
Docking, Binding Free Energy Calculations and In Vitro Characterization of Pyrazine Linked 2-Aminobenzamides as Novel Class I Histone Deacetylase (HDAC) Inhibitors

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
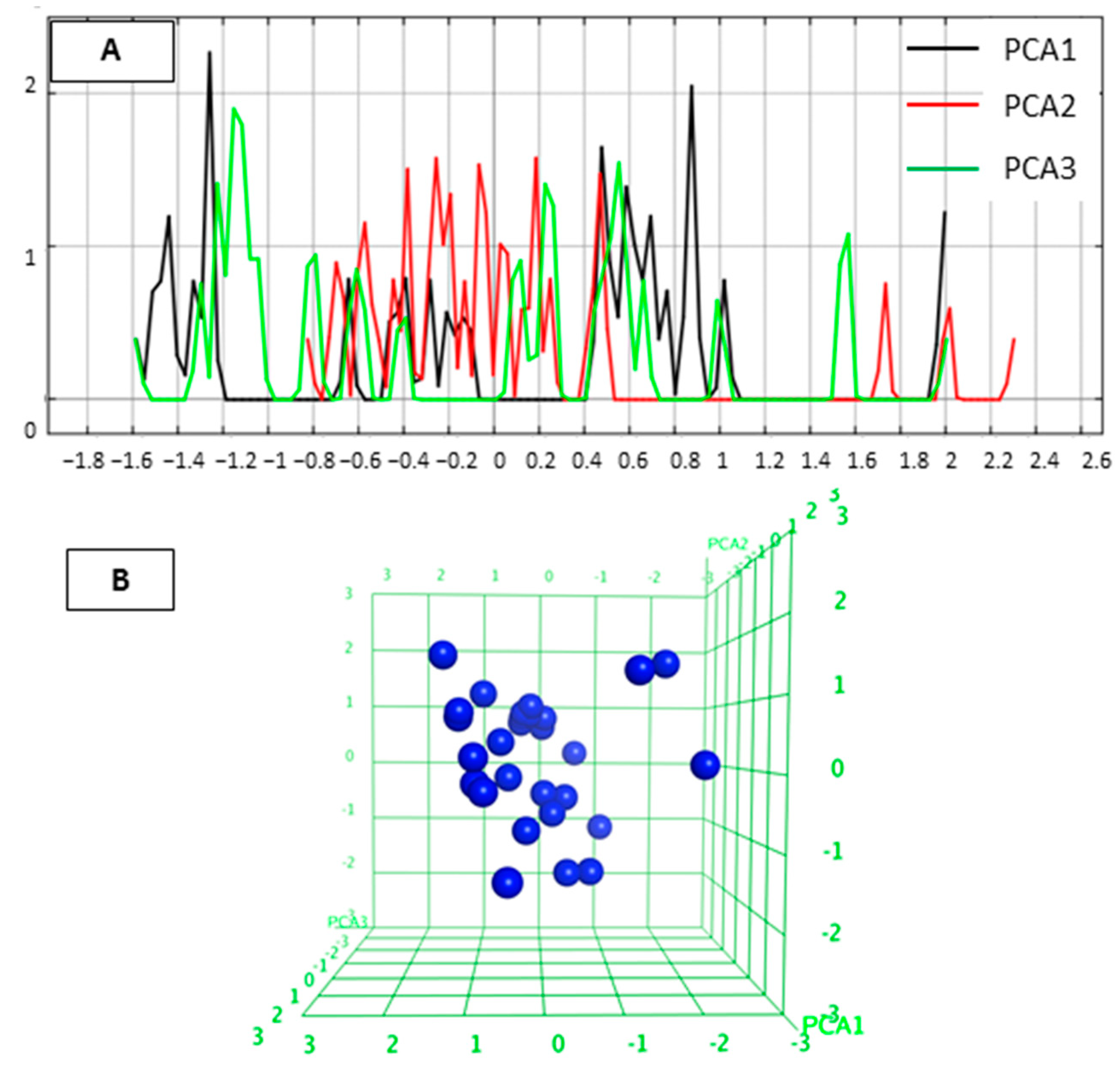
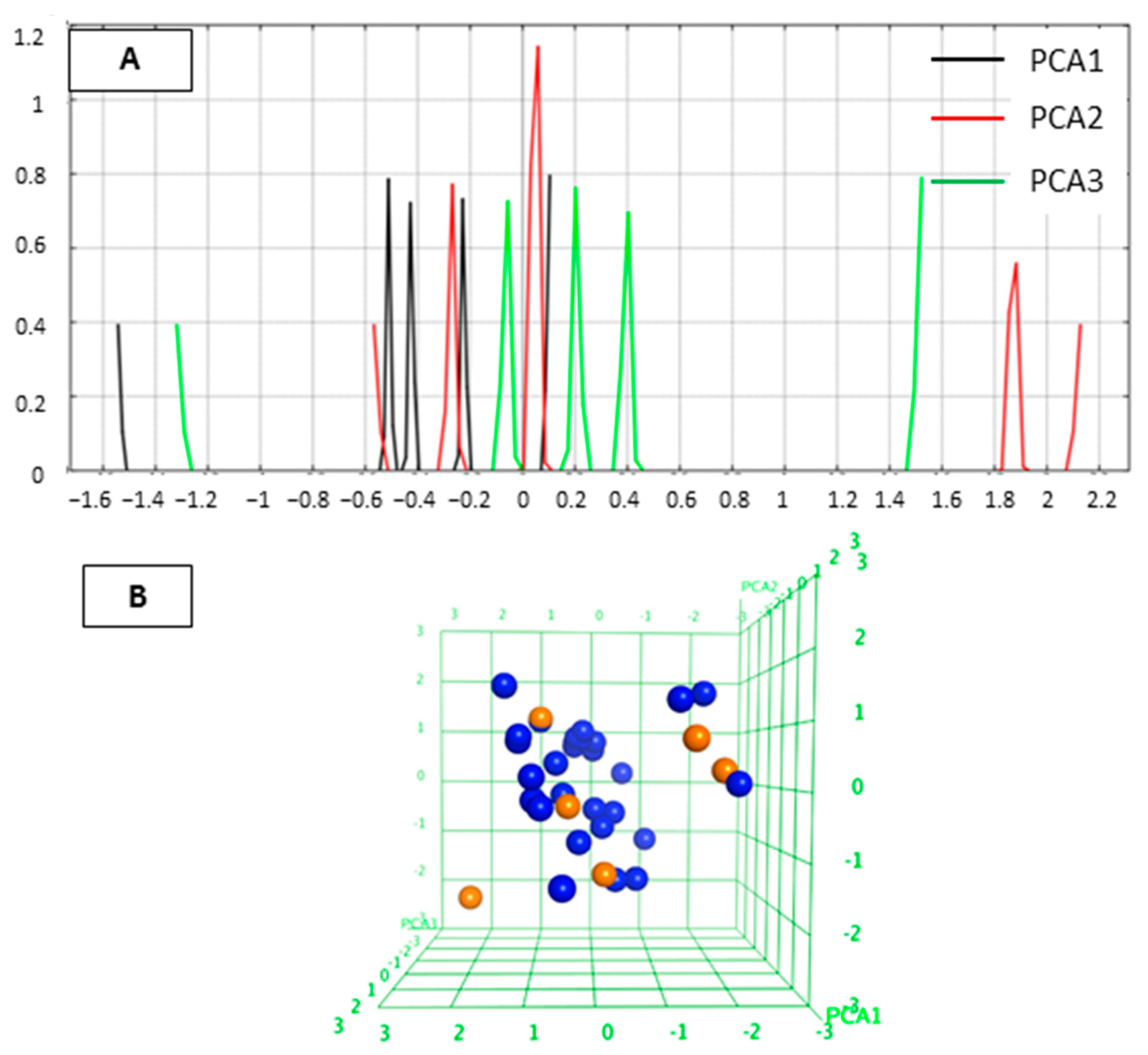
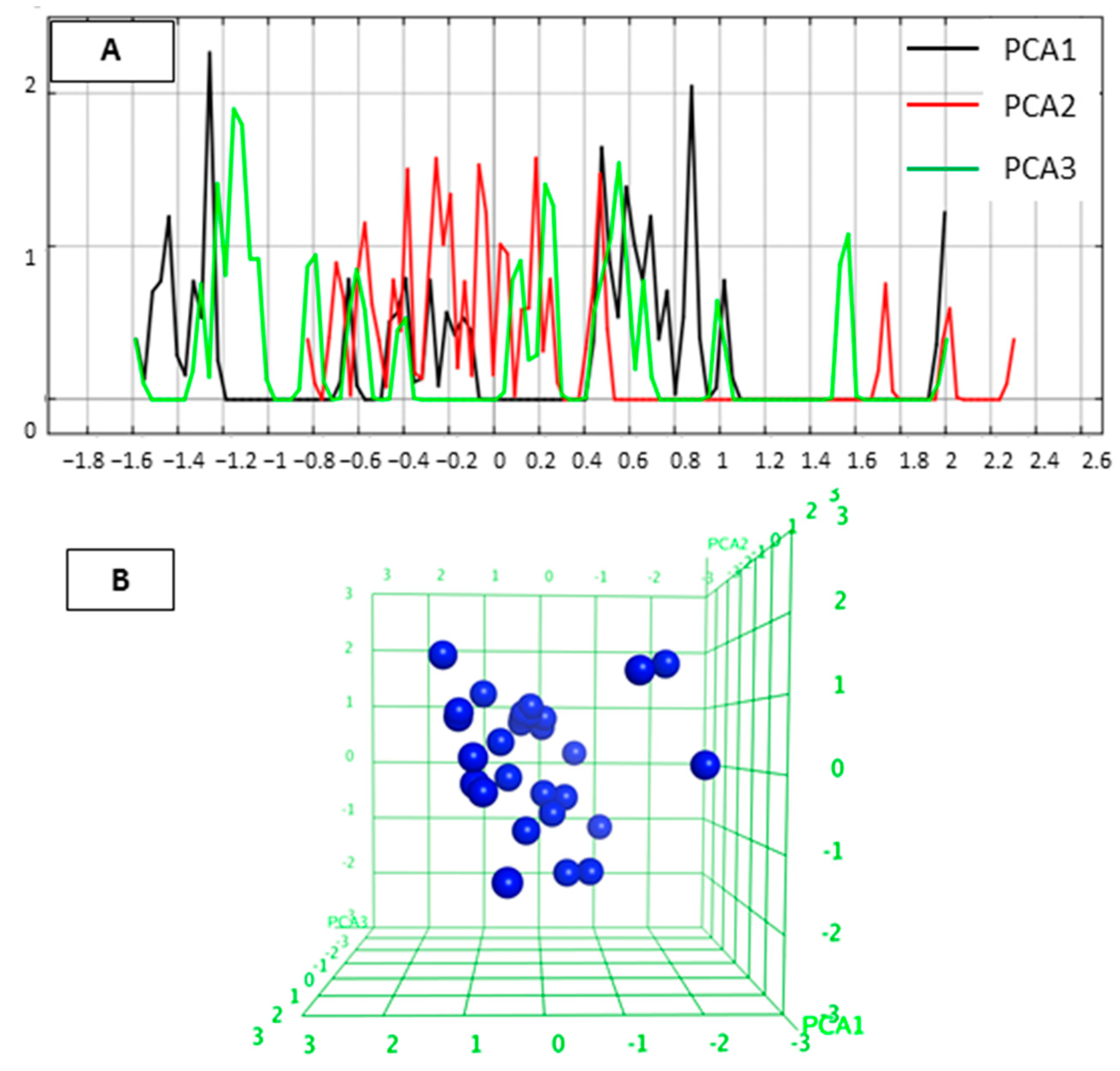
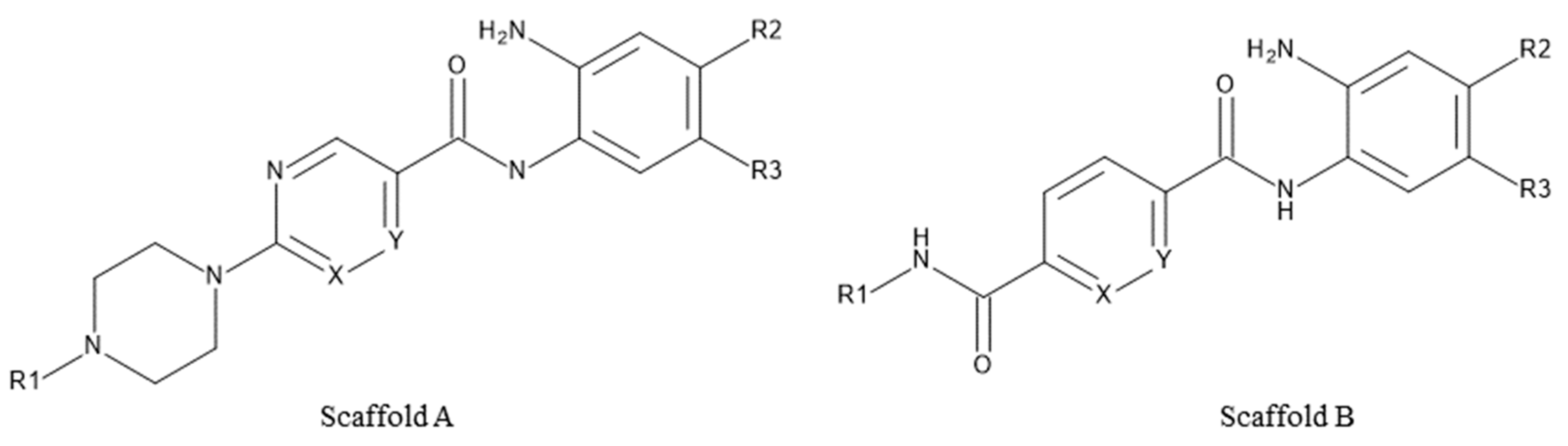
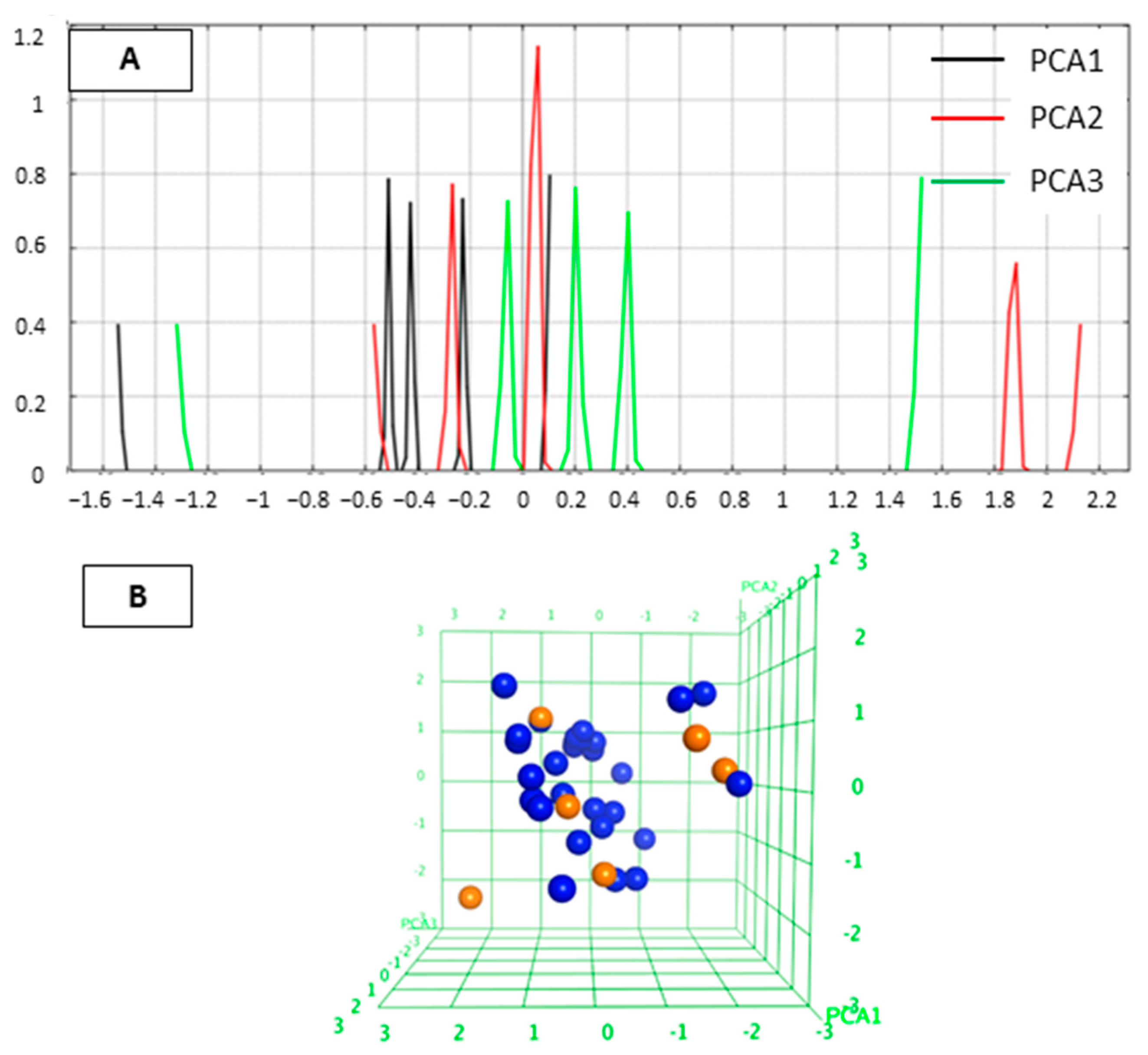
2.1. Diversity Analysis of Studied Dataset
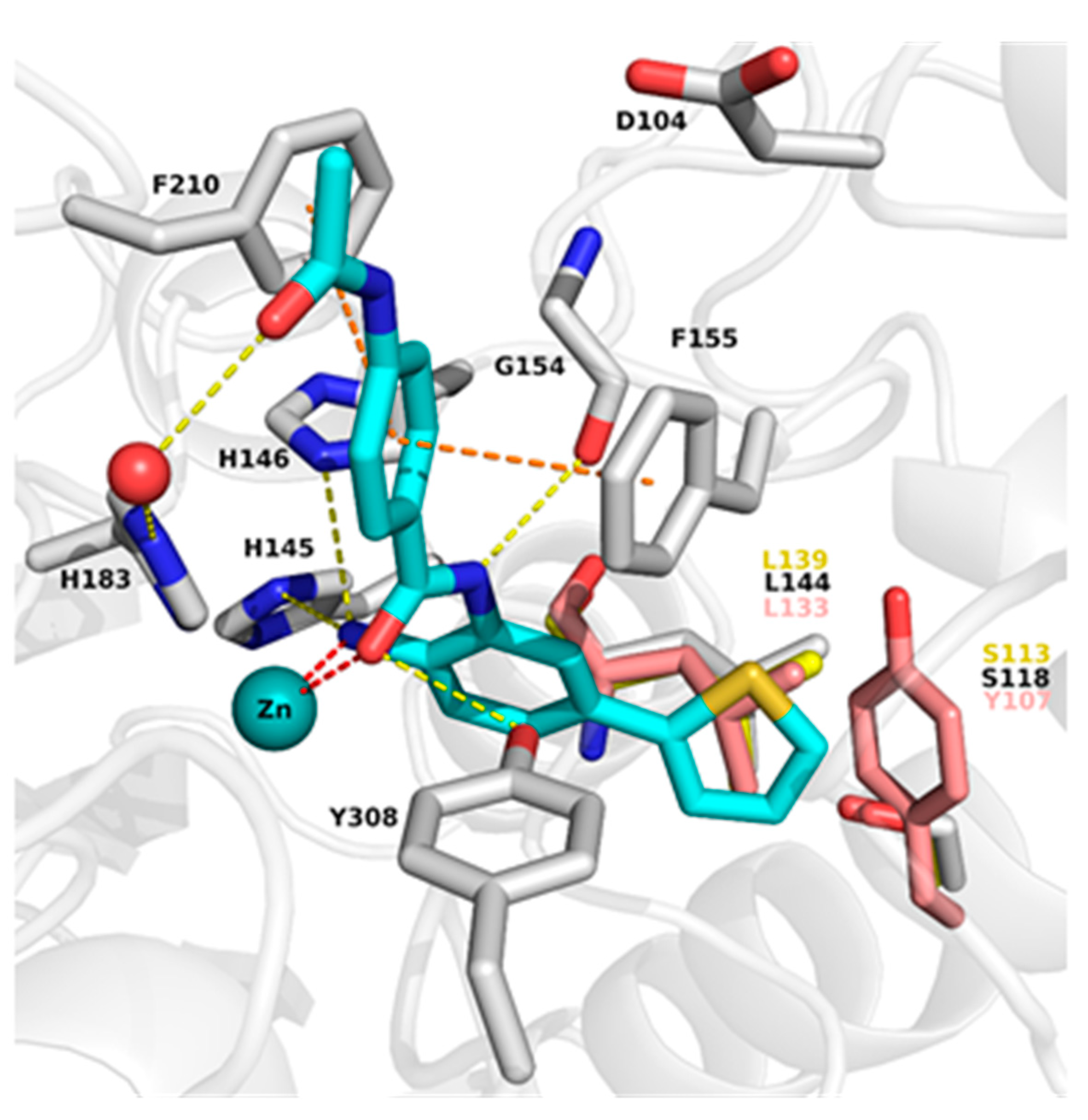
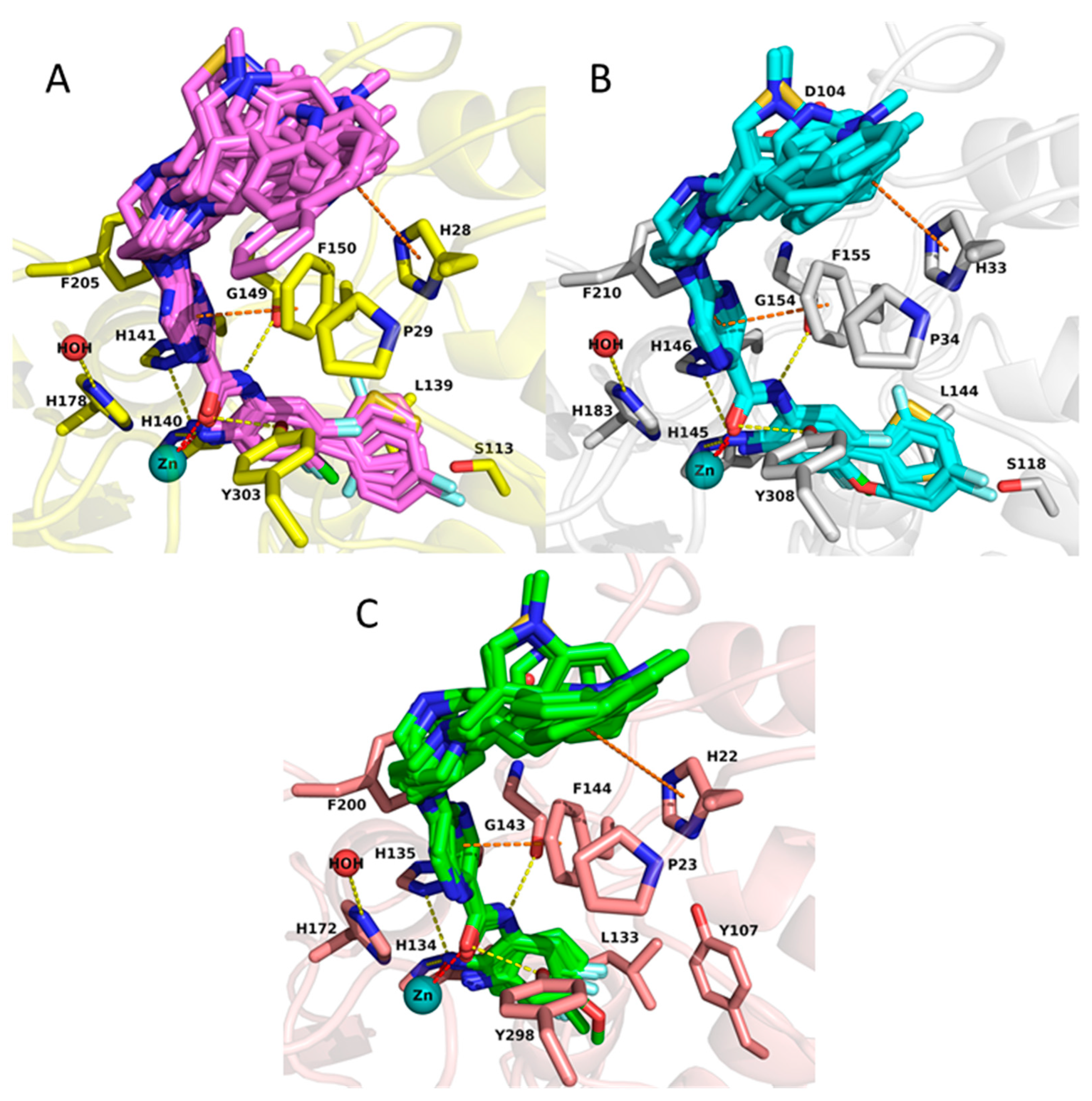
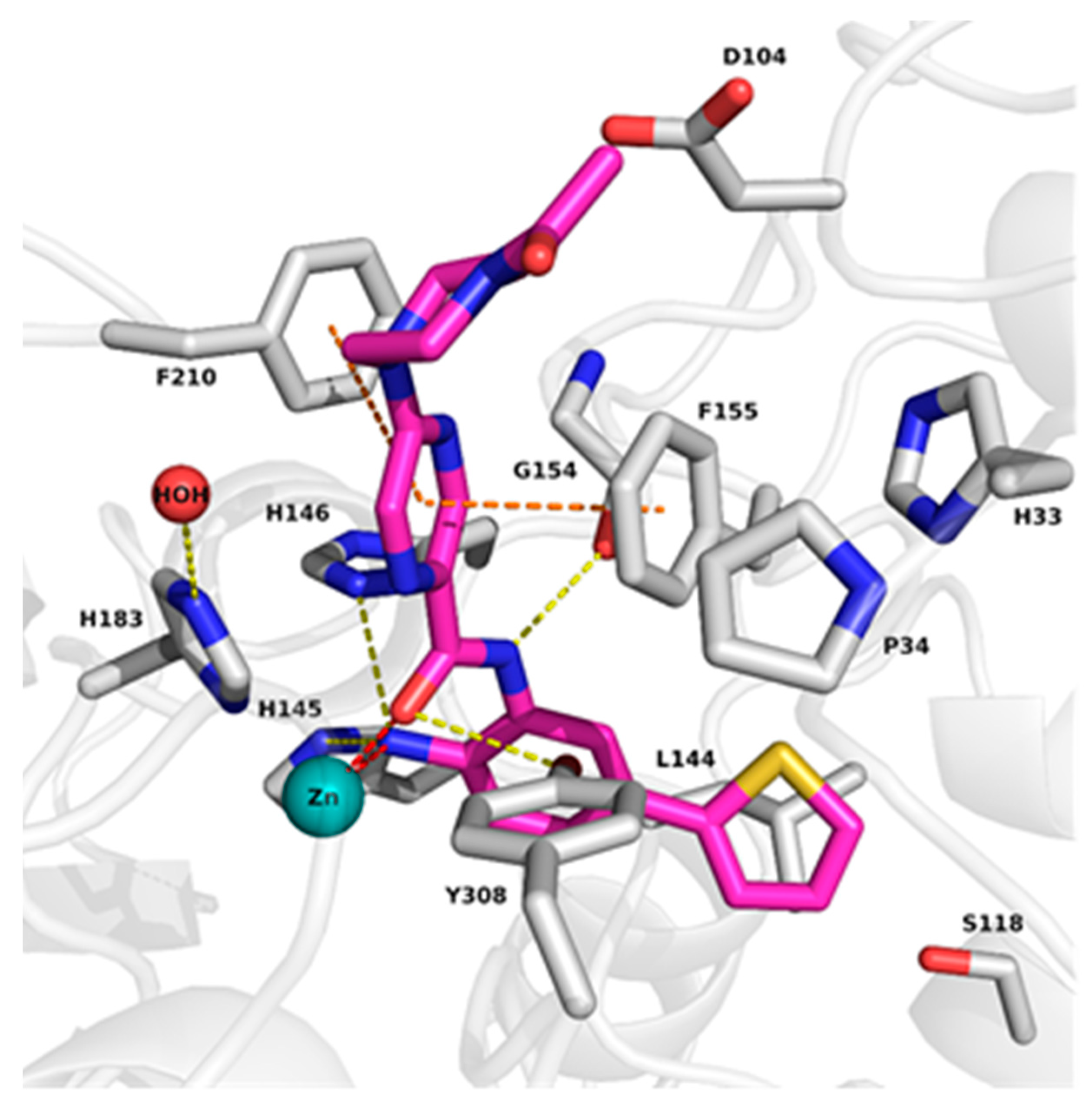
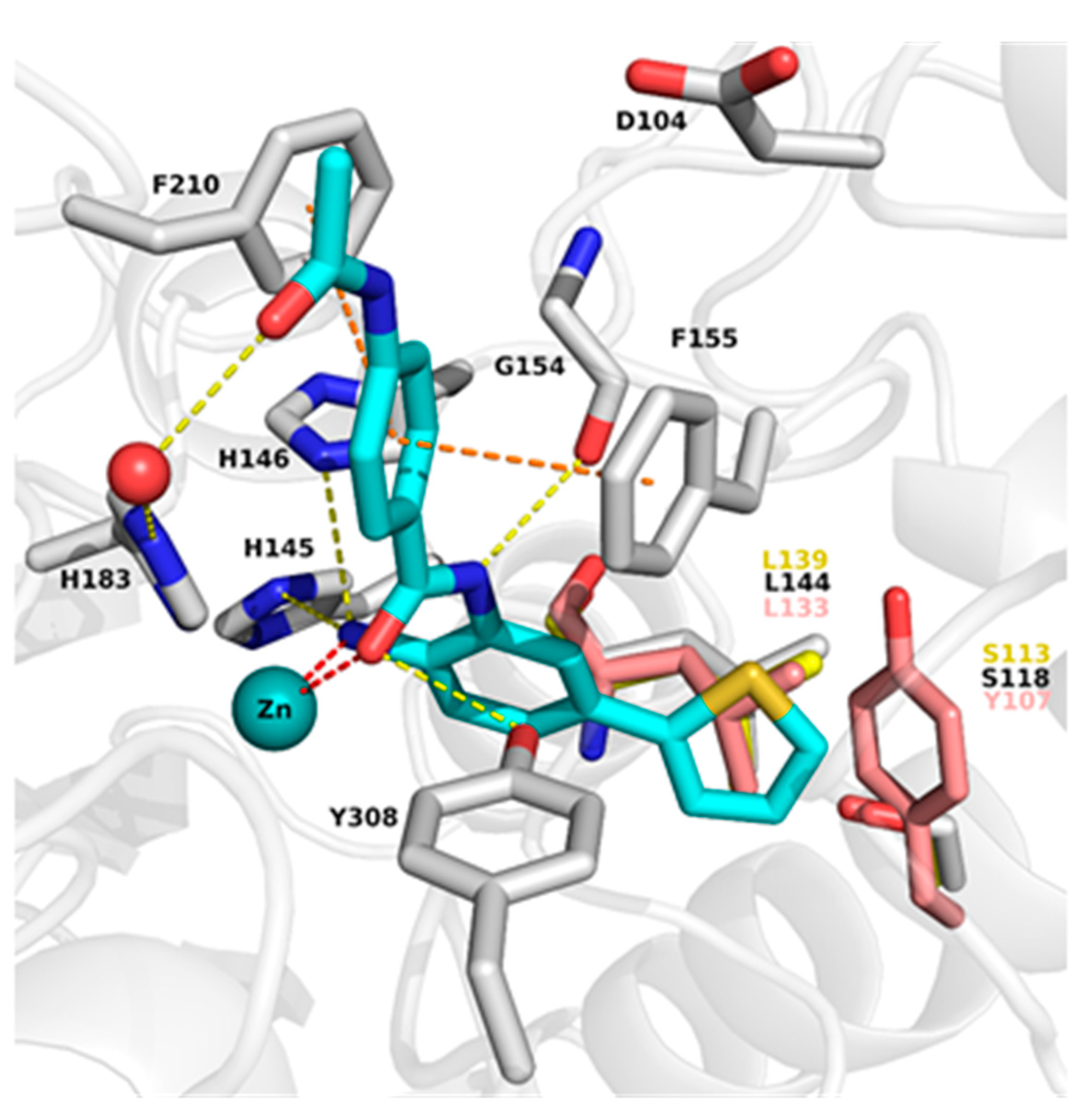
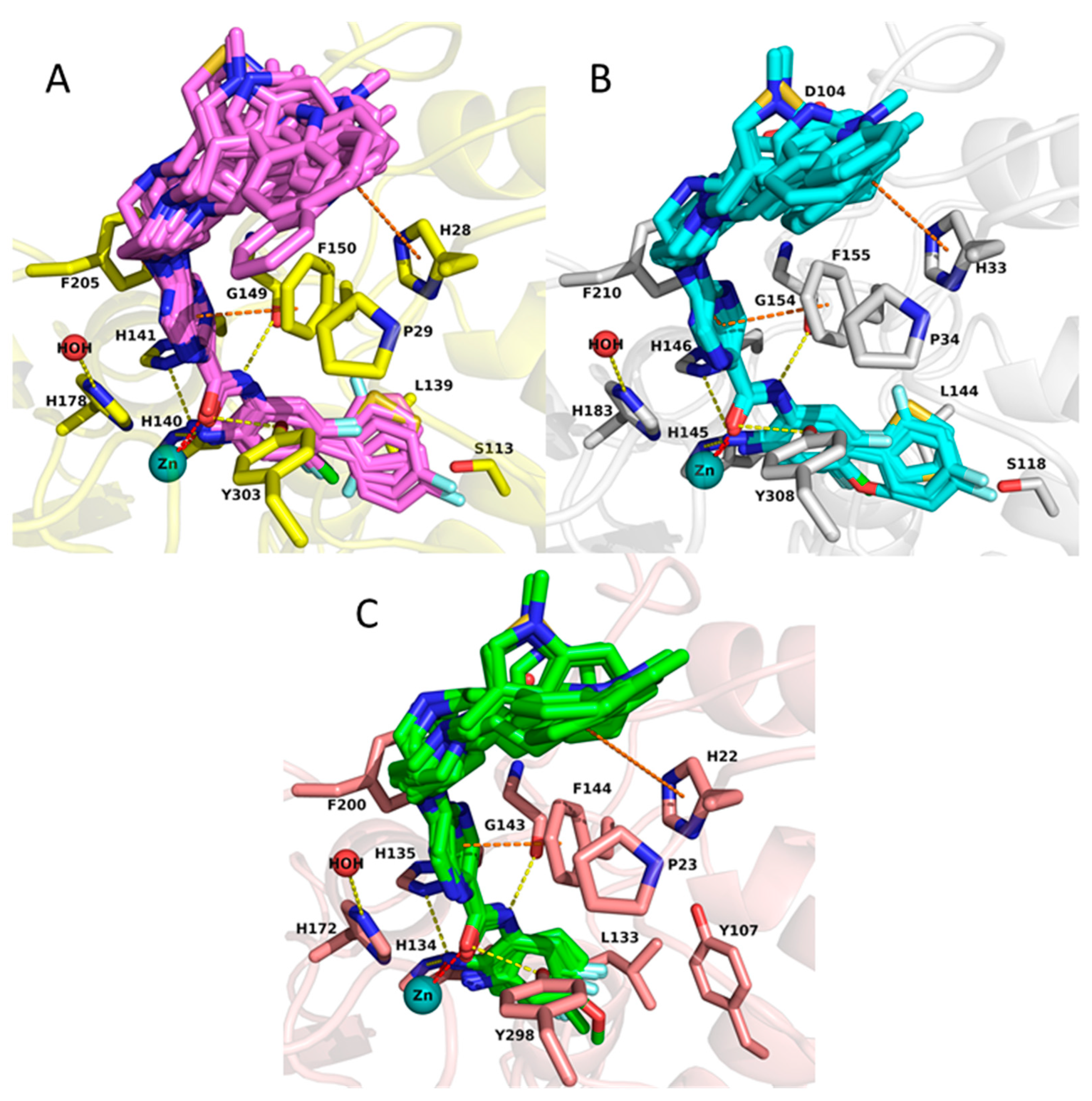
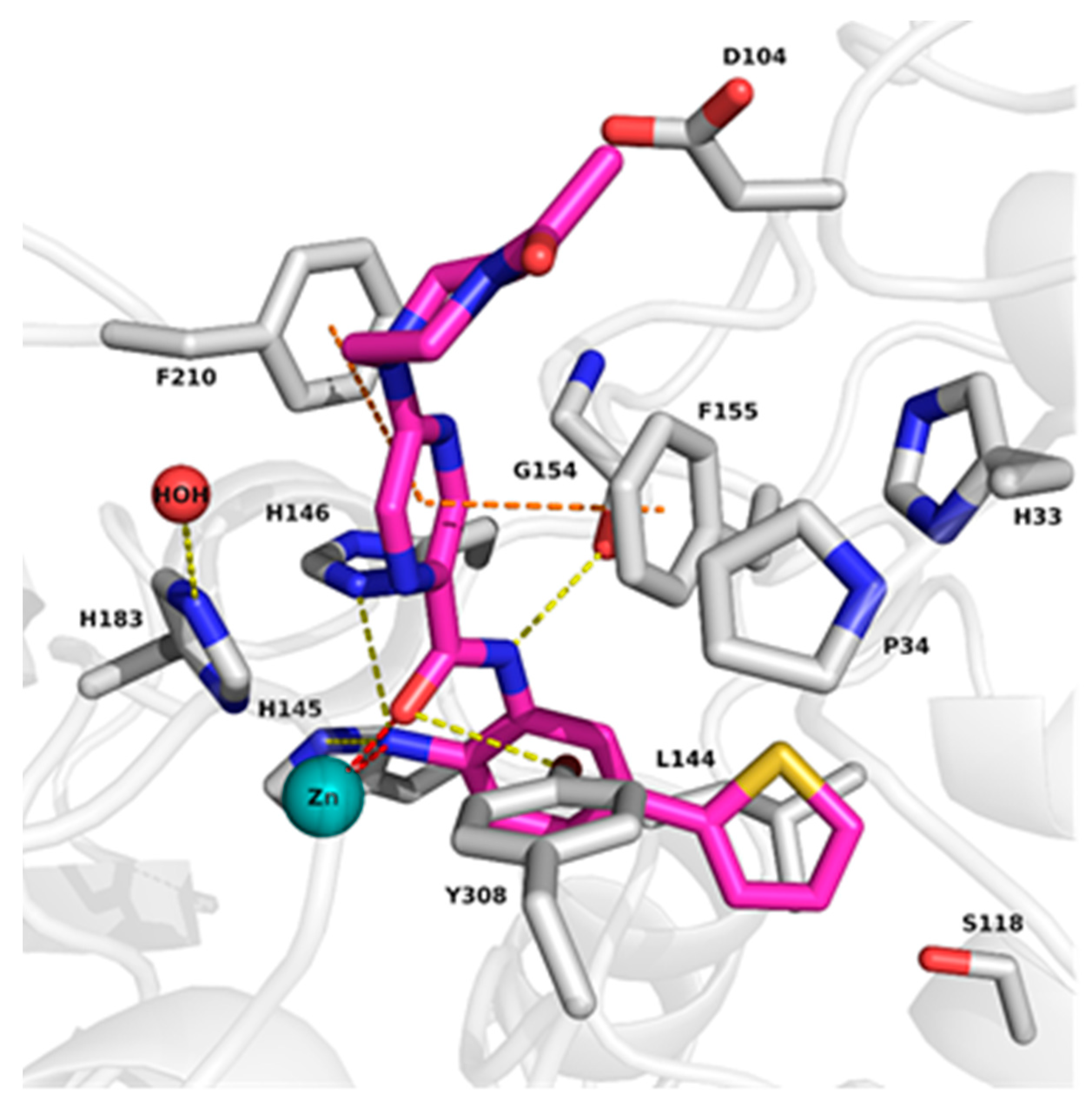
2.2. Analysis of Protein-Inhibitor Complexes
2.3. Docking Results of the Training Set
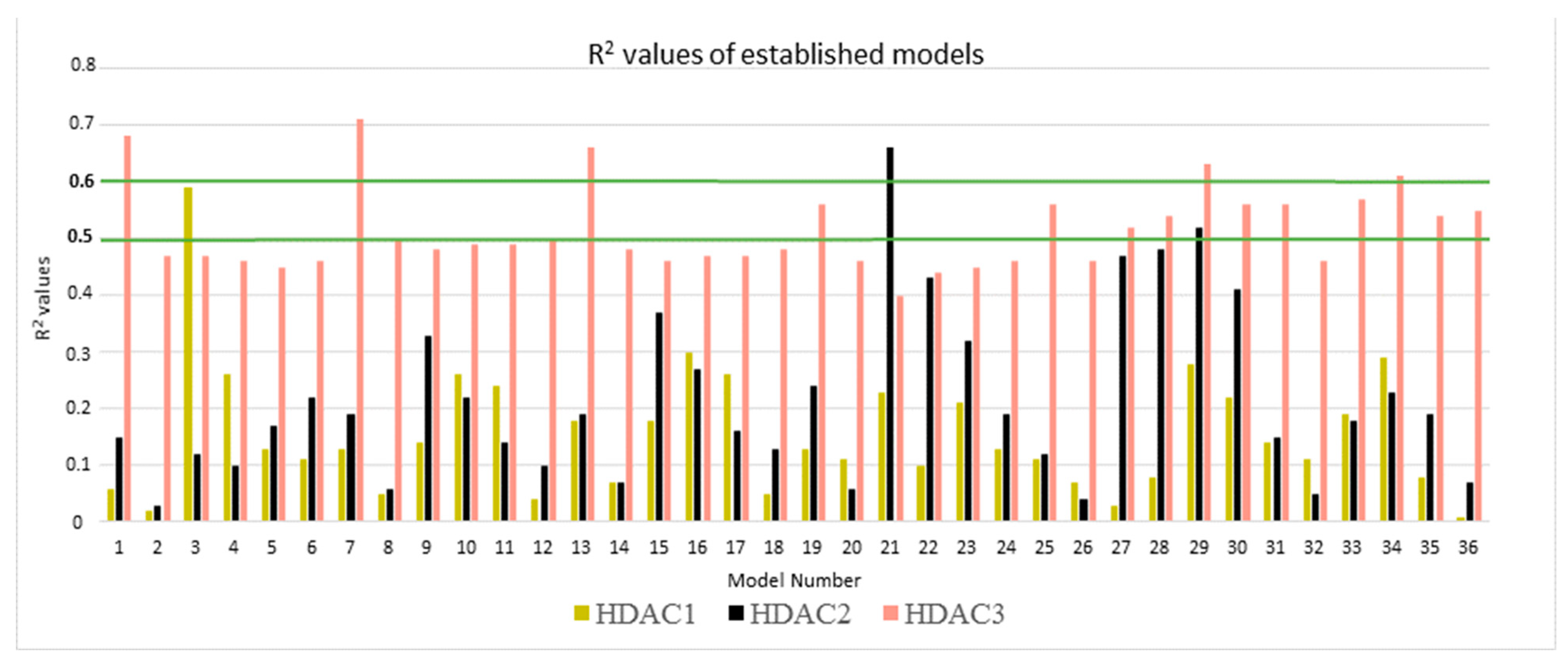
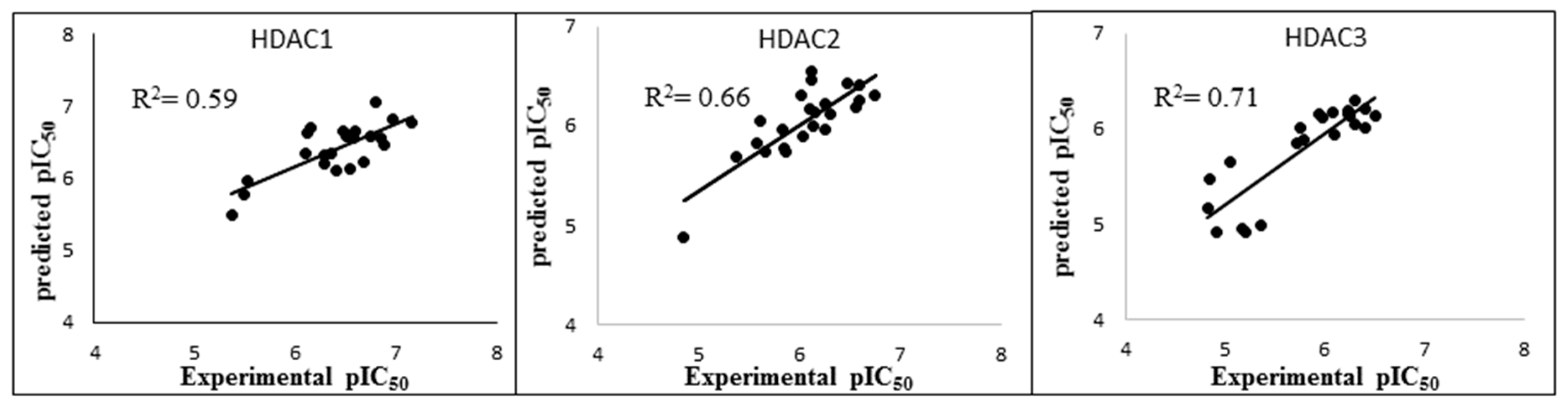
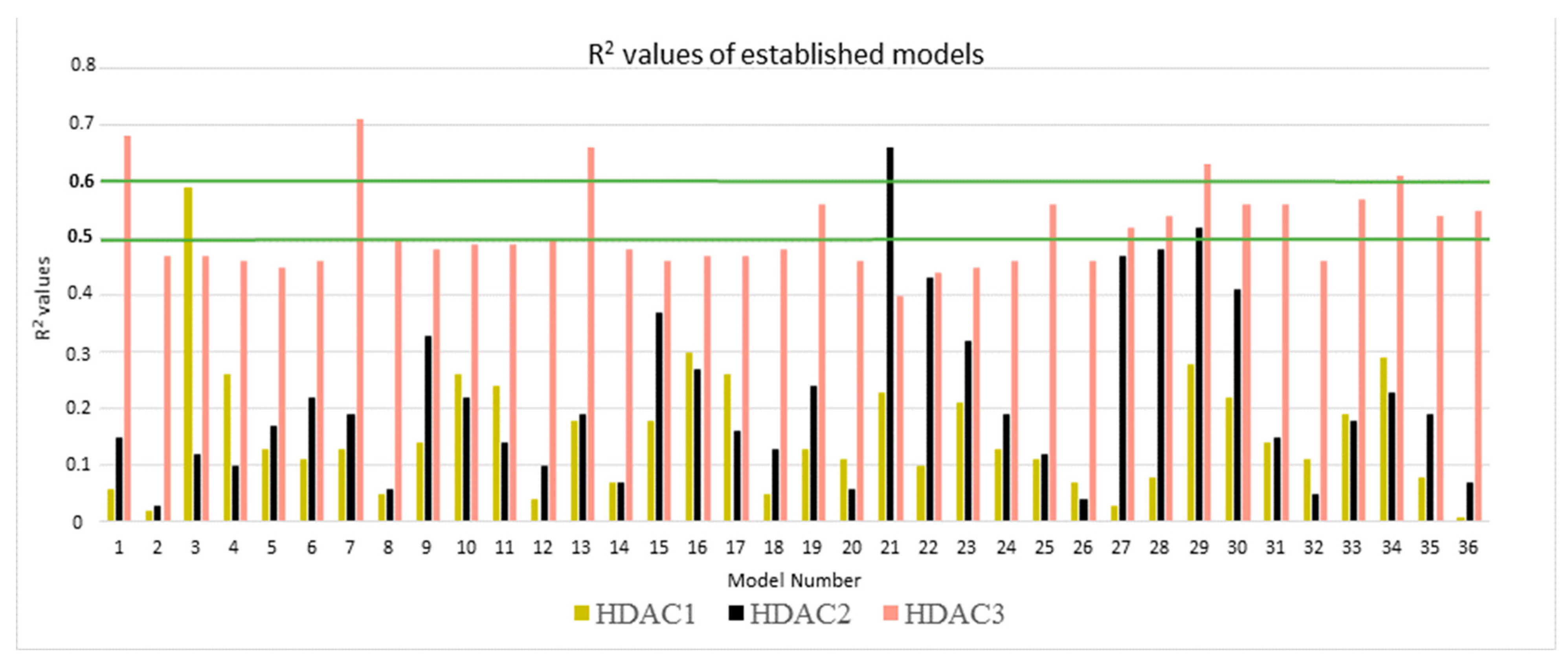
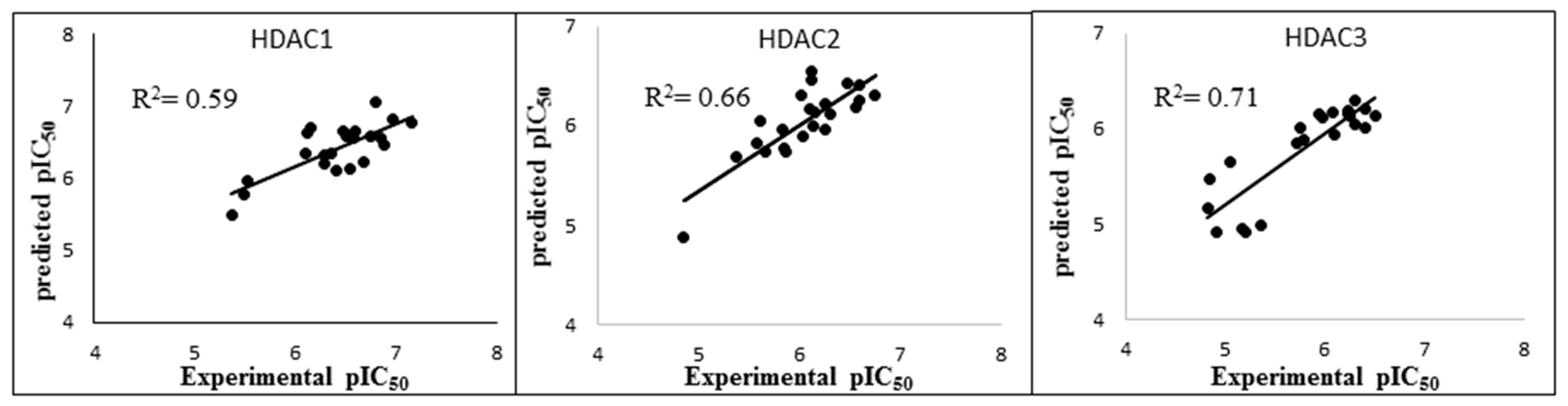
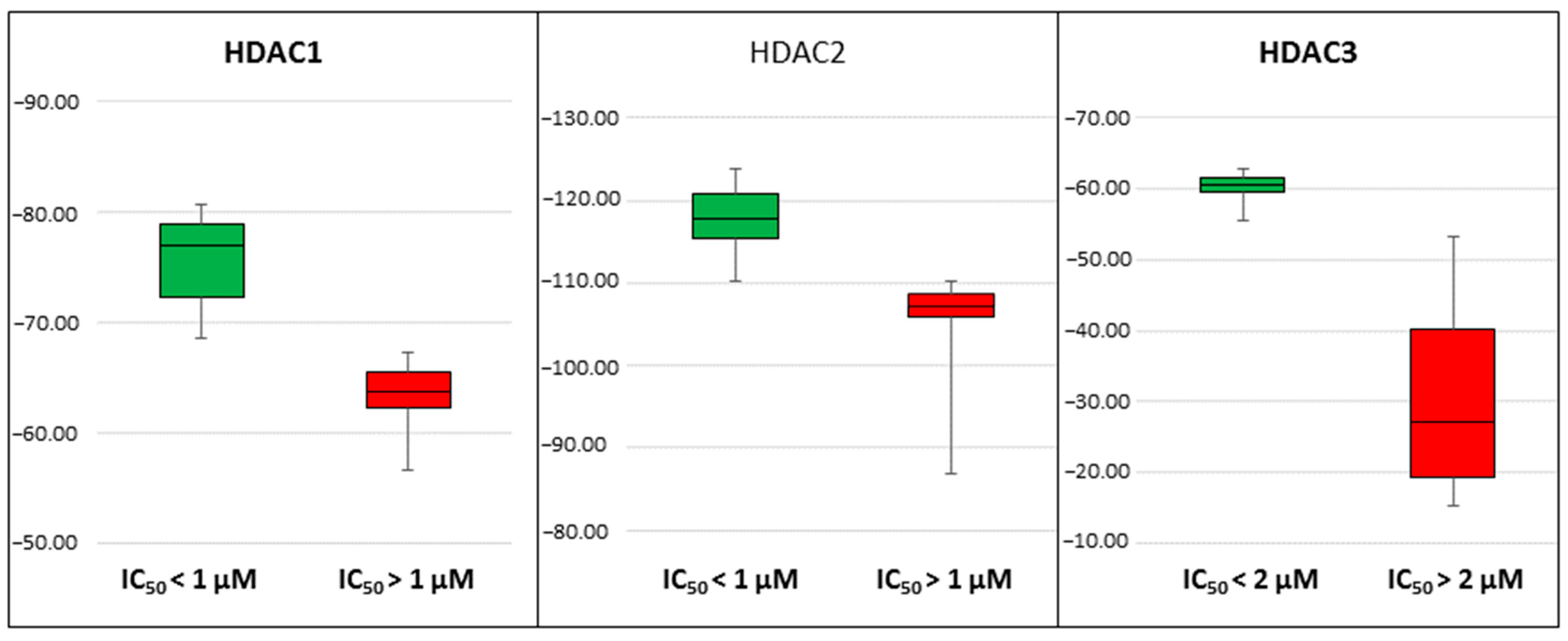
2.4. Rescoring of Docking Poses Using MM-GB/SA and MM-PB/SA
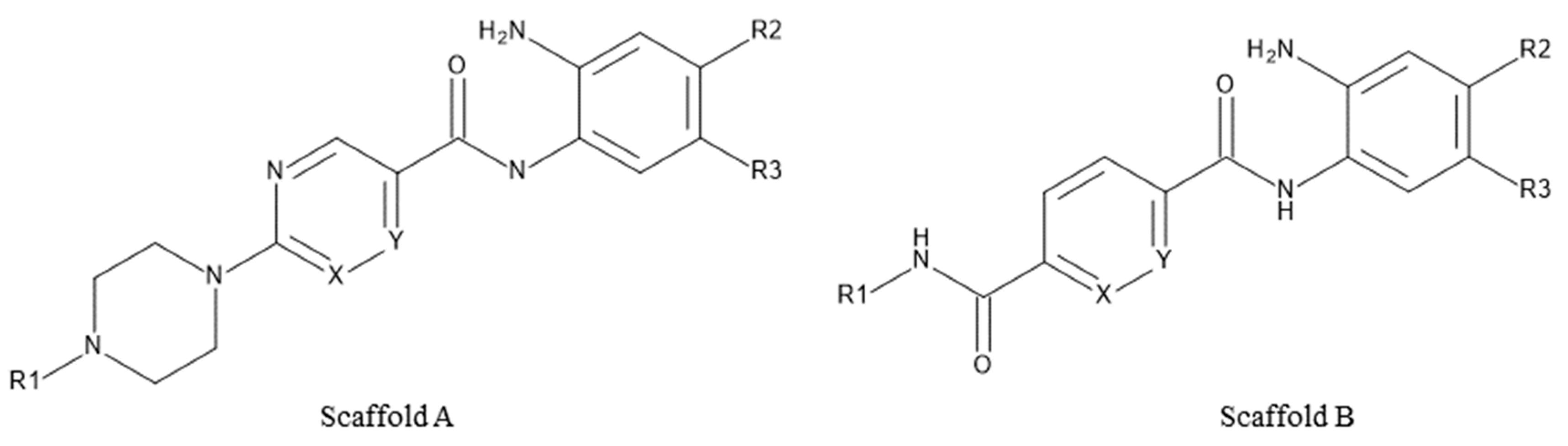
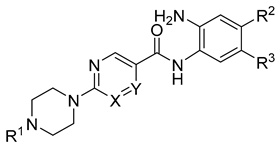
2.5. Evaluation of Novel Designed HDAC1-2-3 Inhibitors
3. Discussion
4. Materials and Methods
4.1. Training Set and Diversity Analysis
4.2. Preparation of Protein-Inhibitor Complexes
4.3. Molecular Docking
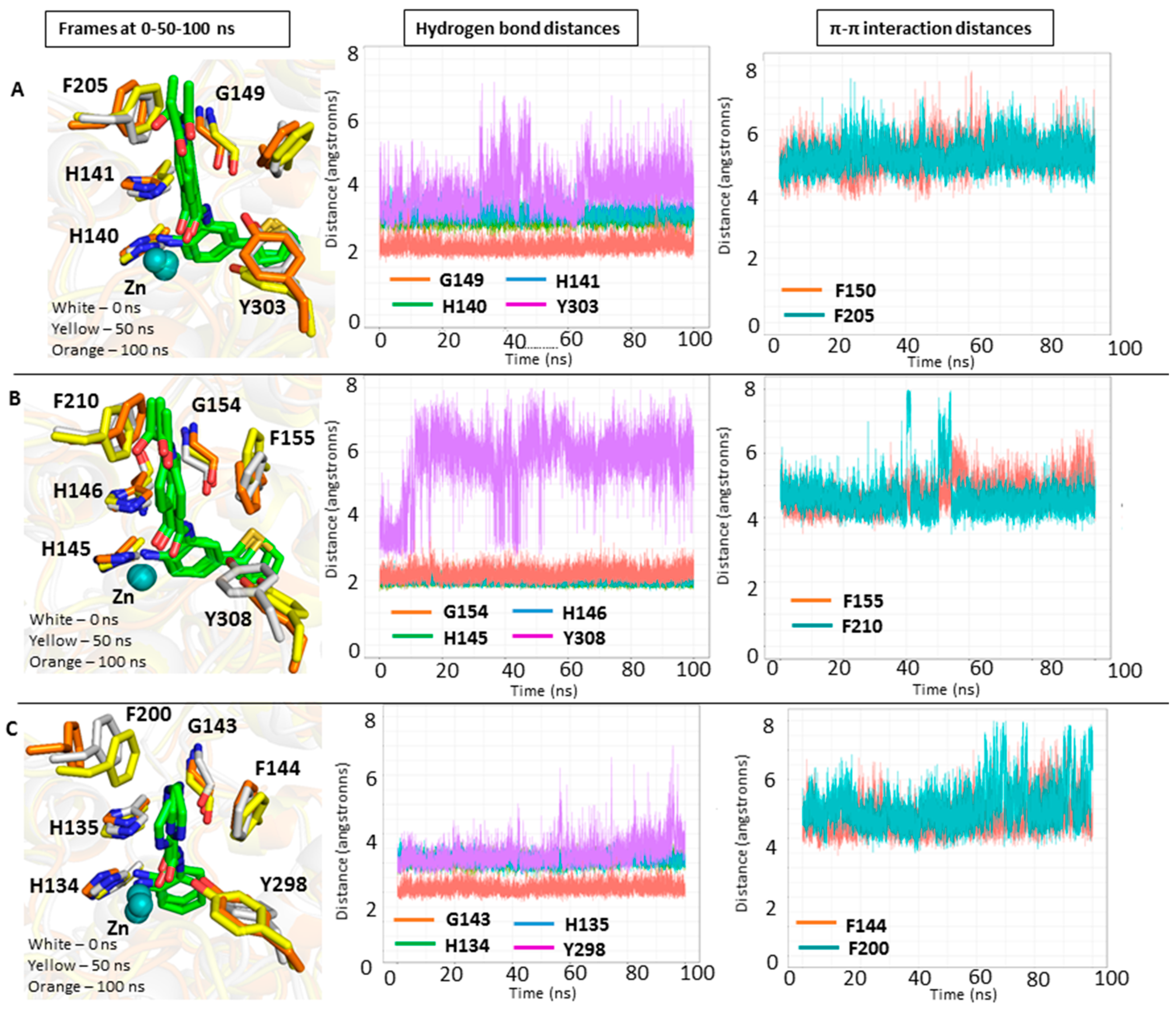
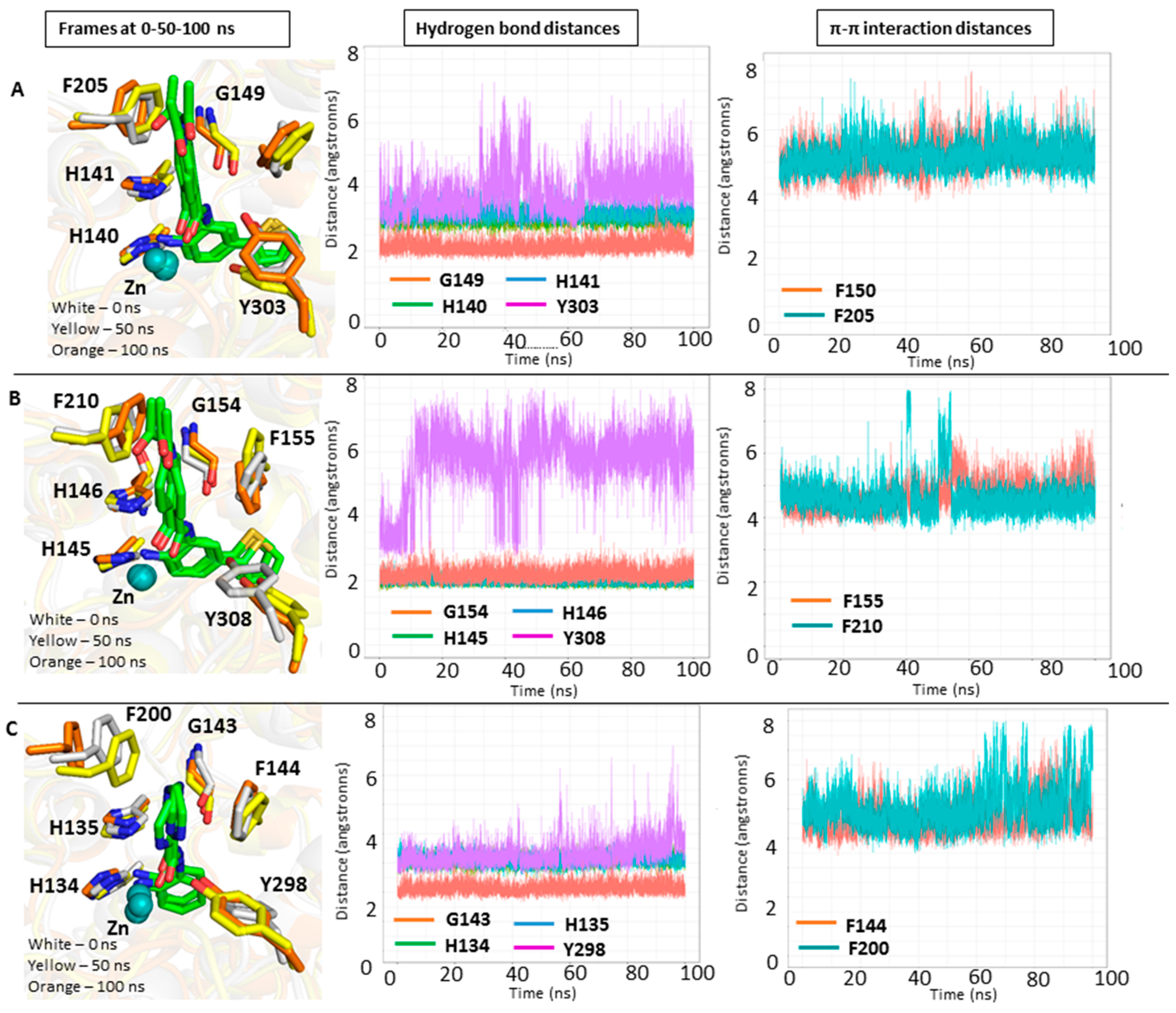
4.4. Molecular Dynamics (MD) Simulations
4.5. Binding Free Energy (BFE) Calculations
4.6. Test Set
4.7. Experimental Data of the Synthesized Compounds
4.8. Biological Assay of the Synthesized Compounds
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Keating, S.T.; Plutzky, J.; El-Osta, A. Epigenetic Changes in Diabetes and Cardiovascular Risk. Circ. Res. 2016, 118, 1706–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro Quiroz, E.; Chavez-Estrada, V.; Macias-Ochoa, K.; Ayala-Navarro, M.F.; Flores-Aguilar, A.S.; Morales-Navarrete, F.; de la Cruz Lopez, F.; Gomez Escorcia, L.; Musso, C.G.; Aroca Martinez, G.; et al. Epigenetic Mechanisms and Posttranslational Modifications in Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2019, 20, 5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felice, C.; Lewis, A.; Armuzzi, A.; Lindsay, J.O.; Silver, A. Review article: Selective histone deacetylase isoforms as potential therapeutic targets in inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2015, 41, 26–38. [Google Scholar] [CrossRef]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef]
- 6. Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies To Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. [Google Scholar] [CrossRef]
- Ning, Z.Q.; Li, Z.; Newman, M.J.; Shan, S.; Wang, X.; Pan, S.; Zhang, J.; Dong, M.; Du, X.; Lu, X. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901–909. [Google Scholar] [CrossRef]
- Dong, M.; Ning, Z.; Xing, P.; Xu, J.; Cao, H.; Dou, G.; Meng, Z.; Shi, Y.; Lu, X.; Feng, F. Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother. Pharmacol. 2012, 69, 1413–1422. [Google Scholar] [CrossRef]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef]
- Ito, T.; Ouchida, M.; Morimoto, Y.; Yoshida, A.; Jitsumori, Y.; Ozaki, T.; Sonobe, H.; Inoue, H.; Shimizu, K. Significant growth suppression of synovial sarcomas by the histone deacetylase inhibitor FK228 in vitro and in vivo. Cancer Lett. 2005, 224, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar] [CrossRef]
- Wagner, F.F.; Weimer, M.; Steinbacher, S.; Schomburg, A.; Reinemer, P.; Gale, J.P.; Campbell, A.J.; Fisher, S.L.; Zhao, W.; Reis, S.A.; et al. Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1, 2) inhibitors. Bioorg. Med. Chem. 2016, 24, 4008–4015. [Google Scholar] [CrossRef]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; Jong, R.D.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.F.; Wiest, O.; Helquist, P.; Lan-Hargest, H.; Wiech, L.N. On the function of the 14 A long internal cavity of histone deacetylase-like protein: Implications for the design of histone deacetylase inhibitors. J. Med. Chem. 2004, 47, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Armen, R.S.; Chen, J.; Brooks, C.L., 3rd. An Evaluation of Explicit Receptor Flexibility in Molecular Docking Using Molecular Dynamics and Torsion Angle Molecular Dynamics. J. Chem. Theory Comput. 2009, 5, 2909–2923. [Google Scholar] [CrossRef] [Green Version]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S7–S26. [Google Scholar] [CrossRef] [Green Version]
- Gilson, M.K.; Zhou, H.X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; capelli, A.; Clarke, B.; Lalonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Karaman, B.; Sippl, W. Docking and binding free energy calculations of sirtuin inhibitors. Eur. J. Med. Chem. 2015, 93, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.S.; Abdelsalam, M.; Zeyn, Y.; Zessin, M.; Mustafa, A.-H.M.; Fischer, M.A.; Zeyen, P.; Sun, P.; Bülbül, E.F.; Vecchio, A.; et al. Synthesis, Molecular Docking and Biological Characterization of Pyrazine Linked 2-Aminobenzamides as New Class I Selective Histone Deacetylase (HDAC) Inhibitors with Anti-Leukemic Activity. Int. J. Mol. Sci. 2022, 23, 369. [Google Scholar] [CrossRef] [PubMed]
- Simoben, C.V.; Ghazy, E.; Zeyen, P.; Darwish, S.; Schmidt, M.; Romier, C.; Robaa, D.; Sippl, W. Binding Free Energy (BFE) Calculations and Quantitative Structure–Activity Relationship (QSAR) Analysis of Schistosoma mansoni Histone Deacetylase 8 (smHDAC8) Inhibitors. Molecules 2021, 26, 2584. [Google Scholar] [CrossRef]
- Slynko, I.; Schmidtkunz, K.; Rumpf, T.; Klaeger, S.; Heinzlmeir, S.; Najar, A.; Metzger, E.; Kuster, B.; Schüle, R.; Jung, M.; et al. Identification of Highly Potent Protein Kinase C-Related Kinase 1 Inhibitors by Virtual Screening, Binding Free Energy Rescoring, and in vitro Testing. ChemMedChem 2016, 11, 2084–2094. [Google Scholar] [CrossRef]
- Wichapong, K.; Rohe, A.; Platzer, C.; Slynko, I.; Erdrmann, F.; Schmidt, M.; Sippl, W. Application of Docking and QM/MM-GBSA Rescoring to Screen for Novel Myt1 Kinase Inhibitors. J. Chem. Inf. Model. 2014, 54, 881–893. [Google Scholar] [CrossRef]
- Brandmaier, S.; Novotarskyi, S.; Sushki, I.; Tetko, I.V. From descriptors to predicted properties: Experimental design by using applicability domain estimation. Altern. Lab. Anim. 2013, 41, 33–47. [Google Scholar] [CrossRef]
- Netzeva, T.I.; Worth, A.P.; Aldenberg, T. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. The report and recommendations of ECVAM Workshop 52. Altern. Lab. Anim. 2005, 33, 155–173. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-1; Schrödinger LLC: New York, NY, USA, 2019; Available online: https://www.schrodinger.com/platform#product-list-collapse (accessed on 1 February 2022).
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, K.; Lupyan, D.; Dahigren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W.R. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 2013, 51, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W.R. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment (MOE); Chemical Computing Group (CCG): Montreal, QC, Canada, 2012; Available online: https://www.chemcomp.com/Products.htm (accessed on 1 February 2022).
- Minami, J.; Suzuki, R.; Mazitschek, R.; Gorgun, G.; Ghosh, B.; Cirstea, D.; Hu, Y.; Mimura, N.; Ohguchi, H.; Cottini, F.; et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014, 28, 680–689. [Google Scholar] [CrossRef] [Green Version]
- Song, L.F.; Lee, T.-S.; Zhu, C.; York, D.M.; Merz, K.M., Jr. Using AMBER18 for Relative Free Energy Calculations. J. Chem. Inf. Model. 2019, 59, 3128–3135. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Lee, M.C.; Duan, Y. Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins 2004, 55, 620–634. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Song, L.F.; Merz, K.M., Jr. Parameterization of highly charged metal ions using the 12-6-4 LJ-type nonbonded model in explicit water. J. Phys. Chem. B 2015, 119, 883–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Sagui, C.; Pedersen, L.G.; Darden, T.A. Towards an accurate representation of electrostatics in classical force fields: Efficient implementation of multipolar interactions in biomolecular simulations. J. Chem. Phys. 2004, 120, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Toukmaji, A.; Sagui, C.; Boar, J.; Datden, T. Efficient particle-mesh Ewald based approah to fixed and induced dipolar interactions. J. Chem. Phys. 2000, 113, 10913. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesan equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Mongan, J.; Simmerling, C.; McCammon, A.; Case, D.A.; Onufriev, A. Generalized Born model with a simple, robust molecular volume correction. J. Chem. Theory Comput. 2007, 3, 156–169. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Feig, M.; Onufriev, A.; Lee, M.S.; Case, D.A.; Brooks, C.L. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 2004, 25, 265–284. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized Models of Aqueous Free Energies of Solvation Based on Pairwise Descreening of Solute Atomic Charges from a Dielectric Medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995, 246, 122–129. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.J.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the Molecular Mechanics/Poisson Boltzmann Surface Area and Molecular Mechanics/Generalized Born Surface Area Methods. II. The Accuracy of Ranking Poses Generated From Docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, R.N.; Wold, S.; Westgard, J.O. Principal component analysis: An alternative to “referee” methods in method comparison studies. Anal. Chem. 1975, 47, 1824–1829. [Google Scholar] [CrossRef]
- Liu, J.; Yu, Y.; Kelly, J.; Sha, D.; Alhassan, A.B.; Yu, Q.; Maletic, M.M.; Duffy, J.L.; Klein, D.J.; Holloway, M.K.; et al. Discoveery of highly selective and potent HDAC3 inhibitors based on a 2-substituted benzamide zinc binding group. ACS Med. Chem. Lett. 2020, 11, 2476–2483. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. No. | Substituents | IC50 (µM) or % Inhibition at Given Concentration | ||||||
|---|---|---|---|---|---|---|---|---|
| X | Y | R1 | R2 | R3 | HDAC1 | HDAC2 | HDAC3 | |
| 19a | CH | N | 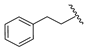 | H | H | 0.51 ± 0.05 | 0.80 ± 0.07 | 1.12 ± 0.07 |
| 19b | CH | N | 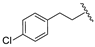 | H | H | 26% @ 2 µM | 30% @ 2 µM | 65% @ 2 µM |
| 19c | N | CH | 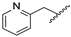 | H | H | 34% @ 2 µM | 20% @ 2 µM | 27% @ 2 µM |
| 19d | CH | N |  | H | H | 0.52 ± 0.07 | 1.43 ± 0.08 | 1.06 ± 0.04 |
| 19e | CH | N | 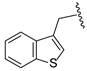 | H | H | 0.21 ± 0.07 | 0.71 ± 0.04 | 0.84 ± 0.03 |
| 19f | CH | N | 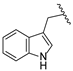 | H | H | 0.13 ± 0.01 | 0.28 ± 0.01 | 0.31 ± 0.01 |
| 19g | CH | N | 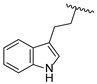 | H | H | 0.31 ± 0.03 | 0.96 ± 0.05 | 0.49 ± 0.06 |
| 19h | CH | N | 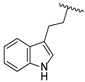 | F | F | 0.81 ± 0.07 | 0.74 ± 0.03 | 0.57 ± 0.02 |
| 19i | CH | N | 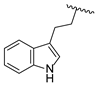 | Cl | H | 3.0 ± 0.2 | 2.7 ± 0.2 | 1.9 ± 0.1 |
| 19j | N | CH |  | H | H | 0.45 ± 0.06 | 0.93 ± 0.04 | 1.75 ± 0.06 |
| 19k | CH | N |  | H | H | 0.14 ± 0.02 | 0.56 ± 0.04 | 0.59 ± 0.03 |
| 19l | CH | N |  | F | F | 0.29 ± 0.03 | 0.56 ± 0.02 | 0.81 ± 0.05 |
| 19m | CH | N |  | F | H | 0.40 ± 0.06 | 1.48 ± 0.19 | 0.40 ± 0.02 |
| 19n | N | CH | CH3 | H | H | 5% @ 1 µM | 7% @ 1 µM | 13% @ 1 µM |
| 19o | CH | N | CH3 | H | H | 27% @ 1 µM | 15% @ 1 µM | 30% @ 1 µM |
| 21a | CH | N |  | H | 2-Thienyl | 0.26 ± 0.01 | 2.47 ± 0.22 | 0% @ 1 µM |
| 21b | CH | N |  | H | 4-F-C6H4 | 0.70 ± 0.08 | 0.77 ± 0.06 | 0% @ 1 µM |
| 21c | CH | N | 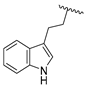 | H | 2-F-C6H4 | 0.76 ± 0.07 | 0.76 ± 0.04 | 15 ± 1 |
| 23a | CH | N | H | F | H | 3.30 ± 0.18 | 2.17 ± 0.18 | 0.40 ± 0.01 |
| 23b | CH | N | 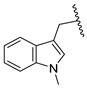 | H | F | 0.27 ± 0.03 | 0.50 ± 0.03 | 0.50 ± 0.02 |
| 23c | CH | N | 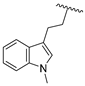 | H | F | 0.33 ± 0.02 | 1.37 ± 0.08 | 0.59 ± 0.04 |
| 25a | CH | N | H | Cl | H | 0% @ 1 µM | 0% @ 1 µM | 8.7 ± 0.4 |
| 25b | CH | N | H | F | F | 4.3 ± 0.3 | 4.2 ± 0.15 | 1.6 ± 0.1 |
| 27a | CH | N | H | H | CF3 | 0% @ 1 µM | 0% @ 1 µM | 0% @ 1 µM |
| 27b | CH | N | H | CH3 | H | 0% @ 1 µM | 0% @ 1 µM | 0% @ 1 µM |
| 27c | CH | N | H | OCH3 | H | 20.0 ± 1.0 | 14.0 ± 2.0 | 14.0 ± 1.0 |
| 29a | CH | N | CH3 | H | 3-Thienyl | 0.11 ± 0.01 | 0.18 ± 0.06 | 4.4 ± 0.1 |
| 29b | CH | N | H | H | 2-Thienyl | 0.07 ± 0.01 | 0.26 ± 0.01 | 6.1 ± 0.7 |
| 29c | CH | N | H | H | 4-F-C6H4 | 0.16 ± 0.03 | 0.34 ± 0.01 | 6.7 ± 0.5 |
| 29d | CH | N | CH3 | H | 2-F-C6H4 | 0.18 ± 0.01 | 0.26 ± 0.07 | 12.0 ± 1.0 |
| CI994 | -- | -- | -- | -- | -- | 37% @ 1 µM | 36% @ 1 µM | 32% @ 1 µM |
| RGFP-966 | -- | -- | -- | -- | -- | 16 ± 2 | 11 ± 1 | 1.3 ± 0.1 |
| MS-275 | -- | -- | -- | -- | -- | 0.93 ± 0.1 | 0.95 ± 0.03 | 1.8 ± 0.1 |
| Mocetinostat | -- | -- | -- | -- | -- | 0.33 ± 0.04 | 0.34 ± 0.01 | 0.93 ± 0.05 |
| Protein | N | Model Number | Solvation Model | Frame | R2 | RMSE | Q2LOO | QMSE |
|---|---|---|---|---|---|---|---|---|
| HDAC1 | 22 | 3 | GB1 | MD1-50 | 0.59 | 0.29 | 0.51 | 0.32 |
| HDAC2 | 23 | 21 | GB8 | MD1-50 | 0.66 | 0.24 | 0.60 | 0.26 |
| HDAC3 | 22 | 7 | GB2 | Emin1 | 0.71 | 0.29 | 0.65 | 0.32 |
| Cpd. No. | Scaffold | Substituents | ||||
|---|---|---|---|---|---|---|
| X | Y | R1 | R2 | R3 | ||
| 30a | A | N | CH | −CH3 | H | 2-Thienyl |
| 30b | A | CH | N | −CH3 | H | 2-Thienyl |
| 30c | A | CH | N | 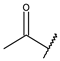 | H | 2-Thienyl |
| 30d | A | CH | N | 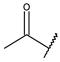 | H | H |
| 31a | B | CH | CH | 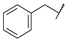 | F | H |
| 31b | B | CH | CH | 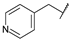 | F | H |
| 31c | B | CH | CH |  | F | H |
| Cpd. No. | Scaffold | Experimental HDAC1 pIC50 | Predicted HDAC1 pIC50) | Difference Experimental—Predicted HDAC1 pIC50 | Experimental HDAC2 pIC50 | Predicted HDAC2 pIC50 | Difference Experimental—Predicted HDAC2 pIC50 | Experimental HDAC3 pIC50 | Predicted HDAC3 pIC50 |
|---|---|---|---|---|---|---|---|---|---|
| 30a | A | 6.49 | 6.55 | 0.06 | 6.21 | 6.00 | 0.21 | 8% @1 µM 38% @10 μM | 5.06 |
| 30b | A | 7.40 | 6.31 | 1.09 | 6.10 | 6.24 | 0.14 | 6% @1 µM 35% @10 μM | 5.01 |
| 30c | A | 7.72 | 6.50 | 1.22 | 5.96 | 5.95 | 0.01 | 4% @1 µM 30% @10 μM | 4.98 |
| 30d | A | 5.72 | 5.70 | 0.02 | 4.62 | 5.89 | 1.27 | 15% @1 µM 72% @10 μM | 5.98 |
| 31a | B | 4% @1 µM 31% @10 µM | 5.58 | 10% @1 µM 37% @10 µM | 5.52 | 21% @1 µM 65% @10 µM | 5.81 | ||
| 31b | B | 0% @1 µM 27% @10 µM | 5.77 | 13% @1 µM 30% @10 µM | 5.29 | 19% @1 µM 59% @10 µM | 5.80 | ||
| 31c | B | 6% @1 µM 36% @10 µM | 5.68 | 14% @1 µM 51% @10 µM | 5.52 | 25% @1 µM 66% @10 µM | 5.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bülbül, E.F.; Melesina, J.; Ibrahim, H.S.; Abdelsalam, M.; Vecchio, A.; Robaa, D.; Zessin, M.; Schutkowski, M.; Sippl, W. Docking, Binding Free Energy Calculations and In Vitro Characterization of Pyrazine Linked 2-Aminobenzamides as Novel Class I Histone Deacetylase (HDAC) Inhibitors. Molecules 2022, 27, 2526. https://doi.org/10.3390/molecules27082526
Bülbül EF, Melesina J, Ibrahim HS, Abdelsalam M, Vecchio A, Robaa D, Zessin M, Schutkowski M, Sippl W. Docking, Binding Free Energy Calculations and In Vitro Characterization of Pyrazine Linked 2-Aminobenzamides as Novel Class I Histone Deacetylase (HDAC) Inhibitors. Molecules. 2022; 27(8):2526. https://doi.org/10.3390/molecules27082526
Chicago/Turabian StyleBülbül, Emre F., Jelena Melesina, Hany S. Ibrahim, Mohamed Abdelsalam, Anita Vecchio, Dina Robaa, Matthes Zessin, Mike Schutkowski, and Wolfgang Sippl. 2022. "Docking, Binding Free Energy Calculations and In Vitro Characterization of Pyrazine Linked 2-Aminobenzamides as Novel Class I Histone Deacetylase (HDAC) Inhibitors" Molecules 27, no. 8: 2526. https://doi.org/10.3390/molecules27082526
APA StyleBülbül, E. F., Melesina, J., Ibrahim, H. S., Abdelsalam, M., Vecchio, A., Robaa, D., Zessin, M., Schutkowski, M., & Sippl, W. (2022). Docking, Binding Free Energy Calculations and In Vitro Characterization of Pyrazine Linked 2-Aminobenzamides as Novel Class I Histone Deacetylase (HDAC) Inhibitors. Molecules, 27(8), 2526. https://doi.org/10.3390/molecules27082526