
In Silico Targeting of Fascin Protein for Cancer Therapy: Benchmarking, Virtual Screening and Molecular Dynamics Approaches
 , and
, and
Abstract
1. Introduction
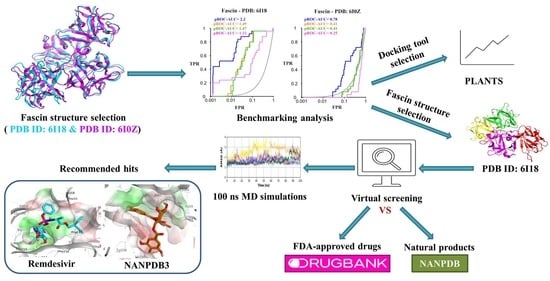
2. Results and Discussion
2.1. Selection of Fascin Actives for Decoys Generation
2.2. Selection of Representative PDB Structure(s) for Fascin
2.3. Benchmarking
2.4. Prospective Virtual Screening
2.5. Molecular Dynamics Simulation
3. Materials and Methods
3.1. Preparation of Protein Structures
3.2. Preparation of Small Molecules of DEKOIS 2.0 Benchmark Set, DrugBank FDA-Approved Drugs, and NANPDB Molecules
3.3. Docking Experiments
3.3.1. Benchmarking
3.3.2. Virtual Screening of DrugBank FDA-Approved Drugs and NANPDB Molecules
3.4. pROC and pROC-Chemotype Calculations
3.5. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| DEKOIS | Demanding Evaluation Kits for Objective In silico Screening |
| SBVS | structure-based virtual screening |
| EF | Enrichment factor |
| LOA | Level of activity |
| TOD | Type of data |
| RMSD | Root mean square deviation |
| PLANTS | Protein-Ligand ANT System |
| MOE | Molecular operating environment |
| SPORES | Structure Protonation and Recognition System |
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M.; Lia, A. Fascin. Commun. Integr. Biol. 2010, 3, 263–270. [Google Scholar] [CrossRef]
- Flemban, A.; Qualtrough, D. The potential role of hedgehog signaling in the luminal/basal phenotype of breast epithelia and in breast cancer invasion and metastasis. Cancers 2015, 7, 1863–1884. [Google Scholar] [CrossRef] [PubMed]
- Alqathama, A. BRAF in malignant melanoma progression and metastasis: Potentials and challenges. Am. J. Cancer Res. 2020, 10, 1114. [Google Scholar]
- Jayo, A.; Parsons, M. Fascin: A key regulator of cytoskeletal dynamics. Int. J. Biochem. Cell Biol. 2010, 42, 1614–1617. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kim, D.J.; Adams, J.C. The roles of fascins in health and disease. J. Pathol. 2011, 224, 289–300. [Google Scholar] [CrossRef]
- Zhang, F.R.; Tao, L.H.; Shen, Z.Y.; Lv, Z.; Xu, L.Y.; Li, E.M. Fascin expression in human embryonic, fetal, and normal adult tissue. J. Histochem. Cytochem. 2008, 56, 193–199. [Google Scholar] [CrossRef]
- Tan, V.Y.; Lewis, S.J.; Adams, J.C.; Martin, R.M. Association of Fascin-1 with mortality, disease progression and metastasis in carcinomas: A systematic review and meta-analysis. BMC Med. 2013, 11, 52. [Google Scholar] [CrossRef]
- Rodrigues, P.C.; Sawazaki-Calone, I.; de Oliveira, C.E.; Macedo, C.C.S.; Dourado, M.R.; Cervigne, N.K.; Miguel, M.C.; do Carmo, A.F.; Lambert, D.W.; Graner, E.; et al. Fascin promotes migration and invasion and is a prognostic marker for oral squamous cell carcinoma. Oncotarget 2017, 8, 74736–74754. [Google Scholar] [CrossRef]
- Kane, R.E. Preparation and purification of polymerized actin from sea urchin egg extracts. J. Cell Biol. 1975, 66, 305–315. [Google Scholar] [CrossRef]
- Cant, K.; Cooley, L. Single amino acid mutations in drosophila fascin disrupt actin bundling function in vivo. Genetics 1996, 143, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Herrera-Sosa, H.; Otto, J.; Bryan, J. Cloning and expression of a murine fascin homolog from mouse brain. J. Biol. Chem. 1995, 270, 10764–10770. [Google Scholar] [CrossRef] [PubMed]
- Holthuis, J.C.M.; Schoonderwoert, V.T.G.; Martens, G.J.M. A vertebrate homolog of the actin-bundling protein fascin. Biochim. Biophys. Acta—Gene Struct. Expr. 1994, 1219, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Duh, F.M.; Latif, F.; Weng, Y.; Geil, L.; Modi, W.; Stackhouse, T.; Matsumura, F.; Duan, D.R.; Linehan, W.M.; Lerman, M.I.; et al. CDNA cloning and expression of the human homolog of the sea urchin fascin and drosophila singed genes which encodes an actin-bundling protein. DNA Cell Biol. 1994, 13, 821–827. [Google Scholar] [CrossRef]
- Yamashiro-Matsumura, S.; Matsumura, F. Purification and Characterization of an F-Actin-Bundling 55-Kilodalton Protein from HeLa Cells. J. Biol. Chem. 1985, 260, 5087–5097. [Google Scholar] [CrossRef]
- Murzin, A.G.; Lesk, A.M.; Chothia, C. β-Trefoil Fold: Patterns of Structure and Sequence in the Kunitz Inhibitors Interleukins-1β and 1α and Fibroblast Growth Factors. J. Mol. Biol. 1992, 223, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Russell, R.B. Identification of Distant Homologues of Fibroblast Growth Factors Suggests a Common Ancestor for All β-Trefoil Proteins. J. Mol. Biol. 2000, 302, 1041–1047. [Google Scholar] [CrossRef]
- Yang, S.; Huang, F.K.; Huang, J.; Chen, S.; Jakoncic, J.; Leo-Macias, A.; Diaz-Avalos, R.; Chen, L.; Zhang, J.J.; Huang, X.Y. Molecular Mechanism of Fascin Function in Filopodial Formation. J. Biol. Chem. 2013, 288, 274–284. [Google Scholar] [CrossRef]
- Aramaki, S.; Mayanagi, K.; Jin, M.; Aoyama, K.; Yasunaga, T. Filopodia Formation by Crosslinking of F-Actin with Fascin in Two Different Binding Manners. Cytoskeleton 2016, 73, 365–374. [Google Scholar] [CrossRef]
- Ono, S.; Yamakita, Y.; Yamashiro, S.; Matsudaira, P.T.; Gnarra, J.R.; Obinata, T.; Matsumura, F. Identification of an Actin Binding Region and a Protein Kinase C Phosphorylation Site on Human Fascin*. J. Biol. Chem. 1997, 272, 2527–2533. [Google Scholar] [CrossRef]
- Chen, L.; Yang, S.; Jakoncic, J.; Zhang, J.J.; Huang, X.Y. Migrastatin analogues target fascin to block tumour metastasis. Nature 2010, 464, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.K.; Han, S.; Xing, B.; Huang, J.; Liu, B.; Bordeleau, F.; Reinhart-King, C.A.; Zhang, J.J.; Huang, X.Y. Targeted Inhibition of Fascin Function Blocks Tumour Invasion and Metastatic Colonization. Nat. Commun. 2015, 6, 7465. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Huang, J.; Liu, B.; Xing, B.; Bordeleau, F.; Reinhart-King, C.A.; Li, W.; Zhang, J.J.; Huang, X.Y. Improving Fascin Inhibitors to Block Tumor Cell Migration and Metastasis. Mol. Oncol. 2016, 10, 966–980. [Google Scholar] [CrossRef]
- Huang, J.; Dey, R.; Wang, Y.; Jakoncic, J.; Kurinov, I.; Huang, X.Y. Structural Insights into the Induced-Fit Inhibition of Fascin by a Small-Molecule Inhibitor. J. Mol. Biol. 2018, 430, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Alburquerque-González, B.; Bernabé-García, M.; Montoro-García, S.; Bernabé-García, Á.; Rodrigues, P.C.; Ruiz Sanz, J.; López-Calderón, F.F.; Luque, I.; Nicolas, F.J.; Cayuela, M.L.; et al. New Role of the Antidepressant Imipramine as a Fascin1 Inhibitor in Colorectal Cancer Cells. Exp. Mol. Med. 2020, 52, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Alburquerque-gonzález, B.; Bernabé-garcía, Á.; Bernabé-garcía, M.; Ruiz-sanz, J.; López-calderón, F.F.; Gonnelli, L.; Banci, L.; Peña-garcía, J.; Luque, I.; Nicolás, F.J.; et al. The FDA-Approved Antiviral Raltegravir Inhibits Fascin1-Dependent Invasion of Colorectal Tumor Cells In Vitro and In Vivo. Cancers 2021, 13, 861. [Google Scholar] [CrossRef]
- Francis, S.; Croft, D.; Schüttelkopf, A.W.; Parry, C.; Pugliese, A.; Cameron, K.; Claydon, S.; Drysdale, M.; Gardner, C.; Gohlke, A.; et al. Structure-Based Design, Synthesis and Biological Evaluation of a Novel Series of Isoquinolone and Pyrazolo[4,3-c]Pyridine Inhibitors of Fascin 1 as Potential Anti-Metastatic Agents. Bioorg. Med. Chem. Lett. 2019, 29, 1023. [Google Scholar] [CrossRef]
- Lin, L.; Lin, K.; Wu, X.; Liu, J.; Cheng, Y.; Xu, L.Y.; Li, E.M.; Dong, G. Potential Inhibitors of Fascin From A Database of Marine Natural Products: A Virtual Screening and Molecular Dynamics Study. Front. Chem. 2021, 9, 829. [Google Scholar] [CrossRef]
- Xue, B.; Chaddha, M.; Elasbali, A.M.; Zhu, Z.; Jairajpuri, D.S.; Alhumaydhi, F.A.; Mohammad, T.; Al Abdulmonem, W.; Sharaf, S.E.; Hassan, M.I. Death-Associated Protein Kinase 3 Inhibitors Identified by Virtual Screening for Drug Discovery in Cancer and Hypertension. Omi. A J. Integr. Biol. 2022, 26, 404–413. [Google Scholar] [CrossRef]
- Eweas, A.F.; Osman, H.E.H.; Naguib, I.A.; Abourehab, M.A.S.; Abdel-Moneim, A.S. Virtual Screening of Repurposed Drugs as Potential Spike Protein Inhibitors of Different SARS-CoV-2 Variants: Molecular Docking Study. Curr. Issues Mol. Biol. 2022, 44, 3018–3029. [Google Scholar] [CrossRef]
- Ashraf, G.M.; DasGupta, D.; Alam, M.Z.; Baeesa, S.S.; Alghamdi, B.S.; Anwar, F.; Alqurashi, T.M.A.; Sharaf, S.E.; Al Abdulmonem, W.; Alyousef, M.A.; et al. Inhibition of Microtubule Affinity Regulating Kinase 4 by Metformin: Exploring the Neuroprotective Potential of Antidiabetic Drug through Spectroscopic and Computational Approaches. Molecules 2022, 27, 4652. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, M.K.; Suriya, U.; Rungrotmongkol, T.; Karuppasamy, R. In Silico Screening of Available Drugs Targeting Non-Small Cell Lung Cancer Targets: A Drug Repurposing Approach. Pharmaceutics 2022, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- García-sosa, A.T.; Maran, U. Combined Naïve Bayesian, Chemical Fingerprints and Molecular Docking Classifiers to Model and Predict Androgen Receptor Binding Data for Environmentally- and Health-Sensitive Substances. Int. J. Mol. Sci. 2021, 22, 6695. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.C.; Bermeo, A.E.C.; Urgilés, V.E.V.; León, M.R.M.; Arévalo, J.M.C. An In-Silico Evaluation of Anthraquinones as Potential Inhibitors of DNA Gyrase B of Mycobacterium Tuberculosis. Microorganisms 2022, 10, 2434. [Google Scholar] [CrossRef]
- Ghufran, M.; Khan, H.A.; Ullah, M.; Ghufran, S.; Ayaz, M.; Siddiq, M.; ul Hassan, S.S.; Bungau, S. In Silico Strategies for Designing of Peptide Inhibitors of Oncogenic K-Ras G12V Mutant: Inhibiting Cancer Growth and Proliferation. Cancers 2022, 14, 4884. [Google Scholar] [CrossRef]
- Xiang, M.; Cao, Y.; Fan, W.; Chen, L.; Mo, Y. Computer-Aided Drug Design: Lead Discovery and Optimization. Comb. Chem. High Throughput Screen. 2012, 15, 328–337. [Google Scholar] [CrossRef]
- Bauer, M.R.; Ibrahim, T.M.; Vogel, S.M.; Boeckler, F.M. Evaluation and Optimization of Virtual Screening Workflows with DEKOIS 2.0—A Public Library of Challenging Docking Benchmark Sets. J. Chem. Inf. Model. 2013, 53, 1447–1462. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; Bauer, M.R.; Boeckler, F.M. Applying DEKOIS 2.0 in Structure-Based Virtual Screening to Probe the Impact of Preparation Procedures and Score Normalization. J. Cheminform. 2015, 7, 21. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.; Li, L.; Cao, J.; Guo, Y.; Wu, Y.; Gao, W. Fascin Actin-Bundling Protein 1 in Human Cancer: Promising Biomarker or Therapeutic Target? Mol. Ther.—Oncolytics 2021, 20, 240–264. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, New Empirical Halogen Bond Scoring Function, into AutoDock Vina. J. Cheminform. 2016, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.M.; Bauer, M.R.; Boeckler, F.M. DEKOIS: Demanding Evaluation Kits for Objective in Silico Screening—A Versatile Tool for Benchmarking Docking Programs and Scoring Functions. J. Chem. Inf. Model. 2011, 51, 2650–2665. [Google Scholar] [CrossRef]
- Boeckler, F.M.; Bauer, M.R.; Ibrahim, T.M.; Vogel, S.M. Use of DEKOIS 2.0 to Gain Insights for Virtual Screening. J. Cheminform. 2014, 6, O24. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.M.; Bauer, M.R.; Boeckler, F.M. Probing the Impact of Protein and Ligand Preparation Procedures on Chemotype Enrichment in Structure-Based Virtual Screening Using DEKOIS 2.0 Benchmark Sets. J. Cheminform. 2014, 6, P19. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; Bauer, M.R.; Dörr, A.; Veyisoglu, E.; Boeckler, F.M. PROC-Chemotype Plots Enhance the Interpretability of Benchmarking Results in Structure-Based Virtual Screening. J. Chem. Inf. Model. 2015, 55, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; Moumbock, A.F.A.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A Resource for Natural Products from Northern African Sources. J. Nat. Prod. 2017, 80, 2067–2076. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir Is a Direct-Acting Antiviral That Inhibits RNA-Dependent RNA Polymerase from Severe Acute Respiratory Syndrome Coronavirus 2 with High Potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef]
- Tevaarwerk, A.J.; Kolesar, J.M. Lapatinib: A Small-Molecule Inhibitor of Epidermal Growth Factor Receptor and Human Epidermal Growth Factor Receptor-2 Tyrosine Kinases Used in the Treatment of Breast Cancer. Clin. Ther. 2009, 31, 2332–2348. [Google Scholar] [CrossRef]
- Dicpinigaitis, P.V.; Gayle, Y.E. Effect of the Second-Generation Antihistamine, Fexofenadine, on Cough Reflex Sensitivity and Pulmonary Function. Br. J. Clin. Pharmacol. 2003, 56, 501. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razik, A.F.; Nassar, M.I.; Elshamy, A.I.; Kubacy, T.M.; Hegazy, M.E.F.; Ibrahim, N.; Le Lamer, A.C.; Farrag, A.R.H. A New Cytotoxic Ceramide from Heteroxenia Ghardaqensis and Protective Effect of Chloroform Extract against Cadmium Toxicity in Rats. Arab. J. Chem. 2016, 9, 649–655. [Google Scholar] [CrossRef]
- Darwish, F.M.M.; Reinecke, M.G. Ecdysteroids and other constituents from Sida spinosa L. Phytochemistry 2003, 62, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.A. Unique phenolic carboxylic acids from Sanguisorba minor. Phytochemistry 2003, 63, 433–436. [Google Scholar] [CrossRef]
- Pandey, P.; Rane, J.S.; Chatterjee, A.; Kumar, A.; Khan, R.; Prakash, A.; Ray, S. Targeting SARS-CoV-2 Spike Protein of COVID-19 with Naturally Occurring Phytochemicals: An in Silico Study for Drug Development. J. Biomol. Struct. Dyn. 2021, 39, 6306–6316. [Google Scholar] [CrossRef]
- Pandey, P.; Prasad, K.; Prakash, A.; Kumar, V. Insights into the Biased Activity of Dextromethorphan and Haloperidol towards SARS-CoV-2 NSP6: In Silico Binding Mechanistic Analysis. J. Mol. Med. 2020, 98, 1659–1673. [Google Scholar] [CrossRef]
- Abdelkader, A.; Elzemrany, A.A.; El-Nadi, M.; Elsabbagh, S.A.; Shehata, M.A.; Eldehna, W.M.; El-Hadidi, M.; Ibrahim, T.M. In-Silico Targeting of SARS-CoV-2 NSP6 for Drug and Natural Products Repurposing. Virology 2022, 573, 96–110. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); V. 2019.01; Chemical Computing Group Inc.: Montreal, QC, Canada; Available online: https://www.chemcomp.com/ (accessed on 19 October 2022).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Ten Brink, T.; Exner, T.E. Influence of Protonation, Tautomeric, and Stereoisomeric States on Protein-Ligand Docking Results. J. Chem. Inf. Model. 2009, 49, 1535–1546. [Google Scholar] [CrossRef]
- Ten Brink, T.; Exner, T.E. PKa Based Protonation States and Microspecies for Protein-Ligand Docking. J. Comput. Aided. Mol. Des. 2010, 24, 935–942. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner; Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. [Google Scholar] [CrossRef]
- Clark, R.D.; Webster-Clark, D.J. Managing Bias in ROC Curves. J. Comput. Aided. Mol. Des. 2008, 22, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.Q.; Baase, W.A.; Weaver, L.H.; Matthews, B.W.; Shoichet, B.K. A Model Binding Site for Testing Scoring Functions in Molecular Docking. J. Mol. Biol. 2002, 322, 339–355. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 4.6.0; Schrödinger, LLC: New York, NY, USA; Available online: https://pymol.org (accessed on 19 October 2022).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1998, 81, 3684. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1998, 52, 7182. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1998, 98, 10089. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 14631472. [Google Scholar] [CrossRef]
- Huang, J.; Mackerell, A.D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for Bridging Protein Sequence Evolution and Structural Dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Sedeh, R.S.; Fedorov, A.A.; Fedorov, E.V.; Ono, S.; Matsumura, F.; Almo, S.C.; Bathe, M. Structure, Evolutionary Conservation, and Conformational Dynamics of Homo Sapiens Fascin-1, an F-Actin Crosslinking Protein. J. Mol. Biol. 2010, 400, 589–604. [Google Scholar] [CrossRef]
- Jansen, S.; Collins, A.; Yang, C.; Rebowski, G.; Svitkina, T.; Dominguez, R. Mechanism of Actin Filament Bundling by Fascin. J. Biol. Chem. 2011, 286, 30087–30096. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold/Cluster | Structure | Chemical Name | IC50 (µM) | Kd (µM) | Ref. |
|---|---|---|---|---|---|
| Indazole /Cluster 1 |  | 2-methyl-N-(1-(4-(trifluoromethyl)benzyl)-1H-indazol-3-yl)furan-3-carboxamide | 0.2 | nd | [23] |
 | 4-methyl-N-(1-(4-(trifluoromethyl)benzyl)-1H-indazol-3-yl)isoxazole-5-carboxamide | 0.19 | nd | [24] | |
| N-Phenylacetamide /Cluster 2 |  | N-(2,4-dichlorophenyl)-N-methylacetamide | nd | 92 | [27] |
| Pyrazolo[3,4-d]pyrimidin-4-one /Cluster 3 |  | 1-[(3~{R})-1,1-bis(oxidanylidene)thiolan-3-yl]-5-[(3,4-dichlorophenyl)methyl]pyrazolo [3,4-d]pyrimidin-4-one | 67.6 | 29.5 | |
| Isoquinolone /Cluster 4 |  | ~{N}-(1-methylpyrazol-4-yl)-1-oxidanylidene-2-(phenylmethyl)isoquinoline-4-carboxamide | 67.9 | 29.3 | |
 | 2-[(4-chlorophenyl)methyl]-~{N}-(1-methylpyrazol-4-yl)-1-oxidanylidene-isoquinoline-4-carboxamide | 4.6 | 2.7 | ||
 | 2-[(3-chlorophenyl)methyl]-~{N}-(1-methylpyrazol-4-yl)-1-oxidanylidene-isoquinoline-4-carboxamide | 11.4 | 7.6 | ||
 | 2-[(3,4-dichlorophenyl)methyl]-~{N}-(1-methylpyrazol-4-yl)-1-oxidanylidene-isoquinoline-4-carboxamide | 1.3 | 1.5 | ||
 | 2-(4-fluorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-1,2-dihydroisoquinoline-4-carboxamide | nd | 2.7 | ||
 | 2-(3-fluorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-1,2-dihydroisoquinoline-4-carboxamide | nd | 29.2 | ||
 | 2-(3-chloro-4-fluorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-1,2-dihydroisoquinoline-4-carboxamide | 2.1 | 1.2 | ||
 | 2-(4-fluoro-3-isocyanobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-1,2-dihydroisoquinoline-4-carboxamide | 8.6 | 6.6 | ||
 | 2-(2,4-difluorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-1,2-dihydroisoquinoline-4-carboxamide | nd | 46 | ||
| Naphthyridone /Cluster 5 |  | 2-[(3,4-dichlorophenyl)methyl]-~{N}-(1-methylpyrazol-4-yl)-1-oxidanylidene-6-piperidin-4-yl-2,7-naphthyridine-4-carboxamide | 0.51 | 0.25 | |
 | 2-(3,4-dichlorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-6-(pyrrolidin-1-yl)-1,2-dihydro-2,7-naphthyridine-4-carboxamide | nd | 1.03 | ||
 | 2-(3,4-dichlorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-6-(piperazin-1-yl)-1,2-dihydro-2,7-naphthyridine-4-carboxamide | 0.63 | 0.58 | ||
 | 2-(3,4-dichlorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-1,2-dihydro-2,6-naphthyridine-4-carboxamide | 5.3 | 1.6 | ||
 | 2-(3,4-dichlorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-1-oxo-1,2-dihydro-2,7-naphthyridine-4-carboxamide | 3.8 | 1.3 | ||
 | 2-(3,4-dichlorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-6-(methylamino)-1-oxo-1,2-dihydro-2,7-naphthyridine-4-carboxamide | nd | 1.1 | ||
 | 2-(3,4-dichlorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-6-(N-methylmethylsulfonamido)-1-oxo-1,2-dihydro-2,7-naphthyridine-4-carboxamide | nd | 2 | ||
| Pyrazolo[4,3-c]pyridine /Cluster 6 |  | 5-(3,4-dichlorobenzyl)-4-oxo-1-(piperidin-4-yl)-N-(pyridin-4-yl)-4,5-dihydro-1H-pyrazolo [4,3-c]pyridine-7-carboxamide | 0.24 | 0.09 | |
 | 5-(3,4-dichlorobenzyl)-N-(1-methyl-1H-pyrazol-4-yl)-4-oxo-4,5-dihydro-1H-pyrazolo [4,3-c]pyridine-7-carboxamide | 10.4 | 10 | ||
 | 5-(3,4-dichlorobenzyl)-1-(1,1-dioxidotetrahydrothiophen-3-yl)-N-(1-methyl-1H-pyrazol-4-yl)-4-oxo-4,5-dihydro-1H-pyrazolo [4,3-c]pyridine-7-carboxamide | 0.6 | 0.6 | ||
 | 5-(3,4-dichlorobenzyl)-1-(1,1-dioxidotetrahydrothiophen-3-yl)-4-oxo-N-(pyridin-4-yl)-4,5-dihydro-1H-pyrazolo [4,3-c]pyridine-7-carboxamide | 0.33 | 0.27 | ||
| Pyridone /Cluster 7 |  | 5-amino-1-(3,4-dichlorobenzyl)-6-oxo-N-(1-(pyrrolidin-3-yl)-1H-pyrazol-4-yl)-1,6-dihydropyridine-3-carboxamide | <100 | 21 |
| Docking Rank | Drug | Docking Score | M.wt. | Drugbank ID | Status |
|---|---|---|---|---|---|
| 1 | Remdesivir | −124.43 | 602.59 | DB14761 | Approved; investigational |
| 2 | Lapatinib | −119.231 | 581.06 | DB01259 | Approved; investigational |
| 3 | Fexofenadine | −119.101 | 501.66 | DB00950 | Approved; investigational |
| 4 | Latanoprost | −118.59 | 432.59 | DB00654 | Approved; investigational |
| 5 | Almitrine | −118.311 | 477.55 | DB01430 | Approved |
| 6 | Fulvestrant | −116.406 | 606.78 | DB00947 | Approved; investigational |
| 7 | Travoprost | −116.314 | 500.55 | DB00287 | Approved |
| 8 | Indinavir | −115.639 | 613.79 | DB00224 | Approved |
| 9 | Vilazodone | −114.94 | 441.52 | DB06684 | Approved |
| 10 | Oxetacaine | −114.377 | 467.65 | DB12532 | Approved; investigational |
| 11 | Bimatoprost | −113.888 | 415.57 | DB00905 | Approved; investigational |
| 12 | Imatinib | −113.493 | 493.603 | DB00619 | Approved |
| 13 | Dopexamine | −113.319 | 356.502 | DB12313 | Approved; investigational |
| 14 | Doconexent | −113.105 | 328.488 | DB03756 | Approved; investigational |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, H.H.A.; Ismail, M.I.; Abourehab, M.A.S.; Boeckler, F.M.; Ibrahim, T.M.; Arafa, R.K. In Silico Targeting of Fascin Protein for Cancer Therapy: Benchmarking, Virtual Screening and Molecular Dynamics Approaches. Molecules 2023, 28, 1296. https://doi.org/10.3390/molecules28031296
Hassan HHA, Ismail MI, Abourehab MAS, Boeckler FM, Ibrahim TM, Arafa RK. In Silico Targeting of Fascin Protein for Cancer Therapy: Benchmarking, Virtual Screening and Molecular Dynamics Approaches. Molecules. 2023; 28(3):1296. https://doi.org/10.3390/molecules28031296
Chicago/Turabian StyleHassan, Heba H. A., Muhammad I. Ismail, Mohammed A. S. Abourehab, Frank M. Boeckler, Tamer M. Ibrahim, and Reem K. Arafa. 2023. "In Silico Targeting of Fascin Protein for Cancer Therapy: Benchmarking, Virtual Screening and Molecular Dynamics Approaches" Molecules 28, no. 3: 1296. https://doi.org/10.3390/molecules28031296
APA StyleHassan, H. H. A., Ismail, M. I., Abourehab, M. A. S., Boeckler, F. M., Ibrahim, T. M., & Arafa, R. K. (2023). In Silico Targeting of Fascin Protein for Cancer Therapy: Benchmarking, Virtual Screening and Molecular Dynamics Approaches. Molecules, 28(3), 1296. https://doi.org/10.3390/molecules28031296