
Synthesis of Enantiomerically Enriched Protected 2-Amino-, 2,3-Diamino- and 2-Amino-3-Hydroxypropylphosphonates


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
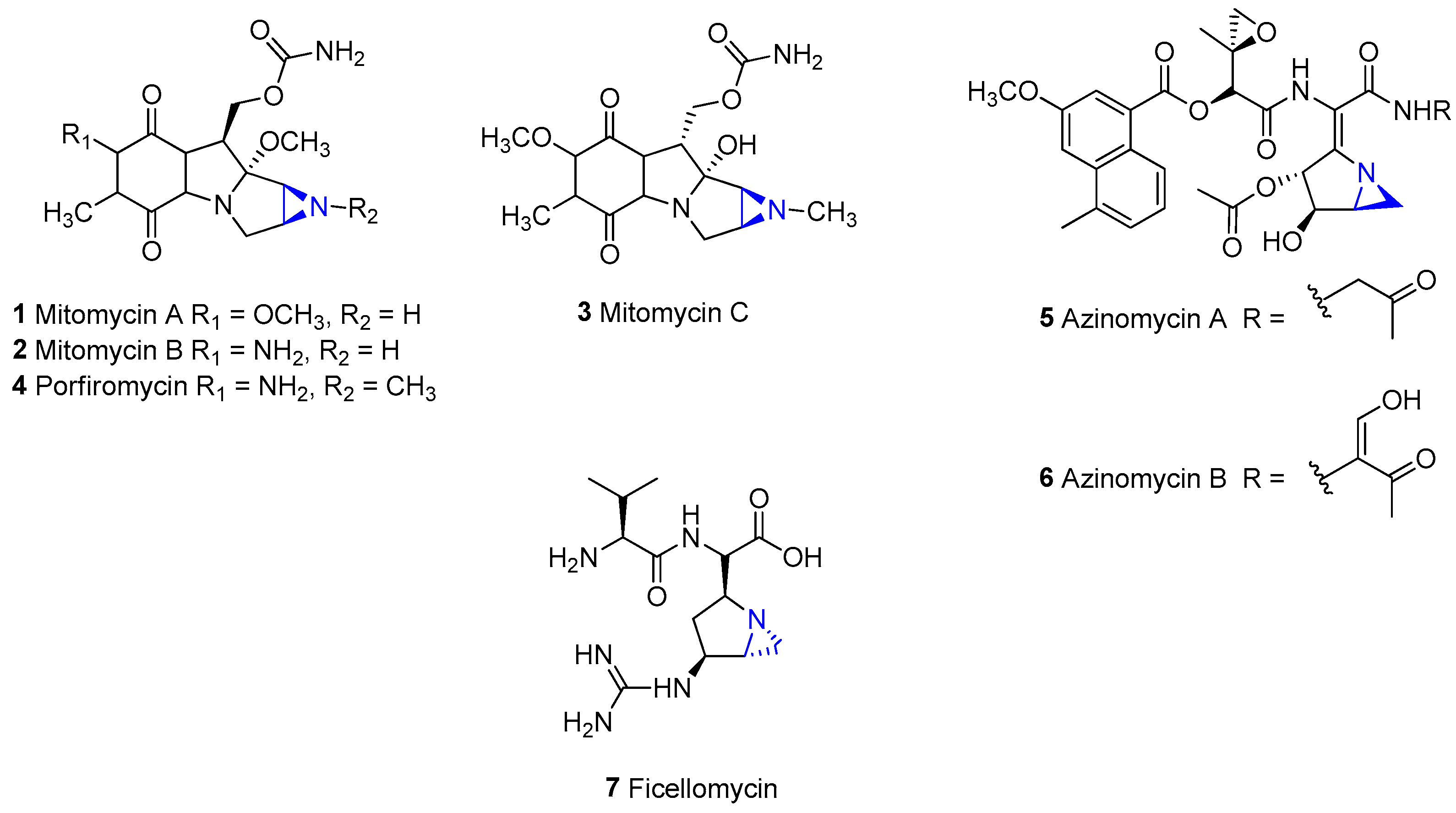
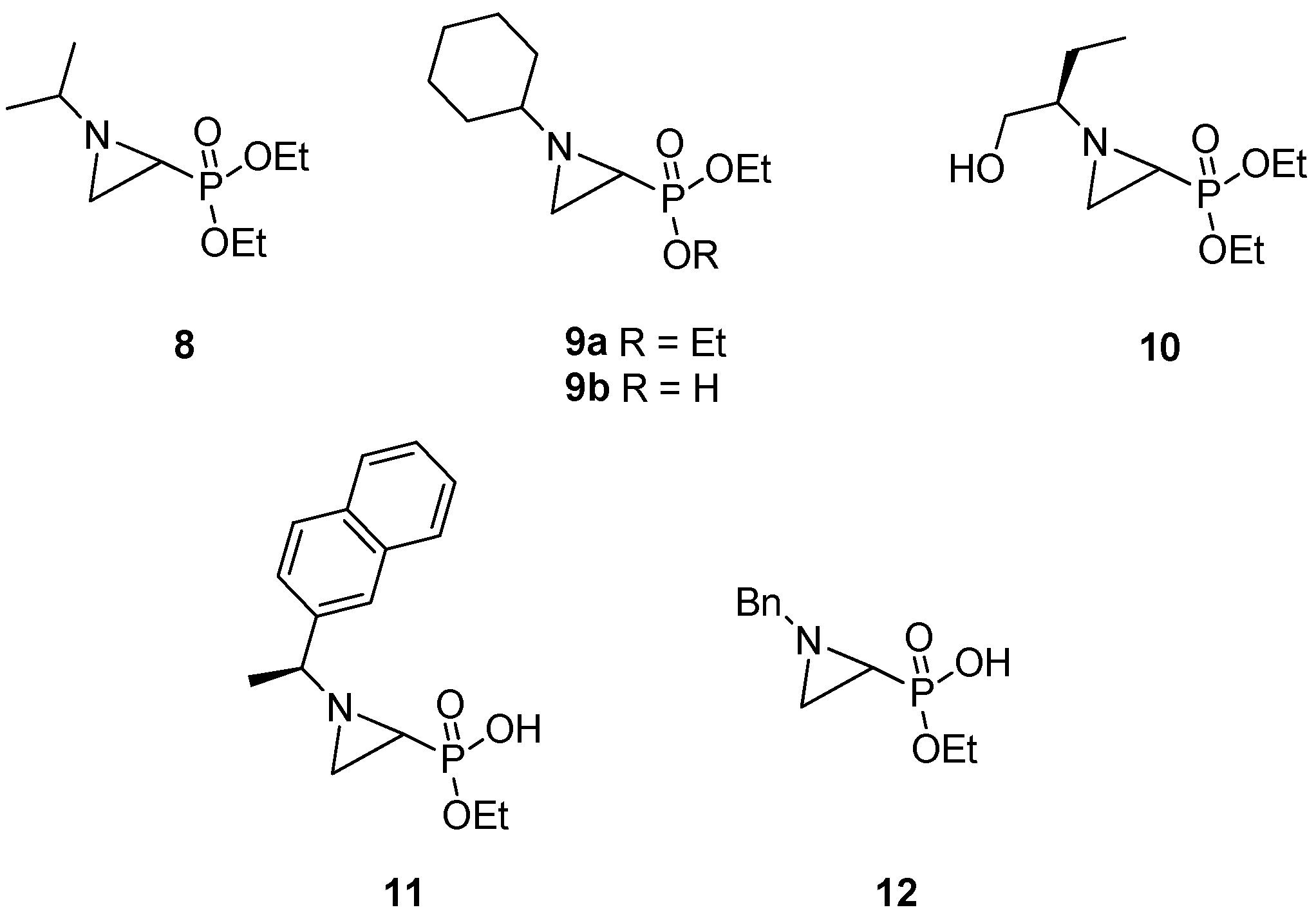
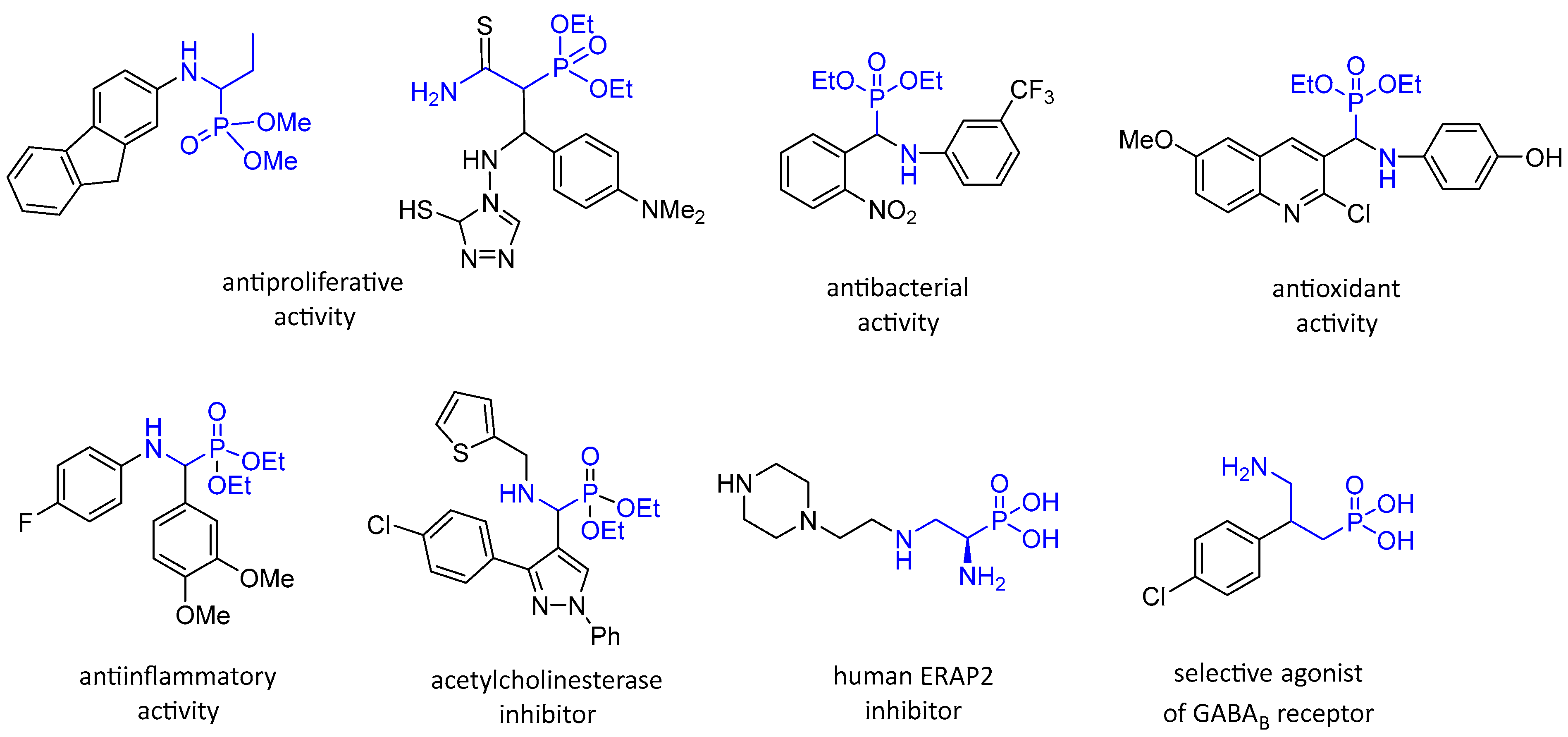
1. Introduction
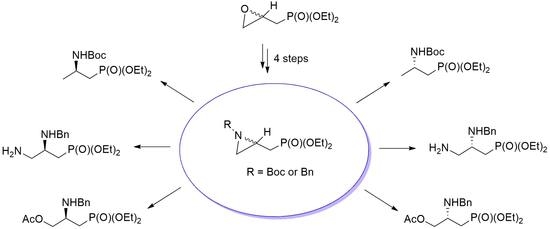
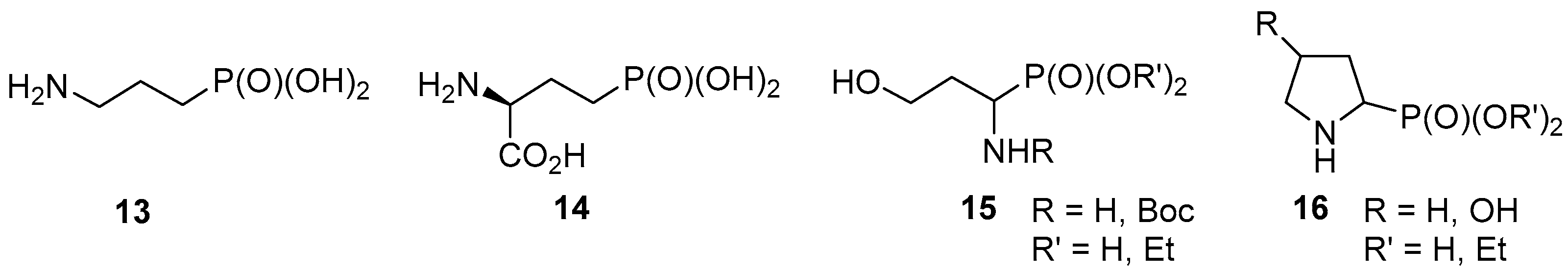
2. Results and Discussion
3. Materials and Methods
3.1. General Information
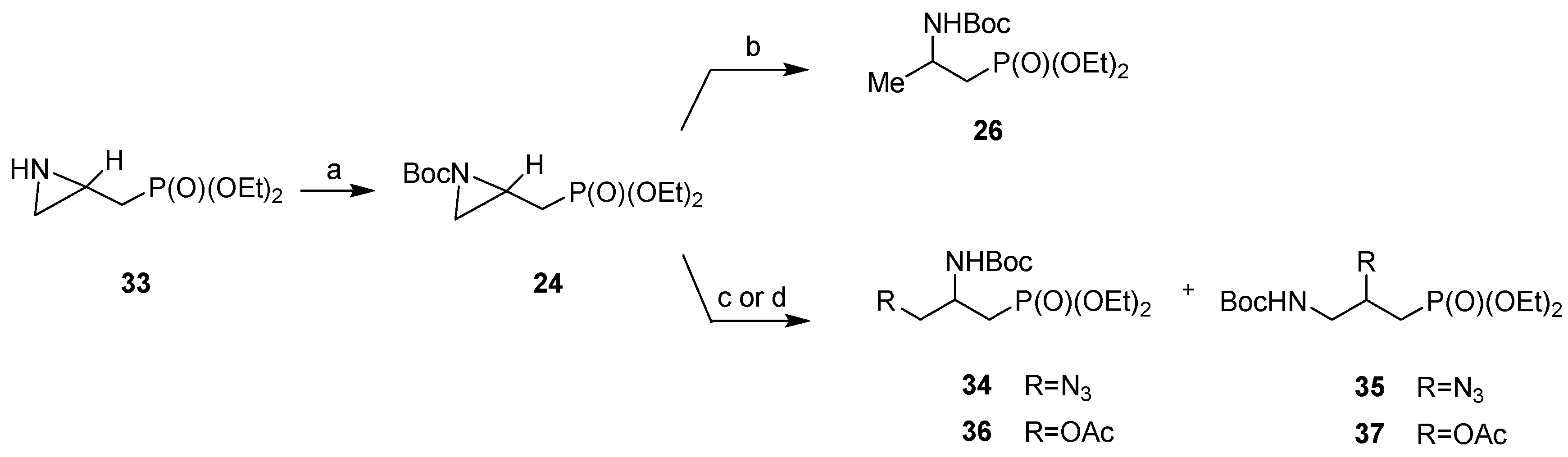
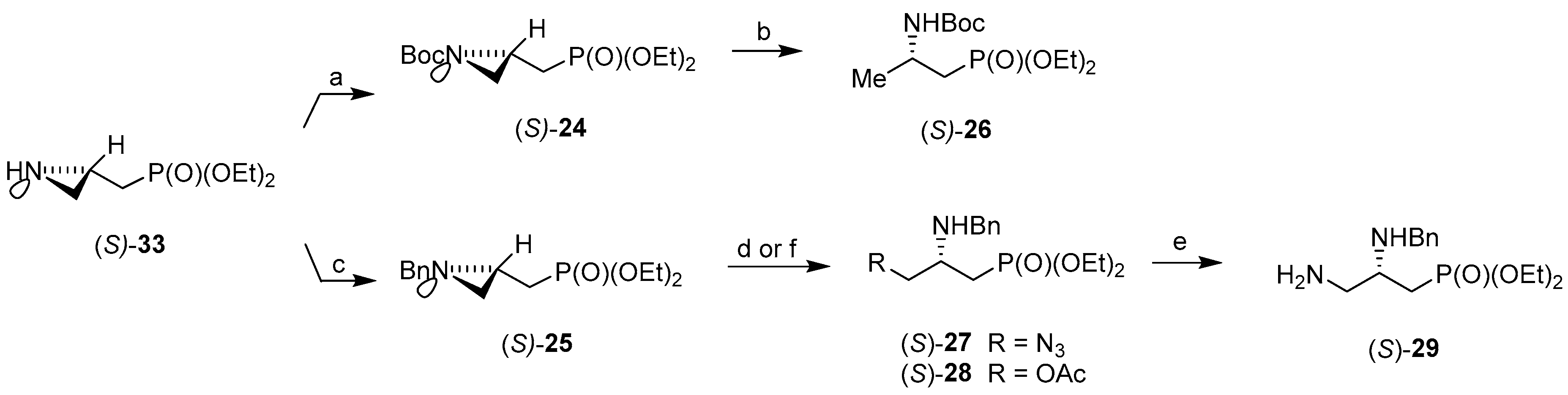
3.2. General Procedure for the Synthesis of Diethyl N-[(tert-Butoxycarbonyl)aziridin-2-yl]methylphosphonates (RS)-24, (R)-24 and (S)-24
3.3. General Procedure for Benzylation of (RS)-33, (R)-33 and (S)-33
3.4. General Procedures for the Synthesis of (RS)-26, (R)-26 and (S)-26
3.5. Ring-Opening Reaction of Diethyl N-[(tert-Butoxycarbonyl)aziridin-2-yl]methylphosphonate (RS)-24 with Trimethylsilyl Azide
3.6. Ring-Opening of Diethyl N-[(tert-Butoxycarbonyl)aziridin-2-yl]methylphosphonate (RS)-24 with Acetic Acid
3.7. General Procedure for the Ring-Opening Reaction of Diethyl N-(Benzylaziridin-2-yl]methylphosphonates (RS)-25, (R)-25 and (S)-25 with Trimethylsilyl Azide
3.8. General Procedure for the Synthesis of Diethyl 2,3-Diaminopropylphosphonates (RS)-29, (R)-29 and (S)-29
3.9. General Procedure for the Ring-Opening Reaction of Diethyl N-(Benzylaziridin-2-yl)methylphosphonates (RS)-25, (R)-25 and (S)-25 with Acetic Acid
3.10. General Procedure for the Ring-Opening Reaction of 2,3-Epoxypropylphosphonates (RS)-30, (S)-30 and (R)-30 with (R)-1-Phenylethylamine (Determination of ee)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Yudin, A.K. Aziridines and Epoxides in Organic Synthesis; Wiley-VCH Verlag GmbH & Co. KGaA: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Kametani, T.; Honda, T. Application of Aziridines to the Synthesis of natural Products. Adv. Heterocycl. Chem. 1986, 39, 181–236. [Google Scholar] [CrossRef]
- Tanner, D. Chiral Aziridines—Their Synthesis and Use in Stereoselective Transformations. Angew. Chem. Int. Ed. Engl. 1994, 33, 599–619. [Google Scholar] [CrossRef]
- McCoull, W.; Davis, F.A. Recent synthetic applications of chiral aziridines. Synthesis 2000, 2000, 1347–1365. [Google Scholar] [CrossRef]
- Sweeney, J.B. Aziridines: Epoxides’ ugly cousins? Chem. Soc. Rev. 2002, 31, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.E. Nucleophilic ring opening of aziridines. Tetrahedron 2004, 60, 2701–2743. [Google Scholar] [CrossRef]
- Singh, G.S.; D’Hooghe, M.; De Kimpe, N. Synthesis and reactivity of C-heteroatom-substituted aziridines. Chem. Rev. 2007, 107, 2080–2135. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, S.; D’Hooghe, M.; Catak, S.; Eum, H.; Waroquier, M.; Van Speybroeck, V.; De Kimpe, N.; Ha, H.J. Regioselectivity in the ring opening of non-activated aziridines. Chem. Soc. Rev. 2012, 41, 643–665. [Google Scholar] [CrossRef]
- Singh, G.S. Synthetic Aziridines in Medicinal Chemistry: A Mini-Review. Mini-Rev. Med. Chem. 2016, 16, 892–904. [Google Scholar] [CrossRef]
- Singh, G.S. Chapter Four—Advances in synthesis and chemistry of aziridines. Adv. Heterocycl. Chem. 2019, 129, 245–335. [Google Scholar] [CrossRef]
- Qadir, T.; Amin, A.; Sarkar, D.; Sharma, P.K. A Review on Recent Advances in the Synthesis of Aziridines and their Applications in Organic Synthesis. Curr. Org. Chem. 2021, 25, 1868–1893. [Google Scholar] [CrossRef]
- Hata, T.; Sano, Y.; Sugawara, R.; Matsumae, A.; Kanamori, K.; Shima, T.; Hoshi, T. Mitomycin, a New Antibiotic from Streptomyces. I. J. Antibiot. 1956, 9, 141–146. [Google Scholar] [CrossRef]
- Sugawara, R.; Hata, T. Mitomycin, a New Antibiotic from Streptomyces. II. Description of the Strain. J. Antibiot. 1956, 9, 147–151. [Google Scholar] [CrossRef]
- Evans, J.S.; Gray, J.E.; Musser, E.A. Porfiromycin Antitumor and Toxicopathologic Studies. Antibiot. Chemother. 1961, 11, 445–453. [Google Scholar]
- Ishizeki, S.; Ohtsuka, M.; Irinoda, K.; Kukita, K.I.; Nagaoka, K.; Nakashima, T. Azinomycin-A and Azinomycin-B, New Antitumor Antibiotics. III. Antitumor Activity. J. Antibiot. 1987, 40, 60–65. [Google Scholar] [CrossRef]
- Argoudelis, A.D.; Reusser, F.; Whaley, H.A.; Baczynskyj, L.; Mizsak, S.A.; Wnuk, R.J. Antibiotics Produced by Streptomyces-Ficellus. I. Ficellomycin. J. Antibiot. 1976, 29, 1001–1006. [Google Scholar] [CrossRef]
- Ham, G.E. Activated Aziridines. I. Reaction of Anilines with O-Ethyl-N,N-Ethyleneurethane. Mechanism + Hammett p-Constant. J. Org. Chem. 1964, 29, 3052–3055. [Google Scholar] [CrossRef]
- Dogan, O.; Babiz, H.; Gozen, A.G.; Budak, S. Synthesis of 2-aziridinyl phosphonates by modified Gabriel-Cromwell reaction and their antibacterial activities. Eur. J. Med. Chem. 2011, 46, 2485–2489. [Google Scholar] [CrossRef]
- Khan, R.; Ulusan, S.; Banerjee, S.; Dogan, O. Synthesis, Characterization and Evaluation of Cytotoxic Activities of Novel Aziridinyl Phosphonic Acid Derivatives. Chem. Biodivers. 2019, 16, e1900375. [Google Scholar] [CrossRef]
- Ordonez, M.; Rojas-Cabrera, H.; Cativiela, C. An overview of stereoselective synthesis of alpha-aminophosphonic acids and derivatives. Tetrahedron 2009, 65, 17–49. [Google Scholar] [CrossRef]
- Viveros-Ceballos, J.L.; Ordonez, M.; Sayago, F.J.; Cativiela, C. Stereoselective Synthesis of -Amino-C-phosphinic Acids and Derivatives. Molecules 2016, 21, 1141. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable Potential of the alpha-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Vigot, R.; Batini, C. Purkinje cell inhibitory responses to 3-APPA (3-aminopropylphosphonic acid) in RAT cerebellar slices. Neurosci. Res. 1999, 34, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ma, D. Conformationally Constrained Analogues of L-Glutamate as Subtype-Selective Modulators of Metabotropic Glutamate Receptors. Bioorg. Chem. 1999, 27, 20–34. [Google Scholar] [CrossRef]
- Chaton, C.T.; Rodriguez, E.S.; Reed, W.W.; Li, J.; Kenner, C.W.; Korotkov, K.V. Structural analysis of mycobacterial homoserine transacetylases central to methionine biosynthesis reveals druggable active site. Sci. Rep. 2019, 9, 20267. [Google Scholar] [CrossRef]
- Dinér, P.; Amedjkouh, M. Aminophosphonates as organocatalysts in the direct asymmetric aldol reaction: Towards syn selectivity in the presence of Lewis bases. Org. Biomol. Chem. 2006, 4, 2091–2096. [Google Scholar] [CrossRef]
- Boduszek, B.; Oleksyszyn, J.; Kam, C.-M.; Selzler, J.; Smith, R.E.; Powers, J.C. Dipeptide Phosphonates as Inhibitors od Dipeptidyl Peptidase IV. J. Med. Chem. 1994, 37, 3969–3976. [Google Scholar] [CrossRef]
- Huang, R.Z.; Wang, C.Y.; Li, J.F.; Yao, G.Y.; Pan, Y.M.; Ye, M.Y.; Wang, H.S.; Zhang, Y. Synthesis, antiproliferative and apoptosis-inducing effects of novel asiatic acid derivatives containing alpha-aminophosphonates. RSC Adv. 2016, 6, 62890–62906. [Google Scholar] [CrossRef]
- Jin, L.; Zhang, B.; Hua, S.X.; Ji, M.; Huang, X.C.; Huang, R.Z.; Wang, H.S. Glycyrrhetinic acid derivatives containing aminophosphonate ester species as multidrug resistance reversers that block the NF-kappa B pathway and cell proliferation. Bioorg. Med. Chem. Lett. 2018, 28, 3700–3707. [Google Scholar] [CrossRef]
- Abdou, W.M.; Ganoub, N.A.; Sabry, E. Design, synthesis and pharmacological screening of beta-amino-, thiadiazole/thiadiazine-phosphonate based triazole motifs as antimicrobial/cytotoxic agents. Acta Pharm. 2014, 64, 267–284. [Google Scholar] [CrossRef]
- Huang, X.C.; Wang, M.; Pan, Y.M.; Tian, X.Y.; Wang, H.S.; Zhang, Y. Synthesis and antitumor activities of novel alpha-aminophosphonates dehydroabietic acid derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 5283–5289. [Google Scholar] [CrossRef]
- Rasal, S.A.; Dhavan, P.P.; Jadhav, B.L.; Shimpi, N.G. Synthesis of new alpha-aminophosphonates using nanoscale nickel-based metal-organic framework as a heterogeneous catalyst and their antibacterial activity. Appl. Organomet. Chem. 2019, 34, e5317. [Google Scholar] [CrossRef]
- Poola, S.; Nagaripati, S.; Tellamekala, S.; Chintha, V.; Kotha, P.; Yagani, J.R.; Golla, N.; Cirandur, S.R. Green synthesis, antibacterial, antiviral and molecular docking studies of alpha-aminophosphonates. Synth. Commun. 2020, 50, 2655–2672. [Google Scholar] [CrossRef]
- Shashikumar, N.D. Preparation of New alpha-Aminophosphonate Derivatives by Kabachnik-Fields Reaction Using a Recyclable Catalyst. J. Chem. 2013, 2013, 240381. [Google Scholar] [CrossRef]
- Xu, Y.S.; Yan, K.; Song, B.; Xu, G.F.; Yang, S.; Xue, W.; Hu, D.Y.; Lu, P.P.; Ouyang, G.P.; Jin, L.H.; et al. Synthesis and antiviral bioactivities of alpha-aminophosphonates containing alkoxyethyl moieties. Molecules 2006, 11, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Ji, R.J.; Wang, H.; Li, S.Y.; Zhang, G.P. Discovery of Novel alpha-Aminophosphonates with Hydrazone as Potential Antiviral Agents Combined with Active Fragment and Molecular Docking. Front. Chem. 2022, 10, 911453. [Google Scholar] [CrossRef]
- Tekale, S.U.; Kauthale, S.S.; Shaikh, R.U.; Marathe, R.P.; Nawale, R.B.; Pawar, R.P. Aluminium nitride catalyzed solvent-free synthesis of some novel biologically active alpha-aminophosphonates. J. Iran. Chem. Soc. 2014, 11, 717–724. [Google Scholar] [CrossRef]
- Aissa, R.; Guezane-Lakoud, S.; Gali, L.; Toffano, M.; Ignaczak, A.; Adamiak, M.; Merabet-Khelassi, M.; Guillot, R.; Aribi-Zouioueche, L. New promising generation of phosphates alpha-aminophosphonates: Design, synthesis, in-vitro biological evaluation and computational study. J. Mol. Struct. 2022, 1247, 131336. [Google Scholar] [CrossRef]
- Bazine, I.; Cheraiet, Z.; Bensegueni, R.; Bensouici, C.; Boukhari, A. Synthesis, antioxidant and anticholinesterase activities of novel quinoline-aminophosphonate derivatives. J. Heterocycl. Chem. 2020, 57, 2139–2149. [Google Scholar] [CrossRef]
- Reddy, K.M.K.; Santhisudha, S.; Mohan, G.; Peddanna, K.; Rao, C.A.; Reddy, C.S. Nano Gd2O3 catalyzed synthesis and anti-oxidant activity of new alpha-aminophosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 933–938. [Google Scholar] [CrossRef]
- Shaik, Y.H.; Chintha, V.; Gundluru, M.; Sarva, S.; Cirandur, S.R. An efficient nano-FGT catalyzed green synthesis of alpha-aminophosphonates and evaluation of their antioxidant, anti-inflammatory activity and molecular docking studies. Synth. Commun. 2022, 52, 129–144. [Google Scholar] [CrossRef]
- Shaikh, S.; Dhavan, P.; Singh, P.; Uparkar, J.; Vaidya, S.P.; Jadhav, B.L.; Ramana, M.M.V. Design, synthesis and biological evaluation of novel antipyrine based alpha-aminophosphonates as anti-Alzheimer and anti-inflammatory agent. J. Biomol. Struct. Dyn. 2023, 41, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Dhavan, P.; Pavale, G.; Ramana, M.M.V.; Jadhav, B.L. Design, synthesis and evaluation of pyrazole bearing α-aminophosphonate derivatives as potential acetylcholinesterase inhibitors against Alzheimer’s disease. Bioorg. Chem. 2020, 96, 103589. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Dhavan, P.; Singh, P.; Uparkar, J.; Vaidya, S.P.; Jadhav, B.L.; Ramana, M.V. Synthesis of carbazole based alpha-aminophosphonate derivatives: Design, molecular docking and in vitro cholinesterase activity. J. Biomol. Struct. Dyn. 2020, 40, 4801–4814. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Dhavan, P.; Ramana, M.M.V.; Jadhav, B.L. Design, synthesis and evaluation of new chromone-derived aminophosphonates as potential acetylcholinesterase inhibitor. Mol. Divers. 2020, 25, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Wanat, W.; Talma, M.; Pawełczak, M.; Kafarski, P. Phosphonic Acid Analogues of Phenylglycine as Inhibitors of Aminopeptidases: Comparison of Porcine Aminopeptidase N, Bovine Leucine Aminopeptidase, Tomato Acidic Leucine Aminopeptidase and Aminopeptidase from Barley Seeds. Pharmaceuticals 2019, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Weglarz-Tomczak, E.; Staszewska, K.; Talma, M.; Mucha, A. Enantiomeric alpha, beta-diaminoethylphosphonic acids as potent inhibitors of aminopeptidases-stereoselective synthesis and biological activity. Tetrahedron Lett. 2016, 57, 4812–4814. [Google Scholar] [CrossRef]
- Sreelakshmi, P.; Santhisudha, S.; Reddy, G.R.; Subbarao, Y.; Peddanna, K.; Apparao, C.; Reddy, C.S. Nano-Cuo-Au-catalyzed solvent-free synthesis of -aminophosphonates and evaluation of their antioxidant and -glucosidase enzyme inhibition activities. Synth. Commun. 2018, 48, 1148–1163. [Google Scholar] [CrossRef]
- Wroblewski, A.E.; Drozd, J. Synthesis of four enantiomers of 2-acetamido-1-hydroxypropylphosphonates. Tetrahedron Asymmetry 2007, 18, 1134–1141. [Google Scholar] [CrossRef]
- Wroblewski, A.E.; Drozd, J. Synthesis of four enantiomers of 2,3-di(N-Boc-amino)-1-hydroxypropylphosphonates. Tetrahedron Asymmetry 2009, 20, 2240–2246. [Google Scholar] [CrossRef]
- Glowacka, I.E.; Piotrowska, D.G.; Wroblewski, A.E.; Trocha, A. Synthesis of the four enantiomers of diethyl 1,2-di(N-Boc-amino) propylphosphonates. Tetrahedron Asymmetry 2017, 28, 1602–1607. [Google Scholar] [CrossRef]
- Trocha, A.; Piotrowska, D.G.; Glowacka, I.E. Synthesis of Enantiomerically Pure N-Boc-Protected 1,2,3-Triaminopropylphosphonates and 1,2-Diamino-3-Hydroxypropylphosphonates. Molecules 2019, 24, 3857. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.E. Synthesis of enantiomerically pure diethyl (R)- and (S)-2-hydroxy-3-(1,2,3-triazol-1-yl)propylphosphonates. Tetrahedron Asymmetry 2009, 20, 2270–2278. [Google Scholar] [CrossRef]
- Nesterov, V.V.; Kolodiazhnyi, O.I. Efficient method for the asymmetric reduction of alpha- and beta-ketophosphonates. Tetrahedron 2007, 63, 6720–6731. [Google Scholar] [CrossRef]
- Wroblewski, A.E.; Drozd, J. Synthesis of four stereoisomers of protected 1,2-epiimino-3-hydroxypropylphosphonates. Tetrahedron Asymmetry 2011, 22, 200–206. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Wroblewski, A.E. Towards Convenient Precursors for alpha-Phosphonylated Aziridinium Ions. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 998–1016. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Wróblewski, A.E. Stereoselective synthesis of β-functionalized α-aminophosphonates via aziridinium ions. Tetrahedron 2003, 59, 8405–8410. [Google Scholar] [CrossRef]
- D’Hooghe, M.; Van Brabandt, W.; De Kimpe, N. New synthesis of propargylic amines from 2-(bromomethyl)aziridines. Intermediacy of 3-bromoazetidinium salts. J. Org. Chem. 2004, 69, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, A.E.; Halajewska-Wosik, A. Towards enantiomeric 2,3-epoxypropylphosphonates. Tetrahedron Asymmetry 2000, 11, 2053–2055. [Google Scholar] [CrossRef]
- Gryshkun, E.V.; Nesterov, V.; Kolodyazhnyi, O.I. Enantioselective reduction of ketophosphonates using adducts of chiral natural acids with sodium borohydride. Arkivoc 2012, 2012, 100–117. [Google Scholar] [CrossRef]
- Wroblewski, A.E.; Halajewska-Wosik, A. Synthesis of novel enantiomerically pure C-3-symmetric trialkanolamine ligand containing phosphoryl groups. Synthesis 2006, 2006, 989–994. [Google Scholar] [CrossRef]
- Griffin, C.E.; Kundu, S.K. Phosphonic Acids and Esters. 20. Preparation and Ring-Opening Reactions of Alpha, Beta-Epoxyalkylphosphonates and Beta-Gamma- Epoxyalkylphosphonates. Proton Magnetic Resonance Spectra of Vicinally Substituted Ethyl- and Propylphosphonates. J. Org. Chem. 1969, 34, 1532–1539. [Google Scholar] [CrossRef]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trocha, A.; Piotrowska, D.G.; Głowacka, I.E. Synthesis of Enantiomerically Enriched Protected 2-Amino-, 2,3-Diamino- and 2-Amino-3-Hydroxypropylphosphonates. Molecules 2023, 28, 1466. https://doi.org/10.3390/molecules28031466
Trocha A, Piotrowska DG, Głowacka IE. Synthesis of Enantiomerically Enriched Protected 2-Amino-, 2,3-Diamino- and 2-Amino-3-Hydroxypropylphosphonates. Molecules. 2023; 28(3):1466. https://doi.org/10.3390/molecules28031466
Chicago/Turabian StyleTrocha, Aleksandra, Dorota G. Piotrowska, and Iwona E. Głowacka. 2023. "Synthesis of Enantiomerically Enriched Protected 2-Amino-, 2,3-Diamino- and 2-Amino-3-Hydroxypropylphosphonates" Molecules 28, no. 3: 1466. https://doi.org/10.3390/molecules28031466
APA StyleTrocha, A., Piotrowska, D. G., & Głowacka, I. E. (2023). Synthesis of Enantiomerically Enriched Protected 2-Amino-, 2,3-Diamino- and 2-Amino-3-Hydroxypropylphosphonates. Molecules, 28(3), 1466. https://doi.org/10.3390/molecules28031466