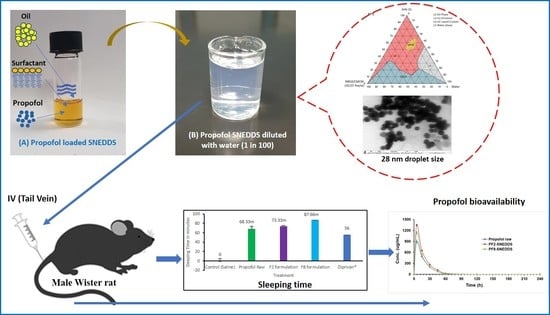
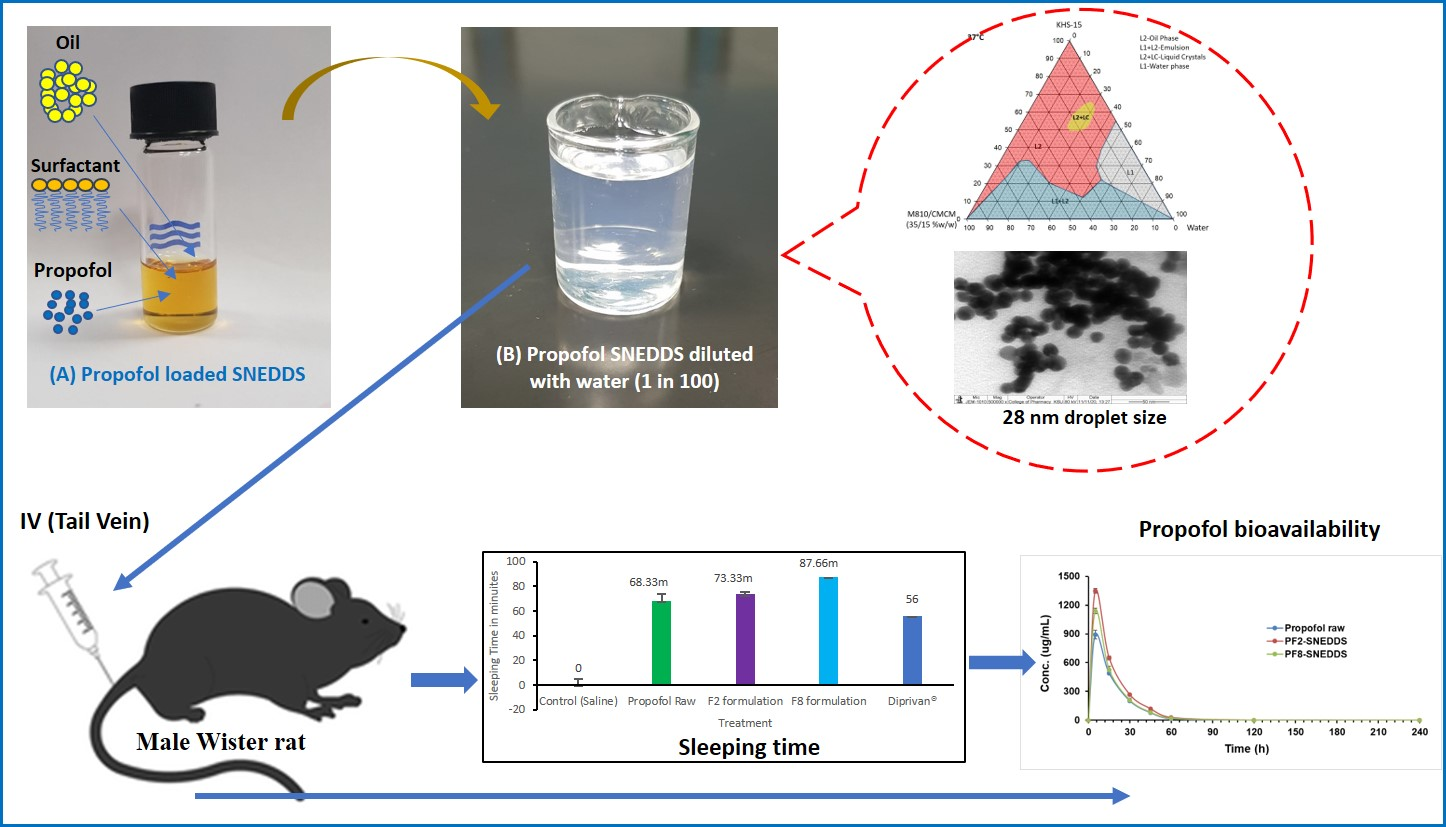
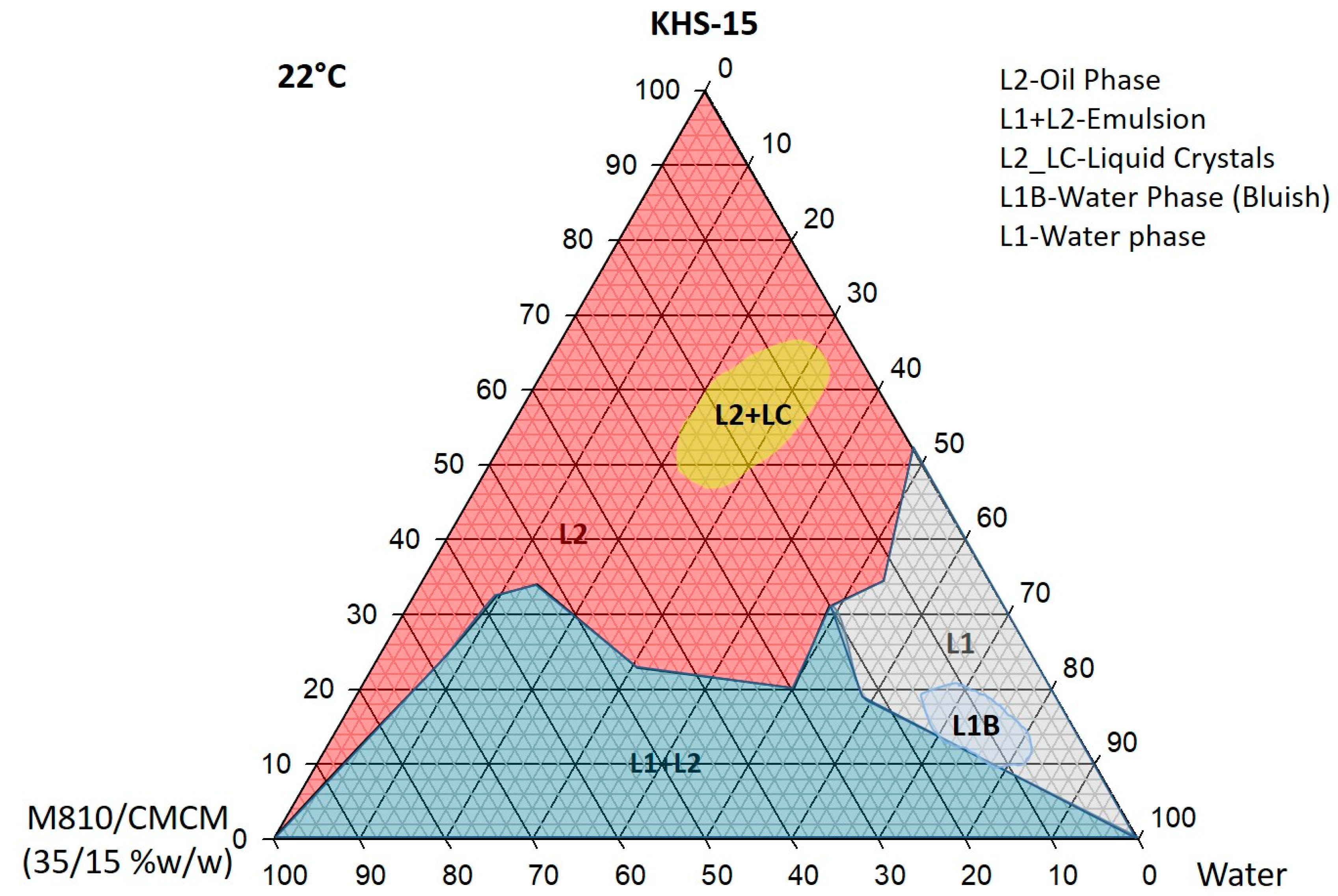
Figure 1.
Ternary phase diagram of M810:CMCM (7:3)/HCO-30/water systems at 22 °C, in which L1 denotes a clear aqueous phase, L2 a transparent oily phase, L1 + L2 a milky emulsion, L2 + LC an oily phase with liquid crystals, and L1B a water continuous bluish phase.
Figure 1.
Ternary phase diagram of M810:CMCM (7:3)/HCO-30/water systems at 22 °C, in which L1 denotes a clear aqueous phase, L2 a transparent oily phase, L1 + L2 a milky emulsion, L2 + LC an oily phase with liquid crystals, and L1B a water continuous bluish phase.
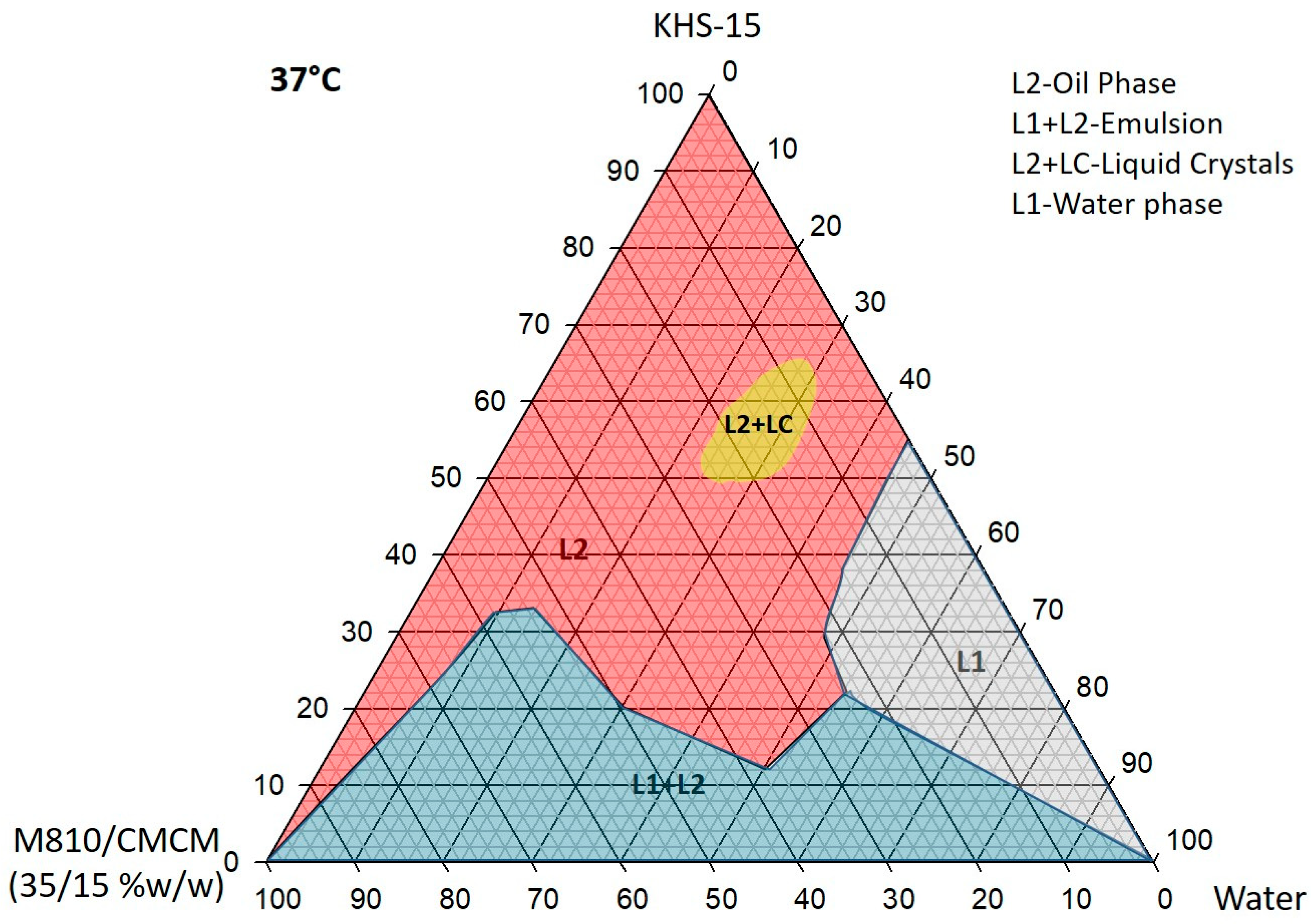
Figure 2.
Ternary phase diagram of M810:CMCM (7:3)/HCO-30/water systems at 37 °C, in which L1 denotes a clear aqueous phase, L2 a transparent oily phase, L1 + L2 a milky emulsion, and L2 + LC an oily phase with liquid crystals.
Figure 2.
Ternary phase diagram of M810:CMCM (7:3)/HCO-30/water systems at 37 °C, in which L1 denotes a clear aqueous phase, L2 a transparent oily phase, L1 + L2 a milky emulsion, and L2 + LC an oily phase with liquid crystals.
Figure 3.
Appearances of the drug-free lipid-based formulations after aqueous dilution with water for the optimization of the representative SNEDDS. Formulations are F1: M810/CMCM/KELP (35/15/50); F2: M810/CMCM/KHS15 (35/15/50); F3: ZRO/CMCM/KELP (35/15/50); F4: ZRO/CMCM/KHS15 (35/15/50); F5: ZRO/CMCM/T80 (35/15/50); F6: CO/CMCM/KELP (35/15/50); F7: ZRO/M35-1/KELP (35/15/50); and F8: ZRO/M35-1/KHS15 (35/15/50) %w/w/w, respectively.
Figure 3.
Appearances of the drug-free lipid-based formulations after aqueous dilution with water for the optimization of the representative SNEDDS. Formulations are F1: M810/CMCM/KELP (35/15/50); F2: M810/CMCM/KHS15 (35/15/50); F3: ZRO/CMCM/KELP (35/15/50); F4: ZRO/CMCM/KHS15 (35/15/50); F5: ZRO/CMCM/T80 (35/15/50); F6: CO/CMCM/KELP (35/15/50); F7: ZRO/M35-1/KELP (35/15/50); and F8: ZRO/M35-1/KHS15 (35/15/50) %w/w/w, respectively.
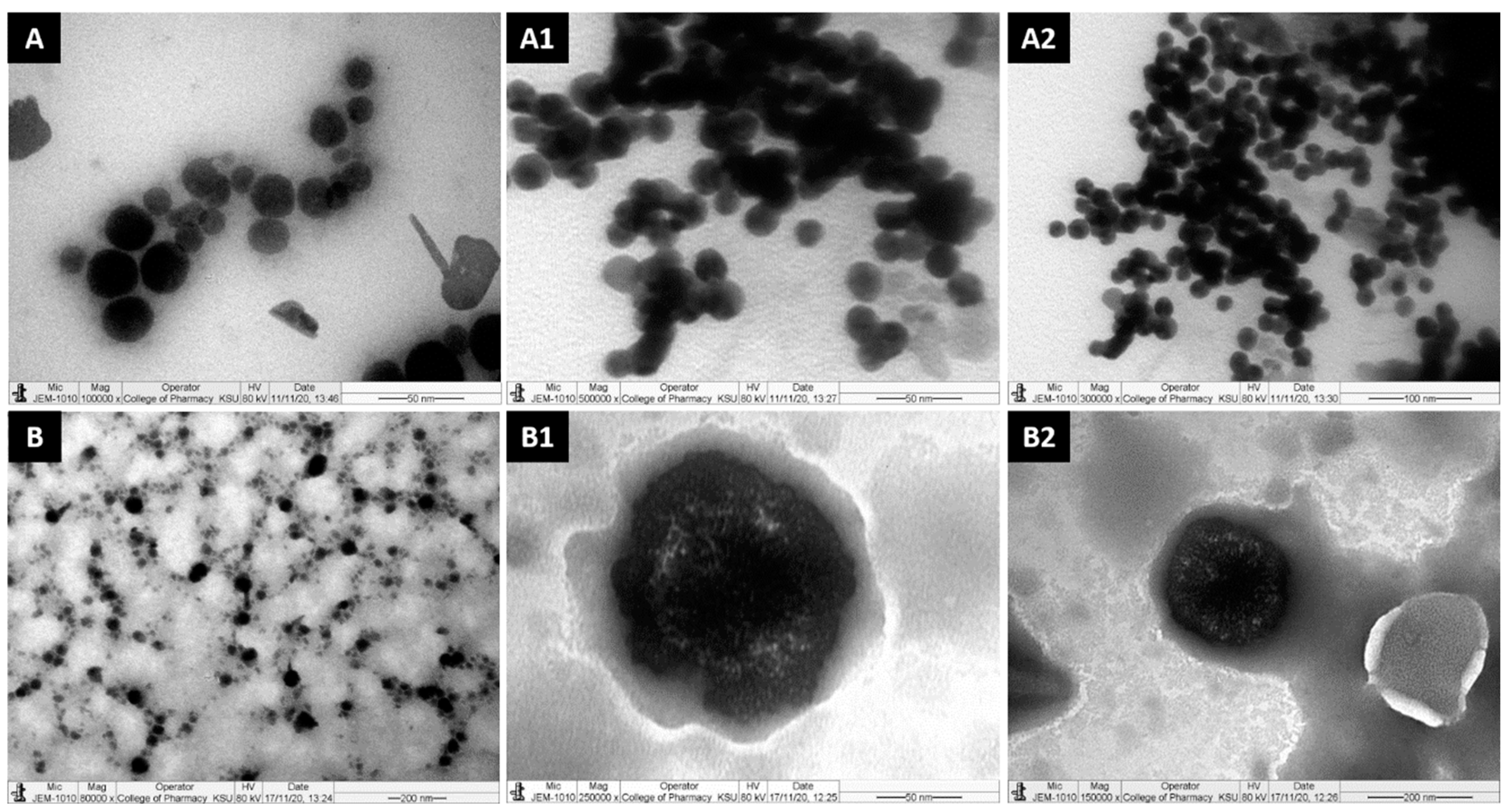
Figure 4.
TEM micrographs of propofol-loaded representative formulations. (A,A1,A2) represent images of the droplet sizes of the PF2-SNEDDS at a 50–100 nm scale, and (B,B1,B2) represent images of the droplet sizes of the PF8-SNEDDS at a 50–200 nm scale.
Figure 4.
TEM micrographs of propofol-loaded representative formulations. (A,A1,A2) represent images of the droplet sizes of the PF2-SNEDDS at a 50–100 nm scale, and (B,B1,B2) represent images of the droplet sizes of the PF8-SNEDDS at a 50–200 nm scale.
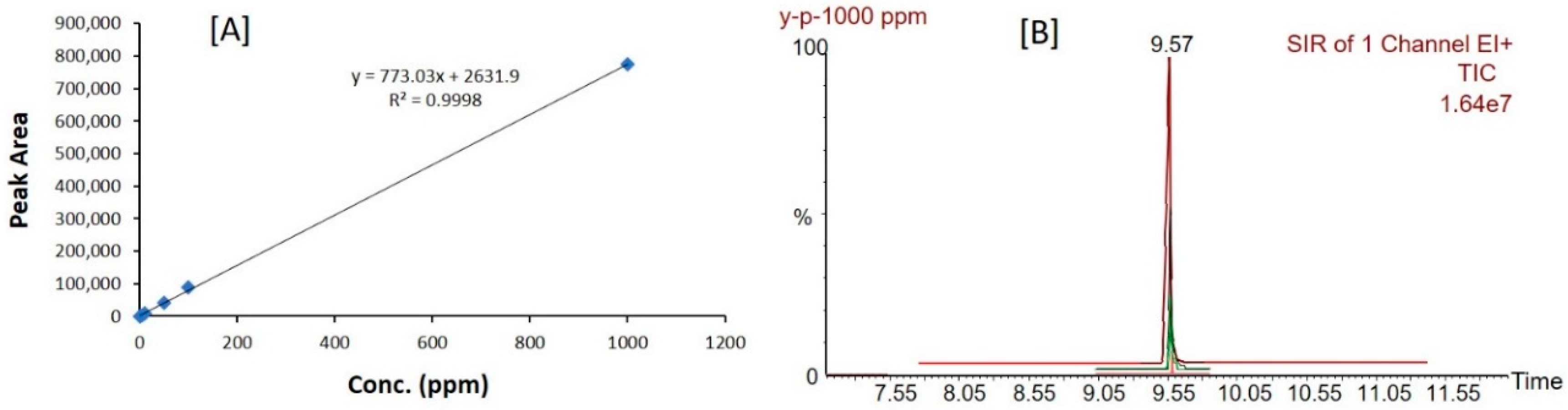
Figure 5.
Calibration curve (A) and peaks (B) of propofol constructed by GC/MS sample analysis.
Figure 5.
Calibration curve (A) and peaks (B) of propofol constructed by GC/MS sample analysis.
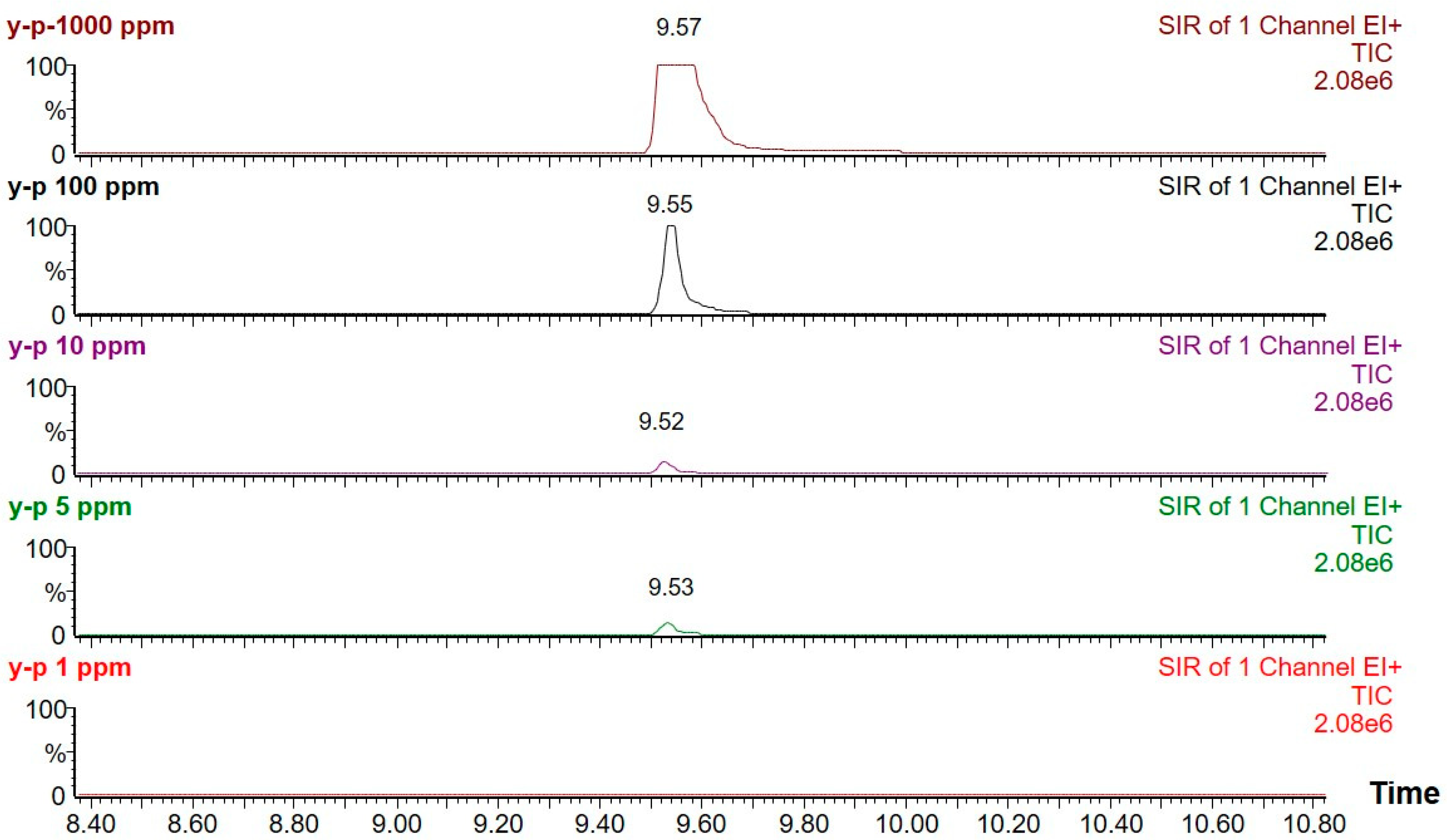
Figure 6.
The chromatograms (TIC) of calibration standard (1000 ppm, 100 ppm, 10 ppm, 5 ppm, 1 ppm) using optimized GC-MS conditions.
Figure 6.
The chromatograms (TIC) of calibration standard (1000 ppm, 100 ppm, 10 ppm, 5 ppm, 1 ppm) using optimized GC-MS conditions.
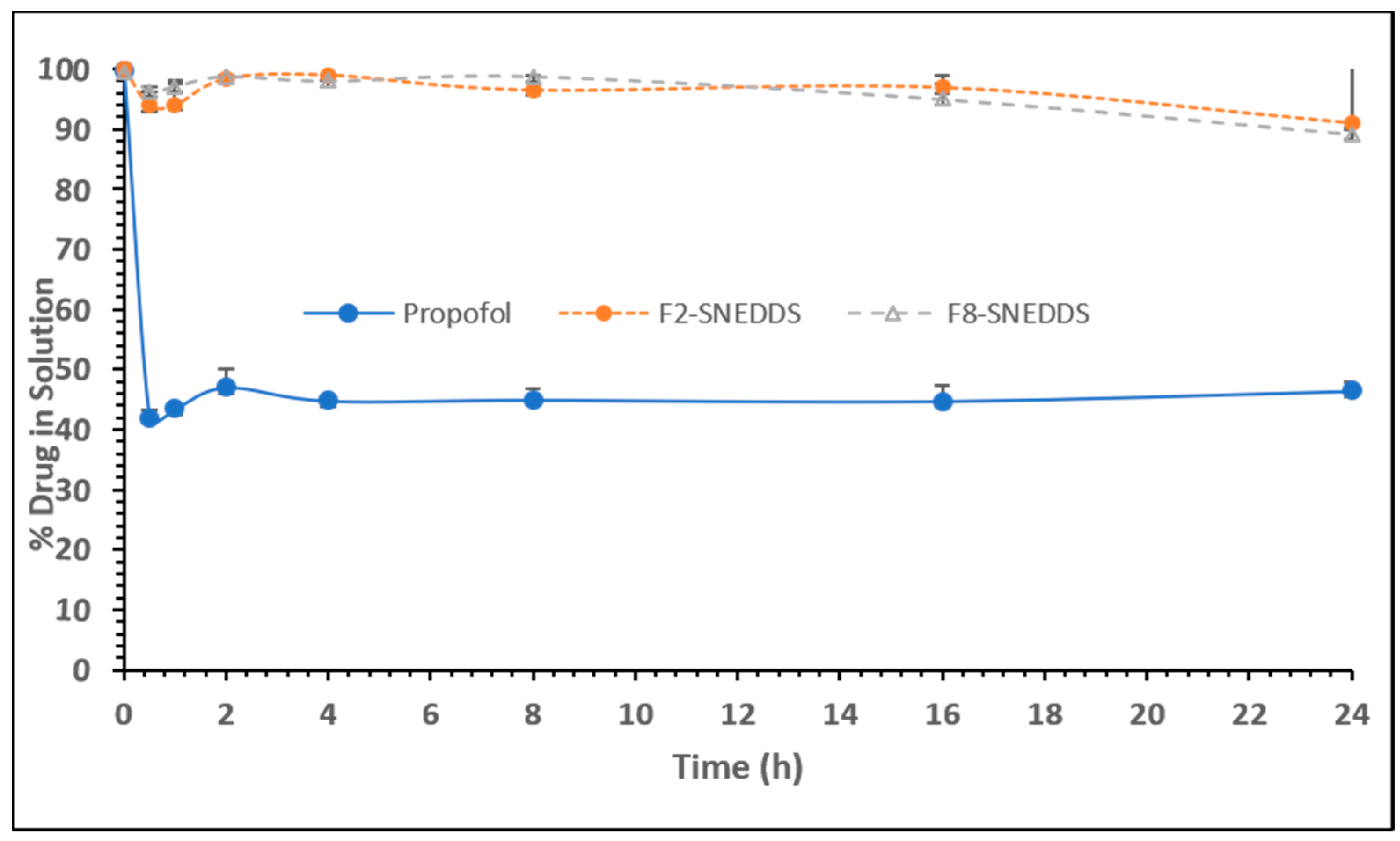
Figure 7.
Percentage of propofol remaining in solution during 24 h after a dynamic dispersion study carried out with representative SNEDDS formulations. The formulations are F2-SNEDDS, M810/CMCM/KHS15 (35/15/50), and F8-SNEDDS, ZRO/M35-1/KHS15 (35/15/50) %w/w/w, respectively. Data are expressed as mean ± SD, n = 3.
Figure 7.
Percentage of propofol remaining in solution during 24 h after a dynamic dispersion study carried out with representative SNEDDS formulations. The formulations are F2-SNEDDS, M810/CMCM/KHS15 (35/15/50), and F8-SNEDDS, ZRO/M35-1/KHS15 (35/15/50) %w/w/w, respectively. Data are expressed as mean ± SD, n = 3.
Figure 8.
Appearances of the propofol-loaded F2 (PF2-SNEDDS) and F8 (PF8-SNEDDS) lipid-based formulations after six months of storage conditions at 37 °C. The formulations were diluted with water. “D denotes drug-loaded”. The left images represent at 0 months and the right images at 6 months.
Figure 8.
Appearances of the propofol-loaded F2 (PF2-SNEDDS) and F8 (PF8-SNEDDS) lipid-based formulations after six months of storage conditions at 37 °C. The formulations were diluted with water. “D denotes drug-loaded”. The left images represent at 0 months and the right images at 6 months.
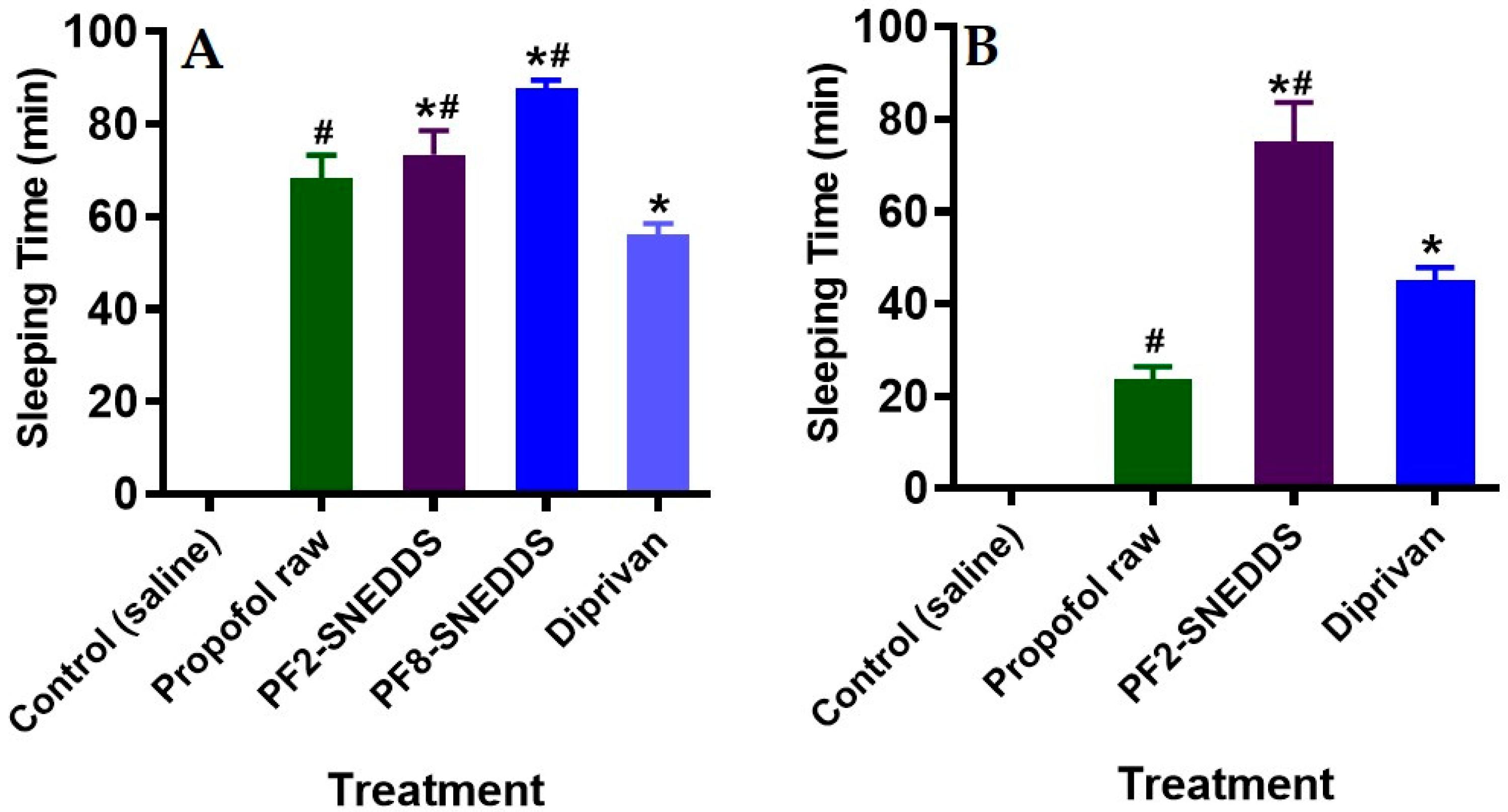
Figure 9.
Measurement of propofol-induced sleeping time in rats (n = 6). (A) An equivalent dose of 10 mg/kg was administered intravenously to all the treatment groups (propofol raw, propofol-loaded formulation PF2-SNEDDS, PF8-SNEDDS, and marketed drug Diprivan®). (B) The dose (propofol raw, propofol-loaded formulation PF2-SNEDDS, and marketed drug Diprivan®) was administered intraperitoneally (IP). All results are presented as the average ± SEM. The asterisk and hash symbols (above the bars) indicate significant differences between samples * p < 0.05 (Propofol raw); # p < 0.05 (Diprivan®).
Figure 9.
Measurement of propofol-induced sleeping time in rats (n = 6). (A) An equivalent dose of 10 mg/kg was administered intravenously to all the treatment groups (propofol raw, propofol-loaded formulation PF2-SNEDDS, PF8-SNEDDS, and marketed drug Diprivan®). (B) The dose (propofol raw, propofol-loaded formulation PF2-SNEDDS, and marketed drug Diprivan®) was administered intraperitoneally (IP). All results are presented as the average ± SEM. The asterisk and hash symbols (above the bars) indicate significant differences between samples * p < 0.05 (Propofol raw); # p < 0.05 (Diprivan®).
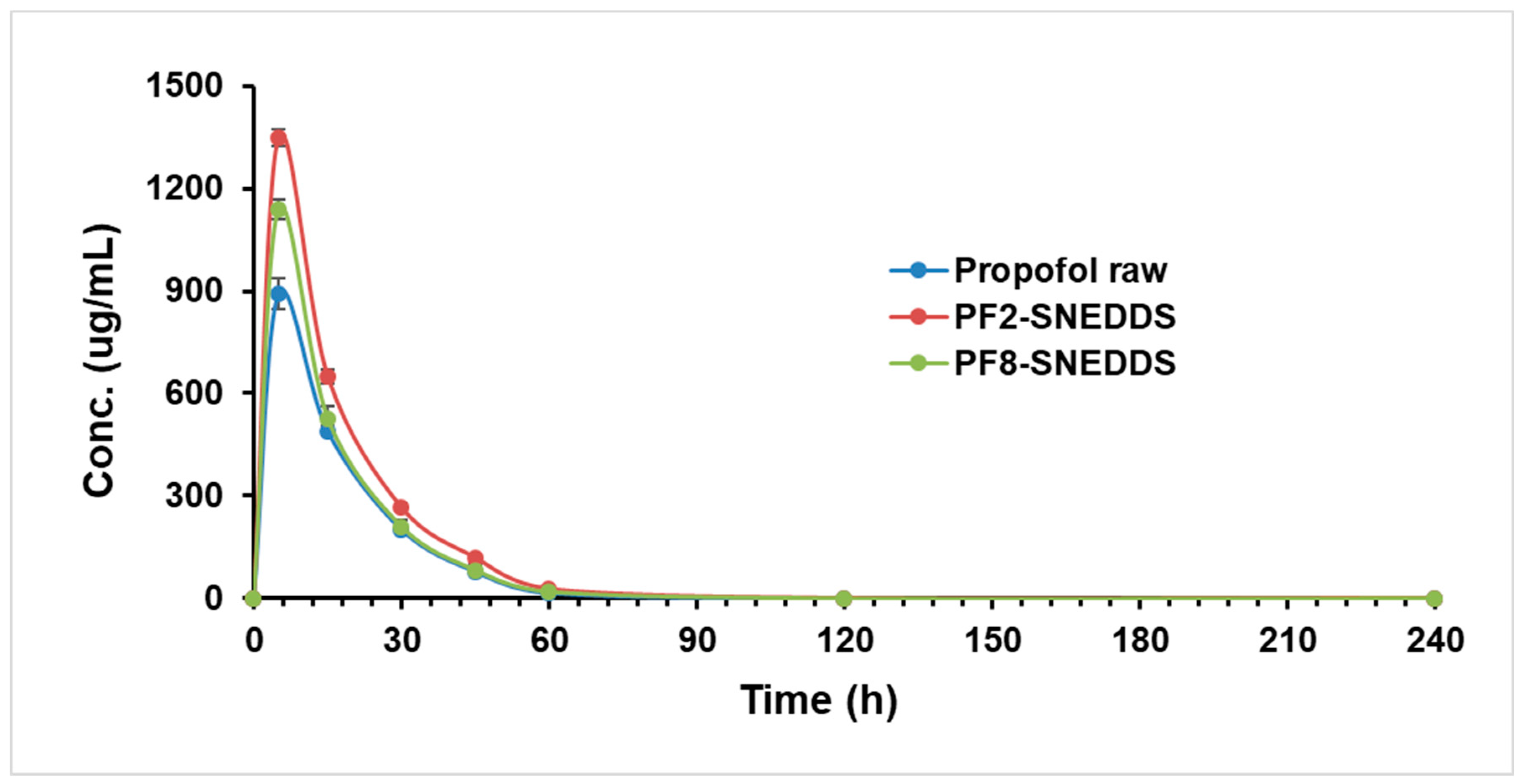
Figure 10.
Plasma concentration–time profiles of propofol SNEDDS formulations PF2, PF8, and raw propofol (STD) after a single IV administration to rats at a dose equivalent to 10 mg/kg propofol (mean ± SEM, n = 6).
Figure 10.
Plasma concentration–time profiles of propofol SNEDDS formulations PF2, PF8, and raw propofol (STD) after a single IV administration to rats at a dose equivalent to 10 mg/kg propofol (mean ± SEM, n = 6).
Table 1.
The composition of the formulations developed for propofol. The systems show the composition of excipients used in each formulation at different ratios (%w/w).
Table 1.
The composition of the formulations developed for propofol. The systems show the composition of excipients used in each formulation at different ratios (%w/w).
| F. No. | M810 | ZRO | CO | CMCM | M35-1 | KELP | KHS15 | T80 | Total% |
|---|
| F1 | 35 | | | 15 | | 50 | | | 100 |
| F2 | 35 | | | 15 | | | 50 | | 100 |
| F3 | | 35 | | 15 | | 50 | | | 100 |
| F4 | | 35 | | 15 | | | 50 | | 100 |
| F5 | | 35 | | 15 | | | | 50 | 100 |
| F6 | | | 35 | 15 | | 50 | | | 100 |
| F7 | | 35 | | | 15 | 50 | | | 100 |
| F8 | | 35 | | | 15 | | 50 | | 100 |
Table 2.
Performance of lipid-based formulations on the basis of homogeneity, appearance, and self-emulsification capability.
Table 2.
Performance of lipid-based formulations on the basis of homogeneity, appearance, and self-emulsification capability.
| Formulation | Compositions (%w/w) | Homogeneity | Appearance |
|---|
| F1 | M810/CMCM/KELP (35/15/50) | Yes | Transparent |
| F2 | M810/CMCM/KHS15 (35/15/50) | Yes | Transparent |
| F3 | ZRO/CMCM/KELP (35/15/50) | Yes | Turbid |
| F4 | ZRO/CMCM/KHS15 (35/15/50) | Yes | Bluish |
| F5 | ZRO/CMCM/T80 (35/15/50) | Yes | Hazy |
| F6 | CO/CMCM/KELP (35/15/50) | No | Turbid |
| F7 | ZRO/M35-1/KELP (35/15/50) | Yes | Bluish |
| F8 | ZRO/M35-1/KHS15 (35/15/50) | Yes | Transparent |
Table 3.
Mean particle size and PDI values of the different propofol-loaded liquid SNEDDS formulations. Data are expressed as mean ± SD, n = 3.
Table 3.
Mean particle size and PDI values of the different propofol-loaded liquid SNEDDS formulations. Data are expressed as mean ± SD, n = 3.
| Formulation | Particle Size (nm) | PDI | Zeta Potential (mV) |
|---|
| F1 | 28.08 ± 0.21 | 0.089 ± 0.027 | −4.10 ± 0.14 |
| F2 | 28.81 ± 0.28 | 0.112± 0.014 | −11.37 ± 0.70 |
| F3 | 426.2 ± 11.0 | 0.71 ± 0.14 | −29.2 ± 1.20 |
| F4 | 128.0 ± 1.6 | 0.184 ± 0.011 | −29.3 ± 0.60 |
| F5 | 181.26 ± 1.67 | 0.253 ± 0.014 | −28.4 ± 1.10 |
| F6 | 526 ± 140 | 0.602 ± 0.017 | −11.3 ± 0.50 |
| F7 | 130.90 ± 0.70 | 0.258 ± 0.010 | −25.5 ± 1.20 |
| F8 | 44.4 ± 0.20 | 0.176 ± 0.008 | −14.3 ± 0.70 |
Table 4.
The propofol dissolved in the optimized formulations at 50 mg per gram of formulation.
Table 4.
The propofol dissolved in the optimized formulations at 50 mg per gram of formulation.
| Formulation | Compositions (%w/w) | Solubility (mg/g) |
|---|
| PF2-SNEDDS | M810/CMCM/KHS15 [35/15/50] | 50 |
| PF8-SNEDDS | ZRO/M35-1/KHS15 [35/15/50] | 50 |
Table 5.
Effect of dynamic dispersion on the solubilization profiles of propofol (initially 25 mg of propofol was dissolved in water). Data are expressed as mean ± SD, n = 3.
Table 5.
Effect of dynamic dispersion on the solubilization profiles of propofol (initially 25 mg of propofol was dissolved in water). Data are expressed as mean ± SD, n = 3.
| Time (h) | Drug Concentration (mg) |
|---|
| | Propofol | PF2-SNEDDS | PF8-SNEDDS |
|---|
| 0 | 24.97 ± 0.12 | 25.02 ± 0.56 | 24.87 ± 0.99 |
| 0.5 | 10.50 ± 0.28 | 23.49 ± 0.78 | 24.07 ± 0.00 |
| 1 | 10.88 ± 0.00 | 23.54 ± 0.84 | 24.29 ± 0.27 |
| 2 | 11.76 ± 0.75 | 24.61 ± 0.13 | 24.69 ± 2.28 |
| 4 | 11.20 ± 0.05 | 24.75 ± 1.32 | 24.50 ± 0.02 |
| 8 | 11.24 ± 0.49 | 24.13 ± 1.43 | 24.69 ± 0.06 |
| 16 | 11.19 ± 0.66 | 24.24 ± 0.13 | 23.74 ± 1.01 |
| 24 | 11.61 ± 0.35 | 22.78 ± 2.31 | 22.30 ± 0.21 |
Table 6.
Mean particle size, PDI, and zeta potential values of the propofol (PF)-loaded SNEDDS and PF-free SNEDDS formulations at 0 time.
Table 6.
Mean particle size, PDI, and zeta potential values of the propofol (PF)-loaded SNEDDS and PF-free SNEDDS formulations at 0 time.
| Formulation | Particle Size | Zeta Potential | PDI |
|---|
| Without PF | With PF | Without PF | With PF | Without PF | With PF |
|---|
| F2 | 28.81 ± 0.28 | 34.00 ± 0.27 | −11.37 ± 0.70 | −16 ± 1 | 0.112 ± 0.014 | 0.229 ± 0.005 |
| F8 | 44.40 ± 0.20 | 45.70 ± 0.20 | −14.3 ± 0.6 | −15.5 ± 0.9 | 0.176 ± 0.008 | 0.198 ± 0.013 |
Table 7.
Mean particle size, PDI, and zeta potential values of the PF-loaded SNEDDS formulations after 3 months of storage at 4 °C and 37 °C, respectively.
Table 7.
Mean particle size, PDI, and zeta potential values of the PF-loaded SNEDDS formulations after 3 months of storage at 4 °C and 37 °C, respectively.
| Formulations | 4 °C | 37 °C |
|---|
| F2 | F8 | F2 | F8 |
|---|
| Particle size | 36.74 ± 0.1721 | 114.3 ± −2.68 | 40.9 ± 0.40 | 98.82 ± −0.31 |
| PDI | 0.184 ± −0.004 | 0.369 ± −0.014 | 0.260 ± −0.007 | 0.224 ± −0.004 |
| Zeta potential | −10.4 ± −2.03 | −18.6 ± −0.45 | −12.2 ± −2.8 | −10.2 ± −3.6 |
Table 8.
Mean particle size, PDI, and zeta potential values of the PF-loaded SNEDDS formulations after 6 months of storage at 4 °C and 37 °C.
Table 8.
Mean particle size, PDI, and zeta potential values of the PF-loaded SNEDDS formulations after 6 months of storage at 4 °C and 37 °C.
| Formulations | 4 °C | 37 °C |
|---|
| F2 | F8 | F2 | F8 |
|---|
| Particle size | 35.19 ± 0.101 | 85.35 ± −4.52 | 32.36 ± 0.32 | 97.13 ± 1.99 |
| PDI | 0.130 ± −0.009 | 0.206 ± −0.027 | 0.052 ± 0.009 | 0.261 ± 0.011 |
| Zeta potential | −13.1 ± −3.15 | −16.86 ± −1.44 | −11.26 ± 0.45 | −12.10 ± 1.51 |
Table 9.
Comparison of pharmacokinetic parameters of propofol formulation F2-SNEDDS (PF2, M810/CMCM/KHS15; 35/15/50), PF8-SNEDDS (ZRO/M35-1/KHS15; 35/15/50) and raw propofol.
Table 9.
Comparison of pharmacokinetic parameters of propofol formulation F2-SNEDDS (PF2, M810/CMCM/KHS15; 35/15/50), PF8-SNEDDS (ZRO/M35-1/KHS15; 35/15/50) and raw propofol.
| | | Propofol Raw (STD) | PF2-SNEDDS | PF8-SNEDDS |
|---|
| Parameter | Unit | Average | SD | Average | SD | Average | SD |
|---|
| Lambda_z | 1/min | 0.07 | 0.00 | 0.07 | 0.01 | 0.07 | 0.00 |
| t1/2 | min | 9.74 | 0.49 | 10.12 | 0.88 | 9.71 | 0.12 |
| Tmax | min | 5.00 | 0.00 | 5.00 | 0.00 | 5.00 | 0.00 |
| Cmax | μg/mL | 891.44 | 26.05 | 1348.07 | 27.31 | 1138.66 | 44.97 |
| AUC 0-t | μg/mL*min | 20,130.69 | 885.63 | 29,038.38 | 879.13 | 23,866.09 | 800.63 |
| AUC 0-inf_obs | μg/mL*min | 20,354.33 | 946.12 | 29,445.19 | 1057.02 | 24,140.19 | 807.34 |
| MRT 0-inf_obs | min | 14.36 | 0.48 | 14.05 | 0.77 | 13.32 | 0.07 |
| Vz_obs | (mg/kg)/(μg/mL) | 0.01 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 |
| Cl_obs | (mg/kg)/(μg/mL)/min | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Vss_obs | (mg/kg)/(μg/mL) | 0.01 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 |


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}