
Biotransformation of Penindolone, an Influenza A Virus Inhibitor

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
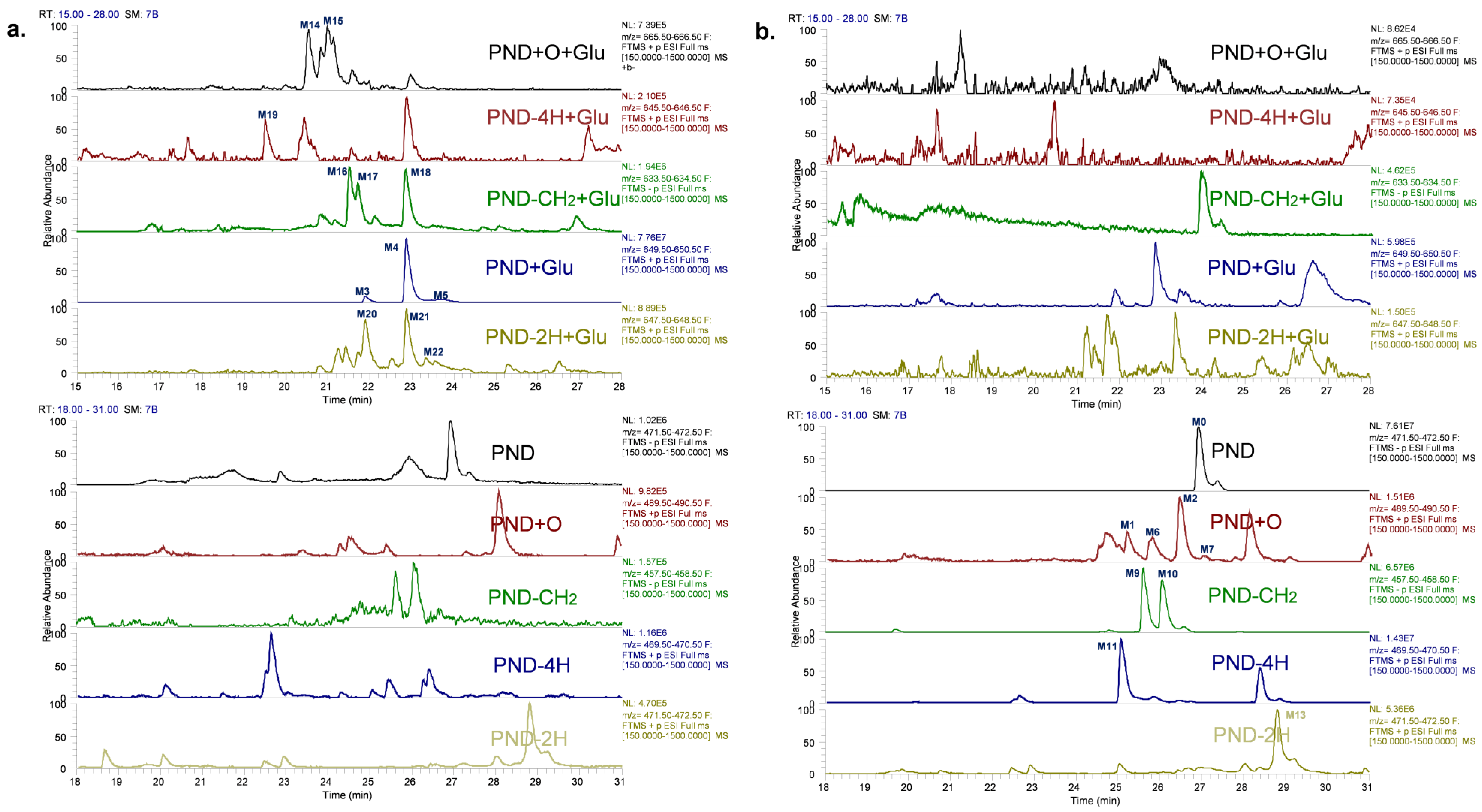
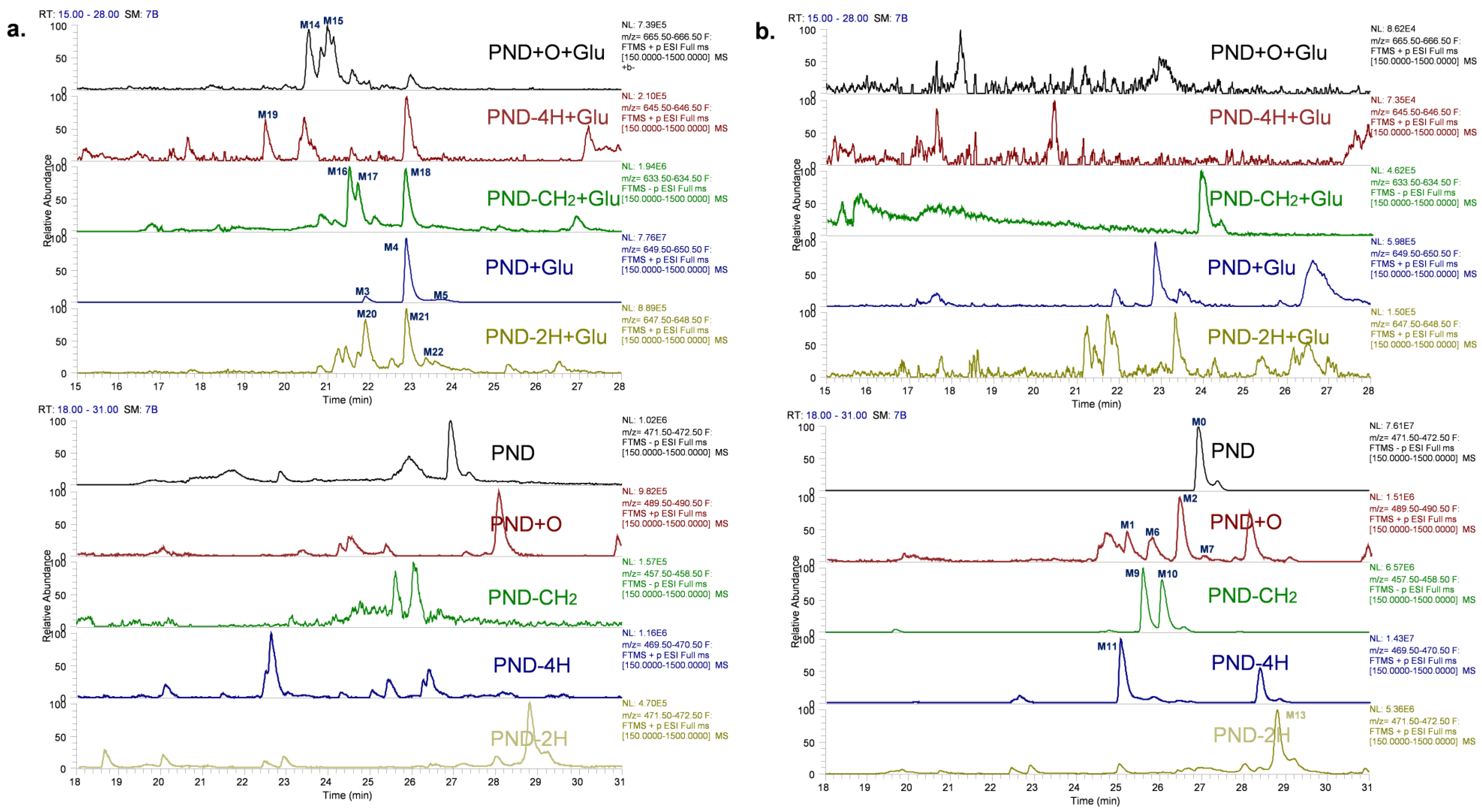
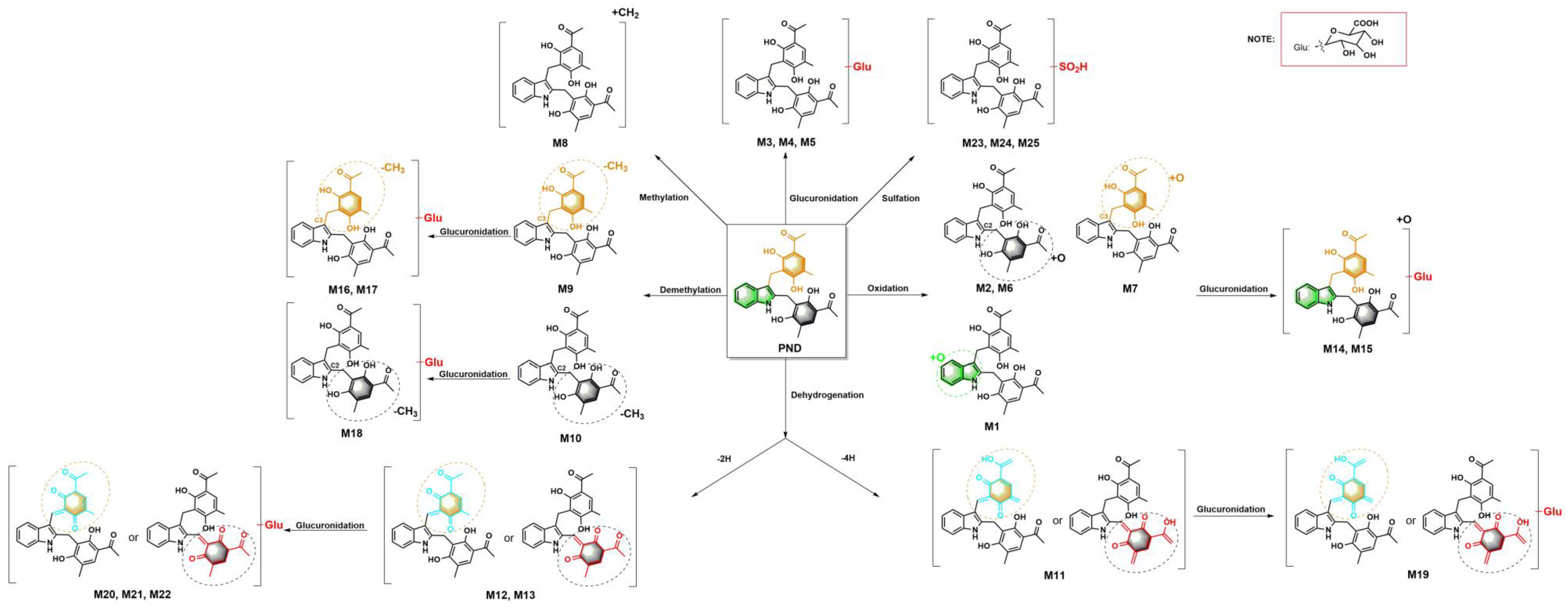
2.1. Characterizing and Profiling the Metabolites of PND in Rats
2.1.1. PND (M0) and Glucuronidation Metabolites (M3, M4 and M5)
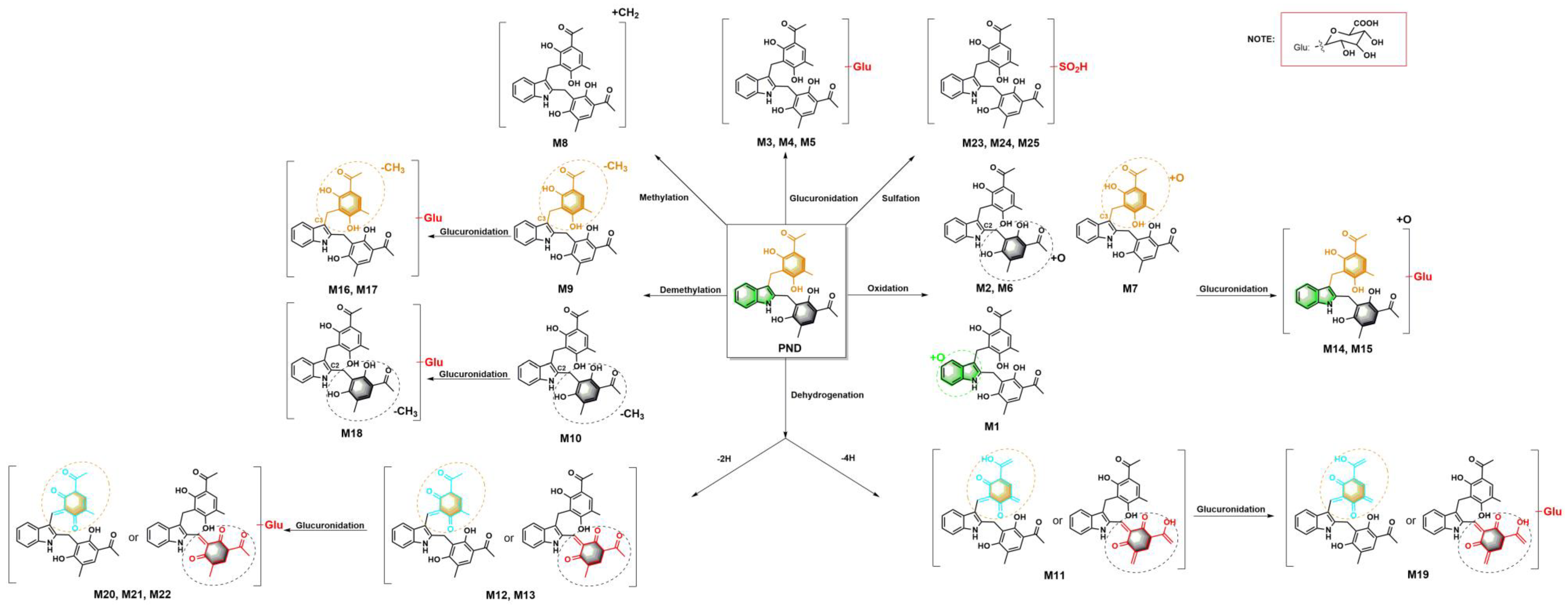
2.1.2. Oxidation Metabolites (M1, M2, M6 and M7) and Oxidation–Glucuronidation Metabolites (M14 and M15)
2.1.3. Methylation Metabolite (M8)
2.1.4. Demethylation Metabolites (M9 and M10) and Demethylation–Glucuronidation Metabolites (M16, M17 and M18)
2.1.5. Dehydrogenation Metabolite (−4H) (M11) and Dehydrogenation–Glucuronidation Metabolite (M19)
2.1.6. Dehydrogenation Metabolites (−2H) (M12 and M13) and Dehydrogenation–Glucuronidation Metabolites (M20, M21 and M22)
2.1.7. Sulfation Metabolites (M23, M24 and M25)
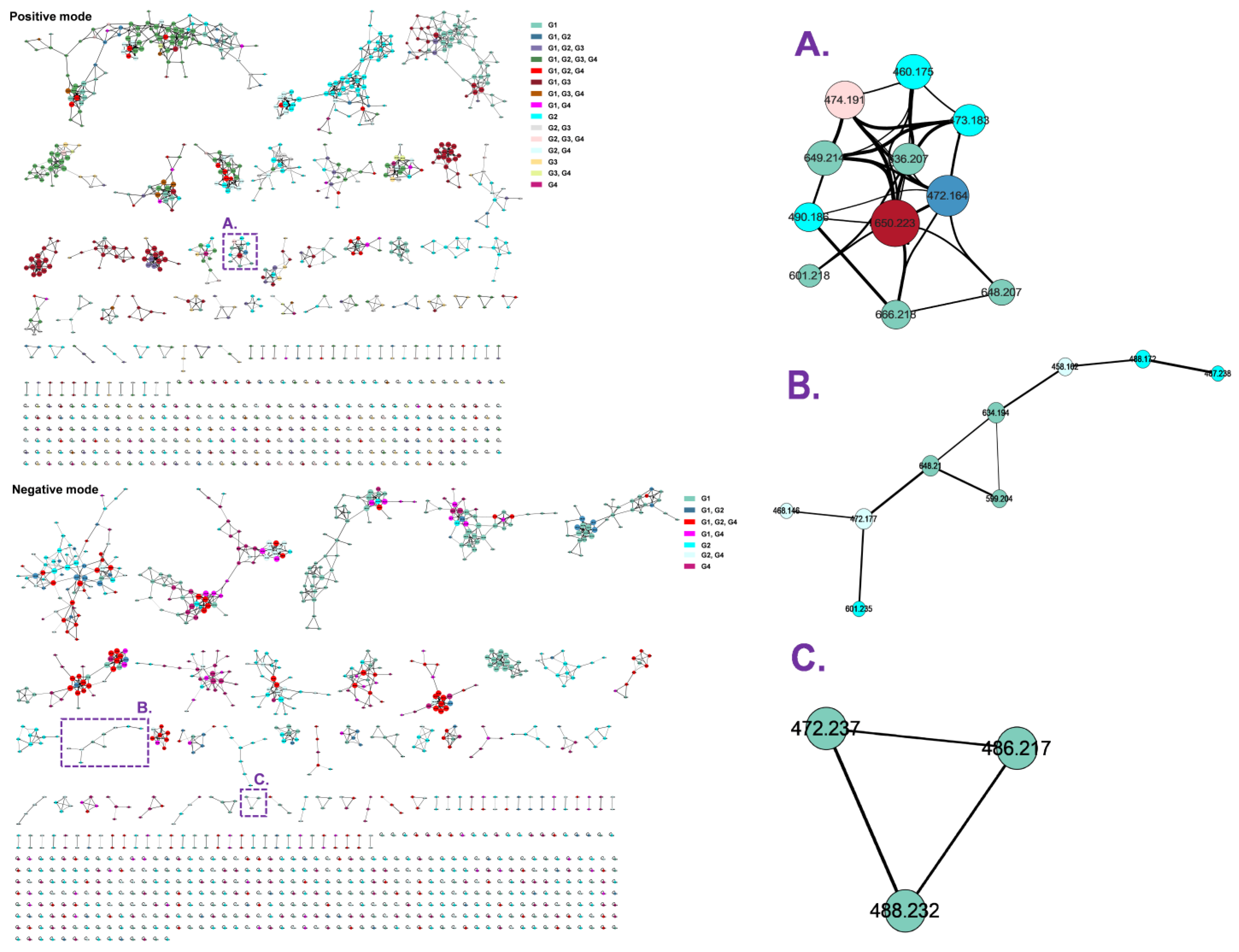
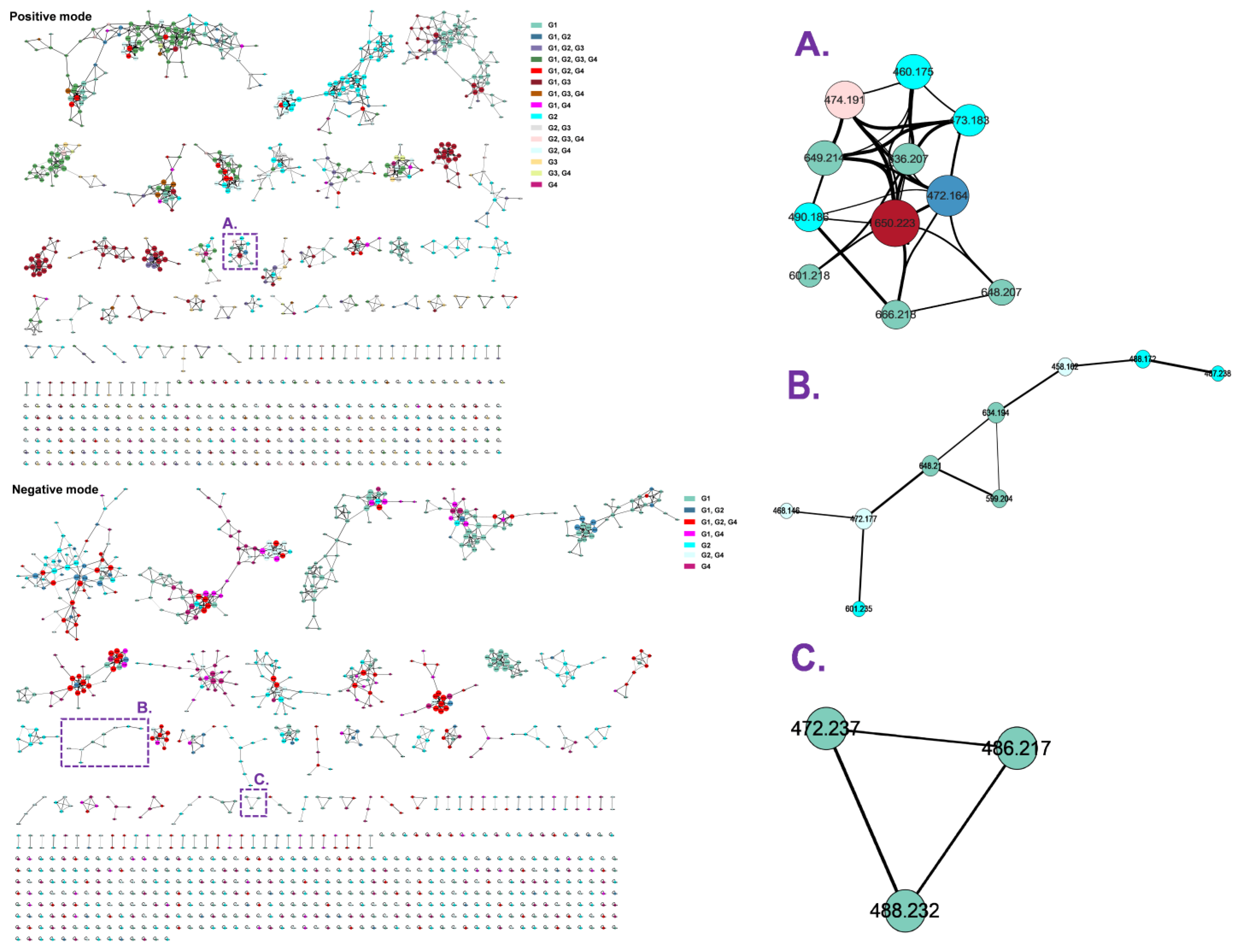
2.2. Molecular Networking
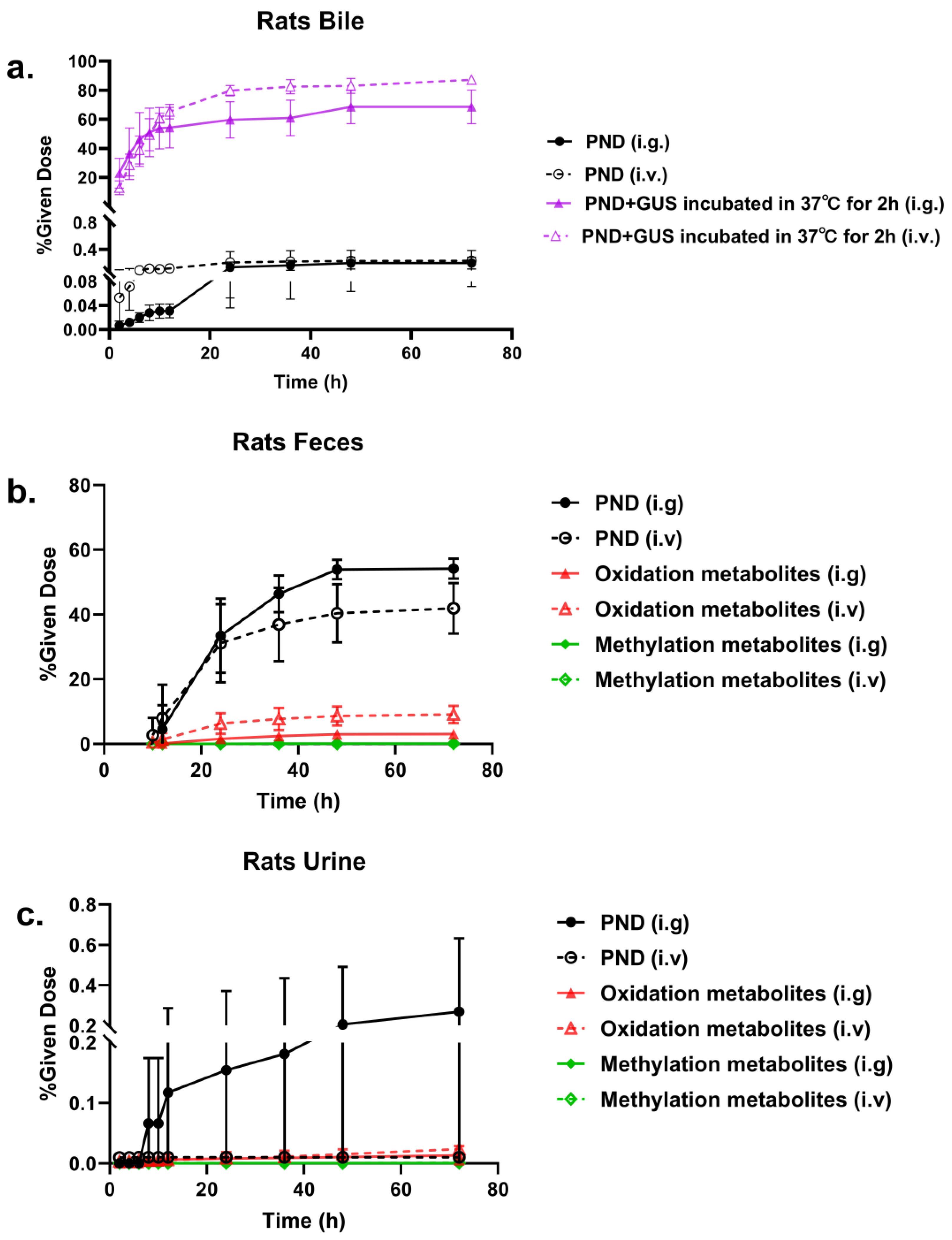
2.3. Metabolites Quantification
2.4. In Vitro Metabolism Studies
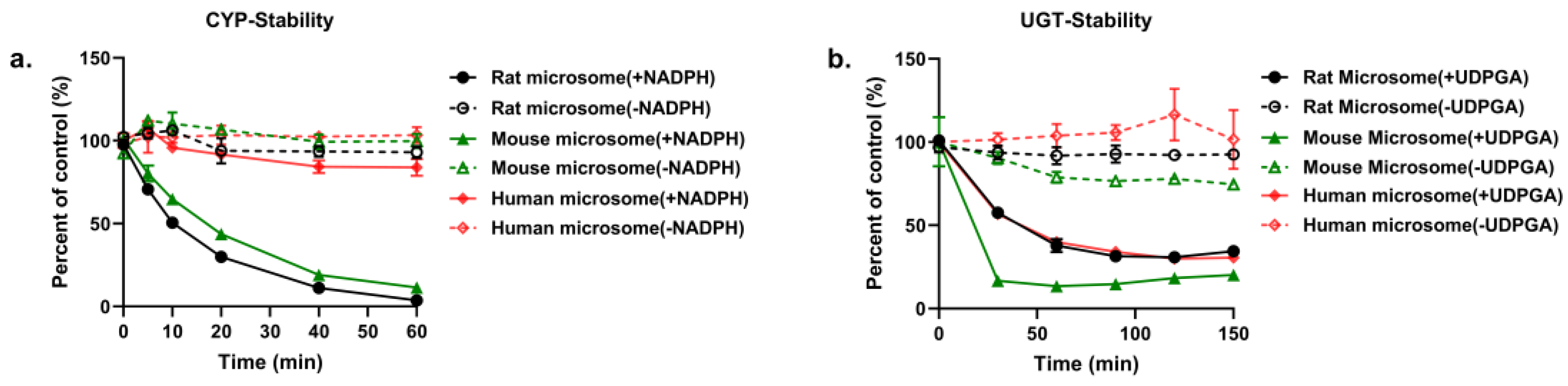
2.4.1. In Vitro Metabolism of PND by Rat, Mouse and Human LMs
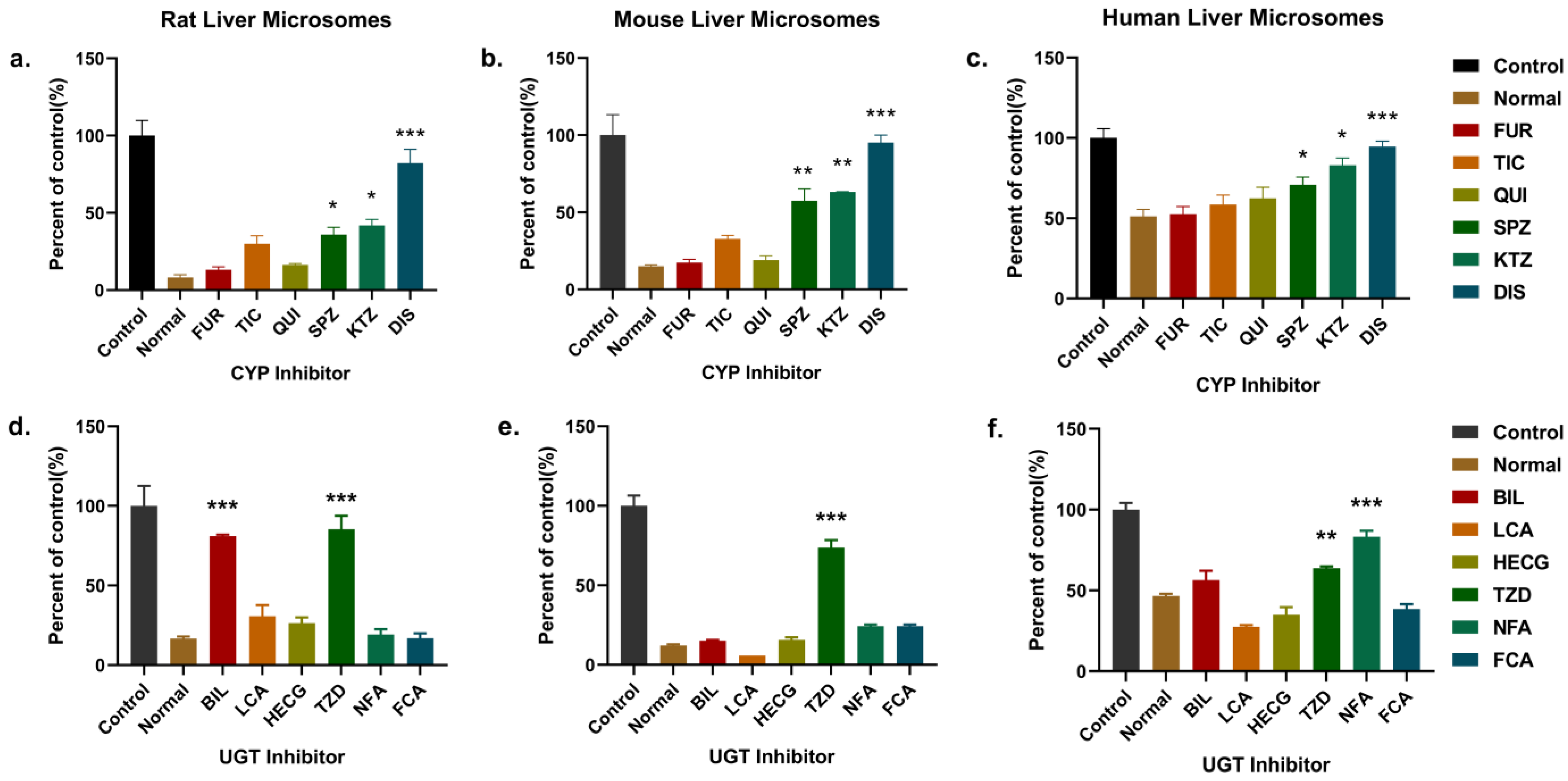
2.4.2. Identification of Enzymes Responsible for the Metabolism of PND in LMs
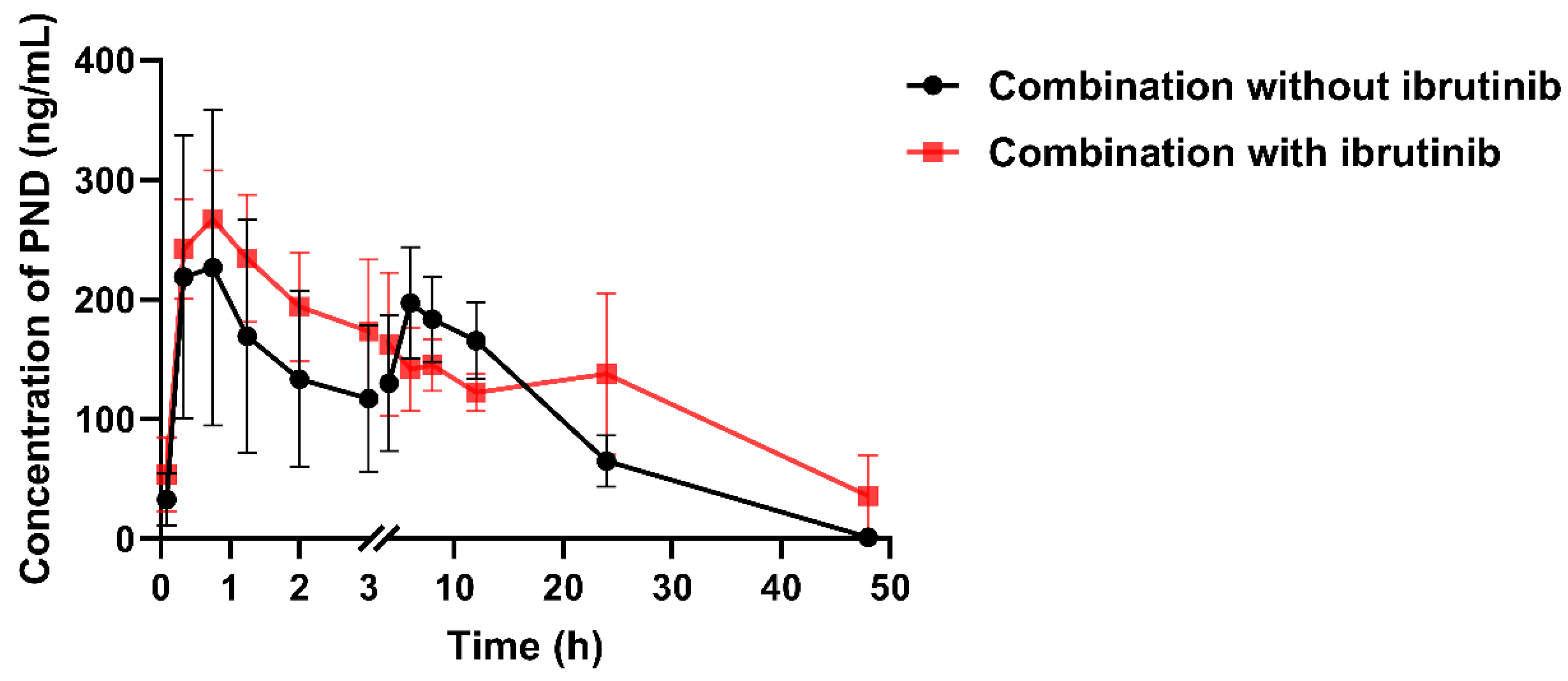
2.5. The Effect of Ibrutinib on PND Pharmacokinetics
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Animals
3.3. Metabolite Profiling
3.3.1. Sample Collection
3.3.2. Identification of the Metabolites of PND by UHPLC–HRMS
3.3.3. Molecular Networking
3.3.4. Determination of PND and Its Metabolites by UPLC–MS/MS
3.4. Liver Microsomal Metabolism
3.4.1. Incubation of PND with LMs
3.4.2. Kinetics of PND Metabolism by Rat, Mouse and Human LMs
3.4.3. Enzymes Inhibition Experiments in LMs
3.5. Pharmacokinetic Profile of PND after Blocking the UGTs Metabolism
3.6. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. WHO Fact Sheet on Influenza (Seasonal) 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 12 January 2023).
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef]
- Han, J.; Perez, J.; Schafer, A.; Cheng, H.; Peet, N.; Rong, L.; Manicassamy, B. Influenza Virus: Small Molecule Therapeutics and Mechanisms of Antiviral Resistance. Curr. Med. Chem. 2018, 25, 5115–5127. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yu, G.; Yu, Y.; Yang, S.; Duan, Z.; Wang, W.; Liu, Y.; Yu, R.; Li, J.; Zhu, T.; et al. Chemoreactive-Inspired Discovery of Influenza A Virus Dual Inhibitor to Block Hemagglutinin-Mediated Adsorption and Membrane Fusion. J. Med. Chem. 2020, 63, 6924–6940. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gai, S.; Han, G.; Jiang, Y.; Li, B.; Li, D.; Yang, S.; Lv, Z. Pharmacokinetics and metabolism of penindolone in rat plasma using liquid chromatography-tandem mass spectrometry. Biomed. Chromatogr. 2022, 36, e5388. [Google Scholar] [CrossRef]
- Luffer-Atlas, D.; Atrakchi, A. A decade of drug metabolite safety testing: Industry and regulatory shared learning. Expert Opin. Drug Metab. Toxicol. 2017, 13, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Fura, A. Role of pharmacologically active metabolites in drug discovery and development. Drug Discov. Today 2006, 11, 133–142. [Google Scholar] [CrossRef] [PubMed]
- US, Food and Drug Administration. Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. 2020. Available online: https://www.fda.gov/media/134582/download (accessed on 12 January 2020).
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Knights, K.M.; Stresser, D.M.; Miners, J.O.; Crespi, C.L. In Vitro Drug Metabolism Using Liver Microsomes. Curr. Protoc. Pharmacol. 2016, 74, 7–8. [Google Scholar] [CrossRef]
- Lucas, A.J.; Sproston, J.L.; Barton, P.; Riley, R.J. Estimating human ADME properties, pharmacokinetic parameters and likely clinical dose in drug discovery. Expert. Opin. Drug Discov. 2019, 14, 1313–1327. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Tatosian, D.A.; Chu, X.; Railkar, R.; Evers, R.; Chavez-Eng, C.; Lutz, R.; Zeng, W.; Yabut, J.; Chan, G.H.; et al. Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin. Pharmacol. Ther. 2017, 101, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Zhu, M. Applications of mass spectrometry in drug metabolism: 50 years of progress. Drug Metab. Rev. 2015, 47, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.S.; Pirhalla, J.L.; Bowers, G.D. Metabolite structure analysis by high-resolution MS: Supporting drug-development studies. Bioanalysis 2013, 5, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Garg, A.; Takwani, H.; Singh, S. Metabolite identification by liquid chromatography-mass spectrometry. TrAC Trends Anal. Chem. 2011, 30, 360–387. [Google Scholar] [CrossRef]
- Von Moltke, L.L.; Manis, M.; Harmatz, J.S.; Poorman, R.; Greenblatt, D.J. Inhibition of acetaminophen and lorazepam glucuronidation in vitro by probenecid. Biopharm. Drug Dispos. 1993, 14, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Boase, S.; Miners, J.O. In vitro-in vivo correlations for drugs eliminated by glucuronidation: Investigations with the model substrate zidovudine. Br. J. Clin. Pharmacol. 2002, 54, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Korprasertthaworn, P.; Chau, N.; Nair, P.C.; Rowland, A.; Miners, J.O. Inhibition of human UDP-glucuronosyltransferase (UGT) enzymes by kinase inhibitors: Effects of dabrafenib, ibrutinib, nintedanib, trametinib and BIBF 1202. Biochem. Pharmacol. 2019, 169, 113616. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Transformations | Formula | MW | RT (min) | Polarity | m/z of Precursor Ion (Error, ppm) | Fragment Ions | Source |
|---|---|---|---|---|---|---|---|---|
| M0 | — | C28H27NO6 | 473.1838 | 27.00 | ESI (−) | 472.1777 (2.32) | 306.1134, 294.1135, 165.0549 | P, B, U, F, |
| ESI (+) | 474.1910 (−0.22) | 308.1276, 296.1278, 179.0701, 161.0591 | ||||||
| M1 | Oxidation | C28H27NO7 | 489.1789 | 24.79 | ESI (−) | 488.1746 (2.05) | 322.1088, 310.1088, 292.0980, 165.0548 | P, U, F |
| ESI (+) | 490.1859 (1.83) | 472.1751, 306.1122, 294.1122, 179.0701, 161.0596 | ||||||
| M2 | Oxidation | C28H27NO7 | 489.1789 | 25.77 | ESI (−) | 488.1720 (0.82) | 322.1087, 310.1088, 292.0983, 165.0549 | P, F |
| ESI (+) | 490.1859 (1.83) | 472.1755, 306.1122, 294.1121, 179.0701, 177.0545, 161.0596 | ||||||
| M3 | Glucuronidation | C34H35NO12 | 649.2171 | 21.93 | ESI (−) | 648.2096 (−0.31) | 472.1769, 306.1138, 294.1140, 165.0550 | P, B |
| ESI (+) | 650.2231 (−0.09) | 474.1902, 308.1275, 296.1280, 179.0701, 161.0597 | ||||||
| M4 | Glucuronidation | C34H35NO12 | 649.2171 | 22.90 | ESI (−) | 648.2097 (0.15) | 472.1773, 306.1139, 294.1140, 165.0550 | P, B |
| ESI (+) | 650.2232 (0.01) | 474.1911, 308.1275, 296.1280, 179.0702, 161.0595 | ||||||
| M5 | Glucuronidation | C34H35NO12 | 649.2171 | 23.59 | ESI (−) | 648.2095 (0.46) | 472.1769, 306.1140, 294.1140, 165.0550 | P, B |
| ESI (+) | 650.2233 (0.19) | 474.1917, 308.1268, 296.1279, 179.0701, 161.0597 | ||||||
| M6 | Oxidation | C28H27NO7 | 489.1789 | 25.21 | ESI (+) | 490.1827 (−4.69) | 306.1120, 294.1122, 179.0701, 177.0545, 161.0593 | P, F |
| M7 | Oxidation | C28H27NO7 | 489.1789 | 26.50 | ESI (+) | 490.1840 (−2.04) | 312.1228, 296.1282, 195.0651, 179.0701, 177.0545, 161.0596 | P, U, F |
| M8 | Methylation | C29H29NO6 | 487.1632 | 27.77 | ESI (−) | 486.1928 (1.23) | 320.1273, 308.1277, 294.1122, 179.0696, 165.0540 | P, F |
| M9 | Demethylation | C27H25NO6 | 459.1682 | 25.69 | ESI (−) | 458.1620 (2.40) | 306.1134, 294.1138, 165.0549, 151.0391 | P, U, F |
| M10 | Demethylation | C27H25NO6 | 459.1682 | 26.17 | ESI (−) | 458.1617 (1.74) | 306.1147, 292.0982, 280.0982, 165.0550, 151.0391 | P, U, F |
| M11 | Dehydrogenation (−4H) | C28H23NO6 | 469.1525 | 25.11 | ESI (+) | 470.1596 (1.27) | 304.0965, 292.0960, 262.0861, 179.0861 | P, U, F |
| M12 | Dehydrogenation (−2H) | C28H25NO6 | 471.1683 | 26.14 | ESI (+) | 472.1750 (−1.06) | 294.1121, 292.0962, 179.0701, 161.0595 | F |
| M13 | Dehydrogenation (−2H) | C28H25NO6 | 471.1683 | 28.86 | ESI (+) | 472.1743 (−2.54) | 294.1120, 292.0964, 179.0701, 161.0595 | F |
| M14 | Oxidation, Glucuronidation | C34H35NO13 | 665.2104 | 20.92 | ESI (+) | 666.2177 (−0.61) | 490.1852, 472.1748, 294.1120, 179.0701, 161.0596 | B |
| M15 | Oxidation, Glucuronidation | C34H35NO13 | 665.2104 | 21.18 | ESI (+) | 666.2176 (−0.70) | 490.1866, 472.1743, 294.1119, 179.0700, 161.0597 | B |
| M16 | Demethylation, Glucuronidation | C33H34NO11 | 635.1435 | 21.56 | ESI (−) | 634.1939 (1.42) | 458.1627, 306.1140, 294.1140, 165.0546, 151.0392 | B |
| M17 | Demethylation, Glucuronidation | C33H34NO11 | 635.1435 | 21.78 | ESI (−) | 634.1940 (1.57) | 458.1616, 306.1137, 294.1140, 165.0522, 151.0391 | B |
| M18 | Demethylation, Glucuronidation | C33H34NO11 | 635.1435 | 22.89 | ESI (−) | 634.1939 (1.42) | 458.1626, 292.0982, 280.0982, 165.0550, 151.0392 | B |
| M19 | Dehydrogenation (−4H), Glucuronidation | C34H31NO12 | 645.1842 | 19.56 | ESI (+) | 646.1926 (1.08) | 470.1595, 304.0964, 292.0964, 262.0858 | B |
| M20 | Dehydrogenation (−2H), Glucuronidation | C34H33NO12 | 647.2117 | 21.96 | ESI (+) | 648.2079 (0.62) | 472.1749, 294.1121, 292.1005, 179.0701 | B |
| M21 | Dehydrogenation (−2H), Glucuronidation | C34H33NO12 | 647.2117 | 22.91 | ESI (+) | 648.2080 (0.52) | 472.1744, 294.1121, 292.0952, 179.0701 | B |
| M22 | Dehydrogenation (−2H), Glucuronidation | C34H33NO12 | 647.2117 | 23.38 | ESI (+) | 648.2079 (0.46) | 472.1754, 294.1123, 292.0978, 179.0701 | B |
| M23 | Sulfation | C28H27NO9S | 553.1414 | 25.21 | ESI (−) | 552.1341 (1.27) | 470.1616, 304.0979, 292.0984, 165.0550 | F |
| M24 | Sulfation | C28H27NO9S | 553.1414 | 25.47 | ESI (−) | 552.1337 (0.54) | 470.1616, 304.0982, 292.0983, 165.0550 | F |
| M25 | Sulfation | C28H27NO9S | 553.1414 | 26.14 | ESI (−) | 552.1341 (1.27) | 470.1616, 304.0981, 292.0984, 165.0550 | F |
| Metabolites | Bile | Urine | Feces | Plasma | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0–4 h | 4–8 h | 8–24 h | 0–2 h | 2–6 h | 6–24 h | 0–24 h | 24–36 h | 36–48 h | 0.33/0.25 h | 3/4 h | 8 h | |
| M1 | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) | Y (i.g) | Y (i.g) | Y (i.v) | Y (i.g, i.v) | Y (i.g, i.v) |
| M2 | N | N | N | N | N | N | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g) | Y (i.v) | Y (i.g, i.v) | Y (i.g, i.v) |
| M3 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g, i.v) | N | N | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) |
| M4 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g, i.v) | N | N | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) |
| M5 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g, i.v) | N | N | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) |
| M6 | N | N | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) | Y (i.v) | Y (i.g, i.v) | Y (i.g, i.v) |
| M7 | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) | Y (i.g) | Y (i.g) | Y (i.v) | Y (i.g, i.v) | Y (i.g, i.v) |
| M8 | N | N | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) | Y (i.v) | Y (i.v) | Y (i.g) |
| M9 | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g) | Y (i.v) | Y (i.v) | N |
| M10 | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g) | Y (i.v) | Y (i.v) | N |
| M11 | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g, i.v) | Y (i.g) | Y (i.g) | Y (i.v) | Y (i.g) | Y (i.g) |
| M12 | N | N | N | N | N | N | Y (i.g) | N | N | N | N | N |
| M13 | N | N | N | N | N | N | Y (i.g) | N | N | N | N | N |
| M14 | Y (i.g) | Y (i.g) | Y (i.g) | N | N | N | N | N | N | N | N | N |
| M15 | Y (i.g) | Y (i.g) | Y (i.g) | N | N | N | N | N | N | N | N | N |
| M16 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g) | N | N | N | N | N | N | N | N | N |
| M17 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g) | N | N | N | N | N | N | N | N | N |
| M18 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g) | N | N | N | N | N | N | N | N | N |
| M19 | Y (i.g) | Y (i.g, i.v) | Y (i.g) | N | N | N | N | N | N | N | N | N |
| M20 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g, i.v) | N | N | N | N | N | N | N | N | N |
| M21 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g, i.v) | N | N | N | N | N | N | N | N | N |
| M22 | Y (i.g, i.v) | Y (i.g, i.v) | Y (i.g, i.v) | N | N | N | N | N | N | N | N | N |
| M23 | N | N | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) | N | N | N |
| M24 | N | N | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) | N | N | N |
| M25 | N | N | N | N | N | N | Y (i.g) | Y (i.g) | Y (i.g) | N | N | N |
| Enzyme Types | LMs | Km (μM) | Vmax [nmol/(min×mg Protein)] | Clint (μL/min) |
|---|---|---|---|---|
| CYPs | Rat | 15.24 | 1.11 | 0.073 |
| Mouse | 16.11 | 1.11 | 0.069 | |
| Human | 97.70 | 1.28 | 0.013 | |
| UGTs | Rat | 46.72 | 1.38 | 0.030 |
| Mouse | 31.61 | 6.25 | 0.198 | |
| Human | 23.36 | 2.08 | 0.089 |
| Parameter | Units | i.g 3.5 mg/kg Mean ± SD | |
|---|---|---|---|
| Control | i.p. Ibrutinib (100 mg/kg) | ||
| Cmax-1 | ng/mL | 229.12 ± 128.48 | 272.11 ± 36.94 * |
| Cmax-2 | ng/mL | 196.93 ± 46.79 | — |
| Tmax-1 | h | 0.61 ± 0.24 | 0.47 ± 0.24 |
| Tmax-2 | h | 6 ± 0 | — |
| t1/2 | h | 4.87 ± 0.30 | 15.82 ± 9.82 * |
| AUC0-∞ | h×ng/mL | 4175.03 ± 1188.72 | 5451.41 ± 2687.37 * |
| Vd | L/kg | 0.88 ± 0.22 | 15.56 ± 6.42 * |
| CLz | L/h/kg | 0.88 ± 0.22 | 0.81 ± 0.51 |
| MRT0-∞ | h | 12.82 ± 0.22 | 23.95 ± 12.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Zheng, K.; Jiang, Y.; Gai, S.; Li, B.; Li, D.; Yang, S.; Lv, Z. Biotransformation of Penindolone, an Influenza A Virus Inhibitor. Molecules 2023, 28, 1479. https://doi.org/10.3390/molecules28031479
Liu S, Zheng K, Jiang Y, Gai S, Li B, Li D, Yang S, Lv Z. Biotransformation of Penindolone, an Influenza A Virus Inhibitor. Molecules. 2023; 28(3):1479. https://doi.org/10.3390/molecules28031479
Chicago/Turabian StyleLiu, Shuai, Keming Zheng, Yilin Jiang, Susu Gai, Bohan Li, Dehai Li, Shuang Yang, and Zhihua Lv. 2023. "Biotransformation of Penindolone, an Influenza A Virus Inhibitor" Molecules 28, no. 3: 1479. https://doi.org/10.3390/molecules28031479
APA StyleLiu, S., Zheng, K., Jiang, Y., Gai, S., Li, B., Li, D., Yang, S., & Lv, Z. (2023). Biotransformation of Penindolone, an Influenza A Virus Inhibitor. Molecules, 28(3), 1479. https://doi.org/10.3390/molecules28031479