

Radiolabelling of Polyclonally Expanded Human Regulatory T Cells (Treg) with 89Zr-oxine for Medium-Term In Vivo Cell Tracking

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
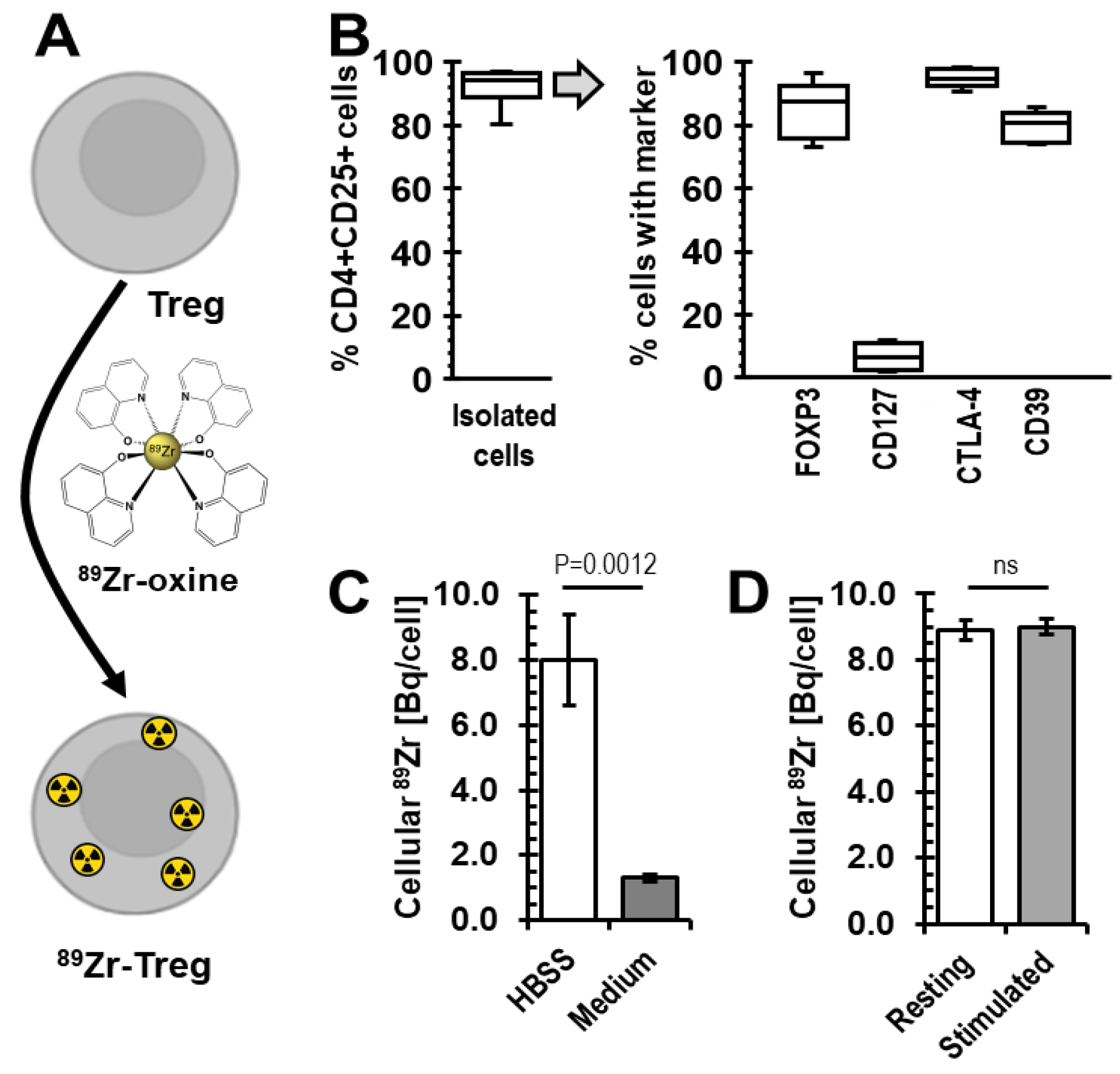
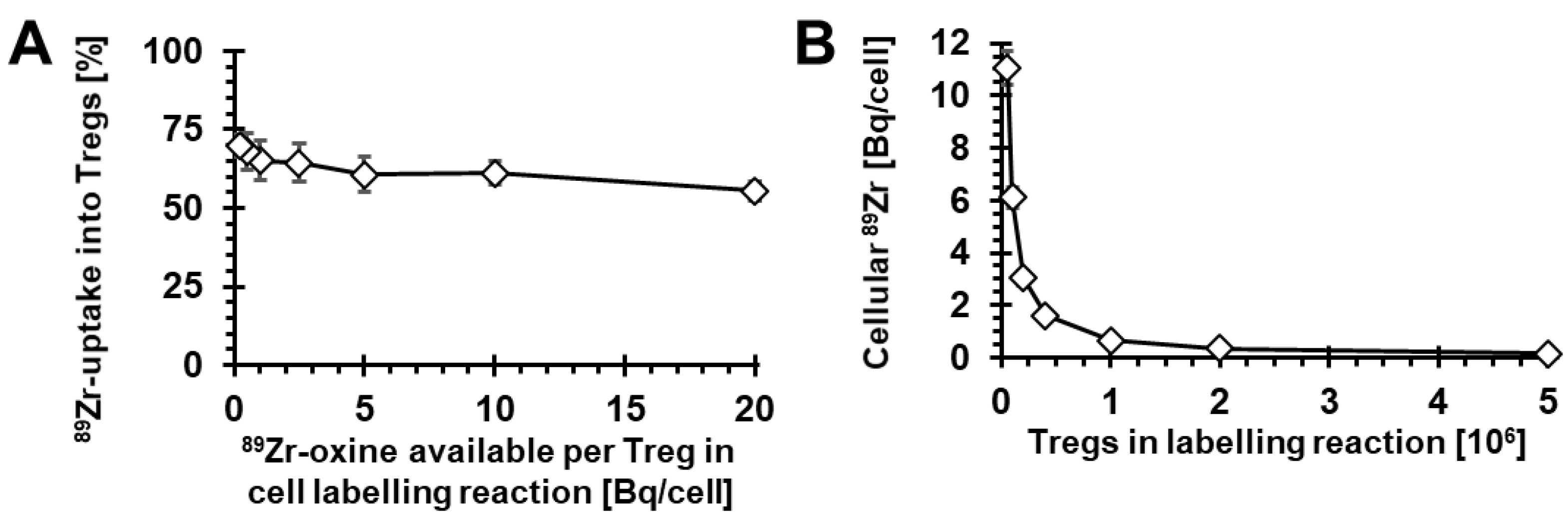
2.1. Radiolabelling of Tregs with 89Zr-oxine
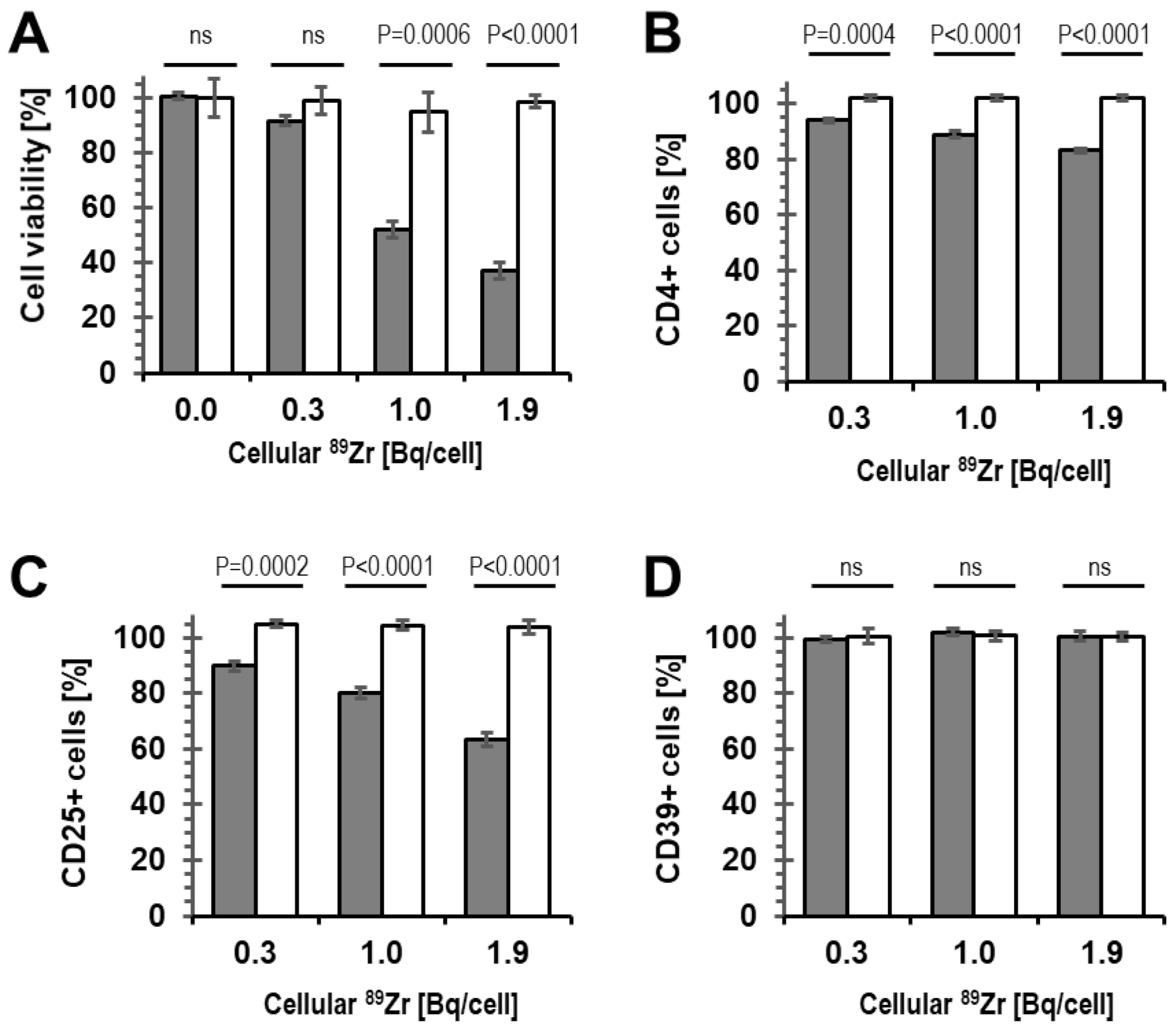
2.2. Viability of 89Zr-labelled Tregs
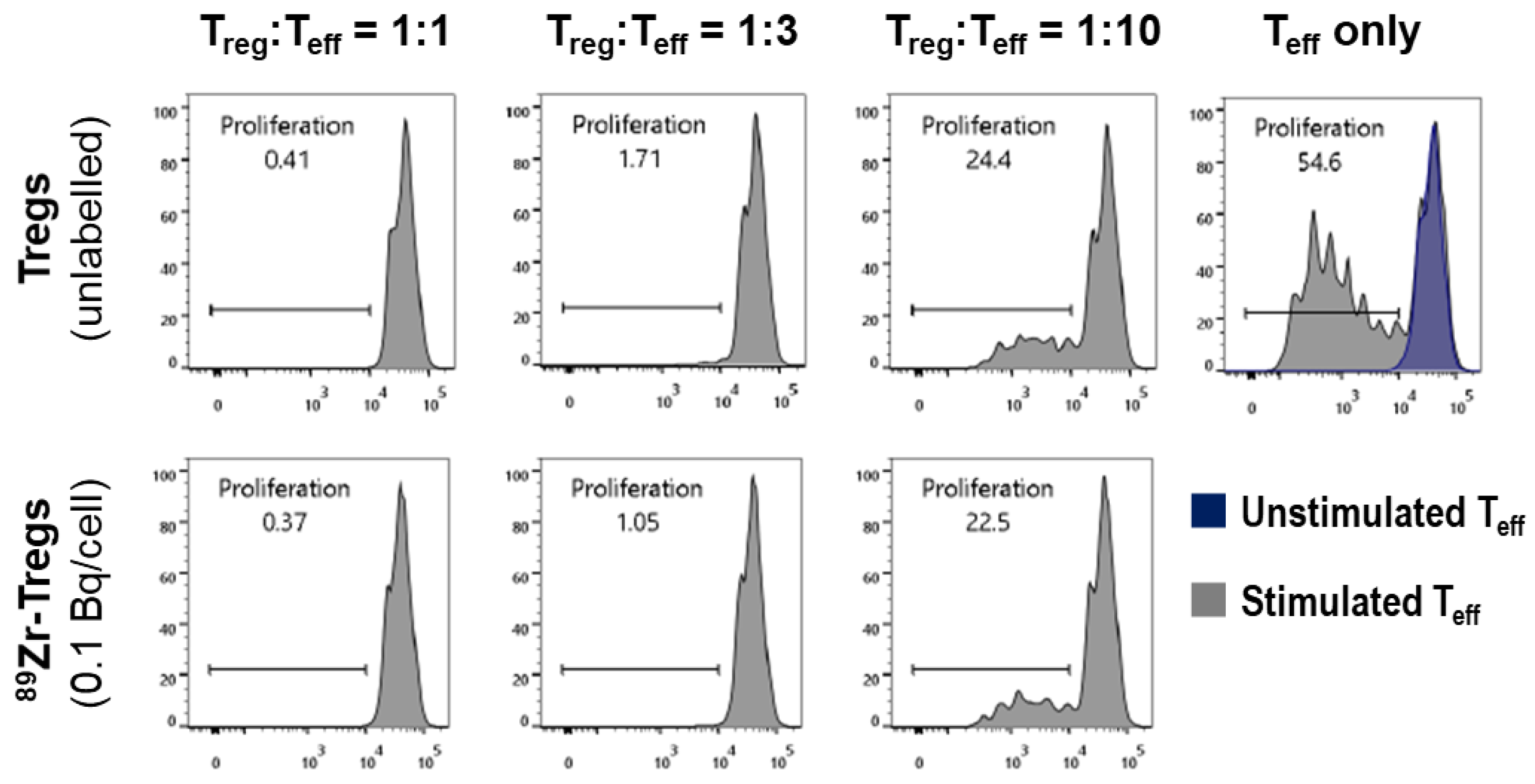
2.3. Phenotype and Suppressive Capacity of 89Zr-labelled Tregs
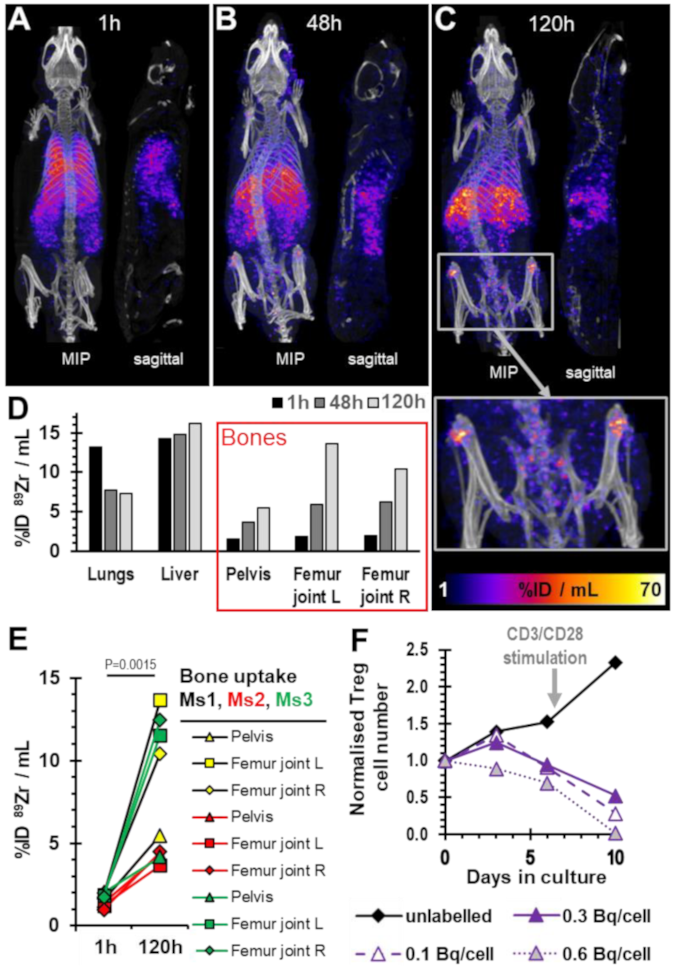
2.4. Survival of 89Zr-labelled Tregs in Immunodeficient Mice
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Katoh, H.; Zheng, P.; Liu, Y. FOXP3: Genetic and epigenetic implications for autoimmunity. J. Autoimmun. 2013, 41, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Miyara, M.; Gorochov, G.; Ehrenstein, M.; Musset, L.; Sakaguchi, S.; Amoura, Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun. Rev. 2011, 10, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.; Zhou, L.; Ma, Y.; Zhou, L.; Liang, T.; Shi, L.; Long, J.; Yuan, D. Regulatory T Cell Plasticity and Stability and Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2020, 58, 52–70. [Google Scholar] [CrossRef]
- Pesenacker, A.M.; Chen, V.; Gillies, J.; Speake, C.; Marwaha, A.K.; Sun, A.; Chow, S.; Tan, R.; Elliott, T.; Dutz, J.P.; et al. Treg gene signatures predict and measure type 1 diabetes trajectory. JCI Insight 2019, 4, e123879. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Trotta, E.; Xu, D. The therapeutic potential of regulatory T cells for the treatment of autoimmune disease. Expert Opin. Ther. Targets 2015, 19, 1091–1103. [Google Scholar] [CrossRef]
- Krystufkova, E.; Sekerkova, A.; Striz, I.; Brabcova, I.; Girmanova, E.; Viklicky, O. Regulatory T cells in kidney transplant recipients: The effect of induction immunosuppression therapy. Nephrol. Dial. Transpl. 2012, 27, 2576–2582. [Google Scholar] [CrossRef]
- Louis, S.; Braudeau, C.; Giral, M.; Dupont, A.; Moizant, F.; Robillard, N.; Moreau, A.; Soulillou, J.P.; Brouard, S. Contrasting CD25hiCD4+T cells/FOXP3 patterns in chronic rejection and operational drug-free tolerance. Transplantation 2006, 81, 398–407. [Google Scholar] [CrossRef]
- Krustrup, D.; Madsen, C.B.; Iversen, M.; Engelholm, L.; Ryder, L.P.; Andersen, C.B. The number of regulatory T cells in transbronchial lung allograft biopsies is related to FoxP3 mRNA levels in bronchoalveolar lavage fluid and to the degree of acute cellular rejection. Transpl. Immunol. 2013, 29, 71–75. [Google Scholar] [CrossRef]
- Sagoo, P.; Ali, N.; Garg, G.; Nestle, F.O.; Lechler, R.I.; Lombardi, G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci. Transl. Med. 2011, 3, 83ra42. [Google Scholar] [CrossRef]
- Putnam, A.L.; Safinia, N.; Medvec, A.; Laszkowska, M.; Wray, M.; Mintz, M.A.; Trotta, E.; Szot, G.L.; Liu, W.; Lares, A.; et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am. J. Transpl. 2013, 13, 3010–3020. [Google Scholar] [CrossRef]
- Trzonkowski, P.; Bieniaszewska, M.; Juscinska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Mysliwska, J.; Hellmann, A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef]
- Brunstein, C.G.; Miller, J.S.; Cao, Q.; McKenna, D.H.; Hippen, K.L.; Curtsinger, J.; Defor, T.; Levine, B.L.; June, C.H.; Rubinstein, P.; et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: Safety profile and detection kinetics. Blood 2011, 117, 1061–1070. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Mysliwiec, M.; Dobyszuk, A.; Grabowska, M.; Techmanska, I.; Juscinska, J.; Wujtewicz, M.A.; Witkowski, P.; Mlynarski, W.; Balcerska, A.; et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care 2012, 35, 1817–1820. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef]
- Safinia, N.; Vaikunthanathan, T.; Fraser, H.; Thirkell, S.; Lowe, K.; Blackmore, L.; Whitehouse, G.; Martinez-Llordella, M.; Jassem, W.; Sanchez-Fueyo, A.; et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget 2016, 7, 7563–7577. [Google Scholar] [CrossRef]
- Afzali, B.; Edozie, F.C.; Fazekasova, H.; Scotta, C.; Mitchell, P.J.; Canavan, J.B.; Kordasti, S.Y.; Chana, P.S.; Ellis, R.; Lord, G.M.; et al. Comparison of regulatory T cells in hemodialysis patients and healthy controls: Implications for cell therapy in transplantation. Clin. J. Am. Soc. Nephrol. 2013, 8, 1396–1405. [Google Scholar] [CrossRef]
- Sawitzki, B.; Harden, P.N.; Reinke, P.; Moreau, A.; Hutchinson, J.A.; Game, D.S.; Tang, Q.; Guinan, E.C.; Battaglia, M.; Burlingham, W.J.; et al. Regulatory cell therapy in kidney transplantation (The ONE Study): A harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet 2020, 395, 1627–1639. [Google Scholar] [CrossRef]
- Ashmore-Harris, C.; Iafrate, M.; Saleem, A.; Fruhwirth, G.O. Non-invasive Reporter Gene Imaging of Cell Therapies, including T Cells and Stem Cells. Mol. Ther. 2020, 28, 1392–1416. [Google Scholar] [CrossRef]
- Sharif-Paghaleh, E.; Leech, J.; Sunassee, K.; Ali, N.; Sagoo, P.; Lechler, R.I.; Smyth, L.A.; Lombardi, G.; Mullen, G.E. Monitoring the efficacy of dendritic cell vaccination by early detection of (99m) Tc-HMPAO-labelled CD4(+) T cells. Eur. J. Immunol. 2014, 44, 2188–2191. [Google Scholar] [CrossRef]
- Iafrate, M.; Fruhwirth, G.O. How non-invasive in vivo cell tracking supports the development and translation of cancer immunotherapies. Front. Physiol. 2020, 11, 154. [Google Scholar] [CrossRef]
- Roca, M.; de Vries, E.F.; Jamar, F.; Israel, O.; Signore, A. Guidelines for the labelling of leucocytes with (111)In-oxine. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Oo, Y.H.; Ackrill, S.; Cole, R.; Jenkins, L.; Anderson, P.; Jeffery, H.C.; Jones, N.; Jeffery, L.E.; Lutz, P.; Wawman, R.E.; et al. Liver homing of clinical grade Tregs after therapeutic infusion in patients with autoimmune hepatitis. JHEP Rep. 2019, 1, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Charoenphun, P.; Meszaros, L.K.; Chuamsaamarkkee, K.; Sharif-Paghaleh, E.; Ballinger, J.R.; Ferris, T.J.; Went, M.J.; Mullen, G.E.; Blower, P.J. [(89)Zr]oxinate4 for long-term in vivo cell tracking by positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.B.; Roberts, M.; Bluchel, C.G.; Odell, R.A. Zirconium: Biomedical and nephrological applications. Asaio J. 2010, 56, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Weist, M.R.; Starr, R.; Aguilar, B.; Chea, J.; Miles, J.; Poku, E.; Gerdts, E.; Yang, X.; Priceman, S.; Forman, S.; et al. Positron emission tomography of adoptively transferred chimeric antigen receptor T cells with Zirconium-89 oxine. J. Nucl. Med. 2018, 59, 1531–1537. [Google Scholar] [CrossRef]
- Man, F.; Lim, L.; Volpe, A.; Gabizon, A.; Shmeeda, H.; Draper, B.; Parente-Pereira, A.C.; Maher, J.; Blower, P.J.; Fruhwirth, G.O.; et al. In vivo PET Tracking of (89)Zr-Labeled Vgamma9Vdelta2 T Cells to Mouse Xenograft Breast Tumors Activated with Liposomal Alendronate. Mol. Ther. 2019, 27, 219–229. [Google Scholar] [CrossRef]
- Sato, N.; Wu, H.; Asiedu, K.O.; Szajek, L.P.; Griffiths, G.L.; Choyke, P.L. (89)Zr-Oxine Complex PET Cell Imaging in Monitoring Cell-based Therapies. Radiology 2015, 275, 490–500. [Google Scholar] [CrossRef]
- Jacob, J.; Nadkarni, S.; Volpe, A.; Peng, Q.; Tung, S.L.; Hannen, R.F.; Mohseni, Y.R.; Scotta, C.; Marelli-Berg, F.M.; Lechler, R.I.; et al. Spatiotemporal in vivo tracking of polyclonal human regulatory T cells (Tregs) reveals a role for innate immune cells in Treg transplant recruitment. Mol. Ther. Methods Clin. Dev. 2021, 20, 324–336. [Google Scholar] [CrossRef]
- Fraser, H.; Safinia, N.; Grageda, N.; Thirkell, S.; Lowe, K.; Fry, L.J.; Scotta, C.; Hope, A.; Fisher, C.; Hilton, R.; et al. A Rapamycin-Based GMP-Compatible Process for the Isolation and Expansion of Regulatory T Cells for Clinical Trials. Mol. Ther. Methods Clin. Dev. 2018, 8, 198–209. [Google Scholar] [CrossRef]
- Boardman, D.A.; Philippeos, C.; Fruhwirth, G.O.; Ibrahim, M.A.; Hannen, R.F.; Cooper, D.; Marelli-Berg, F.M.; Watt, F.M.; Lechler, R.I.; Maher, J.; et al. Expression of a Chimeric Antigen Receptor Specific for Donor HLA Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am. J. Transpl. 2017, 17, 931–943. [Google Scholar] [CrossRef]
- Abou, D.S.; Ku, T.; Smith-Jones, P.M. In vivo biodistribution and accumulation of 89Zr in mice. Nucl. Med. Biol. 2011, 38, 675–681. [Google Scholar] [CrossRef]
- Sato, N.; Stringaris, K.; Davidson-Moncada, J.K.; Reger, R.; Adler, S.S.; Dunbar, C.; Choyke, P.L.; Childs, R.W. In vivo Tracking of Adoptively Transferred Natural Killer Cells in Rhesus Macaques Using (89)Zirconium-Oxine Cell Labeling and PET Imaging. Clin. Cancer Res. 2020, 26, 2573–2581. [Google Scholar] [CrossRef]
- Liu, S.; Sun, X.; Luo, J.; Zhu, H.; Yang, X.; Guo, Q.; Song, Y.; Sun, X. Effects of radiation on T regulatory cells in normal states and cancer: Mechanisms and clinical implications. Am. J. Cancer Res. 2015, 5, 3276–3285. [Google Scholar]
- Severin, G.W.; Engle, J.W.; Barnhart, T.E.; Nickles, R.J. 89Zr radiochemistry for positron emission tomography. Med. Chem. 2011, 7, 389–394. [Google Scholar] [CrossRef]
- Diocou, S.; Volpe, A.; Jauregui-Osoro, M.; Boudjemeline, M.; Chuamsaamarkkee, K.; Man, F.; Blower, P.J.; Ng, T.; Mullen, G.E.D.; Fruhwirth, G.O. [(18)F]tetrafluoroborate-PET/CT enables sensitive tumor and metastasis in vivo imaging in a sodium iodide symporter-expressing tumor model. Sci. Rep. 2017, 7, 946. [Google Scholar] [CrossRef]
- Volpe, A.; Lang, C.; Lim, L.; Man, F.; Kurtys, E.; Ashmore-Harris, C.; Johnson, P.; Skourti, E.; de Rosales, R.T.M.; Fruhwirth, G.O. Spatiotemporal PET Imaging Reveals Differences in CAR-T Tumor Retention in Triple-Negative Breast Cancer Models. Mol. Ther. 2020, 28, 2271–2285. [Google Scholar] [CrossRef]
- Volpe, A.; Man, F.; Lim, L.; Khoshnevisan, A.; Blower, J.; Blower, P.J.; Fruhwirth, G.O. Radionuclide-fluorescence Reporter Gene Imaging to Track Tumor Progression in Rodent Tumor Models. J. Vis. Exp. 2018, 133, e57088. [Google Scholar]
- Minn, I.; Huss, D.J.; Ahn, H.H.; Chinn, T.M.; Park, A.; Jones, J.; Brummet, M.; Rowe, S.P.; Sysa-Shah, P.; Du, Y.; et al. Imaging CAR T cell therapy with PSMA-targeted positron emission tomography. Sci. Adv. 2019, 5, eaaw5096. [Google Scholar] [CrossRef]
- Gawne, P.J.; Pinto, S.M.A.; Nielsen, K.M.; Keeling, G.P.; Pereira, M.M.; de Rosales, R.T. Microwave-assisted synthesis of [(52)Mn]Mn-porphyrins: Applications in cell and liposome radiolabelling. Nucl. Med. Biol. 2022, 114–115, 6–17. [Google Scholar] [CrossRef]
- Luca, A. Decay data evaluation project: Evaluation of 52 Mn and 52m Mn nuclear decay data. In Proceedings of the ND 2016: International Conference on Nuclear Data for Science and Technology, Bruges, Belgium, 11–16 September 2016; Volume 146, p. 08003. [Google Scholar]
- Fruhwirth, G.O.; Diocou, S.; Blower, P.J.; Ng, T.; Mullen, G.E. A whole-body dual-modality radionuclide optical strategy for preclinical imaging of metastasis and heterogeneous treatment response in different microenvironments. J. Nucl. Med. 2014, 55, 686–694. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacob, J.; Volpe, A.; Peng, Q.; Lechler, R.I.; Smyth, L.A.; Lombardi, G.; Fruhwirth, G.O. Radiolabelling of Polyclonally Expanded Human Regulatory T Cells (Treg) with 89Zr-oxine for Medium-Term In Vivo Cell Tracking. Molecules 2023, 28, 1482. https://doi.org/10.3390/molecules28031482
Jacob J, Volpe A, Peng Q, Lechler RI, Smyth LA, Lombardi G, Fruhwirth GO. Radiolabelling of Polyclonally Expanded Human Regulatory T Cells (Treg) with 89Zr-oxine for Medium-Term In Vivo Cell Tracking. Molecules. 2023; 28(3):1482. https://doi.org/10.3390/molecules28031482
Chicago/Turabian StyleJacob, Jacinta, Alessia Volpe, Qi Peng, Robert I. Lechler, Lesley A. Smyth, Giovanna Lombardi, and Gilbert O. Fruhwirth. 2023. "Radiolabelling of Polyclonally Expanded Human Regulatory T Cells (Treg) with 89Zr-oxine for Medium-Term In Vivo Cell Tracking" Molecules 28, no. 3: 1482. https://doi.org/10.3390/molecules28031482
APA StyleJacob, J., Volpe, A., Peng, Q., Lechler, R. I., Smyth, L. A., Lombardi, G., & Fruhwirth, G. O. (2023). Radiolabelling of Polyclonally Expanded Human Regulatory T Cells (Treg) with 89Zr-oxine for Medium-Term In Vivo Cell Tracking. Molecules, 28(3), 1482. https://doi.org/10.3390/molecules28031482