
Semisynthetic Sesquiterpene Lactones Generated by the Sensibility of Glaucolide B to Lewis and Brønsted–Lowry Acids and Bases: Cytotoxicity and Anti-Inflammatory Activities


 ,
,  ,
,  , and
, and
Abstract
1. Introduction
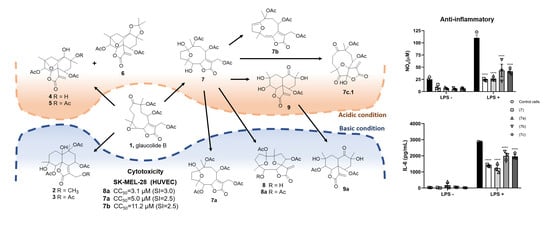
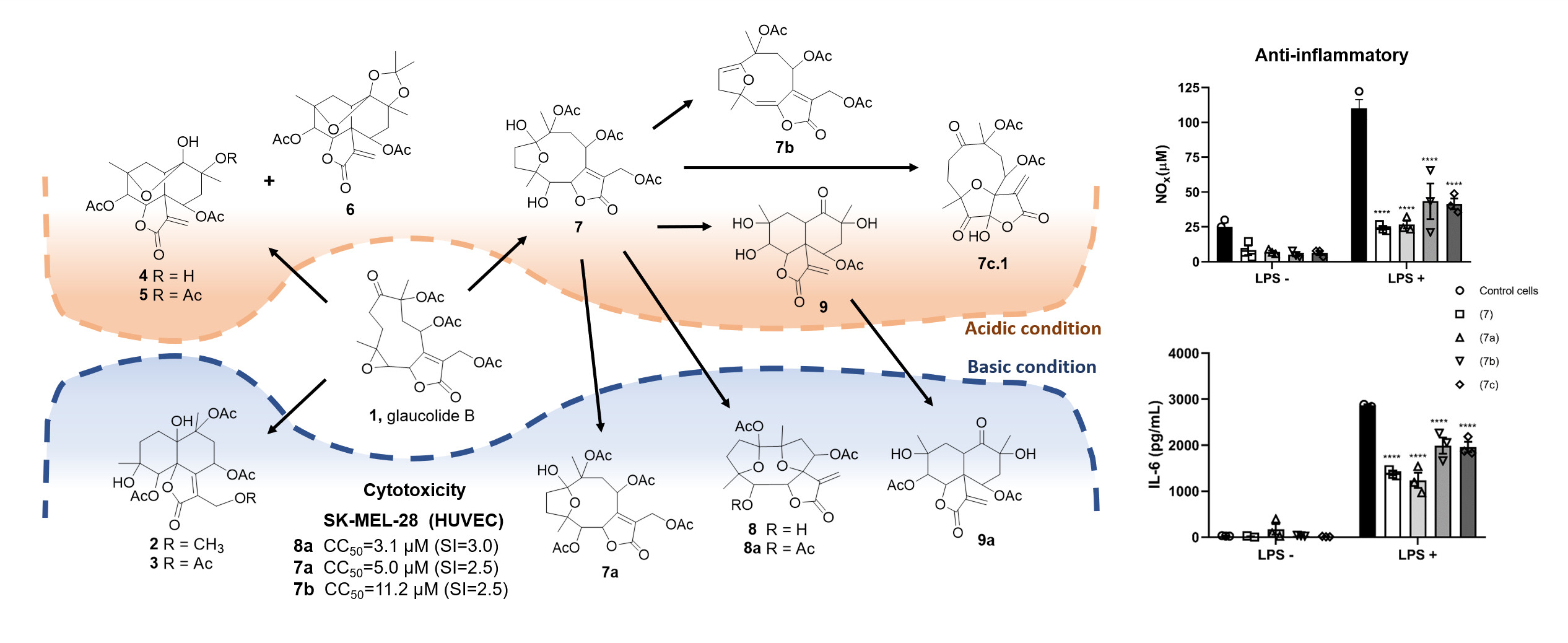
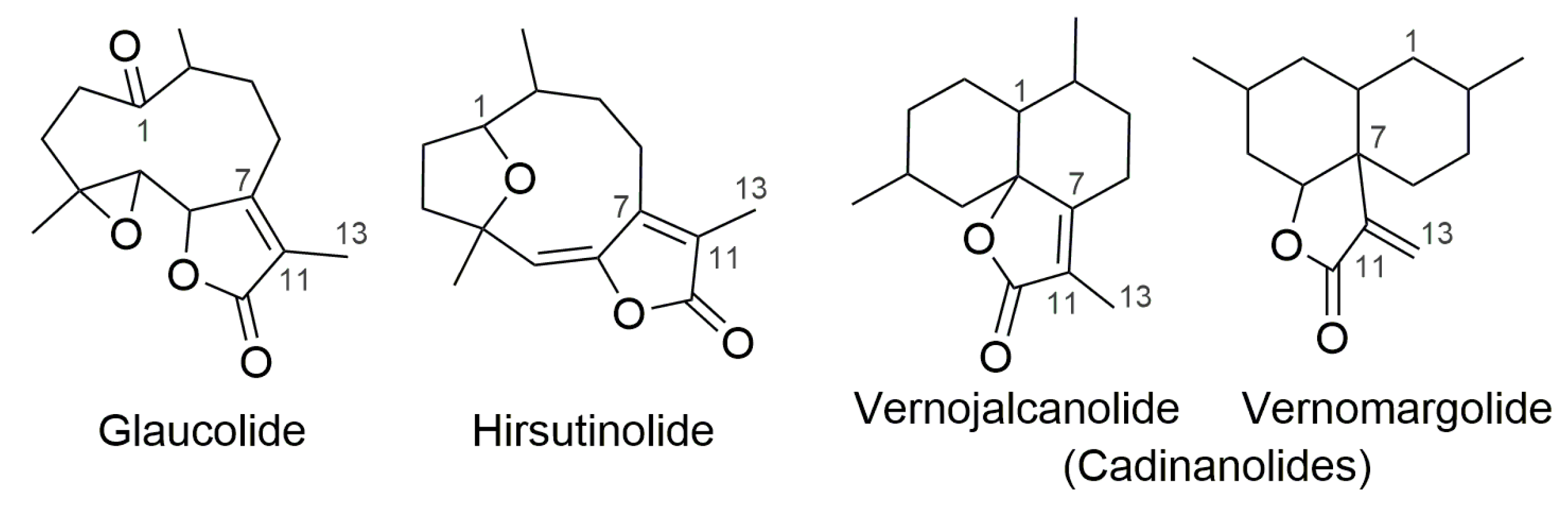
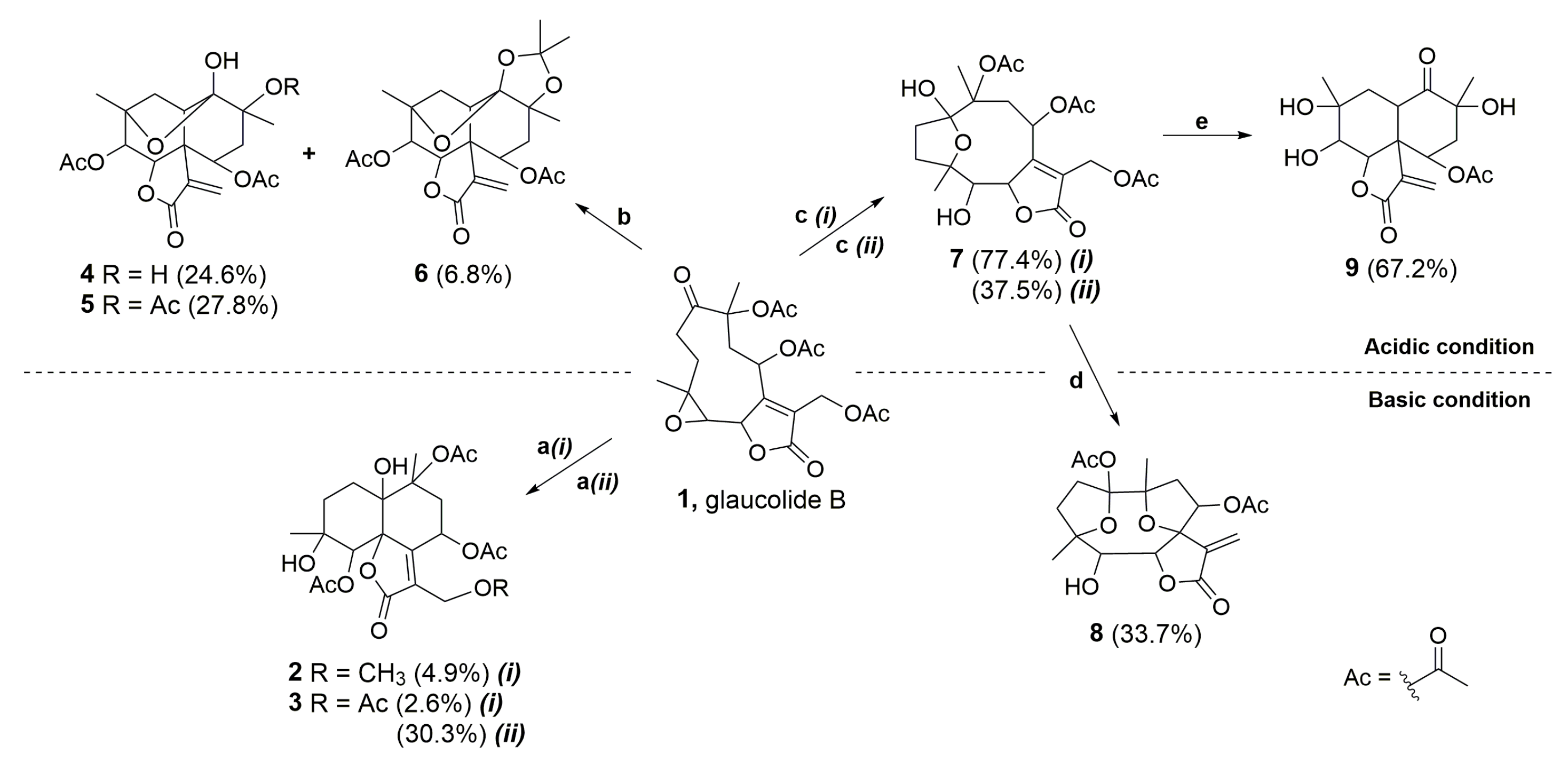
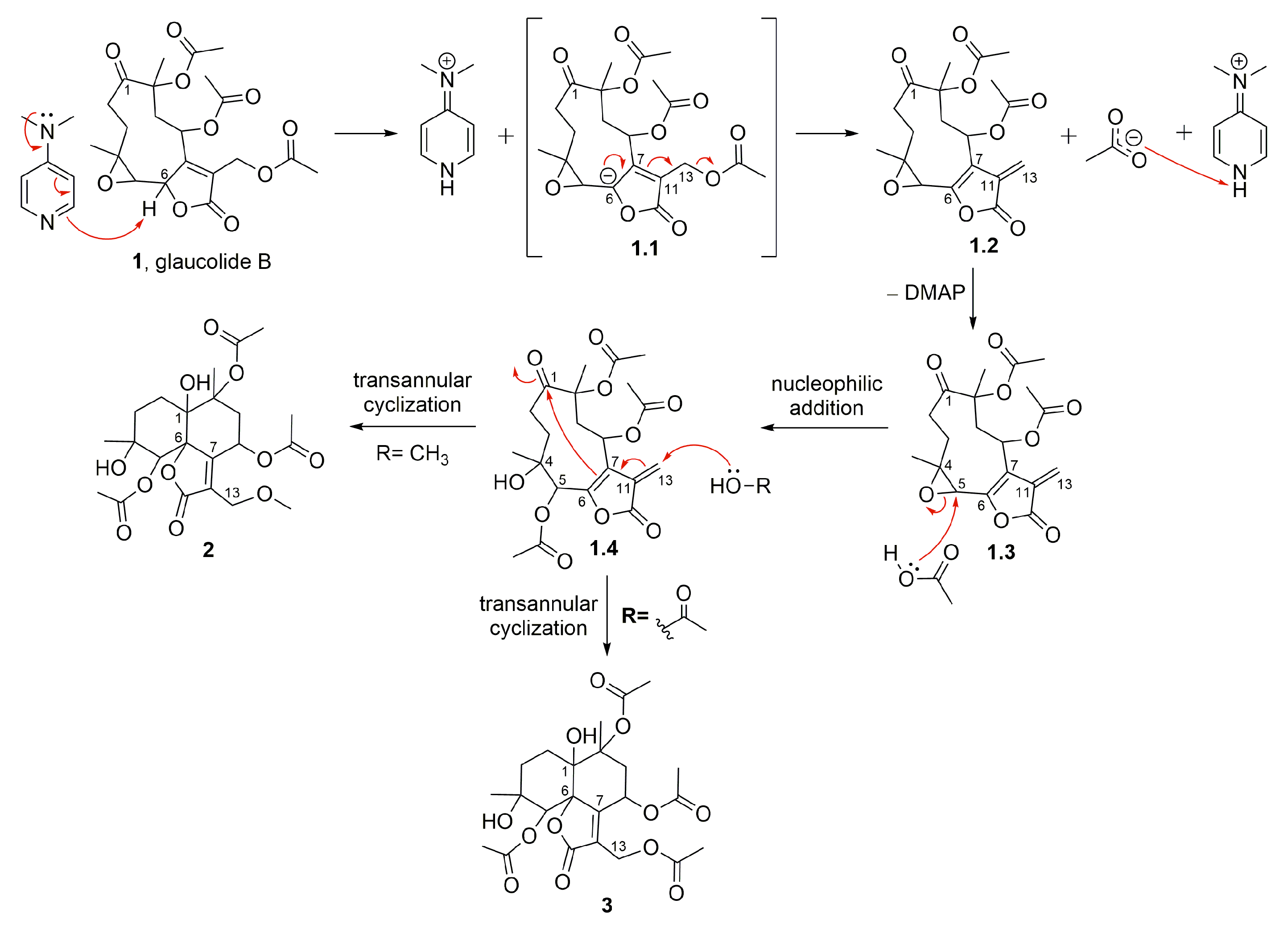
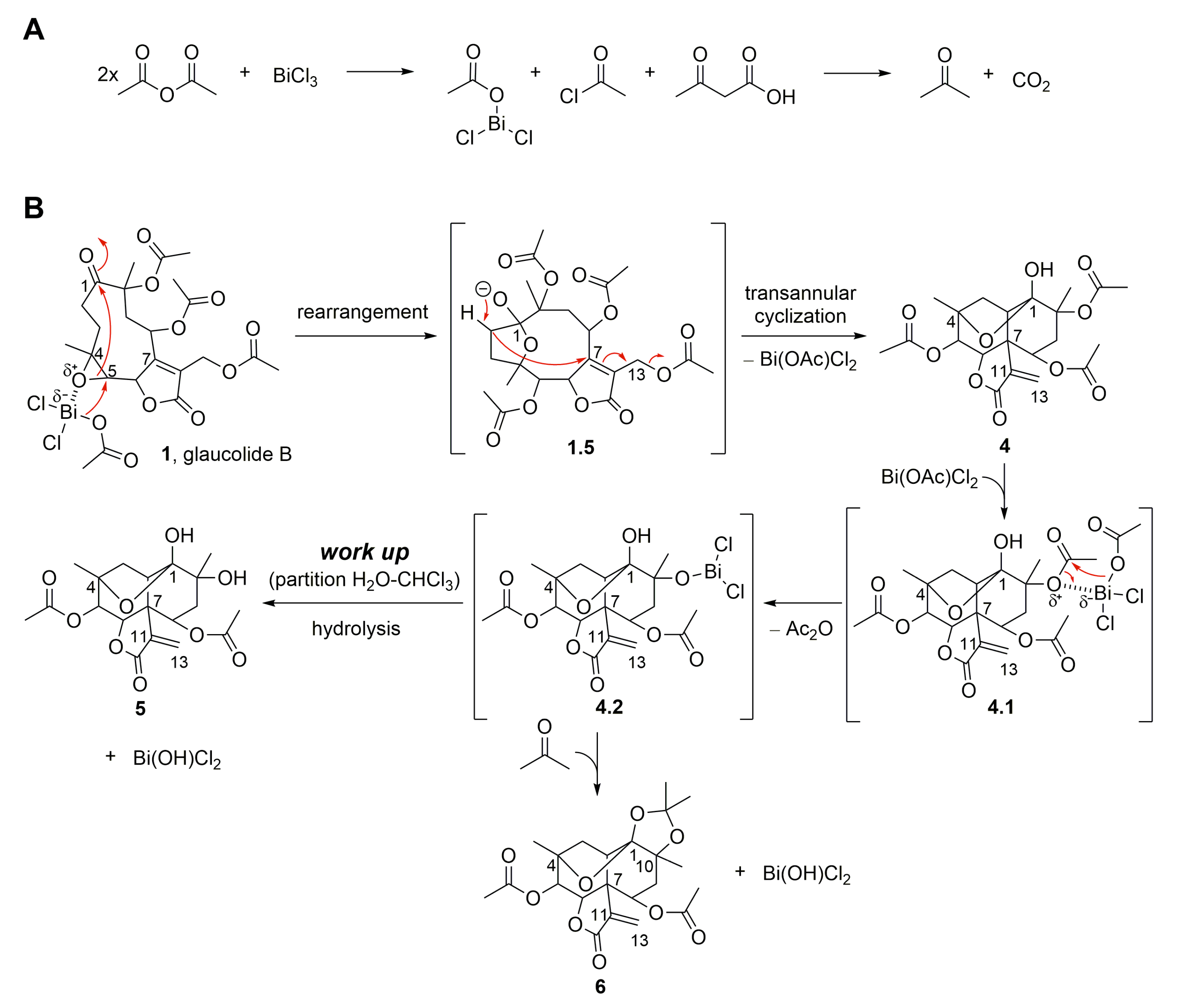
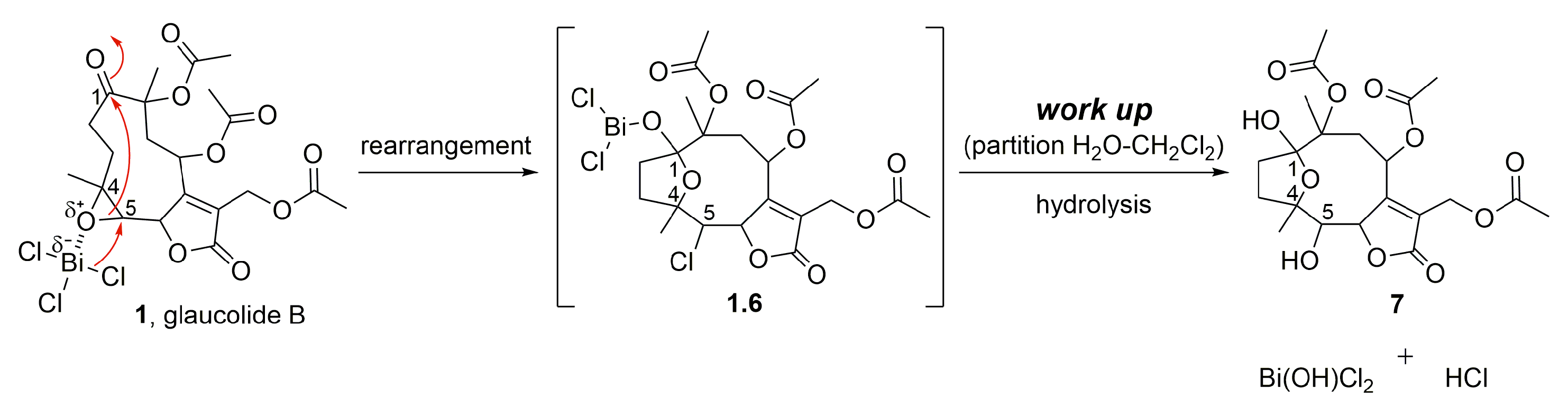
2. Results and Discussion
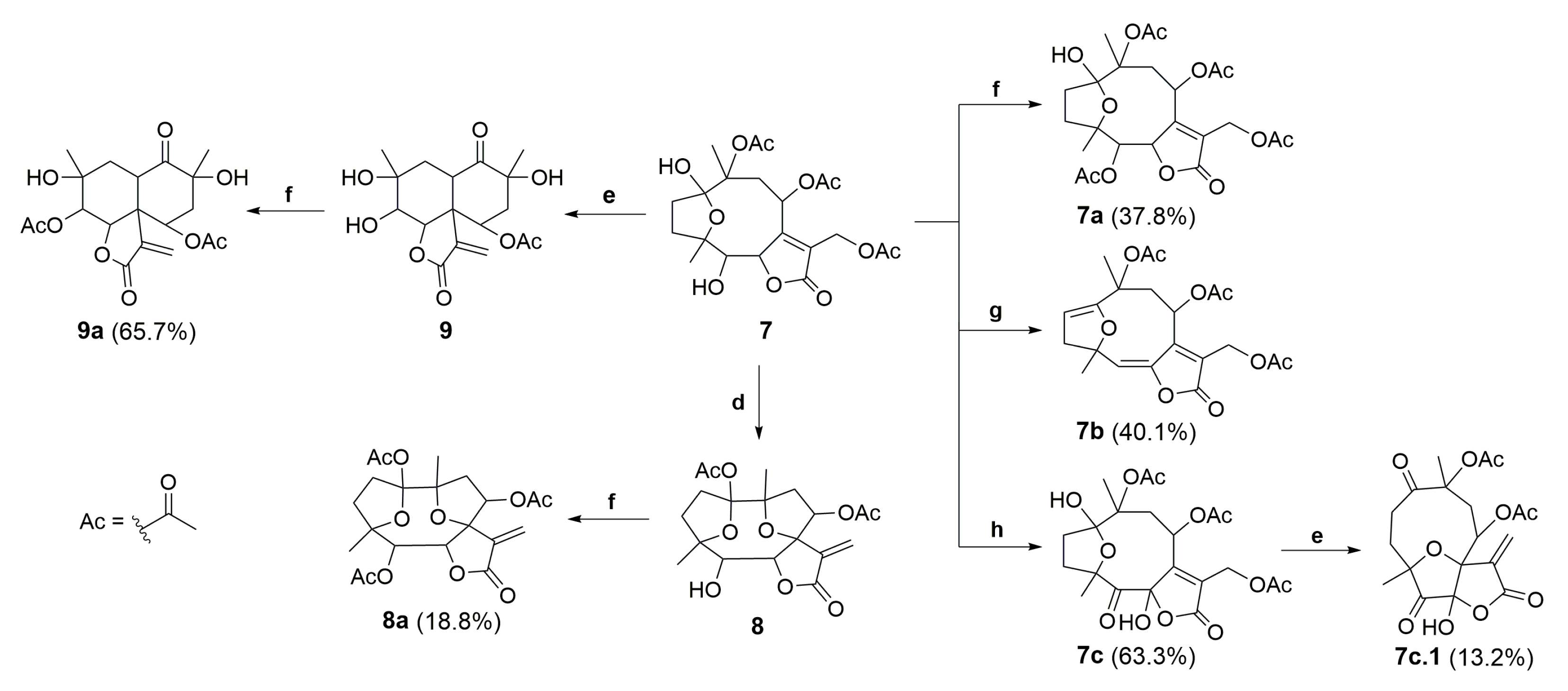
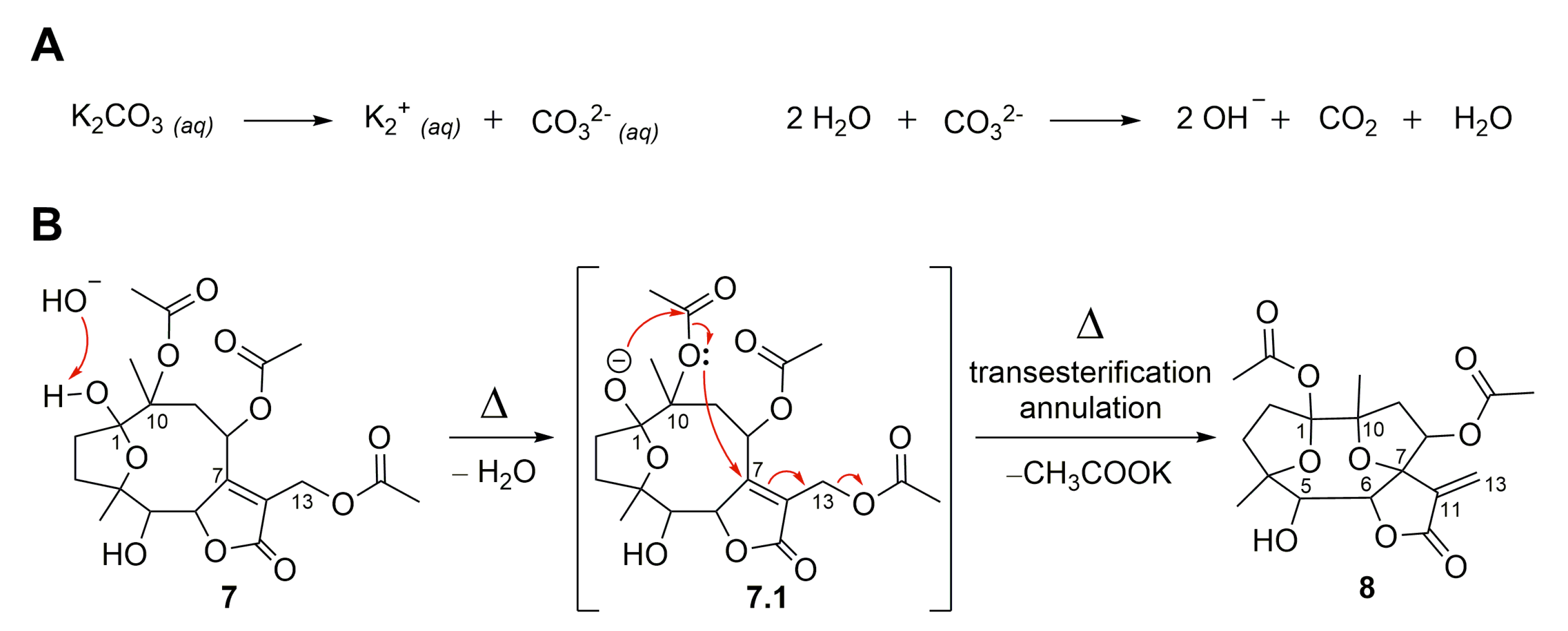
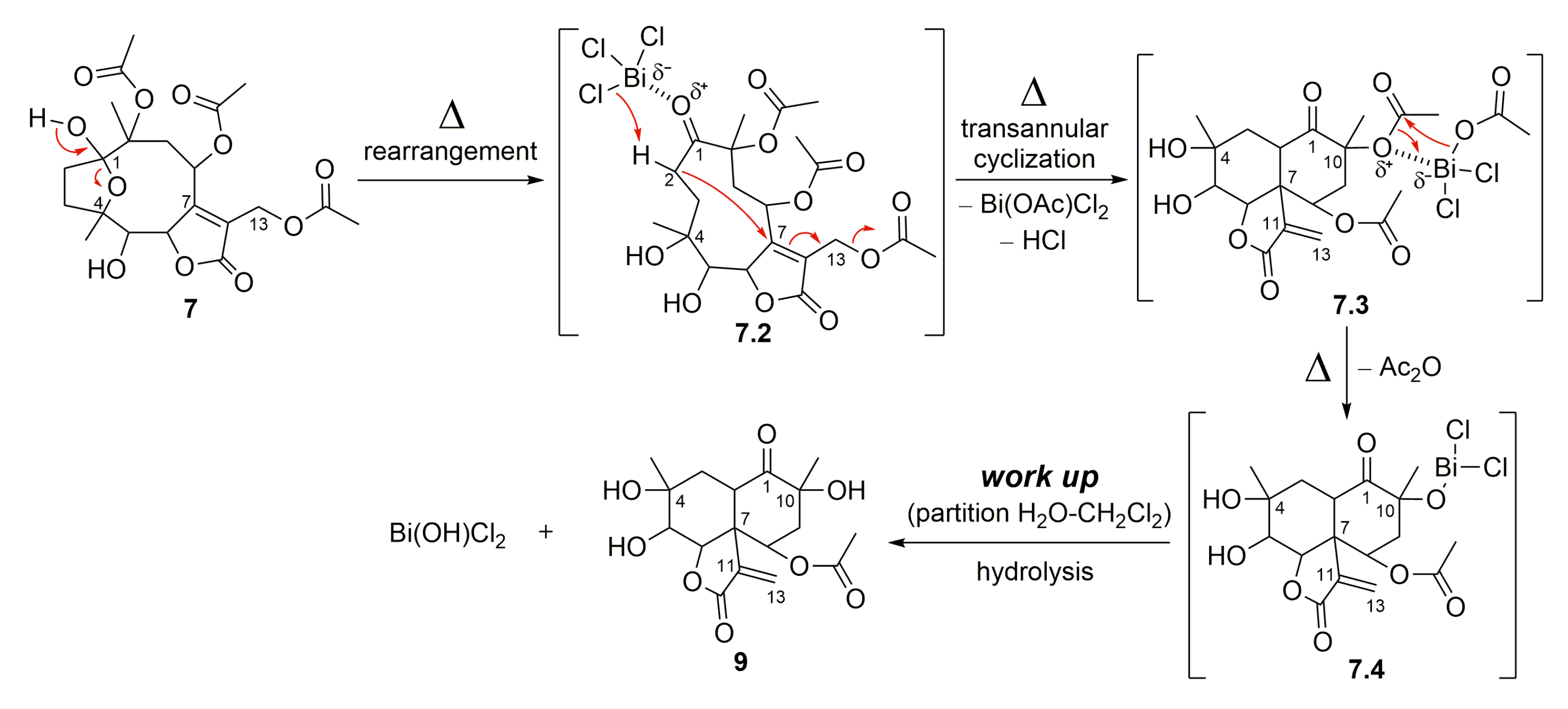
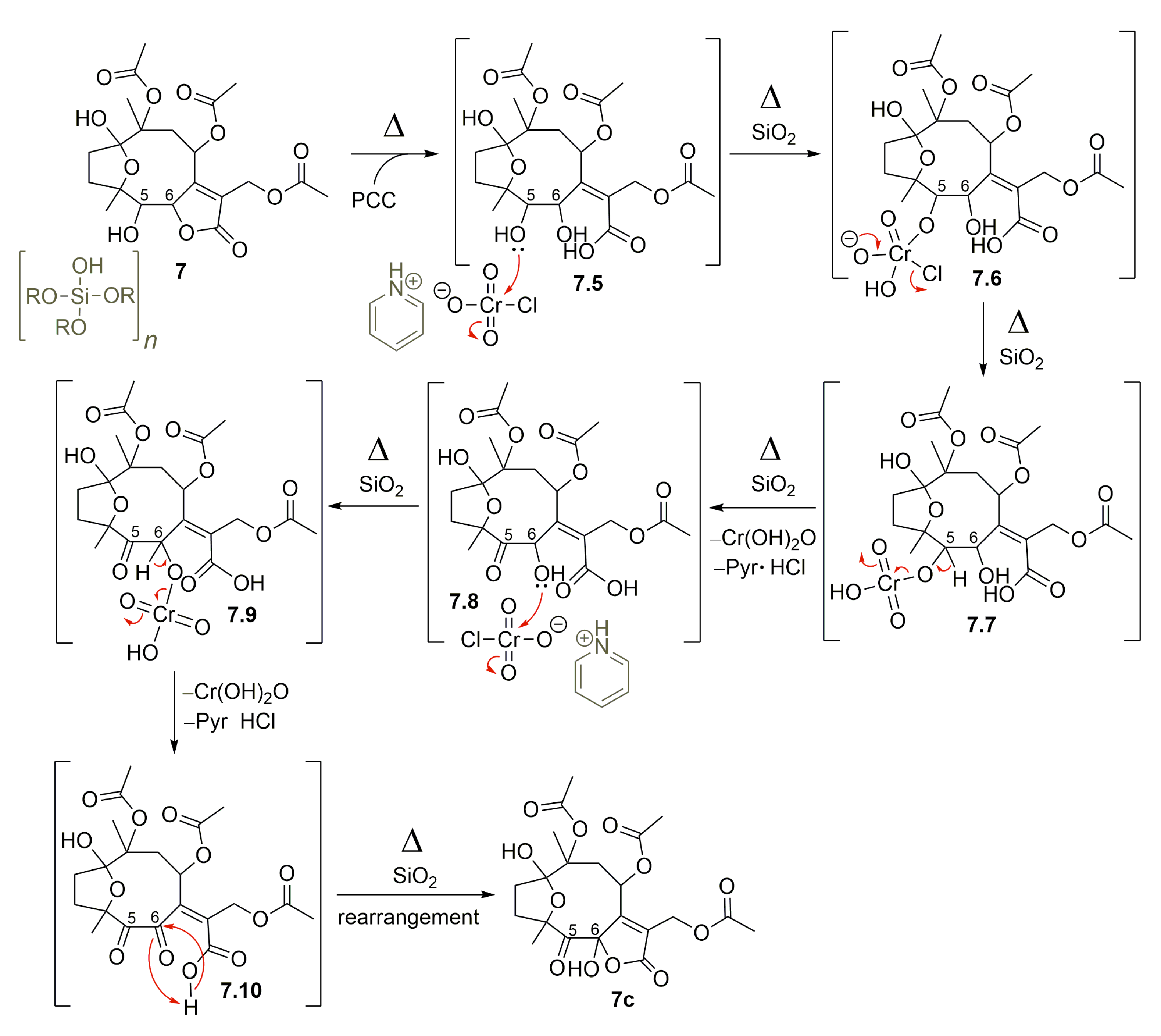
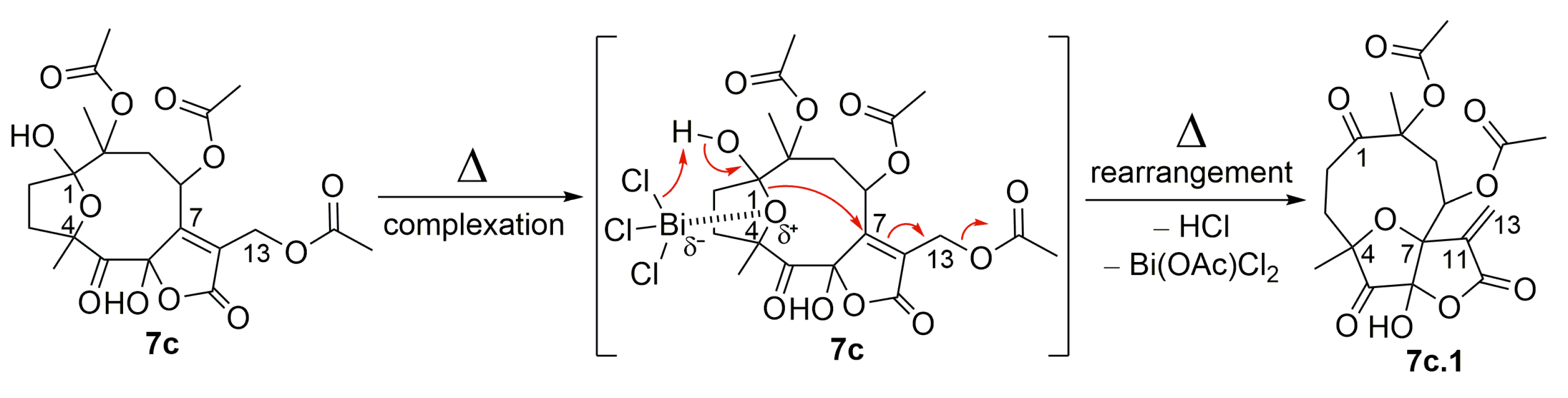
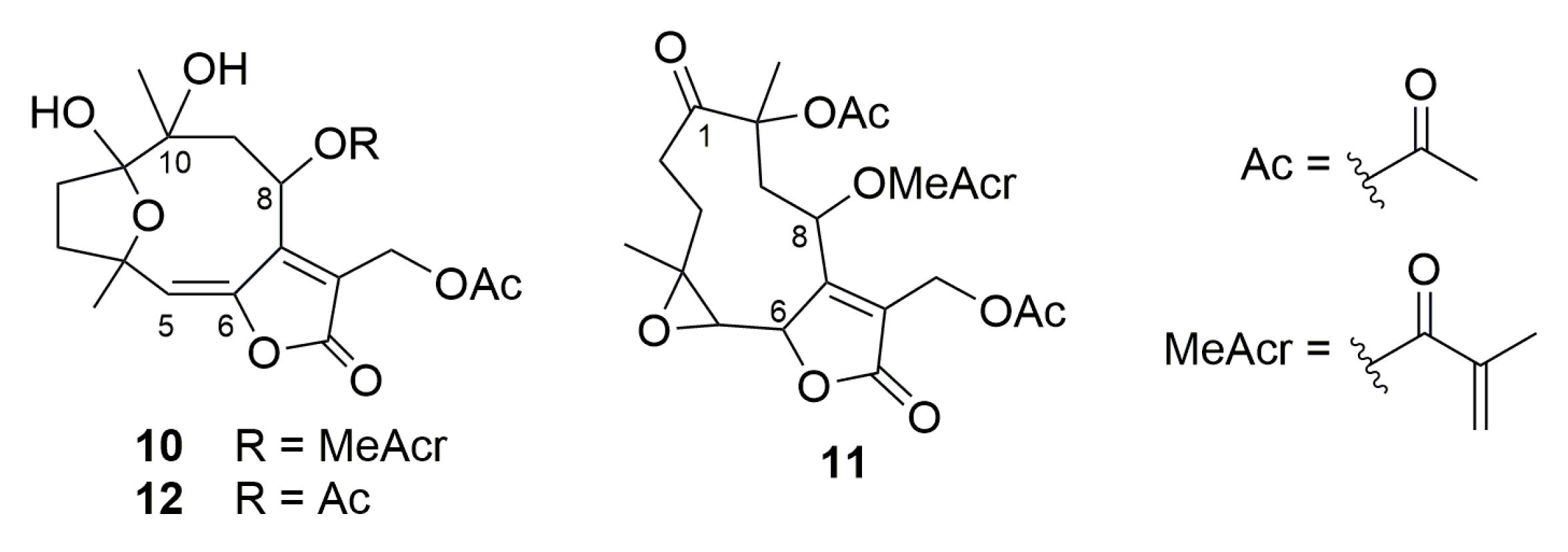
2.1. Semisynthetic Modifications from Glaucolide B
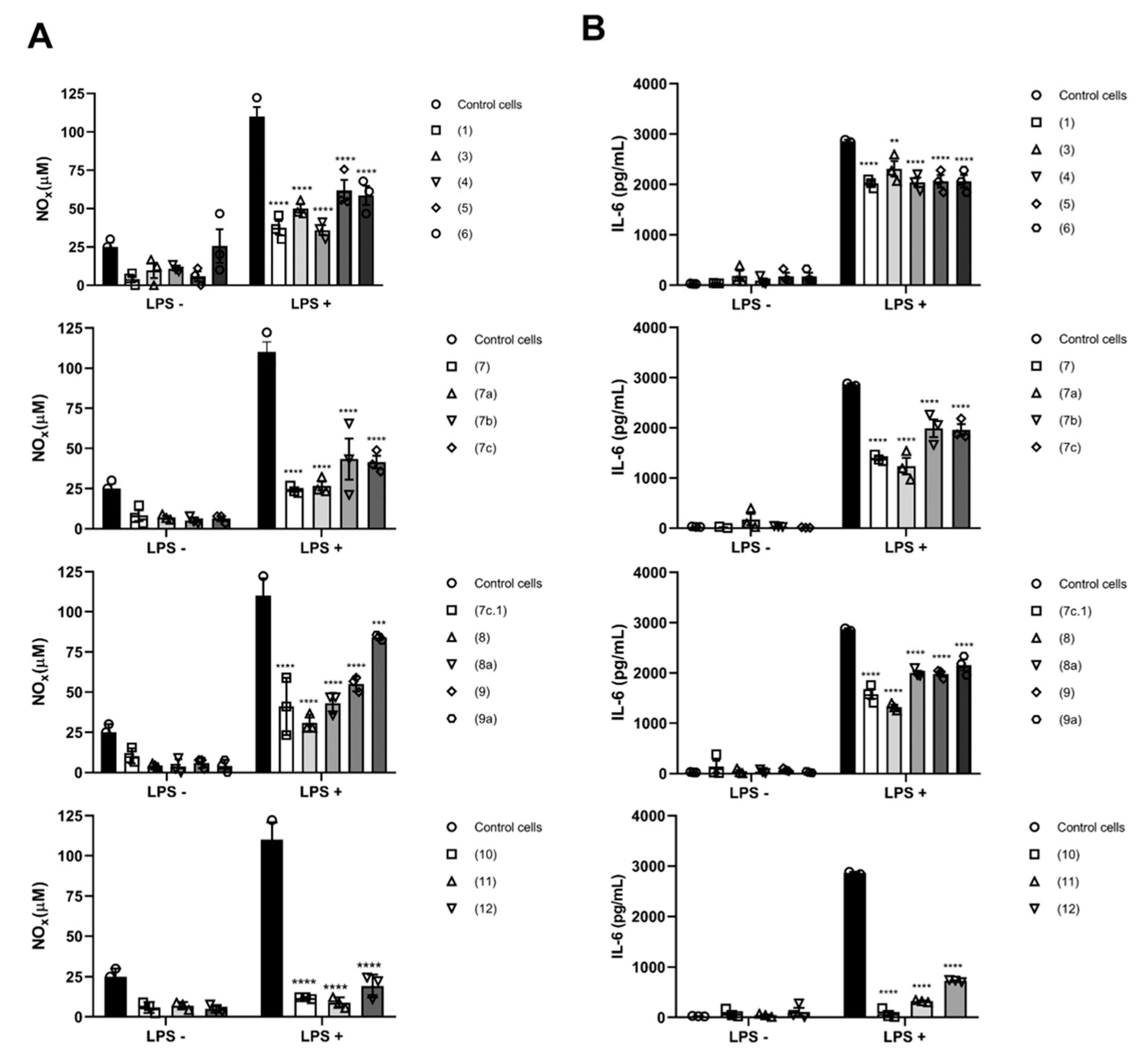
2.2. Biological Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Semisynthetic Derivatives Production and Isolation
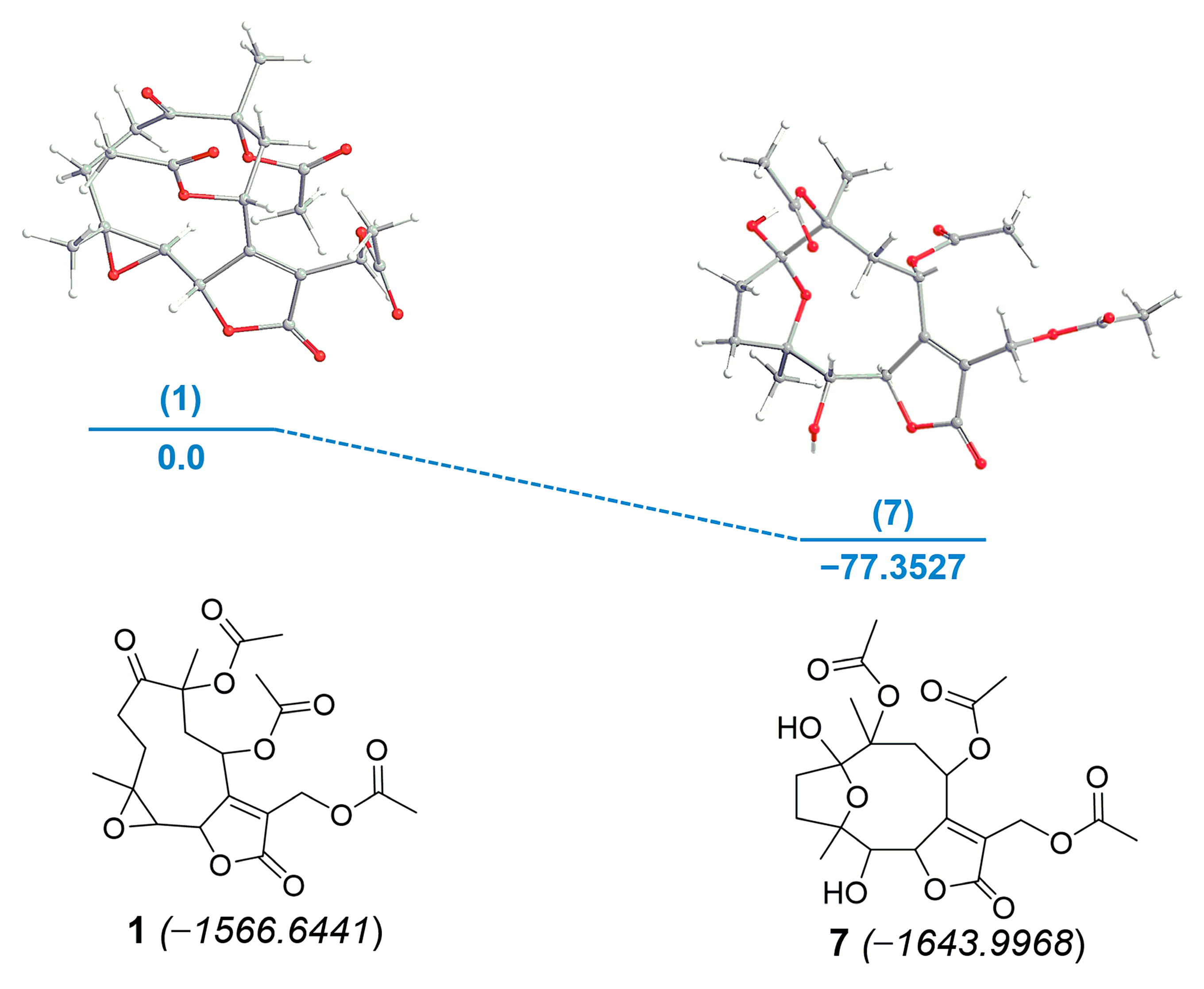
3.3. Computational Calculation
3.4. Biological Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Padilla-Gonzalez, G.F.; dos Santos, F.A.; Da Costa, F.B. Sesquiterpene lactones: More than protective plant compounds with high toxicity. CRC Crit. Rev. Plant Sci. 2016, 35, 18–37. [Google Scholar] [CrossRef]
- Da Costa, F.B.; Terfloth, L.; Gasteiger, J. Sesquiterpene lactone-based classification of three Asteraceae tribes: A study based on self-organizing neural networks applied to chemosystematics. Phytochemistry 2005, 66, 345–353. [Google Scholar] [CrossRef]
- Loeuille, B.; Keeley, S.C.; Pirani, J.R. Systematics and evolution of syncephaly in American Vernonieae (Asteraceae) with emphasis on the Brazilian subtribe Lychnophorinae. Syst. Bot. 2015, 40, 286–298. [Google Scholar] [CrossRef]
- Gallon, M.E.; Jaiyesimi, O.A.; Gobbo-Neto, L. LC-UV-HRMS dereplication of secondary metabolites from Brazilian Vernonieae (Asteraceae) species supported through in-house database. Biochem. Syst. Ecol. 2018, 78, 5–16. [Google Scholar] [CrossRef]
- Jakupovic, J.; Gage, D.A.; Bohlmann, F.; Mabry, T.J. Sesquiterpene lactones from Vernonia marginata. Phytochemistry 1986, 25, 1179–1183. [Google Scholar] [CrossRef]
- Pillay, P.; Vleggaar, R.; Maharaj, V.J.; Smith, P.J.; Lategan, C.A.; Chouteau, F.; Chibale, K. Antiplasmodial hirsutinolides from Vernonia staehelinoides and their utilization towards a simplified pharmacophore. Phytochemistry 2007, 68, 1200–1205. [Google Scholar] [CrossRef]
- Appezzato-da-Gloria, B.; Da Costa, F.B.; da Silva, V.C.; Gobbo-Neto, L.; Rehder, V.L.G.; Hayashi, A.H. Glandular trichomes on aerial and underground organs in Chrysolaena species (Vernonieae—Asteraceae): Structure, ultrastructure and chemical composition. Flora 2012, 207, 878–887. [Google Scholar] [CrossRef]
- Bardón, A.; Montanaro, S.; Catalán, C.A.N.; Diaz, J.G.; Herz, W. Piptocarphols and other constituents of Chrysolaena verbascifolia and Lessingianthus rubricaulis. Phytochemistry 1993, 34, 253–259. [Google Scholar] [CrossRef]
- Borkosky, S.; Bardón, A.; Catalán, C.A.N.; Díaz, J.G.; Herz, W. Glaucolides, hirsutinolides and other sesquiterpene lactones from Vernonanthura pinguis. Phytochemistry 1997, 44, 465–470. [Google Scholar] [CrossRef]
- Buskuhl, H.; de Oliveira, F.L.; Blind, L.Z.; de Freitas, R.A.; Barison, A.; Campos, F.R.; Corilo, Y.E.; Eberlin, M.N.; Caramori, G.F.; Biavatti, M.W. Sesquiterpene lactones from Vernonia scorpioides and their in vitro cytotoxicity. Phytochemistry 2010, 71, 1539–1544. [Google Scholar] [CrossRef]
- Tully, L.E.; Carson, M.S.; McMurry, T.B.H. A novel sesquiterpene lactone from Vernonia erinacea. Tetrahedron Lett. 1987, 28, 5925–5928. [Google Scholar] [CrossRef]
- Martínez, V.M.; Sánchez, F.A.; López, B.G.; Joseph-Nathan, P. Rearrangement of glaucolide A into vernojalcanolide 8-O-methacrylate. Z. Naturforsch. C 1986, 41, 1119–1120. [Google Scholar]
- Bazon, J.N.; Lopes, J.L.C.; Vichnewski, W.; Dias, D.A.; Nagamiti, K.; Cunha, W.R.; Herz, W. Cadinanolides and other constituents from Vernonia fruticulosa and Vernonanthura discolor. Phytochemistry 1997, 44, 1535–1536. [Google Scholar] [CrossRef]
- Martínez-Vázquez, M.; Sepúlveda, S.; Belmont, M.A.; Rubio, M.; Joseph-Nathan, P. The transformation of glaucolide A into cadinanolides and hirsutinolides. J. Nat. Prod. 1992, 55, 884–898. [Google Scholar] [CrossRef]
- Flores-Guzmán, F.; Alvarado-Sansininea, J.J.; López-Muñoz, H.; Escobar, M.L.; Espinosa-Trejo, M.; Tavera-Hernandez, R.; Jiménez-Estrada, M.; Sánchez-Sánchez, L. Antiproliferative, cytotoxic and apoptotic activity of the bentonite transformation of sesquiterpene lactone glaucolide B to 5β-hydroxy-hirsutinolide on tumor cell lines. Eur. J. Pharmacol. 2019, 856, 172406. [Google Scholar] [CrossRef]
- Ramos, A.V.G.; Peixoto, J.L.B.; Cabral, M.R.P.; Amrein, A.M.; Tiuman, T.S.; Cottica, S.M.; Souza, I.M.O.; Ruiz, A.L.T.G.; Foglio, M.A.; Carmo, M.R.B. Chemical constituents, antiproliferative and antioxidant activities of Vernonanthura nudiflora (Less.) H. Rob. aerial parts. J. Braz. Chem. Soc. 2019, 30, 1728–1740. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, X.; Wei, Y.; Wall, M.; Songsak, T.; Wongwiwatthananukit, S.; Chang, L.C. Bioactive sesquiterpene lactones isolated from the whole plants of Vernonia cinerea. J. Nat. Prod. 2019, 82, 2124–2131. [Google Scholar] [CrossRef] [PubMed]
- Chea, A.; Hout, S.; Long, C.; Marcourt, L.; Faure, R.; Azas, N.; Elias, R. Antimalarial activity of sesquiterpene lactones from Vernonia cinerea. Chem. Pharm. Bull. 2006, 54, 1437–1439. [Google Scholar] [CrossRef]
- Youn, U.J.; Park, E.-J.; Kondratyuk, T.P.; Simmons, C.J.; Borris, R.P.; Tanamatayarat, P.; Wongwiwatthananukit, S.; Toyama, O.; Songsak, T.; Pezzuto, J.M. Anti-inflammatory sesquiterpene lactones from the flower of Vernonia cinerea. Bioorg. Med. Chem. Lett. 2012, 22, 5559–5562. [Google Scholar] [CrossRef]
- Kuo, L.-M.Y.; Tseng, P.-Y.; Lin, Y.-C.; Liaw, C.-C.; Zhang, L.-J.; Tsai, K.-C.; Lin, Z.-H.; Ho, H.-O.; Kuo, Y.-H. New hirsutinolide-type sesquiterpenoids from Vernonia cinerea inhibit nitric oxide production in LPS-stimulated RAW 264. 7 cells. Planta Med. 2018, 84, 1348–1354. [Google Scholar]
- Yang, Y.-L.; Chang, S.-M.; Wu, C.-C.; Hsieh, P.-W.; Chen, S.-L.; Chang, F.-R.; Hung, W.-C.; Issa, H.H.; Wu, Y.-C. Cytotoxic sesquiterpene lactones from Pseudoelephantopus spicatus. J. Nat. Prod. 2007, 70, 1761–1765. [Google Scholar] [CrossRef]
- Girardi, C.; Fabre, N.; Paloque, L.; Ramadani, A.P.; Benoit-Vical, F.; González-Aspajo, G.; Haddad, M.; Rengifo, E.; Jullian, V. Evaluation of antiplasmodial and antileishmanial activities of herbal medicine Pseudelephantopus spiralis (Less.) Cronquist and isolated hirsutinolide-type sesquiterpenoids. J. Ethnopharmacol. 2015, 170, 167–174. [Google Scholar] [CrossRef]
- Usuga, N.d.S.J.; Malafronte, N.; Durango, E.J.O.; Braca, A.; De Tommasi, N. Phytochemical investigation of Pseudelephantopus spiralis (Less.) Cronquist. Phytochem. Lett. 2016, 15, 256–259. [Google Scholar] [CrossRef]
- Odonne, G.; Herbette, G.; Eparvier, V.; Bourdy, G.; Rojas, R.; Sauvain, M.; Stien, D. Antileishmanial sesquiterpene lactones from Pseudelephantopus spicatus, a traditional remedy from the Chayahuita Amerindians (Peru). Part III. J. Ethnopharmacol. 2011, 137, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yu, J.; Kinghorn, A.D. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr. Med. Chem. 2016, 23, 2397–2420. [Google Scholar] [CrossRef] [PubMed]
- Quintana, J.; Estévez, F. Recent advances on cytotoxic sesquiterpene lactones. Curr. Pharm. Des. 2018, 24, 4355–4361. [Google Scholar] [CrossRef] [PubMed]
- Sheetal, C.; Mishra, P.K. A review on pharmacognocy of bioactive sesquiterpene lactones. Int. J. Pharmacogn. Phytochem. Res. 2019, 11, 116–121. [Google Scholar]
- Mandal, S.K.; Debnath, U.; Kumar, A.; Thomas, S.; Mandal, S.C.; Choudhury, M.D.; Palit, P. Natural sesquiterpene lactones in the prevention and treatment of inflammatory disorders and cancer: A systematic study of this emerging therapeutic approach based on chemical and pharmacological aspect. Lett. Drug Des. Discov. 2020, 17, 1102–1116. [Google Scholar] [CrossRef]
- Miklossy, G.; Youn, U.J.; Yue, P.; Zhang, M.; Chen, C.-H.; Hilliard, T.S.; Paladino, D.; Li, Y.; Choi, J.; Sarkaria, J.N. Hirsutinolide series inhibit Stat3 activity, alter GCN1, MAP1B, Hsp105, G6PD, vimentin, TrxR1, and importin α-2 expression, and induce antitumor effects against human glioma. J. Med. Chem. 2015, 58, 7734–7748. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Sellier, H.; Rébillard, A.; Guette, C.; Barré, B.; Coqueret, O. How should we define STAT3 as an oncogene and as a potential target for therapy? Jak-Stat 2013, 2, e24716. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- INCA. Estimativa 2020: Incidência de câncer no Brasil; Instituto Nacional de Câncer José Alencar Gomes da Silva, Ministério da Saúde: Rio de Janeiro, Brazil, 2019; pp. 1–120. [Google Scholar]
- De Cicco, P.; Busà, R.; Ercolano, G.; Formisano, C.; Allegra, M.; Taglialatela-Scafati, O.; Ianaro, A. Inhibitory effects of cynaropicrin on human melanoma progression by targeting MAPK, NF-κB, and Nrf-2 signaling pathways in vitro. Phytother. Res. 2021, 35, 1432–1442. [Google Scholar] [CrossRef]
- Rozenblat, S.; Grossman, S.; Bergman, M.; Gottlieb, H.; Cohen, Y.; Dovrat, S. Induction of G2/M arrest and apoptosis by sesquiterpene lactones in human melanoma cell lines. Biochem. Pharmacol. 2008, 75, 369–382. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Liu, M.; Xie, R.; Zang, Y.; Li, J.; Aisa, H.A. Guaianolide sesquiterpene lactones from Achillea millefolium L. Phytochemistry 2021, 186, 112733. [Google Scholar] [CrossRef]
- Da Silva, L.A.L.; Sandjo, L.P.; Fratoni, E.; Moon, Y.J.K.; Dalmarco, E.M.; Biavatti, M.W. A single-step isolation by centrifugal partition chromatography of the potential anti-inflammatory glaucolide B from Lepidaploa chamissonis. J. Chromatogr. A 2019, 1605, 1–8. [Google Scholar] [CrossRef]
- Jakupovic, J.; Schmeda-Hirschmann, G.; Schuster, A.; Zdero, C.; Bohlmann, F.; King, R.M.; Robinson, H.; Pickardt, J. Hirsutinolides, glaucolides and sesquiterpene lactone from Vernonia species. Phytochemistry 1986, 25, 145–158. [Google Scholar] [CrossRef]
- Le Roux, C.; Mandrou, S.; Dubac, J. First catalytic C-acylation of enoxysilanes: An efficient route to β-diketones. J. Org. Chem. 1996, 61, 3885–3887. [Google Scholar] [CrossRef]
- Salvador, J.A.; Silvestre, S.M.; Pinto, R. Bismuth (III) reagents in steroid and terpene chemistry. Molecules 2011, 16, 2884–2913. [Google Scholar] [CrossRef]
- Rodríguez-Hahn, L.; Cárdenas, J.; Maldonado, E.; Ortega, A.; Martínez, M.; Soriano Garcia, M.; Toscano, A. Transannular cyclization of glaucolide A. J. Org. Chem. 1988, 53, 2965–2968. [Google Scholar] [CrossRef]
- Hua, R. Recent advances in bismuth-catalyzed organic synthesis. Curr. Org. Synth. 2008, 5, 1–27. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Pinto, R.; Silvestre, S.M. Recent advances of bismuth (III) salts in organic chemistry: Application to the synthesis of aliphatics, alicyclics, aromatics, amino acids and peptides, terpenes and steroids of pharmaceutical interest. Mini Rev. Org. Chem. 2009, 6, 241–274. [Google Scholar] [CrossRef]
- Salvador, J.A.; Pinto, R.; Silvestre, S.M. Recent advances of bismuth (III) salts in organic chemistry: Application to the synthesis of heterocycles of pharmaceutical interest. Curr. Org. Synth. 2009, 6, 426–470. [Google Scholar] [CrossRef]
- Ondet, P.; Lemière, G.; Duñach, E. Cyclisations catalysed by bismuth (III) triflate. Eur. J. Org. Chem. 2017, 2017, 761–780. [Google Scholar] [CrossRef]
- Catalán, C.A.N.; De Iglesias, D.I.A.; Kavka, J.; Sosa, V.E.; Herz, W. Glaucolides and related sesquiterpene lactones from Vernonia chamaedrys. Phytochemistry 1988, 27, 197–202. [Google Scholar] [CrossRef]
- Mandai, H.; Fujii, K.; Yasuhara, H.; Abe, K.; Mitsudo, K.; Korenaga, T.; Suga, S. Enantioselective acyl transfer catalysis by a combination of common catalytic motifs and electrostatic interactions. Nat. Commun. 2016, 7, 11297. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Held, I.; Kempf, B.; Mayr, H.; Steglich, W.; Zipse, H. The DMAP-catalyzed acetylation of alcohols—A mechanistic study (DMAP= 4-(dimethylamino)pyridine). Chem. Eur. J. 2005, 11, 4751–4757. [Google Scholar] [CrossRef]
- Fischer, C.B.; Xu, S.; Zipse, H. Steric effects in the uncatalyzed and DMAP-catalyzed acylation of alcohols—Quantifying the window of opportunity in kinetic resolution experiments. Chem. Eur. J. 2006, 12, 5779–5784. [Google Scholar] [CrossRef]
- Ouellette, R.J.; Rawn, J.D. Haloalkanes and alcohols: Introduction to nucleophilic substitution and elimination reactions. In Organic Chemistry: Structure, Mechanism, and Synthesis; Ouellette, R.J., Rawn, J.D., Eds.; Elsevier: San Diego, CA, USA, 2014; pp. 287–331. [Google Scholar]
- Bohlmann, F.; Zdero, C.; King, R.M.; Robinson, H. Neue sesquiterpenlactone aus Stokesia laevis. Phytochemistry 1979, 18, 987–989. [Google Scholar] [CrossRef]
- Bohlmann, F.; Mahanta, P.K.; Dutta, L.N. Weitere hirsutinolide aus Vernonia-arten. Phytochemistry 1979, 18, 289–291. [Google Scholar] [CrossRef]
- Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G.-G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar]
- Serasanambati, M.; Chilakapati, S.R. Function of nuclear factor kappa B (NF-kB) in human diseases—A review. South Indian J. Biol. Sci. 2016, 2, 368–387. [Google Scholar] [CrossRef]
- D'Anneo, A.; Carlisi, D.; Lauricella, M.; Emanuele, S.; Di Fiore, R.; Vento, R.; Tesoriere, G. Parthenolide induces caspase-independent and AIF-mediated cell death in human osteosarcoma and melanoma cells. J. Cell. Physiol. 2013, 228, 952–967. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.; Svensson, D.; Oredsson, S.; Sterner, O. Cytotoxic sesquiterpene lactones from Kauna lasiophthalma Griseb. Sci. Pharm. 2014, 82, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, J.-P.; Liu, X.; Tang, J.-J.; Xiang, P.; Ma, X.-M. Semisynthesis, an anti-inflammatory effect of derivatives of 1β-hydroxy alantolactone from Inula britannica. Molecules 2017, 22, 1835. [Google Scholar] [CrossRef]
- Nakagawa-Goto, K.; Chen, J.-Y.; Cheng, Y.-T.; Lee, W.-L.; Takeya, M.; Saito, Y.; Lee, K.-H.; Shyur, L.-F. Novel sesquiterpene lactone analogues as potent anti-breast cancer agents. Mol. Oncol. 2016, 10, 921–937. [Google Scholar] [CrossRef]
- Saroglou, V.; Karioti, A.; Demetzos, C.; Dimas, K.; Skaltsa, H. Sesquiterpene Lactones from Centaurea spinosa and their antibacterial and cytotoxic activities. J. Nat. Prod. 2005, 68, 1404–1407. [Google Scholar] [CrossRef]
- Da Silva, L.A.L.; Sandjo, L.P.; Misturini, A.; Caramori, G.F.; Biavatti, M.W. ESI-QTof-MS characterization of hirsutinolide and glaucolide sesquiterpene lactones: Fragmentation mechanisms and differentiation based on Na+/H+ adducts interactions in complex mixture. J. Mass Spectrom. 2019, 54, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Dirsch, V.M.; Stuppner, H.; Ellmerer-Müller, E.P.; Vollmar, A.M. Structural requirements of sesquiterpene lactones to inhibit LPS-induced nitric oxide synthesis in RAW 264.7 macrophages. Bioorg. Med. Chem. 2000, 8, 2747–2753. [Google Scholar] [CrossRef]
- Han, I.-H.; Song, H.-O.; Ryu, J.-S. IL-6 produced by prostate epithelial cells stimulated with Trichomonas vaginalis promotes proliferation of prostate cancer cells by inducing M2 polarization of THP-1-derived macrophages. PLOS Negl. Trop. Dis. 2020, 14, e0008126. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H.; Bladt, S. Plant Drug Analysis: A Thin Layer Chromatography Atlas, 2nd ed.; Springer: London, UK, 2001. [Google Scholar]
- ChemAxon Marvin 19.1.11.0. Available online: https://www.chemaxon.com (accessed on 27 January 2016).
- ChemAxon Standardizer JChem 16.1.11.0. Available online: https://www.chemaxon.com (accessed on 27 January 2016).
- Imre, G.; Veress, G.; Volford, A.; Farkas, Ö. Molecules from the Minkowski space: An Approach to Building 3D Molecular structures. J. Mol. Struct. THEOCHEM 2003, 666, 51–59. [Google Scholar] [CrossRef]
- Spartan. Model Homepage for Windows. Available online: http://www.wavefun.com/products/windows/SpartanModel/win_model.html (accessed on 27 April 2013).
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Ulam, S. The Monte Carlo method. J. Am. Stat. Assoc. 1949, 44, 335–341. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lin, C.Y.; George, M.W.; Gill, P.M.W. EDF2: A density functional for predicting molecular vibrational frequencies. Aust. J. Chem. 2004, 57, 365–370. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lavogina, D.; Lust, H.; Tahk, M.-J.; Laasfeld, T.; Vellama, H.; Nasirova, N.; Vardja, M.; Eskla, K.-L.; Salumets, A.; Rinken, A. Revisiting the resazurin-based sensing of cellular viability: Widening the application horizon. Biosensors 2022, 12, 196. [Google Scholar] [CrossRef]
- Herath, H.M.T.; Takano-Ishikawa, Y.; Yamaki, K. Inhibitory effect of some flavonoids on tumor necrosis factor-α production in lipopolysaccharide-stimulated mouse macrophage cell line J774. 1. J. Med. Food 2003, 6, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Griess, P. Bemerkungen zu der Abhandlung der HH. Weselky und Benedikt ueber einege Azoverbindungen. Ber. Dtsch. Chem. Ges. 1879, 12, 426–428. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Lines | |||||||
|---|---|---|---|---|---|---|---|---|
| SK-MEL-28 | HUVEC | NCI-H460 | SF295 | |||||
| Viability (%) a | CC50 (μM) | SI | CC50 (μM) | Viability (%) a | CC50 (μM) | Viability (%) a | CC50 (μM) | |
| Glaucolide B (1) | 82.6 ± 4.1 | n.d. | - | n.d. | 85.1 ± 4.6 | n.d. | 88.0 ± 4.8 | n.d. |
| Vernojalcanolide derivative (2) | 87.1 ± 2.6 | n.d. | - | n.d. | 91.8 ± 3.6 | n.d. | 86.8 ± 6.3 | n.d. |
| Vernojalcanolide derivative (3) | 91.1 ± 1.7 | n.d. | - | n.d. | 88.4 ± 3.4 | n.d. | 86.1 ± 2.3 | n.d. |
| Vernomargolide derivative (4) | 96.4 ± 6.9 | n.d. | - | n.d. | 100.2 ± 2.7 | n.d. | 94.7 ± 1.1 | n.d. |
| Vernomargolide derivative (5) | 95.3 ± 4.1 | n.d. | - | n.d. | 100.3 ± 5.5 | n.d. | 95.6 ± 3.9 | n.d. |
| Vernomargolide derivative (6) | 85.9 ± 4.0 | n.d. | - | n.d. | 90.4 ± 1.5 | n.d. | 90.4 ± 3.7 | n.d. |
| Hirsutinolide derivative (7) | 76.2 ± 7.5 | n.d. | - | n.d. | 89.1 ± 3.9 | n.d. | 86.7 ± 3.8 | n.d. |
| Hirsutinolide derivative (7a) | 17.1 ± 3.3 | 5.0 | 2.5 | 12.7 | 74.6 ± 1.3 | n.d. | 86.9 ± 4.3 | n.d. |
| Hirsutinolide derivative (7b) | 16.0 ± 2.6 | 11.2 | 2.5 | 27.8 | 73.2 ± 1.0 | n.d. | 83.3 ± 1.2 | n.d. |
| Hirsutinolide derivative (7c) | 79.2 ± 6.4 | n.d. | - | n.d. | 85.7 ± 1.6 | n.d. | 88.6 ± 4.8 | n.d. |
| Germacranolide derivative (7c.1) | 77.3 ± 0.3 | n.d. | - | n.d. | 88.7 ± 6.7 | n.d. | 87.0 ± 4.7 | n.d. |
| Germacranolide derivative (8) | 66.9 ± 3.5 | n.d. | - | n.d. | 87.3 ± 1.6 | n.d. | 82.0 ± 4.7 | n.d. |
| Germacranolide derivative (8a) | 5.3 ± 0.3 | 3.1 | 3.0 | 9.0 | 53.1 ± 0.0 | n.d. | 67.3 ± 4.0 | n.d. |
| Vernomargolide derivative (9) | 74.5 ± 4.9 | n.d. | - | n.d. | 84.9 ± 0.4 | n.d. | 85.2 ± 2.8 | n.d. |
| Vernomargolide derivative (9a) | 73.8 ± 3.1 | n.d. | - | n.d. | 84.8 ± 0.9 | n.d. | 85.9 ± 5.0 | n.d. |
| Piptocarphin A (10) | 23.1 ± 4.2 | 6.7 | 1.9 | 12.5 | 104.8 ± 1.6 | n.d. | 93.2 ± 3.2 | n.d. |
| Glaucolide A (11) | 56.3 ± 1.4 | 14.6 | 1.4 | 20.4 | 95.1 ± 4.6 | n.d. | 91.8 ± 3.3 | n.d. |
| Diacetylpiptocarphol (12) | 9.0 ± 0.5 | 3.8 | 3.3 | 12.6 | 63.8 ± 6.9 | n.d. | 80.4 ± 3.3 | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, L.A.L.; Sandjo, L.P.; Assunção, L.S.; Prigol, A.N.; de Siqueira, C.D.; Creczynski-Pasa, T.B.; Scotti, M.T.; Scotti, L.; Filippin-Monteiro, F.B.; Biavatti, M.W. Semisynthetic Sesquiterpene Lactones Generated by the Sensibility of Glaucolide B to Lewis and Brønsted–Lowry Acids and Bases: Cytotoxicity and Anti-Inflammatory Activities. Molecules 2023, 28, 1243. https://doi.org/10.3390/molecules28031243
da Silva LAL, Sandjo LP, Assunção LS, Prigol AN, de Siqueira CD, Creczynski-Pasa TB, Scotti MT, Scotti L, Filippin-Monteiro FB, Biavatti MW. Semisynthetic Sesquiterpene Lactones Generated by the Sensibility of Glaucolide B to Lewis and Brønsted–Lowry Acids and Bases: Cytotoxicity and Anti-Inflammatory Activities. Molecules. 2023; 28(3):1243. https://doi.org/10.3390/molecules28031243
Chicago/Turabian Styleda Silva, Layzon A. Lemos, Louis P. Sandjo, Laura S. Assunção, Anne N. Prigol, Carolina D. de Siqueira, Tânia B. Creczynski-Pasa, Marcus T. Scotti, Luciana Scotti, Fabíola B. Filippin-Monteiro, and Maique W. Biavatti. 2023. "Semisynthetic Sesquiterpene Lactones Generated by the Sensibility of Glaucolide B to Lewis and Brønsted–Lowry Acids and Bases: Cytotoxicity and Anti-Inflammatory Activities" Molecules 28, no. 3: 1243. https://doi.org/10.3390/molecules28031243
APA Styleda Silva, L. A. L., Sandjo, L. P., Assunção, L. S., Prigol, A. N., de Siqueira, C. D., Creczynski-Pasa, T. B., Scotti, M. T., Scotti, L., Filippin-Monteiro, F. B., & Biavatti, M. W. (2023). Semisynthetic Sesquiterpene Lactones Generated by the Sensibility of Glaucolide B to Lewis and Brønsted–Lowry Acids and Bases: Cytotoxicity and Anti-Inflammatory Activities. Molecules, 28(3), 1243. https://doi.org/10.3390/molecules28031243