.75 | 0.75 | 0.75 |
| Compound | Decrease in Viral Titre (Δlg) | ||||
|---|---|---|---|---|---|
| 15 Min | 30 Min | 60 Min | 90 Min | 120 Min | |
| Avir-1 | 0 | 1.0 | 2.0 | 2.5 | 2.5 |
| Avir-2 | 0 | 1.0 | 2.0 | 2.25 | 2.25 |
| Avir-3 | 0 | 1.0 | 1.25 | 1.5 | 1.5 |
| Avir-4 | 0.75 | 1.0 | 1.0 | 1.25 | 1.5 |
| 11a + 11b | 0.75 | 1.0 | 1.5 | 2.0 | 2.5 |
| Avir-5 | 0.75 | 1.25 | 1.5 | 1.75 | 2.25 |
| Avir-6 | 0.75 | 1.25 | 2.0 | 2.25 | 2.34 |
| Avir-7 | 0 | 1.0 | 1.75 | 2.25 | 2.5 |
| Avir-8 | 0 | 1.0 | 1.75 | 2.0 | 2.34 |
| Chloroquine | 0.5 | 1.75 | 2.0 | 2.5 | 2.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandinska, M.I.; Burdzhiev, N.T.; Cheshmedzhieva, D.V.; Ilieva, S.V.; Grozdanov, P.P.; Vilhelmova-Ilieva, N.; Nikolova, N.; Lozanova, V.V.; Nikolova, I. Synthesis of Novel 1-Oxo-2,3,4-trisubstituted Tetrahydroisoquinoline Derivatives, Bearing Other Heterocyclic Moieties and Comparative Preliminary Study of Anti-Coronavirus Activity of Selected Compounds. Molecules 2023, 28, 1495. https://doi.org/10.3390/molecules28031495
Kandinska MI, Burdzhiev NT, Cheshmedzhieva DV, Ilieva SV, Grozdanov PP, Vilhelmova-Ilieva N, Nikolova N, Lozanova VV, Nikolova I. Synthesis of Novel 1-Oxo-2,3,4-trisubstituted Tetrahydroisoquinoline Derivatives, Bearing Other Heterocyclic Moieties and Comparative Preliminary Study of Anti-Coronavirus Activity of Selected Compounds. Molecules. 2023; 28(3):1495. https://doi.org/10.3390/molecules28031495
Chicago/Turabian StyleKandinska, Meglena I., Nikola T. Burdzhiev, Diana V. Cheshmedzhieva, Sonia V. Ilieva, Peter P. Grozdanov, Neli Vilhelmova-Ilieva, Nadya Nikolova, Vesela V. Lozanova, and Ivanka Nikolova. 2023. "Synthesis of Novel 1-Oxo-2,3,4-trisubstituted Tetrahydroisoquinoline Derivatives, Bearing Other Heterocyclic Moieties and Comparative Preliminary Study of Anti-Coronavirus Activity of Selected Compounds" Molecules 28, no. 3: 1495. https://doi.org/10.3390/molecules28031495
APA StyleKandinska, M. I., Burdzhiev, N. T., Cheshmedzhieva, D. V., Ilieva, S. V., Grozdanov, P. P., Vilhelmova-Ilieva, N., Nikolova, N., Lozanova, V. V., & Nikolova, I. (2023). Synthesis of Novel 1-Oxo-2,3,4-trisubstituted Tetrahydroisoquinoline Derivatives, Bearing Other Heterocyclic Moieties and Comparative Preliminary Study of Anti-Coronavirus Activity of Selected Compounds. Molecules, 28(3), 1495. https://doi.org/10.3390/molecules28031495