



Coumarin-Based Sulfonamide Derivatives as Potential DPP-IV Inhibitors: Pre-ADME Analysis, Toxicity Profile, Computational Analysis, and In Vitro Enzyme Assay
 , , , , , , , ,
, , , , , , , , 
Abstract
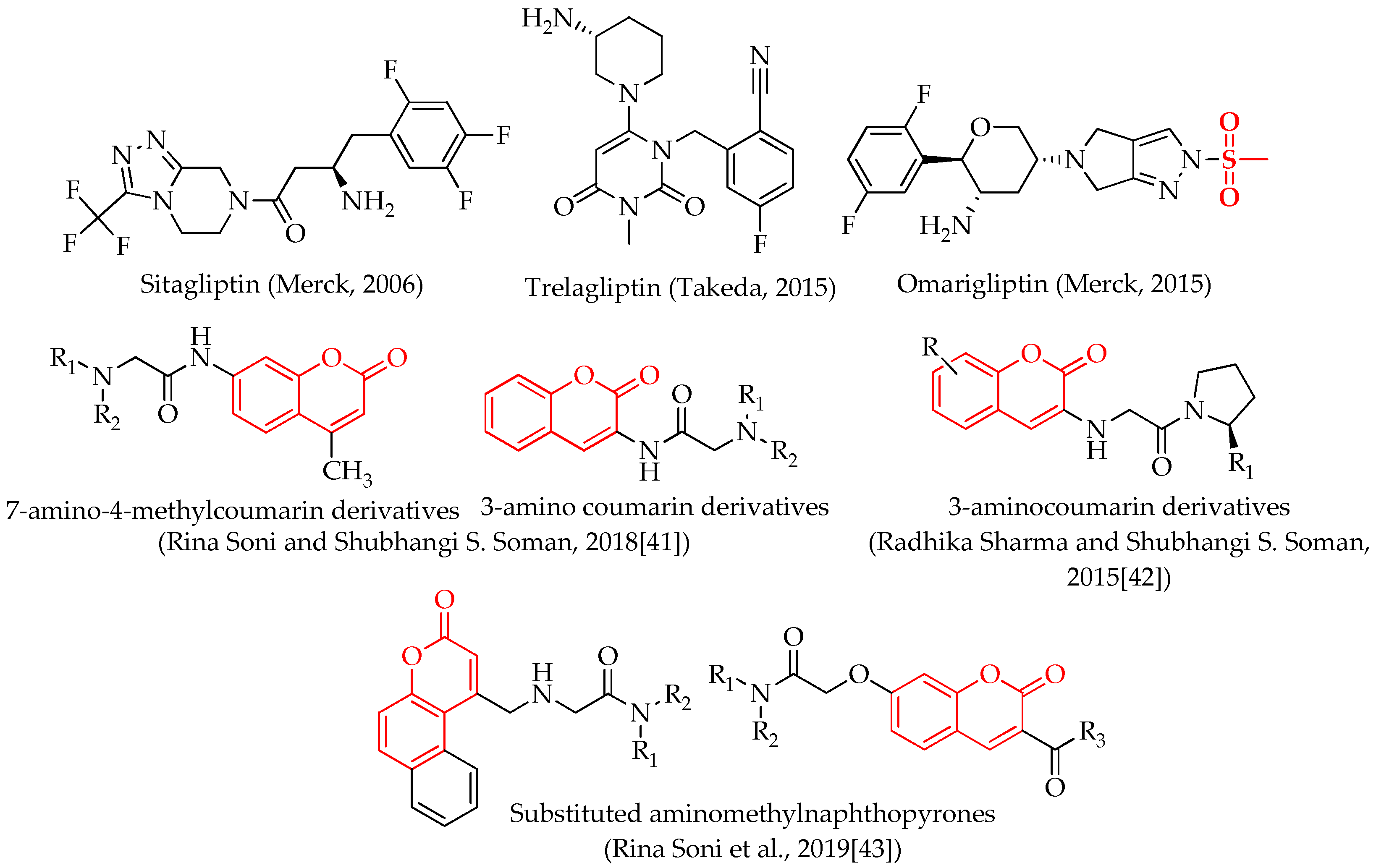
1. Introduction
2. Results and Discussion
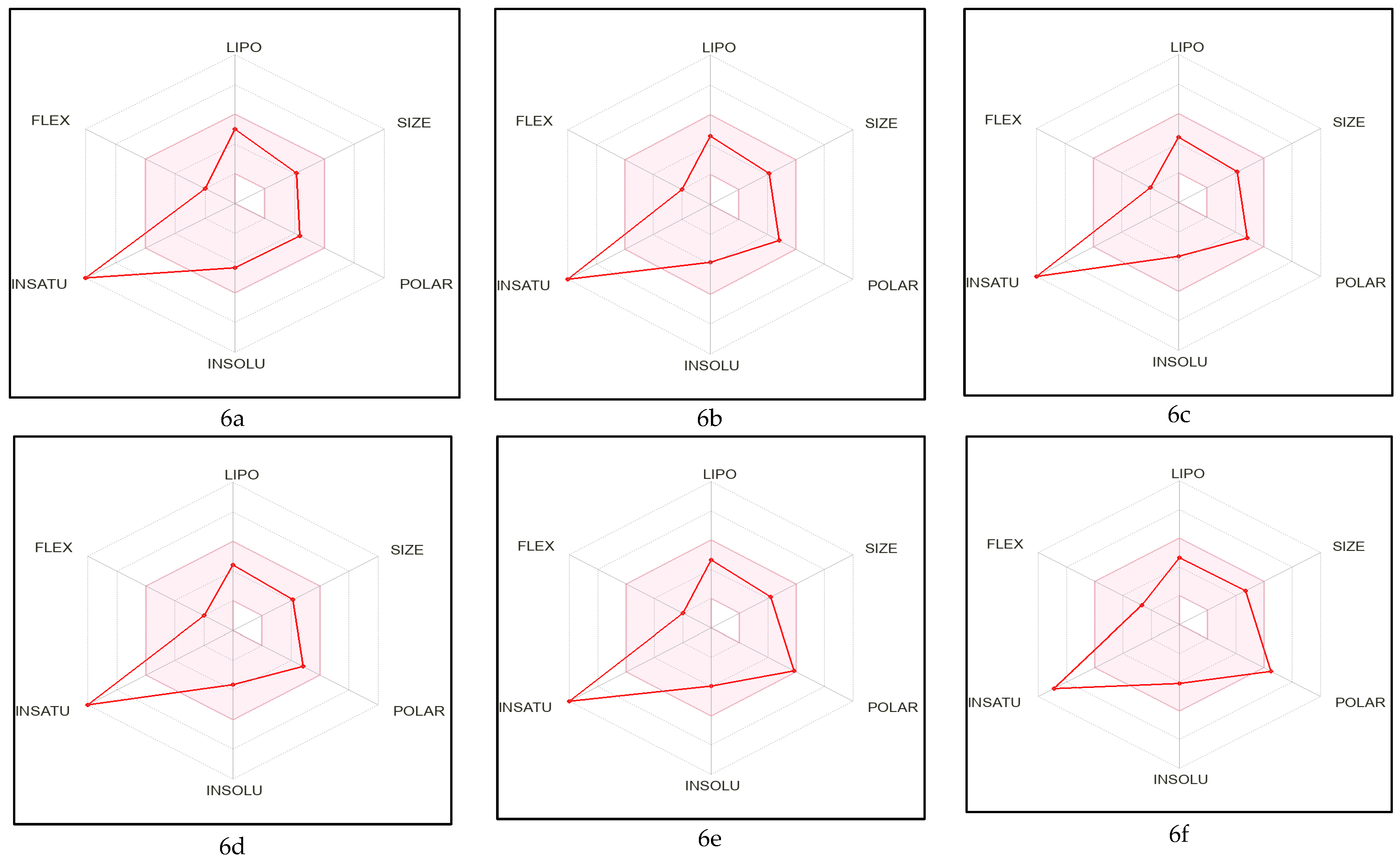
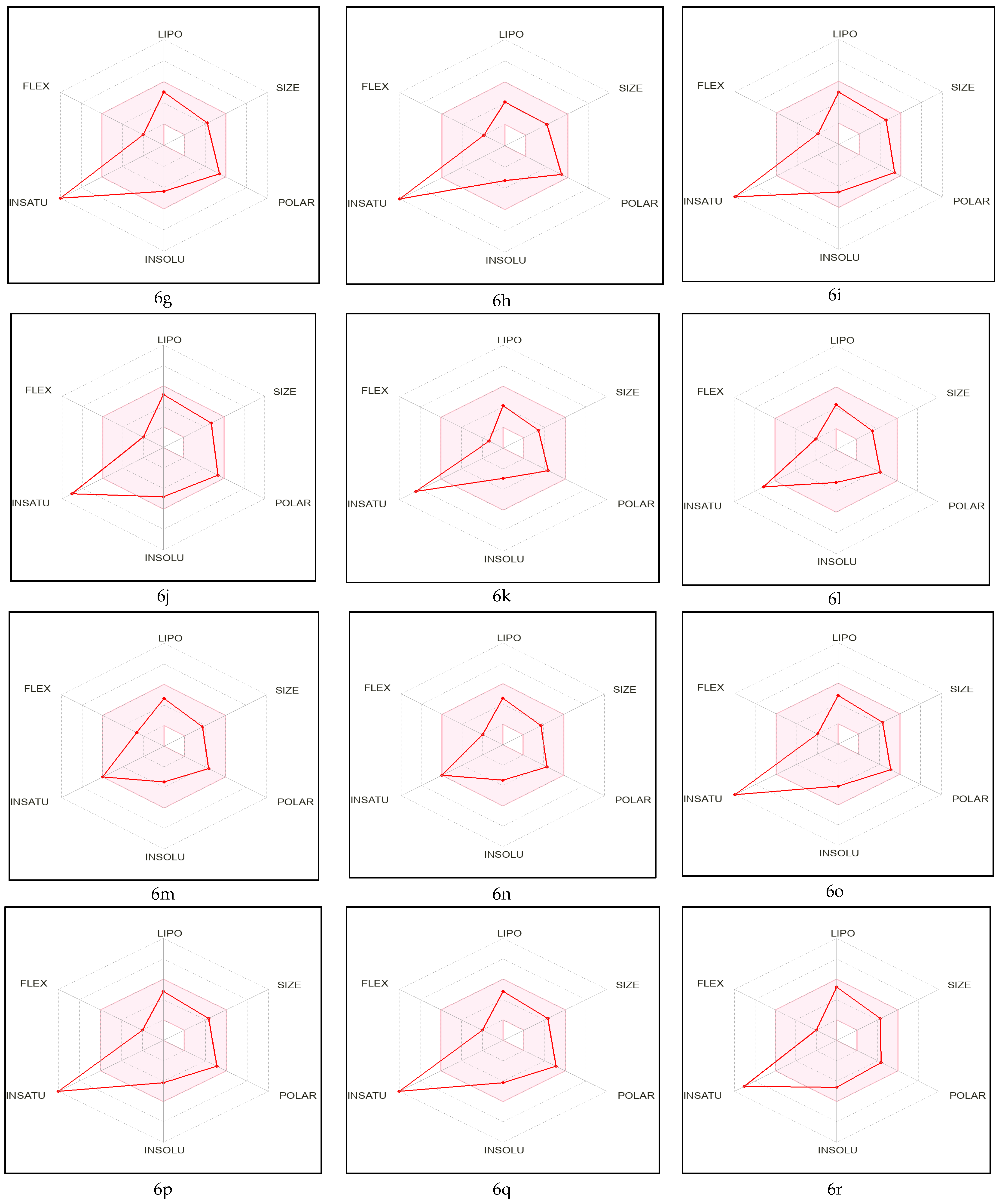
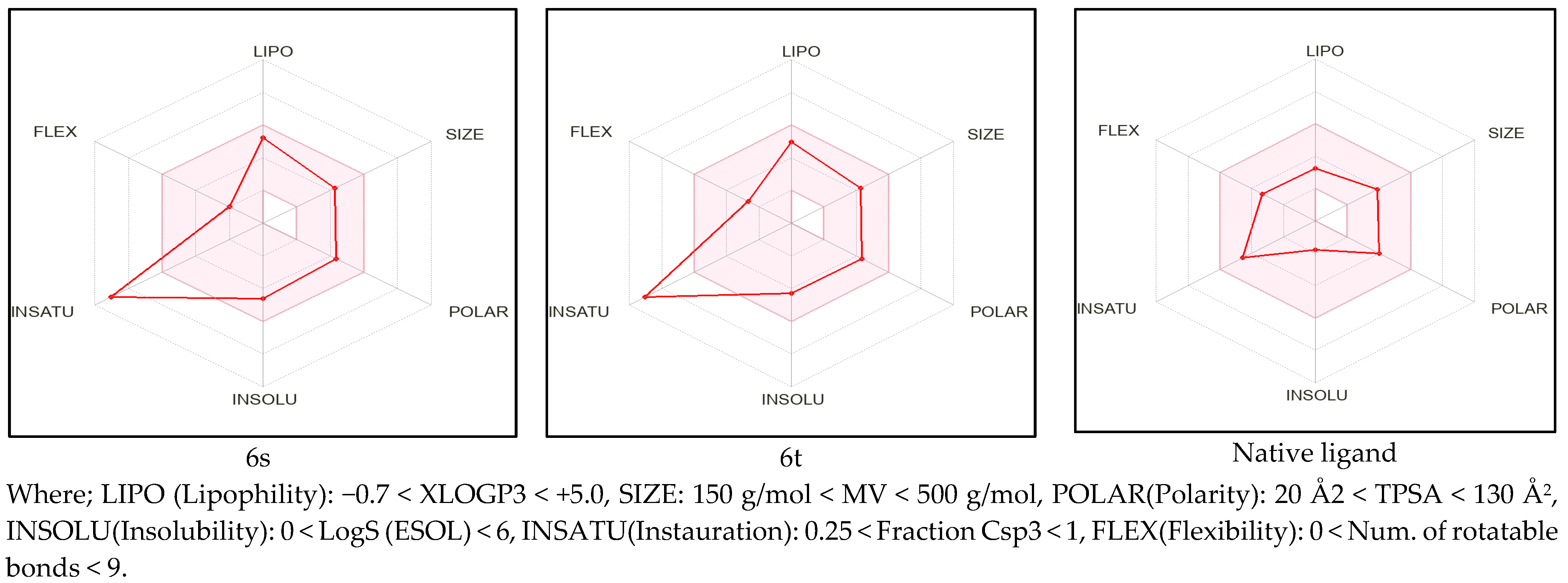
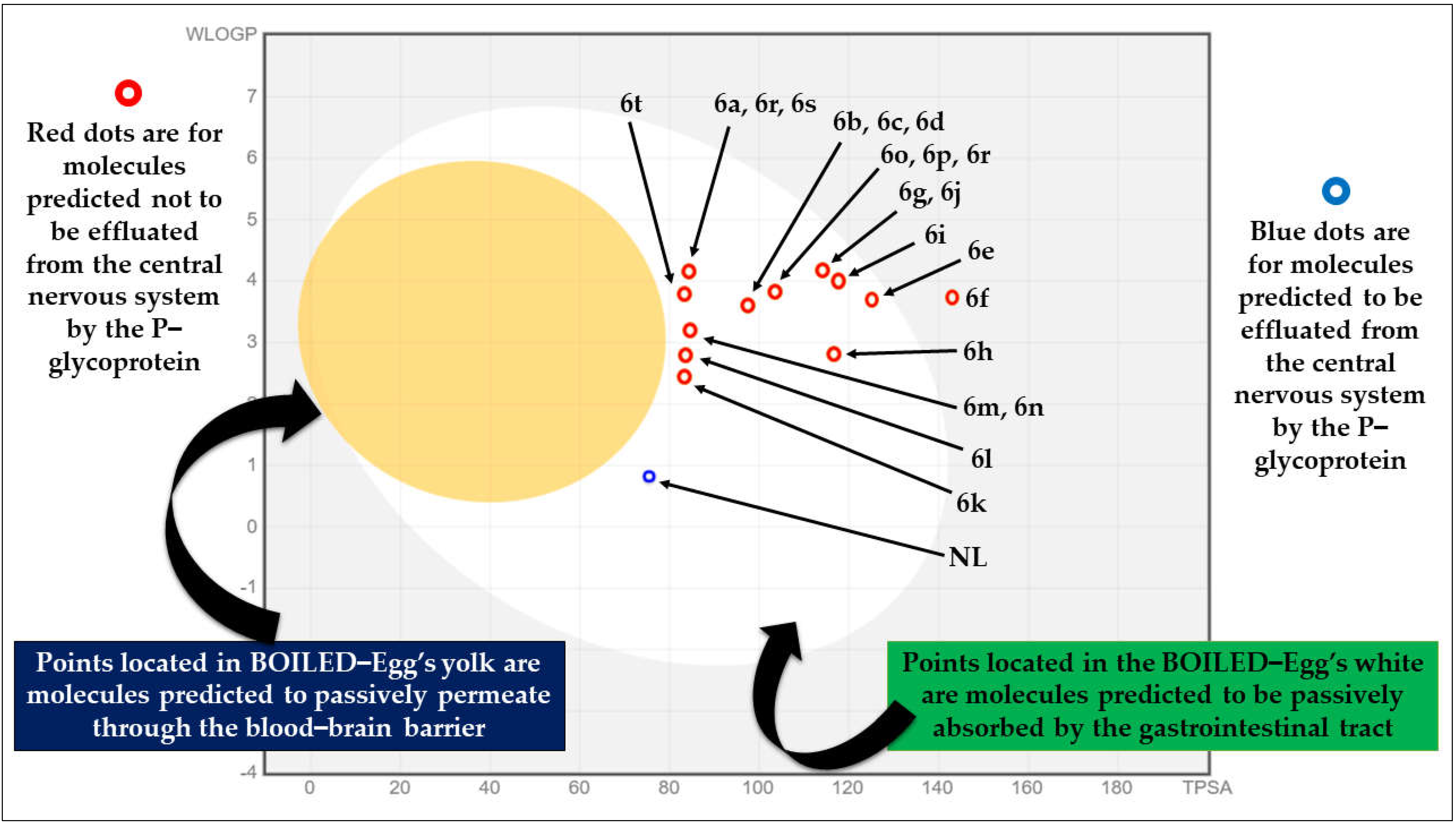
2.1. Pre-ADME Analysis and Toxicity Profile
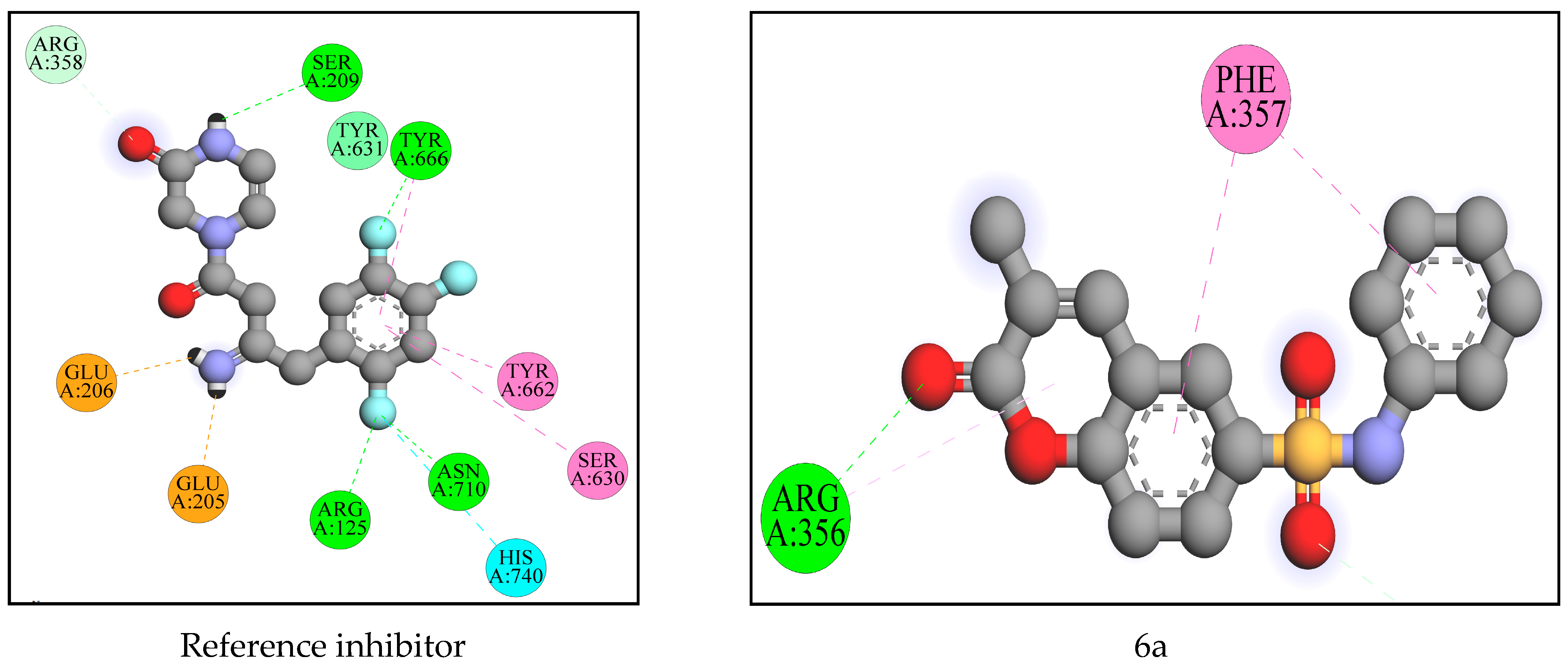
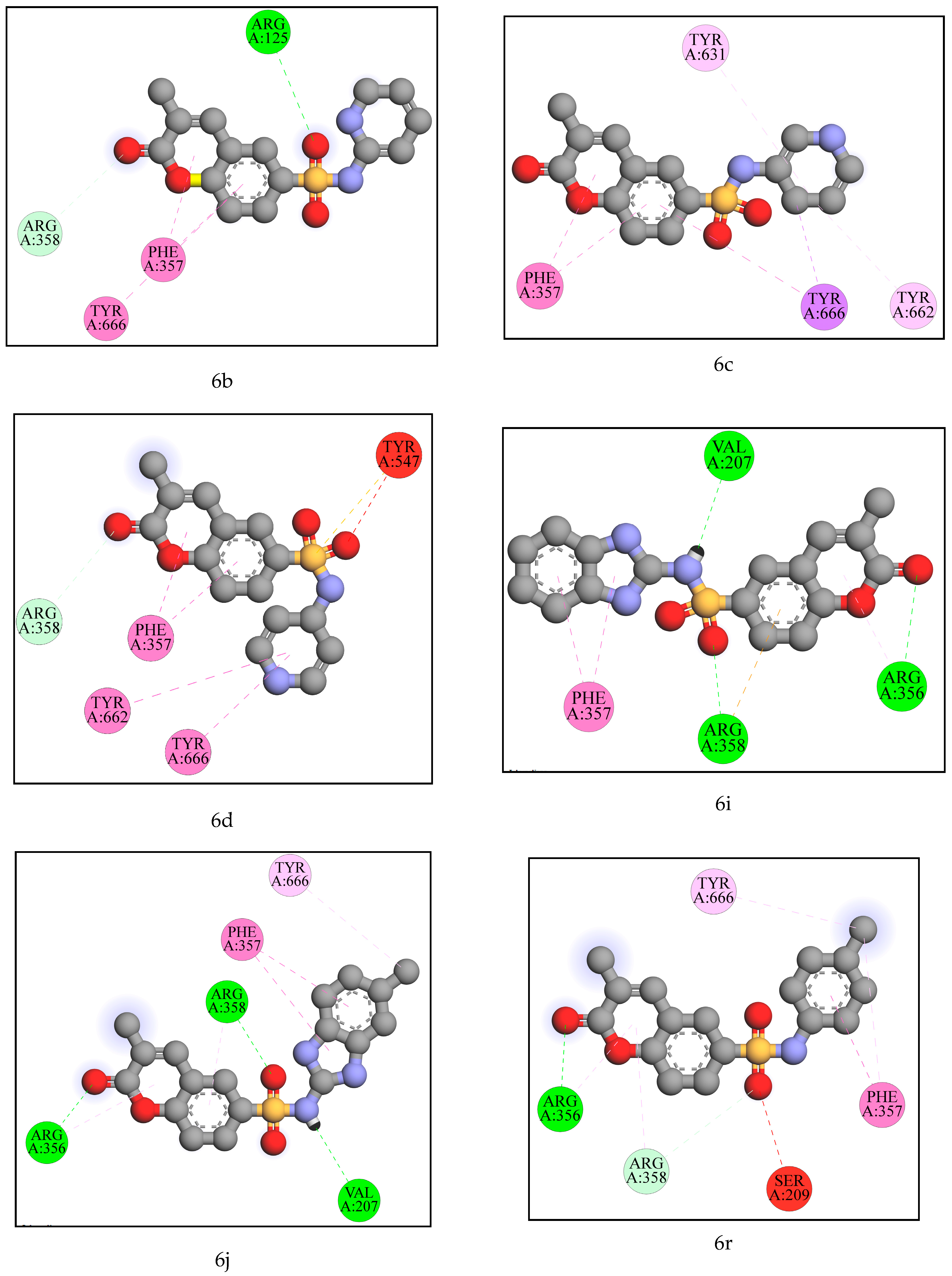
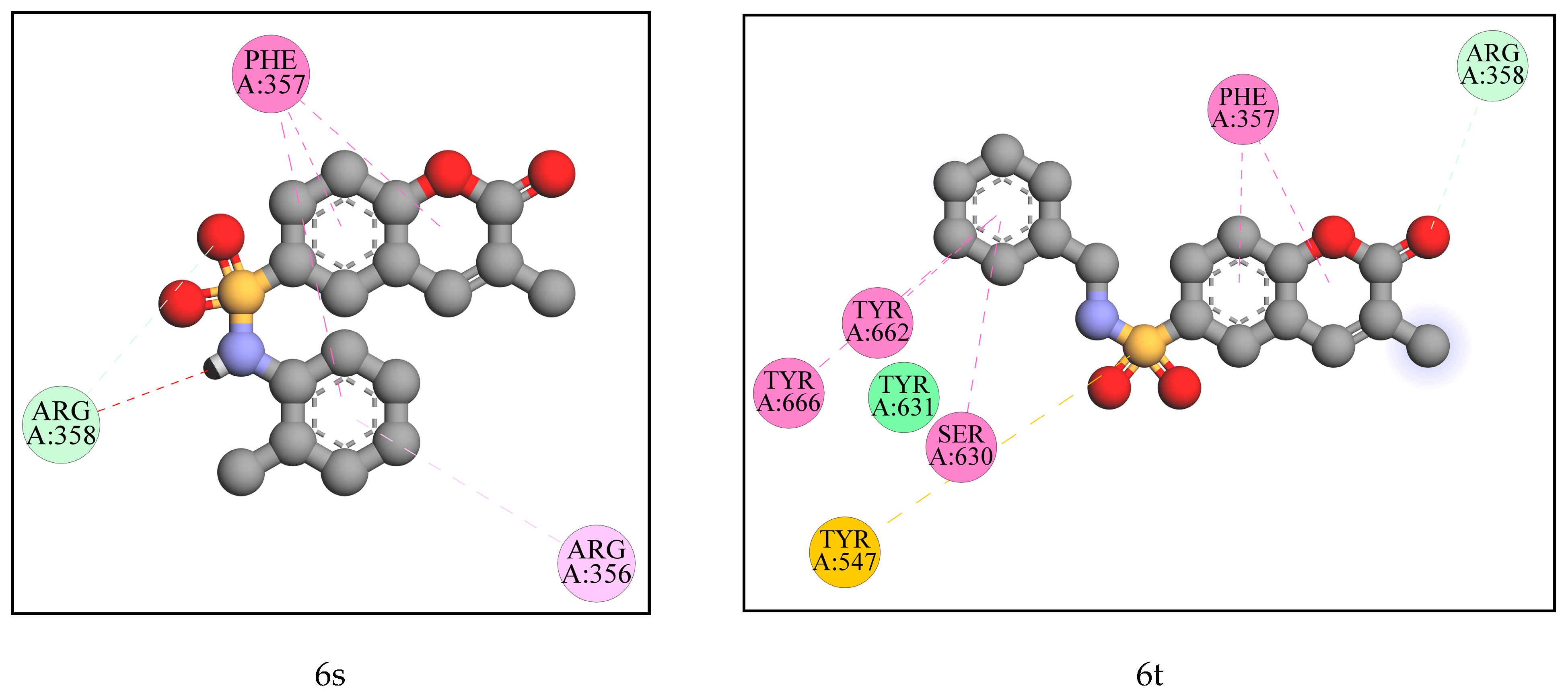
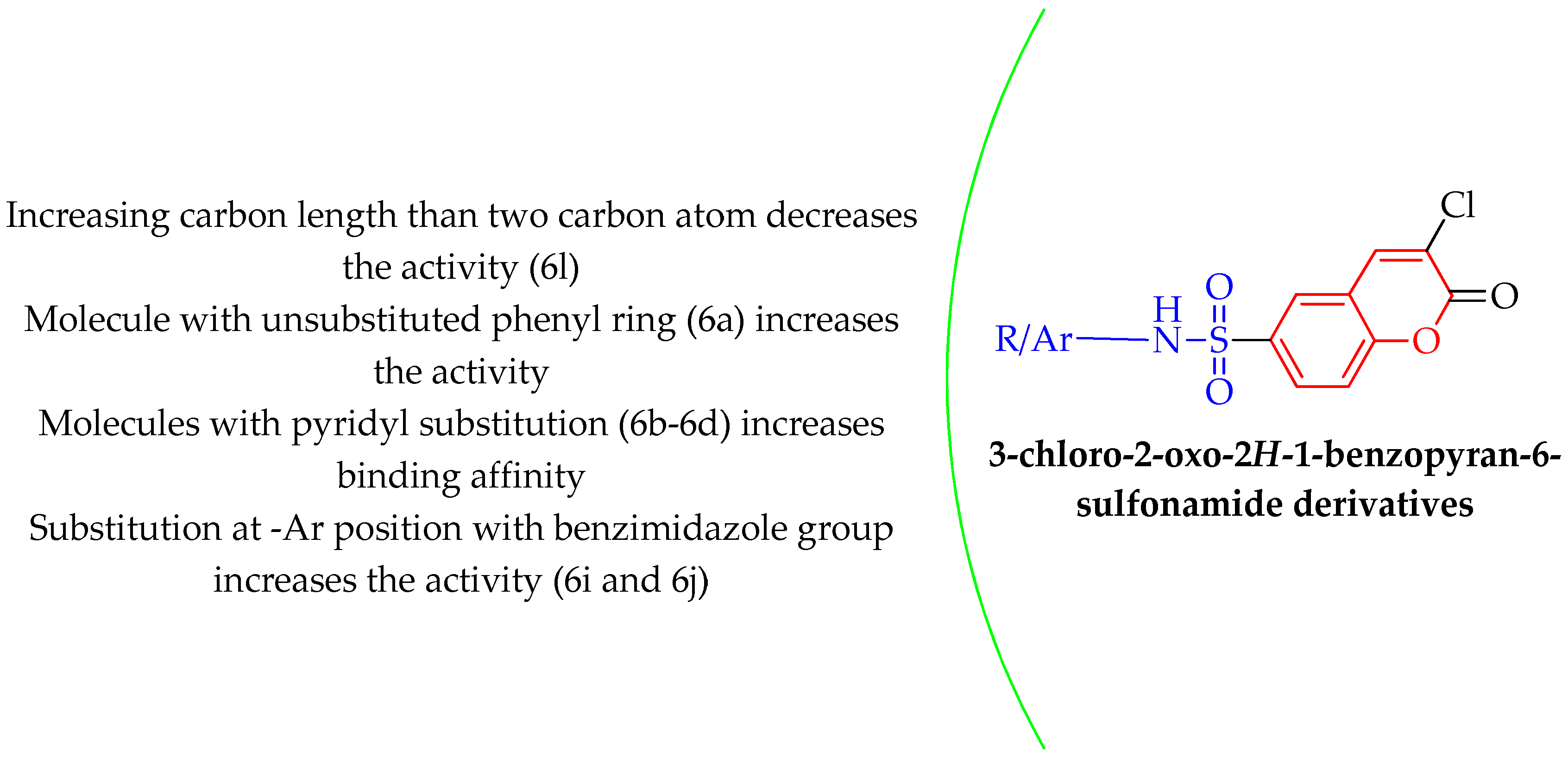
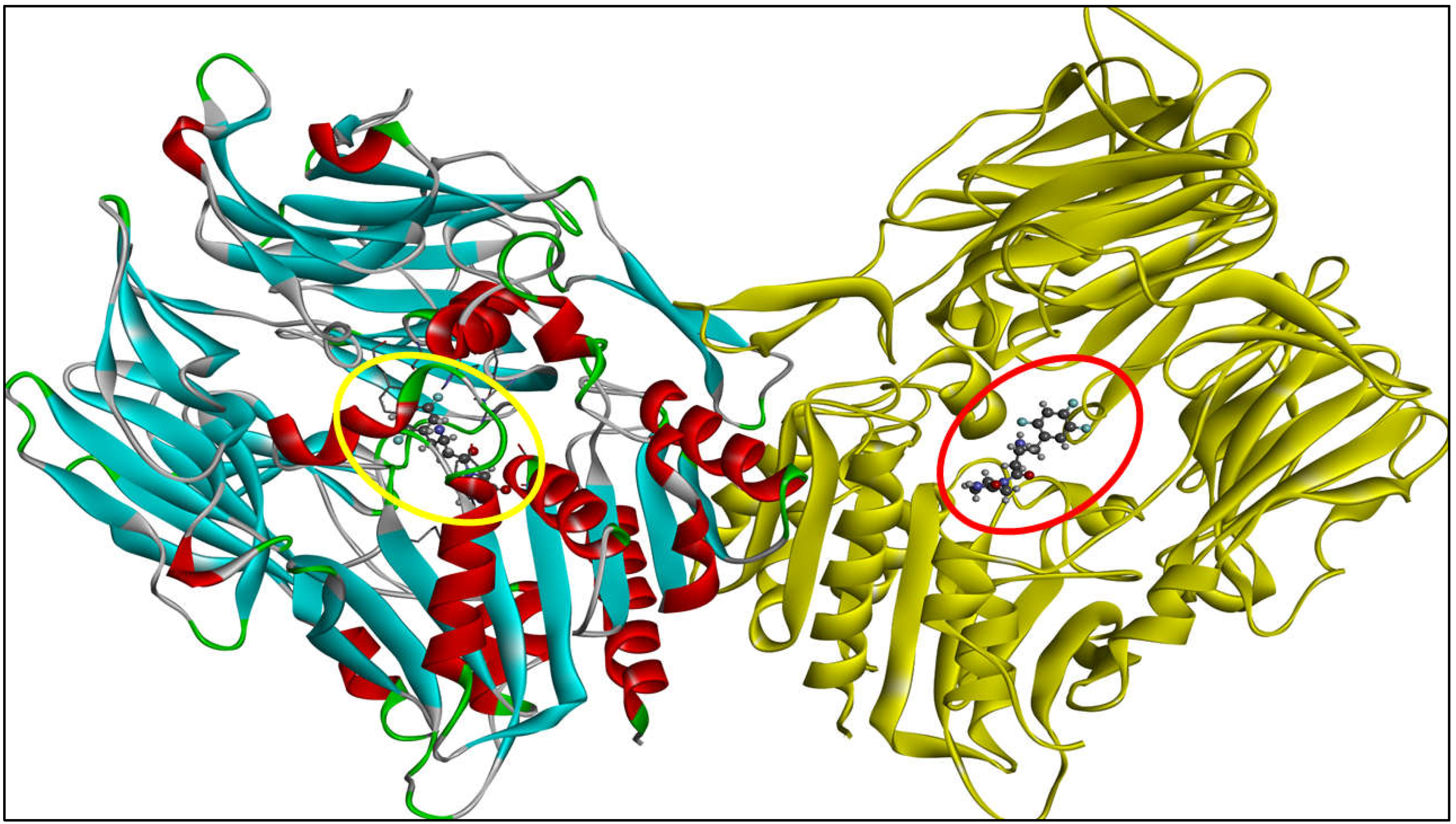
2.2. Computational Analysis
2.3. In Vitro Enzyme Assay
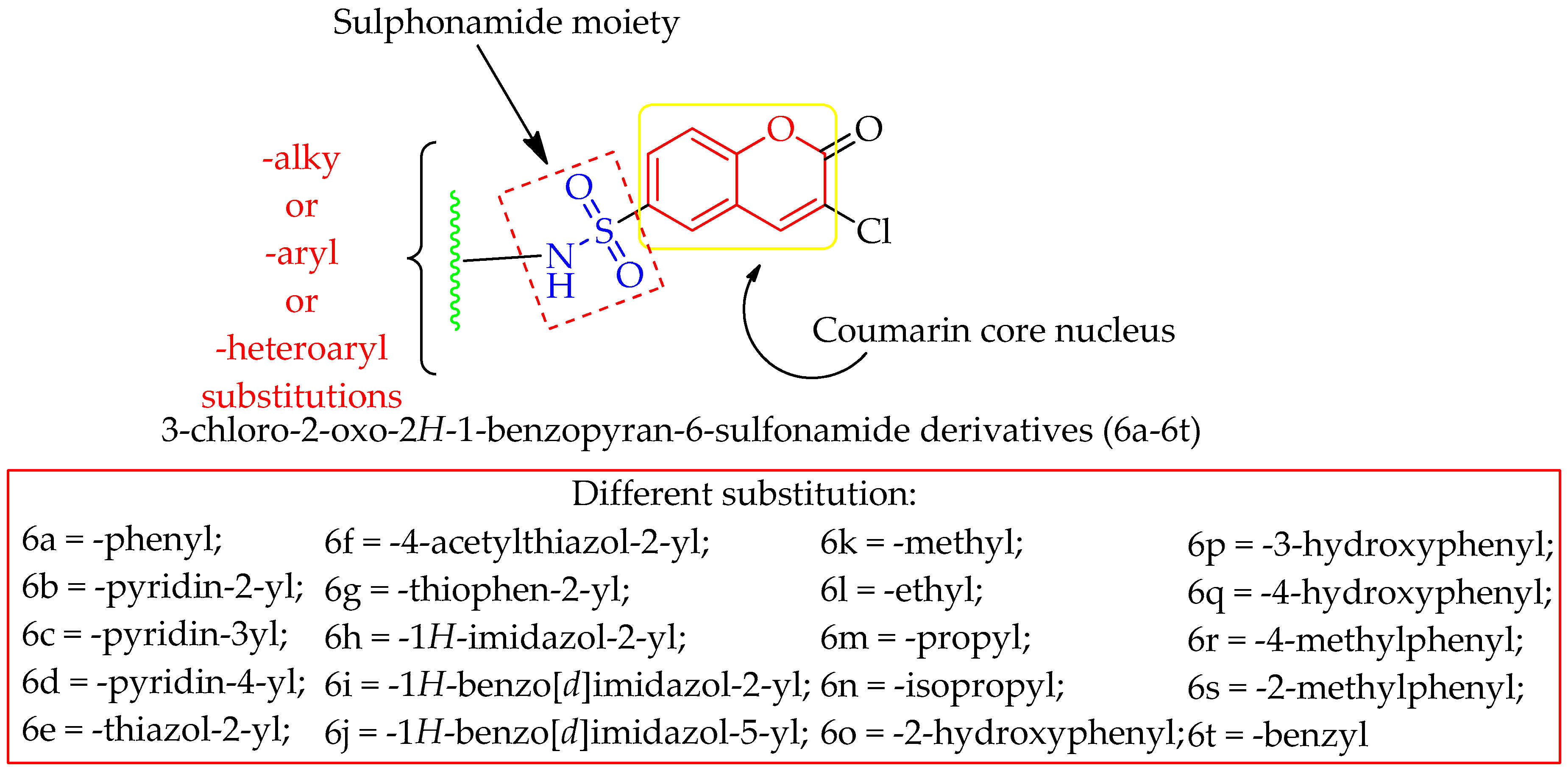
3. Material and Methods
3.1. Pre-ADME Analysis, Toxicity Profile, and Computational Analysis
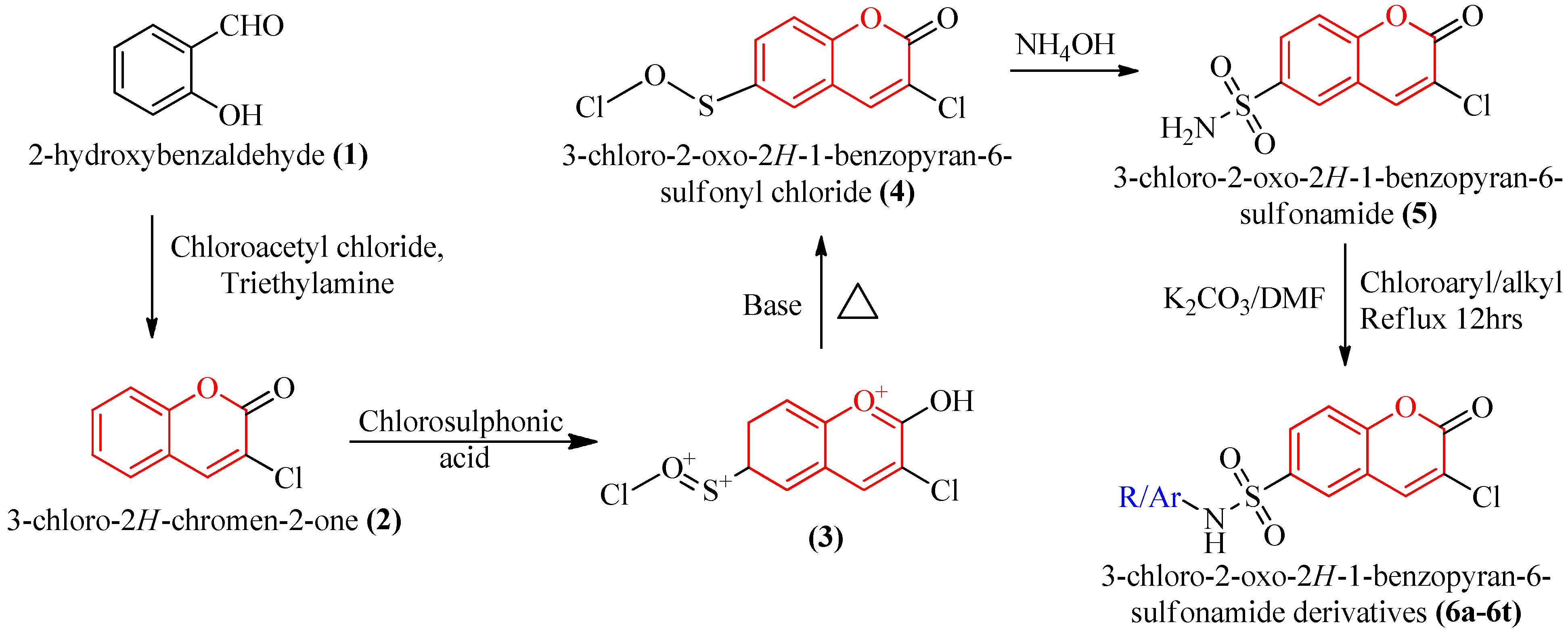
3.2. Chemistry
3.2.1. 6a. [3-Chloro-2-oxo-2H-1-benzopyran-6-(N-phenyl)sulfonamide]
3.2.2. 6b. [3-Chloro-2-oxo-N-(pyridin-2-yl)-2H-chromene-6-sulfonamide]
3.2.3. 6c. [3-Chloro-2-oxo-N-(pyridin-3-yl)-2H-chromene-6-sulfonamide]
3.2.4. 6d. [3-Chloro-2-oxo-N-(pyridin-4-yl)-2H-chromene-6-sulfonamide]
3.2.5. 6i. [3-Chloro-2-oxo-2H-1-benzopyran-6-(N-benzimidazole)sulfonamide]
3.2.6. 6j. [3-Chloro-2-oxo-2H-1-benzopyran-6-(N-(5-methyl)benzimidazole)sulfonamide]
3.2.7. 6r. [3-Chloro-2-oxo-N-(p-tolyl)-2H-chromene-6-sulfonamide]
3.2.8. 6s. [3-Chloro-2-oxo-N-(o-tolyl)-2H-chromene-6-sulfonamide]
3.2.9. 6t. [N-benzyl-3-chloro-2-oxo-2H-chromene-6-sulfonamide]
3.3. In Vitro Enzyme Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gallwitz, B. Clinical Use of DPP-4 Inhibitors. Front. Endocrinol. 2019, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Olokoba, A.B.; Obateru, O.A.; Olokoba, L.B. Type 2 Diabetes Mellitus: A Review of Current Trends. Oman Med. J. 2012, 27, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.Y.; Hsu, T.; Chen, C.T.; Cheng, J.H.; Chiou, M.C.; Huang, C.H.; Tseng, Y.J.; Yeh, T.K.; Huang, C.Y.; Yeh, K.C.; et al. Rational Design and Synthesis of Potent and Long-Lasting Glutamic Acid-Based Dipeptidyl Peptidase IV Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1908–1912. [Google Scholar] [CrossRef] [PubMed]
- Narsimha, S.; Battula, K.S.; Ravinder, M.; Reddy, Y.N.; Nagavelli, V.R. Design, Synthesis and Biological Evaluation of Novel 1,2,3-Triazole-Based Xanthine Derivatives as DPP-4 Inhibitors. J. Chem. Sci. 2020, 132, 59. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Anwar, M.M.; Syam, Y.M.; Nael, M.A.; Ali, H.F.; Motaleb, M.A. Rational Design and Synthesis of New Tetralin-Sulfonamide Derivatives as Potent Anti-Diabetics and DPP-4 Inhibitors: 2D & 3D QSAR, in Vivo Radiolabeling and Bio Distribution Studies. Bioorg. Chem. 2018, 81, 481–493. [Google Scholar] [CrossRef]
- Han, B.; Liu, J.L.; Huan, Y.; Li, P.; Wu, Q.; Lin, Z.Y.; Shen, Z.F.; Yin, D.L.; Huang, H.H. Design, Synthesis and Primary Activity of Thiomorpholine Derivatives as DPP-IV Inhibitors. Chinese Chem. Lett. 2012, 23, 297–300. [Google Scholar] [CrossRef]
- Mohammad, B.D.; Baig, M.S.; Bhandari, N.; Siddiqui, F.A.; Khan, S.L.; Ahmad, Z.; Khan, F.S.; Tagde, P.; Jeandet, P. Heterocyclic Compounds as Dipeptidyl Peptidase-IV Inhibitors with Special Emphasis on Oxadiazoles as Potent Anti-Diabetic Agents. Molecules 2022, 27, 6001. [Google Scholar] [CrossRef]
- Pereira, A.L.E.; dos Santos, G.B.; Franco, M.S.F.; Federico, L.B.; da Silva, C.H.T.P.; Santos, C.B.R. Molecular Modeling and Statistical Analysis in the Design of Derivatives of Human Dipeptidyl Peptidase IV. J. Biomol. Struct. Dyn. 2018, 36, 318–334. [Google Scholar] [CrossRef]
- Patel, B.D.; Bhadada, S.V.; Ghate, M.D. Design, Synthesis and Anti-Diabetic Activity of Triazolotriazine Derivatives as Dipeptidyl Peptidase-4 (DPP-4) Inhibitors. Bioorg. Chem. 2017, 72, 345–358. [Google Scholar] [CrossRef]
- Scheen, A.J. Safety of Dipeptidyl Peptidase-4 Inhibitors for Treating Type 2 Diabetes. Expert Opin. Drug Saf. 2015, 14, 505–524. [Google Scholar] [CrossRef]
- Tella, S.H.; Rendell, M.S. DPP-4 Inhibitors: Focus on Safety. Expert Opin. Drug Saf. 2015, 14, 127–140. [Google Scholar] [CrossRef]
- Gupta, R.; Walunj, S.; Tokala, R.; Parsa, K.; Singh, S.; Pal, M. Emerging Drug Candidates of Dipeptidyl Peptidase IV (DPP IV) Inhibitor Class for the Treatment of Type 2 Diabetes. Curr. Drug Targets 2009, 10, 71–87. [Google Scholar] [CrossRef]
- Lacroix, I.M.E.; Li-Chan, E.C.Y. Food-Derived Dipeptidyl-Peptidase IV Inhibitors as a Potential Approach for Glycemic Regulation—Current Knowledge and Future Research Considerations. Trends Food Sci. Technol. 2016, 54, 1–16. [Google Scholar] [CrossRef]
- Smelcerovic, A.; Miljkovic, F.; Kolarevic, A.; Lazarevic, J.; Djordjevic, A.; Kocic, G.; Anderluh, M. An Overview of Recent Dipeptidyl Peptidase-IV Inhibitors: Linking Their Structure and Physico-Chemical Properties with SAR, Pharmacokinetics and Toxicity. Curr. Top. Med. Chem. 2015, 15, 2342–2372. [Google Scholar] [CrossRef]
- Salvatore, T.; Carbonara, O.; Cozzolino, D.; Torella, R.; Carlo Sasso, F. Adapting the GLP-1-Signaling System to the Treatment of Type 2 Diabetes. Curr. Diabetes Rev. 2007, 3, 15–23. [Google Scholar] [CrossRef]
- Kushwaha, R.N.; Haq, W.; Katti, S.B. Sixteen-Years of Clinically Relevant Dipeptidyl Peptidase-IV (DPP-IV) Inhibitors for Treatment of Type-2 Diabetes: A Perspective. Curr. Med. Chem. 2014, 21, 4013–4045. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, Y.; Liu, T. Recent Advances in Non-Peptidomimetic Dipeptidyl Peptidase 4 Inhibitors: Medicinal Chemistry and Preclinical Aspects. Curr. Med. Chem. 2012, 19, 3982–3999. [Google Scholar] [CrossRef]
- Salvo, F.; Moore, N.; Arnaudl, M.; Robinson, P.; Raschi, E.; De Ponti, F.; Bégaud, B.; Pariente, A. Addition of Dipeptidyl Peptidase-4 Inhibitors to Sulphonylureas and Risk of Hypoglycaemia: Systematic Review and Meta-Analysis. BMJ 2016, 353, i2231. [Google Scholar] [CrossRef]
- Liu, M.; Sun, X.; Zhao, X. Investigating the Contributions of Residues to Dipeptidyl Peptidase-IV Inhibitor Binding by Molecular Dynamics Simulation. Lett. Drug Des. Discov. 2014, 11, 886–893. [Google Scholar] [CrossRef]
- Kumar Verma, S.; Kant Sharma, S.; Thareja, S. Docking Study of Novel Pyrrolidine Derivatives as Potential Dipeptidyl Peptidase-IV (DPP-IV) Inhibitors. Lett. Drug Des. Discov. 2015, 12, 284–291. [Google Scholar] [CrossRef]
- Amuthalakshmi, S.; Anton Smith, A.; Manavalan, R. Modeling Assisted In Silico Design of Ligand Molecule for DPP IV in Type II Diabetes Mellitus. Lett. Drug Des. Discov. 2012, 9, 764–766. [Google Scholar] [CrossRef]
- Gupta, S.; Chaudhary, K.; Raj, U.; Mishra, N. Computational Identification of Inhibitors Against DPP-IV for Checking Type-2 Diabetes. Lett. Drug Des. Discov. 2016, 14, 66–73. [Google Scholar] [CrossRef]
- Mattei, P.; Boehringer, M.; Di Giorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Koçer, B.; Kuhn, B.; Loeffler, B.M.; MacDonald, A.; et al. Discovery of Carmegliptin: A Potent and Long-Acting Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. Bioorganic Med. Chem. Lett. 2010, 20, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Said, S.; Nwosu, A.; Mukherjee, D.; Hernandez, G. Alogliptin; A Review of a New Dipeptidyl Peptidase-4 (DPP-4) Inhibitor for the Treatment of Type 2 Diabetes Mellitus. Cardiovasc. Hematol. Disord. Targets 2014, 14, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, M.; Reutter, W.; Arck, P.; Rose, M.; Klapp, B.F. A Guardian Angel: The Involvement of Dipeptidyl Peptidase IV in Psychoneuroendocrine Function, Nutrition and Immune Defence. Clin. Sci. 2000, 99, 93–104. [Google Scholar] [CrossRef]
- Kirby, M.; Yu, D.M.T.; O’Connor, S.P.; Gorrell, M.D. Inhibitor Selectivity in the Clinical Application of Dipeptidyl Peptidase-4 Inhibition. Clin. Sci. 2010, 118, 31–41. [Google Scholar] [CrossRef]
- Pratley, R.E.; Salsali, A. Inhibition of DPP-4: A New Therapeutic Approach for the Treatment of Type 2 Diabetes. Curr. Med. Res. Opin. 2007, 23, 919–931. [Google Scholar] [CrossRef]
- El-Kaissi, S.; Sherbeeni, S. Pharmacological Management of Type 2 Diabetes Mellitus: An Update. Curr. Diabetes Rev. 2011, 7, 392–405. [Google Scholar] [CrossRef]
- Liang, G.B.; Qian, X.; Biftu, T.; Singh, S.; Gao, Y.D.; Scapin, G.; Patel, S.; Leiting, B.; Patel, R.; Wu, J.; et al. Discovery of New Binding Elements in DPP-4 Inhibition and Their Applications in Novel DPP-4 Inhibitor Design. Bioorganic Med. Chem. Lett. 2008, 18, 3706–3710. [Google Scholar] [CrossRef]
- Wallace, M.B.; Feng, J.; Zhang, Z.; Skene, R.J.; Shi, L.; Caster, C.L.; Kassel, D.B.; Xu, R.; Gwaltney, S.L. Structure-Based Design and Synthesis of Benzimidazole Derivatives as Dipeptidyl Peptidase IV Inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 2362–2367. [Google Scholar] [CrossRef]
- Wu, W.L.; Hao, J.; Domalski, M.; Burnett, D.A.; Pissarnitski, D.; Zhao, Z.; Stamford, A.; Scapin, G.; Gao, Y.D.; Soriano, A.; et al. Discovery of Novel Tricyclic Heterocycles as Potent and Selective DPP-4 Inhibitors for the Treatment of Type 2 Diabetes. ACS Med. Chem. Lett. 2016, 7, 498–501. [Google Scholar] [CrossRef]
- Kaur, J.; Singla, R.; Jaitak, V. In Silico Study of Flavonoids as DPP-4 and α-Glucosidase Inhibitors. Lett. Drug Des. Discov. 2018, 15, 634–642. [Google Scholar] [CrossRef]
- McKeage, K. Trelagliptin: First Global Approval. Drugs 2015, 75, 1161–1164. [Google Scholar] [CrossRef]
- Burness, C.B. Omarigliptin: First Global Approval. Drugs 2015, 75, 1947–1952. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Rao, K.B.; Singh, S.; Modukuri, R.K.; Aruna Teja, G.; Chandasana, H.; Shukla, S.; Bhatta, R.S. Synthesis and Evaluation of New 3-Phenylcoumarin Derivatives as Potential Antidepressant Agents. Bioorganic Med. Chem. Lett. 2014, 24, 4876–4880. [Google Scholar] [CrossRef]
- Husain, A.A.; Husain, A.S.; Gawhale, N.D.; Khan, S.L.; Murlidhar, D.V. An Overview of Natural and Synthetic Coumarin Derivatives as Potential Antidiabetic Agents. J. Pharm. Negat. Results 2022, 13, 739–744. [Google Scholar] [CrossRef]
- Tamene, D.; Endale, M. Antibacterial Activity of Coumarins and Carbazole Alkaloid from Roots of Clausena Anisata. Adv. Pharmacol. Sci. 2019, 2019, 5419854. [Google Scholar] [CrossRef]
- De Souza, S.M.; Delle Monache, F.; Smânia, A. Antibacterial Activity of Coumarins. Zeitschrift Fur Naturforsch. Sect. C J. Biosci. 2005, 60, 693–700. [Google Scholar] [CrossRef]
- Teoh, S.L.; Das, S. Phytochemicals and Their Effective Role in the Treatment of Diabetes Mellitus: A Short Review. Phytochem. Rev. 2018, 17, 1111–1128. [Google Scholar] [CrossRef]
- Galayev, O.; Garazd, Y.; Garazd, M.; Lesyk, R. Synthesis and Anticancer Activity of 6-Heteroarylcoumarins. Eur. J. Med. Chem. 2015, 105, 171–181. [Google Scholar] [CrossRef]
- Soni, R.; Soman, S.S. Design and Synthesis of Aminocoumarin Derivatives as DPP-IV Inhibitors and Anticancer Agents. Bioorg. Chem. 2018, 79, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Soman, S.S. Design, Synthesis and Preliminary Evaluation of 3-Aminocoumarin Derivatives as DPP-IV Inhibitors. Pharmanest 2015, 6, 2679–2684. [Google Scholar]
- Soni, R.; Durgapal, S.D.; Soman, S.S.; Georrge, J.J. Design, Synthesis and Anti-Diabetic Activity of Chromen-2-One Derivatives. Arab. J. Chem. 2019, 12, 701–708. [Google Scholar] [CrossRef]
- Singh, A.K.; Patel, P.K.; Choudhary, K.; Joshi, J.; Yadav, D.; Jin, J.O. Quercetin and Coumarin Inhibit Dipeptidyl Peptidase-IV and Exhibits Antioxidant Properties: In Silico, in Vitro, Ex Vivo. Biomolecules 2020, 10, 207. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.A.; Khan, S.L.; Marathe, R.P.; Nema, N.V. Design, Synthesis, and In Silico Studies of Novel N-(2-Aminophenyl)-2,3- Diphenylquinoxaline-6-Sulfonamide Derivatives Targeting Receptor- Binding Domain (RBD) of SARS-CoV-2 Spike Glycoprotein and Their Evaluation as Antimicrobial and Antimalarial Agents. Lett. Drug Des. Discov. 2021, 18, 915–931. [Google Scholar] [CrossRef]
- Syam, Y.M.; Anwar, M.M.; Abd El-Karim, S.S.; Elseginy, S.A.; Essa, B.M.; Sakr, T.M. New Quinoxaline Compounds as DPP-4 Inhibitors and Hypoglycemics: Design, Synthesis, Computational and Bio-Distribution Studies. RSC Adv. 2021, 11, 36989–37010. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Lim, S.M.; Ramasamy, K.; Vasudevan, M.; Shah, S.A.A.; Selvaraj, M.; Narasimhan, B. Synthesis, Molecular Docking and Biological Evaluation of Bis-Pyrimidine Schiff Base Derivatives. Chem. Cent. J. 2017, 11, 89. [Google Scholar] [CrossRef]
- Akhtar, W.; Khan, M.F.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M.A.; Mehdi, S.H.; Akhter, M.; Alam, M.M. Therapeutic Evolution of Benzimidazole Derivatives in the Last Quinquennial Period. Eur. J. Med. Chem. 2017, 126, 705–753. [Google Scholar] [CrossRef]
- Kamal, A.; Shaik, A.B.; Jain, N.; Kishor, C.; Nagabhushana, A.; Supriya, B.; Bharath Kumar, G.; Chourasiya, S.S.; Suresh, Y.; Mishra, R.K.; et al. Design and Synthesis of Pyrazole-Oxindole Conjugates Targeting Tubulin Polymerization as New Anticancer Agents. Eur. J. Med. Chem. 2015, 92, 501–513. [Google Scholar] [CrossRef]
- Barret, R. Lipinski’s Rule of Five. In Therapeutical Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 97–100. [Google Scholar] [CrossRef]
- Khan, S.; Kale, M.; Siddiqui, F.; Nema, N. Novel Pyrimidine-Benzimidazole Hybrids with Antibacterial and Antifungal Properties and Potential Inhibition of SARS-CoV-2 Main Protease and Spike Glycoprotein. Digit. Chinese Med. 2021, 4, 102–119. [Google Scholar] [CrossRef]
- Krzywinski, M.; Altman, N. Points of Significance: Significance, p Values and t-Tests. Nat. Methods 2013, 10, 1041–1042. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Khan, A.; Unnisa, A.; Sohel, M.; Date, M.; Panpaliya, N.; Saboo, S.G.; Siddiqui, F.; Khan, S. Investigation of Phytoconstituents of Enicostemma Littorale as Potential Glucokinase Activators through Molecular Docking for the Treatment of Type 2 Diabetes Mellitus. Silico Pharmacol. 2022, 10, 1. [Google Scholar] [CrossRef]
- Shntaif, A.H.; Khan, S.; Tapadiya, G.; Chettupalli, A.; Saboo, S.; Shaikh, M.S.; Siddiqui, F.; Amara, R.R. Rational Drug Design, Synthesis, and Biological Evaluation of Novel N-(2-Arylaminophenyl)-2,3-Diphenylquinoxaline-6-Sulfonamides as Potential Antimalarial, Antifungal, and Antibacterial Agents. Digit. Chinese Med. 2021, 4, 290–304. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Accelrys Software Inc. Discovery Studio Modeling Environment, Release 3.1; Accelrys Software Inc.: San Diego, CA, USA, 2012; pp. 98–104. [Google Scholar]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Khan, S.L.; Siddiui, F.A. Beta-Sitosterol: As Immunostimulant, Antioxidant and Inhibitor of SARS-CoV-2 Spike Glycoprotein. Arch. Pharmacol. Ther. 2020, 2, 12–16. [Google Scholar] [CrossRef]
- Chaudhari, R.N.; Khan, S.L.; Chaudhary, R.S.; Jain, S.P.; Siddiqui, F.A. Β-Sitosterol: Isolation from Muntingia Calabura Linn Bark Extract, Structural Elucidation And Molecular Docking Studies As Potential Inhibitor of SARS-CoV-2 Mpro (COVID-19). Asian J. Pharm. Clin. Res. 2020, 13, 204–209. [Google Scholar] [CrossRef]
- Khan, S.L.; Siddiqui, F.A.; Shaikh, M.S.; Nema, N.V.; Shaikh, A.A. Discovery of Potential Inhibitors of the Receptor-Binding Domain (RBD) of Pandemic Disease-Causing SARS-CoV-2 Spike Glycoprotein from Triphala through Molecular Docking. Curr. Chinese Chem. 2022, 2, e220321192390. [Google Scholar] [CrossRef]
- Khan, S.L.; Sonwane, G.M.; Siddiqui, F.A.; Jain, S.P.; Kale, M.A.; Borkar, V.S. Discovery of Naturally Occurring Flavonoids as Human Cytochrome P450 (CYP3A4) Inhibitors with the Aid of Computational Chemistry. Indo Glob. J. Pharm. Sci. 2020, 10, 58–69. [Google Scholar] [CrossRef]
- Vawhal, P.K.; Jadhav, S.B. Design, Synthesis, and Biological Evaluation of 3-Chloro-2-Oxo-N-(Arylcarbamoyl)-2H-1-Benzopyran-6-Sulfonamide Derivatives as Potential DPP-IV Inhibitors. Int. J. Health Sci. 2022, 6, 373–392. [Google Scholar] [CrossRef]
- Khattab, M.; Ragab, F.; Galal, S.; El Diwani, H. Synthesis of 4-(1H-Benzo[d]Imidazol-2-Yl) Aniline Derivatives of Expected Anti-HCV Activity. Int. J. Res. Pharm. Chem. 2012, 2, 937–946. [Google Scholar]
- Guasch, L.; Ojeda, M.J.; González-Abuín, N.; Sala, E.; Cereto-Massagué, A.; Mulero, M.; Valls, C.; Pinent, M.; Ardévol, A.; Garcia-Vallvé, S.; et al. Identification of Novel Human Dipeptidyl Peptidase-IV Inhibitors of Natural Origin (Part I): Virtual Screening and Activity Assays. PLoS ONE 2012, 7, e44971. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules Code | % Inhibition (at Conc.) | IC50 (µM) |
|---|---|---|
| Sitagliptin | 102.6 ± 1.1 (100 µM) | 0.018 ± 0.002 |
| 6a | 93.52 ± 2.02 (250 µM) | 14.52 ± 1.2 |
| 6b | 91.15 ± 0.9 (250 µM) | 15.72 ± 0.98 |
| 6c | 90.34 ± 2.04 (250 µM) | 16.28 ± 2.04 |
| 6d | 92.40 ± 1.11 (250 µM) | 15.19 ± 1.02 |
| 6i | 95.77 ± 1.08 (250 µM) | 10.98 ± 1 |
| 6j | 96.59 ± 0.2 (250 µM) | 10.14 ± 0.02 |
| 6r | 95.44 ± 1.1 (250 µM) | 12.92 ± 0.11 |
| 6s | 89.33 ± 0.9 (250 µM) | 18.29 ± 0.1 |
| 6t | 88.29 ± 2.06 (250 µM) | 17.73 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vawhal, P.K.; Jadhav, S.B.; Kaushik, S.; Panigrahi, K.C.; Nayak, C.; Urmee, H.; Khan, S.L.; Siddiqui, F.A.; Islam, F.; Eftekhari, A.; et al. Coumarin-Based Sulfonamide Derivatives as Potential DPP-IV Inhibitors: Pre-ADME Analysis, Toxicity Profile, Computational Analysis, and In Vitro Enzyme Assay. Molecules 2023, 28, 1004. https://doi.org/10.3390/molecules28031004
Vawhal PK, Jadhav SB, Kaushik S, Panigrahi KC, Nayak C, Urmee H, Khan SL, Siddiqui FA, Islam F, Eftekhari A, et al. Coumarin-Based Sulfonamide Derivatives as Potential DPP-IV Inhibitors: Pre-ADME Analysis, Toxicity Profile, Computational Analysis, and In Vitro Enzyme Assay. Molecules. 2023; 28(3):1004. https://doi.org/10.3390/molecules28031004
Chicago/Turabian StyleVawhal, Pallavi Kishor, Shailaja B. Jadhav, Sumit Kaushik, Kahnu Charan Panigrahi, Chandan Nayak, Humaira Urmee, Sharuk L. Khan, Falak A. Siddiqui, Fahadul Islam, Aziz Eftekhari, and et al. 2023. "Coumarin-Based Sulfonamide Derivatives as Potential DPP-IV Inhibitors: Pre-ADME Analysis, Toxicity Profile, Computational Analysis, and In Vitro Enzyme Assay" Molecules 28, no. 3: 1004. https://doi.org/10.3390/molecules28031004
APA StyleVawhal, P. K., Jadhav, S. B., Kaushik, S., Panigrahi, K. C., Nayak, C., Urmee, H., Khan, S. L., Siddiqui, F. A., Islam, F., Eftekhari, A., Alzahrani, A. R., Azlina, M. F. N., Sarker, M. M. R., & Ibrahim, I. A. A. (2023). Coumarin-Based Sulfonamide Derivatives as Potential DPP-IV Inhibitors: Pre-ADME Analysis, Toxicity Profile, Computational Analysis, and In Vitro Enzyme Assay. Molecules, 28(3), 1004. https://doi.org/10.3390/molecules28031004