

On the Shoulders of Giants—Reaching for Nitrogenase

Abstract
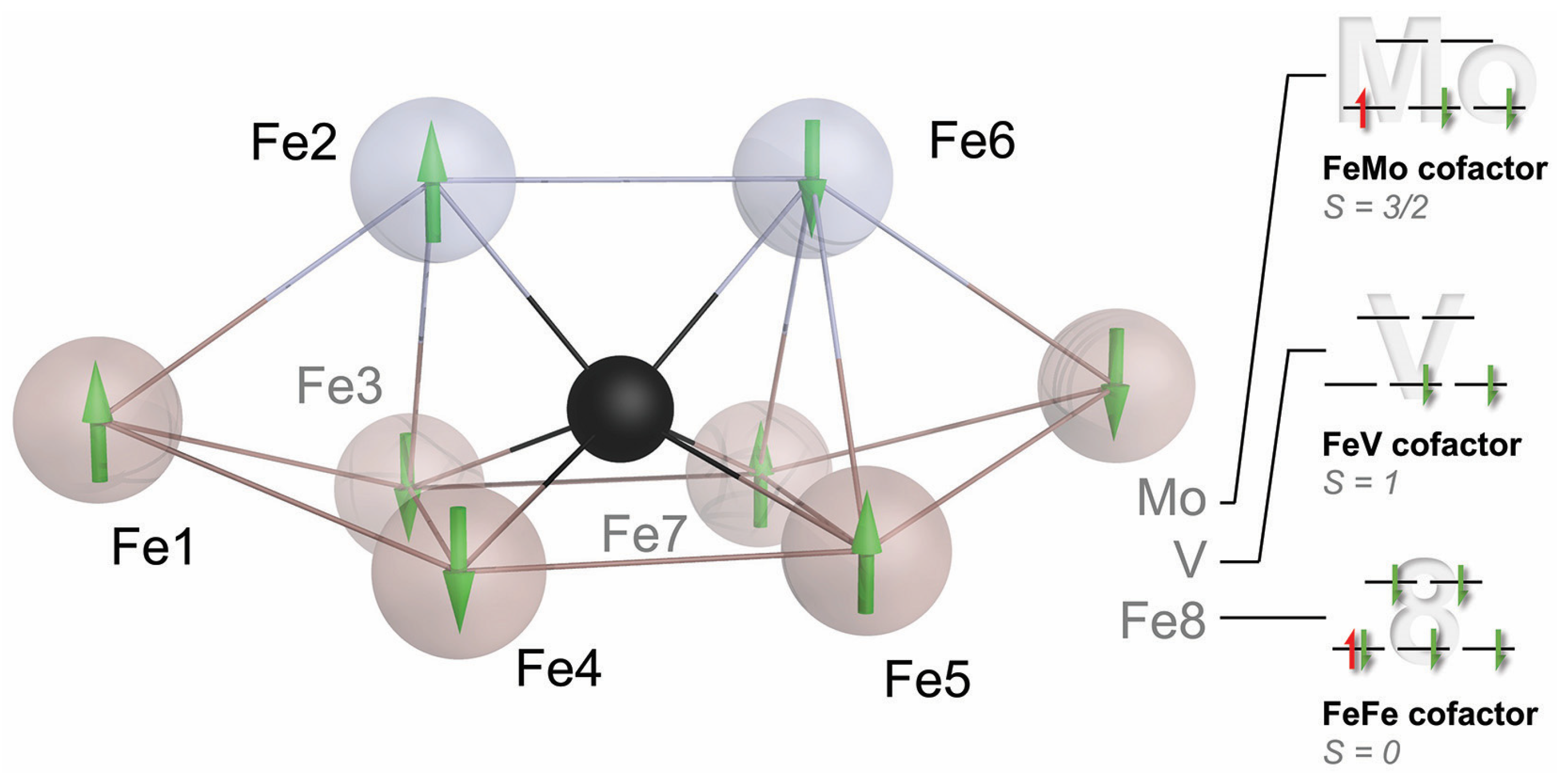
: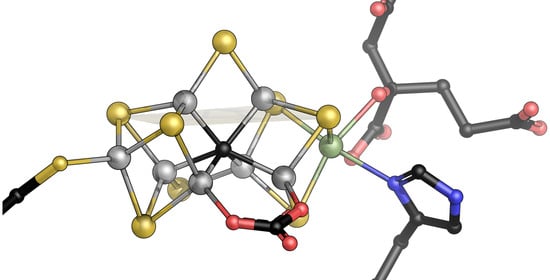
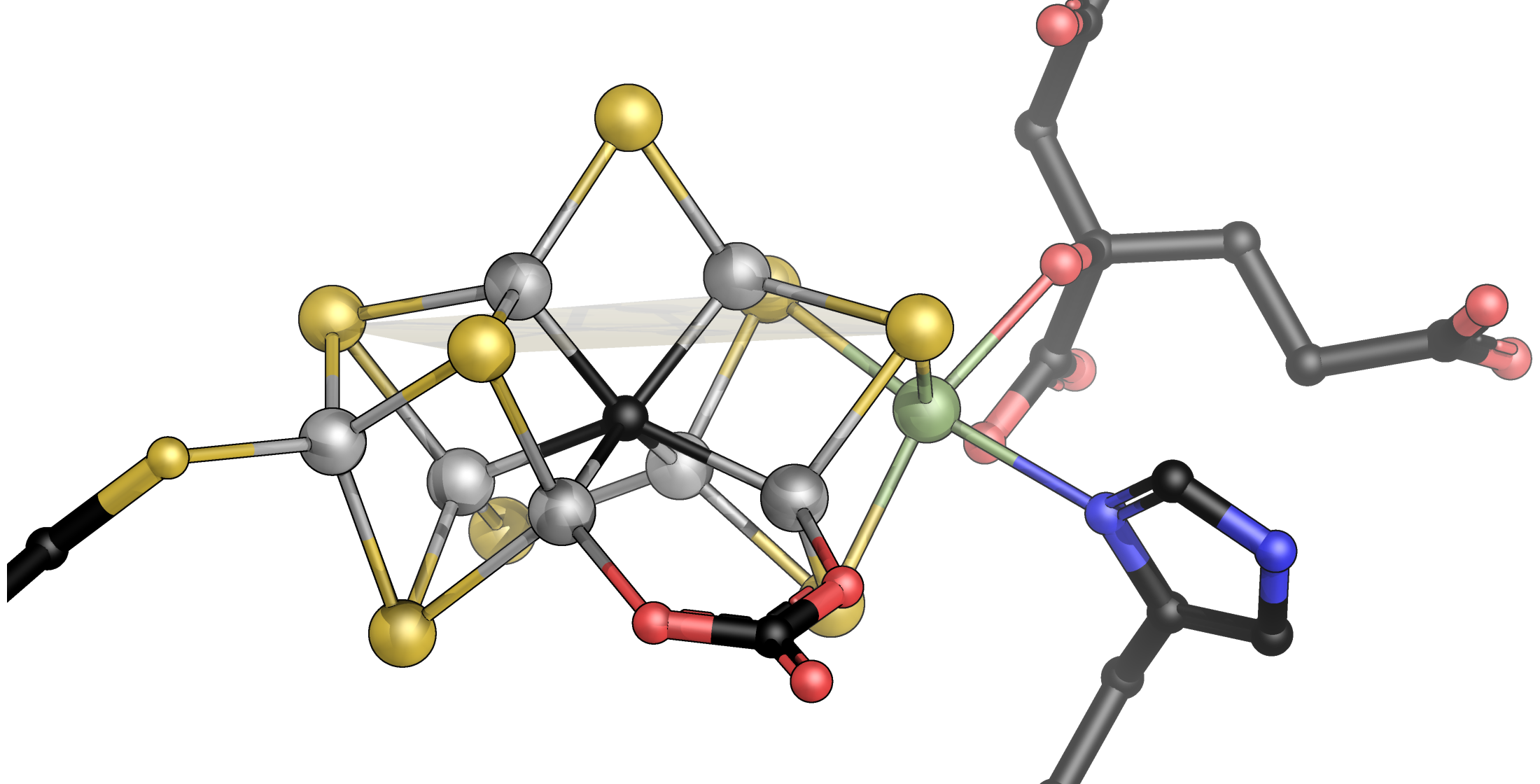{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
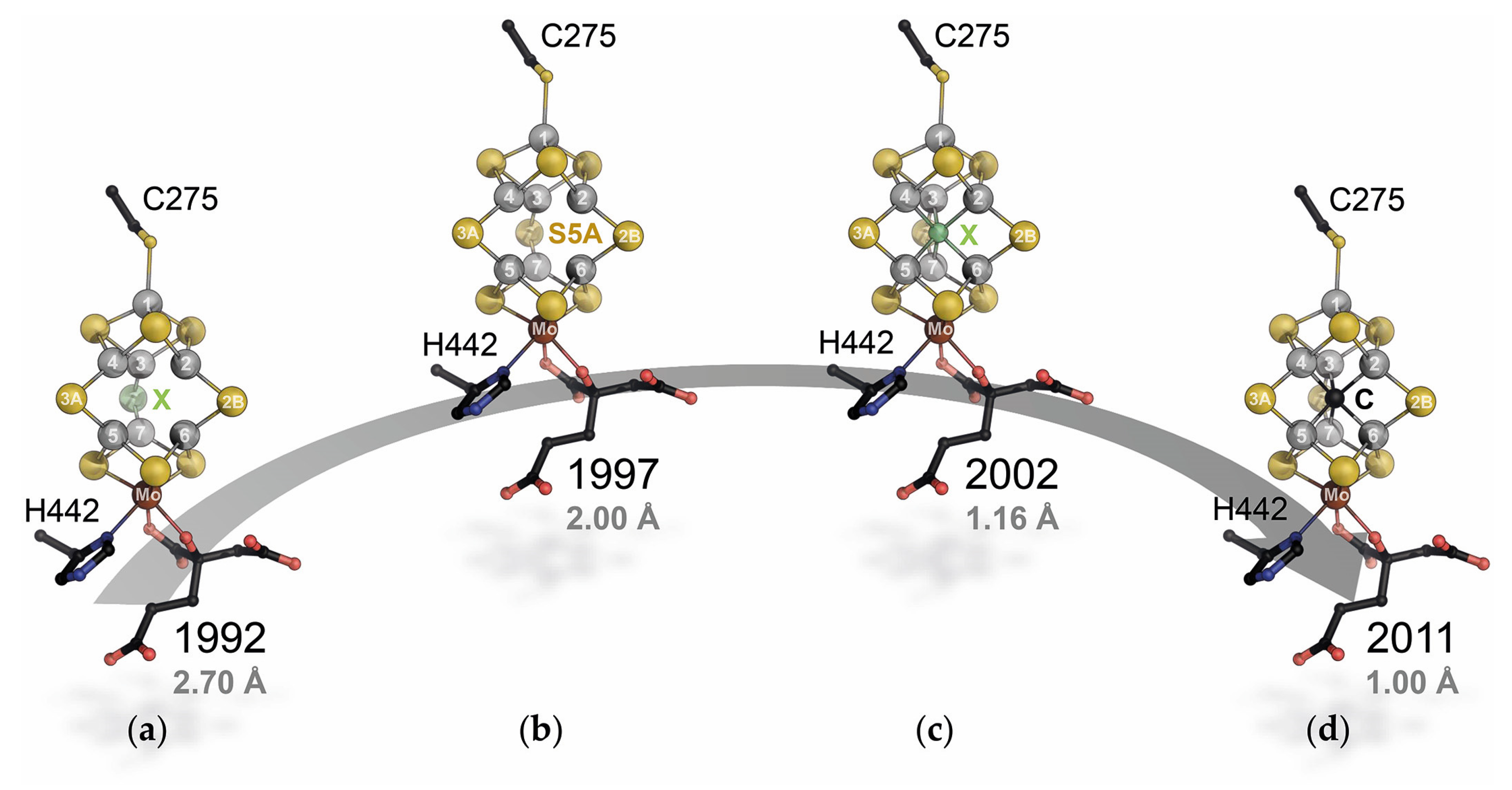
{kind=link}
1. Introduction: You Cannot Fix Nitrogen with an Enzyme
2. Have You Tried to Crystallize the Protein?
3. A Closer Look
4. Episode IV: A New Beginning
5. Family Business: The Three Nitrogenase Isoforms
6. Alternative Substrates: Learning from CO about N2
7. So What about Molybdenum?
8. What Is Next in Nitrogenase Research?
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hellriegel, H.; Willfarth, H. Untersuchungen über die Stickstoffnährung der Gramineen und Leguminosen. Beilageheft Z. Ver. Rübenzucker-Ind. Dtsch. Reichs 1888, 38, 1–234. [Google Scholar]
- Lipman, J.G. Experiments on the transformation and fixation of nitrogen by bacteria. Rep. N. J. Agric. Exper. Stat. 1903, 24, 217–285. [Google Scholar]
- Canfield, D.E.; Glazer, A.N.; Falkowski, P.G. The Evolution and Future of Earth’s Nitrogen Cycle. Science 2010, 330, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production; MIT Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Bagchi, S.N.; Ernst, A.; Böger, P. The Effect of Activated Oxygen Species on Nitrogenase of Anabaena variabilis. Z. Naturforsch. C 1991, 46, 407–415. [Google Scholar] [CrossRef]
- Kim, J.S.; Rees, D.C. Structural Models for the Metal Centers in the Nitrogenase Molybdenum-Iron Protein. Science 1992, 257, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Rees, D.C. Crystallographic Structure and Functional Implications of the Nitrogenase Molybdenum Iron Protein from Azotobacter vinelandii. Nature 1992, 360, 553–560. [Google Scholar]
- Georgiadis, M.M.; Komiya, H.; Chakrabarti, P.; Woo, D.; Kornuc, J.J.; Rees, D.C. Crystallographic Structure of the Nitrogenase Iron Protein from Azotobacter vinelandii. Science 1992, 257, 1653–1659. [Google Scholar] [CrossRef]
- Bulen, W.A.; LeComte, J.R. Nitrogenase System from Azotobacter: 2-Enzyme Requirement for N2 Reduction, ATP-Dependent H2 Evolution and ATP Hydrolysis. Proc. Natl. Acad. Sci. USA 1966, 56, 979–986. [Google Scholar] [CrossRef]
- Deisenhofer, J.; Epp, O.; Miki, K.; Huber, R.; Michel, H. Structure of the Protein Subunits in the Photosynthetic Reaction Center of Rhodopseudomonas viridis at 3 Å Resolution. Nature 1985, 318, 618–624. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 22. [Google Scholar] [CrossRef]
- Messerschmidt, A.; Rossi, A.; Ladenstein, R.; Huber, R.; Bolognesi, M.; Gatti, G.; Marchesini, A.; Petruzzelli, R.; Finazziagro, A. X-Ray Crystal-Structure of the Blue Oxidase Ascorbate Oxidase from Zucchini—Analysis of the Polypeptide Fold and a Model of the Copper Sites and Ligands. J. Mol. Biol. 1989, 206, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, A.; Steigemann, W.; Huber, R.; Lang, G.; Kroneck, P.M.H. X-Ray Crystallographic Characterization of Type-2-Depleted Ascorbate Oxidase from Zucchini. Eur. J. Biochem. 1992, 209, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.K.; Mukund, S.; Kletzin, A.; Adams, M.W.W.; Rees, D.C. Structure of a Hyperthermophilic Tungstopterin Enzyme, Aldehyde Ferredoxin Oxidoreductase. Science 1995, 267, 1463–1469. [Google Scholar] [CrossRef]
- Romao, M.J.; Archer, M.; Moura, I.; Moura, J.J.G.; LeGall, J.; Engh, R.; Schneider, M.; Hof, P.; Huber, R. Crystal-Structure of the Xanthine Oxidase-Related Aldehyde Oxidoreductase from D. gigas. Science 1995, 270, 1170–1176. [Google Scholar] [CrossRef]
- Einsle, O.; Messerschmidt, A.; Stach, P.; Bourenkov, G.P.; Bartunik, H.D.; Huber, R.; Kroneck, P.M.H. Structure of cytochrome c nitrite reductase. Nature 1999, 400, 476–480. [Google Scholar] [CrossRef]
- Peters, J.W.; Stowell, M.H.B.; Soltis, S.M.; Finnegan, M.G.; Johnson, M.K.; Rees, D.C. Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry 1997, 36, 1181–1187. [Google Scholar] [CrossRef]
- Einsle, O.; Tezcan, F.A.; Andrade, S.L.A.; Schmid, B.; Yoshida, M.; Howard, J.B.; Rees, D.C. Nitrogenase MoFe-protein at 1.16 Å resolution: A central ligand in the FeMo-cofactor. Science 2002, 297, 1696–1700. [Google Scholar] [CrossRef]
- Smith, B.E. Nitrogenase reveals its inner secrets. Science 2002, 297, 1654–1655. [Google Scholar] [CrossRef]
- Lancaster, K.M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.L.; Ribbe, M.W.; Neese, F.; Bergmann, U.; DeBeer, S. X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. [Google Scholar] [CrossRef]
- Spatzal, T.; Aksoyoğlu, M.; Zhang, L.M.; Andrade, S.L.A.; Schleicher, E.; Weber, S.; Rees, D.C.; Einsle, O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. [Google Scholar] [CrossRef] [PubMed]
- Tezcan, F.A.; Kaiser, J.T.; Mustafi, D.; Walton, M.Y.; Howard, J.B.; Rees, D.C. Nitrogenase complexes: Multiple docking sites for a nucleotide switch protein. Science 2005, 309, 1377–1380. [Google Scholar] [CrossRef]
- Hu, Y.L.; Corbett, M.C.; Fay, A.W.; Webber, J.A.; Hodgson, K.O.; Hedman, B.; Ribbe, M.W. FeMo cofactor maturation on NifEN. Proc. Natl. Acad. Sci. USA 2006, 103, 17119–17124. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Fay, A.W.; Lee, C.C.; Ribbe, M.W. P-cluster maturation on nitrogenase MoFe protein. Proc. Natl. Acad. Sci. USA 2007, 104, 10424–10429. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Fay, A.W.; Lee, C.C.; Yoshizawa, J.; Ribbe, M.W. Assembly of nitrogenase MoFe protein. Biochemistry 2008, 47, 3973–3981. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.M.; Ludden, P.W. Maturation of nitrogenase: A biochemical puzzle. J. Bacteriol. 2005, 187, 405–414. [Google Scholar] [CrossRef]
- Rubio, L.M.; Hernandez, J.A.; Soboh, B.; Zhao, D.; Igarashi, R.Y.; Curatti, L.; Ludden, P.W. The Role of Nif Proteins in Nitrogenase Maturation. Curr. Plant Sci. Biot. 2008, 42, 325–328. [Google Scholar]
- Lukoyanov, D.; Barney, B.M.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. Connecting nitrogenase intermediates with the kinetic scheme for N2 reduction by a relaxation protocol and identification of the N2 binding state. Proc. Natl. Acad. Sci. USA 2007, 104, 1451–1455. [Google Scholar] [CrossRef]
- Yang, T.C.; Maeser, N.K.; Laryukhin, M.; Lee, H.I.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. The interstitial atom of the nitrogenase FeMo-Cofactor: ENDOR and ESEEM evidence that it is not a nitrogen. J. Am. Chem. Soc. 2005, 127, 12804–12805. [Google Scholar] [CrossRef]
- Lukoyanov, D.; Pelmenschikov, V.; Maeser, N.; Laryukhin, M.; Yang, T.C.; Noodleman, L.; Dean, D.R.; Case, D.A.; Seefeldt, L.C.; Hoffman, B.M. Testing if the interstitial atom, X, of the nitrogenase molybdenum-iron cofactor is N or C: ENDOR, ESEEM, and DFT studies of the S=3/2 resting state in multiple environments. Inorg. Chem. 2007, 46, 11437–11449. [Google Scholar] [CrossRef]
- Wiig, J.A.; Hu, Y.L.; Lee, C.C.; Ribbe, M.W. Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-Cluster. Science 2012, 337, 1672–1675. [Google Scholar] [CrossRef]
- Lowe, D.J.; Thorneley, R.N.F. The Mechanism of Klebsiella pneumoniae Nitrogenase Action—Pre-Steady-State Kinetics of H2 Formation. Biochem. J. 1984, 224, 877–886. [Google Scholar] [CrossRef]
- Lowe, D.J.; Thorneley, R.N.F. The Mechanism of Klebsiella pneumoniae Nitrogenase Action—The Determination of Rate Constants Required for the Simulation of the Kinetics of N2 Reduction and H2 Evolution. Biochem. J. 1984, 224, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Thorneley, R.N.F.; Lowe, D.J. The Mechanism of Klebsiella pneumoniae Nitrogenase Action—Pre-Steady-State Kinetics of an Enzyme-Bound Intermediate in N2 Reduction and of NH3 Formation. Biochem. J. 1984, 224, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Thorneley, R.N.F.; Lowe, D.J. The Mechanism of Klebsiella pneumoniae Nitrogenase Action—Simulation of the Dependences of H2 Evolution Rate on Component-Protein Concentration and Ratio and Sodium Dithionite Concentration. Biochem. J. 1984, 224, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Thorneley, R.N.F.; Lowe, D.J. Kinetics and mechanism of the nitrogenase enzyme system. In Molybdenum Enzymes; Spiro, T.G., Ed.; Wiley-Interscience: New York, NY, USA, 1985; Volume 1, pp. 221–284. [Google Scholar]
- Spatzal, T.; Einsle, O.; Andrade, S.L. Analysis of the Magnetic Properties of Nitrogenase FeMo Cofactor by Single-Crystal EPR Spectroscopy. Angew. Chem. 2013, 52, 10116–10119. [Google Scholar] [CrossRef] [PubMed]
- Einsle, O.; Andrade, S.L.; Dobbek, H.; Meyer, J.; Rees, D.C. Assignment of individual metal redox states in a metalloprotein by crystallographic refinement at multiple X-ray wavelengths. J. Am. Chem. Soc. 2007, 129, 2210–2211. [Google Scholar] [CrossRef]
- Spatzal, T.; Schlesier, J.; Burger, E.M.; Sippel, D.; Zhang, L.M.; Andrade, S.L.A.; Rees, D.C.; Einsle, O. Nitrogenase FeMoco investigated by spatially resolved anomalous dispersion refinement. Nat. Commun. 2016, 7, 10902. [Google Scholar] [CrossRef]
- Björnsson, R.; Lima, F.A.; Spatzal, T.; Weyhermüller, T.; Glatzel, P.; Bill, E.; Einsle, O.; Neese, F.; DeBeer, S. Identification of a spin-coupled Mo(III) in the nitrogenase iron-molybdenum cofactor. Chem. Sci. 2014, 5, 3096–3103. [Google Scholar] [CrossRef]
- Lovell, T.; Li, J.; Liu, T.Q.; Case, D.A.; Noodleman, L. FeMo cofactor of nitrogenase: A density functional study of states MN, MOX, MR, and MI. J. Am. Chem. Soc. 2001, 123, 12392–12410. [Google Scholar] [CrossRef]
- Chatt, J.; Dilworth, J.R.; Richards, R.L.; Sanders, J.R. Chemical Evidence Concerning Function of Molybdenum in Nitrogenase. Nature 1969, 224, 1201. [Google Scholar] [CrossRef] [PubMed]
- Bishop, P.E.; Jarlenski, D.M.L.; Hetherington, D.R. Evidence for an Alternative Nitrogen Fixation System in Azotobacter vinelandii. Proc. Natl. Acad. Sci. USA 1980, 77, 7342–7346. [Google Scholar] [CrossRef]
- Bishop, P.E.; Hawkins, M.E.; Eady, R.R. Nitrogen fixation in molybdenum-deficient continuous culture by a strain of Azotobacter vinelandii carrying a deletion of the structural genes for nitrogenase (nifHDK). Biochem. J. 1986, 238, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Hales, B.J.; Case, E.E.; Morningstar, J.E.; Dzeda, M.F.; Mauterer, L.A. Isolation of a New Vanadium-Containing Nitrogenase from Azotobacter vinelandii. Biochemistry 1986, 25, 7253–7255. [Google Scholar] [CrossRef] [PubMed]
- Chisnell, J.R.; Premakumar, R.; Bishop, P.E. Purification of a 2nd Alternative Nitrogenase from a nifHDK Deletion Strain of Azotobacter vinelandii. J. Bacteriol. 1988, 170, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Setubal, J.C.; dos Santos, P.; Goldman, B.S.; Ertesvag, H.; Espin, G.; Rubio, L.M.; Valla, S.; Almeida, N.F.; Balasubramanian, D.; Cromes, L.; et al. Genome Sequence of Azotobacter vinelandii, an Obligate Aerobe Specialized To Support Diverse Anaerobic Metabolic Processes. J. Bacteriol. 2009, 191, 4534–4545. [Google Scholar] [CrossRef]
- Sippel, D.; Schlesier, J.; Rohde, M.; Trncik, C.; Decamps, L.; Djurdjevic, I.; Spatzal, T.; Andrade, S.L.A.; Einsle, O. Production and isolation of vanadium nitrogenase from Azotobacter vinelandii by molybdenum depletion. J. Biol. Inorg. Chem. 2017, 22, 161–168. [Google Scholar] [CrossRef]
- Sippel, D.; Einsle, O. The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat. Chem. Biol. 2017, 13, 956–960. [Google Scholar] [CrossRef]
- Sippel, D.; Rohde, M.; Netzer, J.; Trncik, C.; Gies, J.; Grunau, K.; Djurdjevic, I.; Decamps, L.; Andrade, S.L.A.; Einsle, O. A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359, 1484–1489. [Google Scholar] [CrossRef]
- Cao, L.L.; Caldararu, O.; Ryde, U. Does the crystal structure of vanadium nitrogenase contain a reaction intermediate? Evidence from quantum refinement. J. Biol. Inorg. Chem. 2020, 25, 847–861. [Google Scholar] [CrossRef]
- Benediktsson, B.; Björnsson, R. Quantum Mechanics/Molecular Mechanics Study of Resting-State Vanadium Nitrogenase: Molecular and Electronic Structure of the Iron-Vanadium Cofactor. Inorg. Chem. 2020, 59, 11514–11527. [Google Scholar] [CrossRef] [PubMed]
- Trncik, C.; Müller, T.; Franke, P.; Einsle, O. Structural analysis of the reductase component AnfH of iron-only nitrogenase from Azotobacter vinelandii. J. Inorg. Biochem. 2022, 227, 111690. [Google Scholar] [CrossRef]
- Trncik, C.; Detemple, F.; Einsle, O. Iron-only Fe-Nitrogenase underscores common catalytic principles in Biological Nitrogen Fixation. Nat. Catal. 2023, 6, 415–424. [Google Scholar] [CrossRef]
- Einsle, O.; Rees, D.C. Structural Enzymology of Nitrogenase Enzymes. Chem. Rev. 2020, 120, 4969–5004. [Google Scholar] [CrossRef] [PubMed]
- Lukoyanov, D.; Khadka, N.; Yang, Z.Y.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. Reductive Elimination of H2 Activates Nitrogenase to Reduce the N-N Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme. J. Am. Chem. Soc. 2016, 138, 10674–10683. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.F.; Lukoyanov, D.A.; Shaw, S.; Compton, P.; Tokmina-Lukaszewska, M.; Bothner, B.; Kelleher, N.; Dean, D.R.; Hoffman, B.M.; Seefeldt, L.C. Mechanism of N2 Reduction Catalyzed by Fe-Nitrogenase Involves Reductive Elimination of H2. Biochemistry 2018, 57, 701–710. [Google Scholar] [CrossRef]
- Harris, D.F.; Lukoyanov, D.A.; Kallas, H.; Trncik, C.; Yang, Z.Y.; Compton, P.; Kelleher, N.; Einsle, O.; Dean, D.R.; Hoffman, B.M.; et al. Mo-, V-, and Fe-Nitrogenases Use a Universal Eight-Electron Reductive-Elimination Mechanism To Achieve N2 Reduction. Biochemistry 2019, 58, 3293–3301. [Google Scholar] [CrossRef]
- Rohde, M.; Sippel, D.; Trncik, C.; Andrade, S.L.A.; Einsle, O. The Critical E4 State of Nitrogenase Catalysis. Biochemistry 2018, 57, 5497–5504. [Google Scholar] [CrossRef]
- Burgess, B.K.; Wherland, S.; Newton, W.E.; Stiefel, E.I. Nitrogenase Reactivity—Insight into the Nitrogen-Fixing Process through Hydrogen-Inhibition and HD-Forming Reactions. Biochemistry 1981, 20, 5140–5146. [Google Scholar] [CrossRef]
- Fisher, K.; Dilworth, M.J.; Newton, W.E. Differential effects on N2 binding and reduction, HD formation, and azide reduction with alpha-195(His)- and alpha-191(Gln)-substituted MoFe proteins of Azotobacter vinelandii nitrogenase. Biochemistry 2000, 39, 15570–15577. [Google Scholar] [CrossRef]
- Harris, D.F.; Yang, Z.Y.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. Kinetic Understanding of N2 Reduction versus H2 Evolution at the E4 (4H) Janus State in the Three Nitrogenases. Biochemistry 2018, 57, 5706–5714. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, M.J. Acetylene Reduction by Nitrogen-Fixing Preparations from Clostridium Pasteurianum. Biochim. Biophys. Acta 1966, 127, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.J.; Eady, R.R.; Thorneley, R.N.F. Electron-Paramagnetic-Resonance Studies on Nitrogenase of Klebsiella pneumoniae—Evidence for Acetylene-Nitrogenase and Ethylene-Nitrogenase Transient Complexes. Biochem. J. 1978, 173, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.C.; Chen, C.H.; Burris, R.H. Inhibition of Nitrogenase-Catalyzed Reductions. Biochim. Biophys. Acta 1973, 292, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Hu, Y.L.; Ribbe, M.W. Vanadium Nitrogenase Reduces CO. Science 2010, 329, 642. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Hu, Y.L.; Ribbe, M.W. Insights into Hydrocarbon Formation by Nitrogenase Cofactor Homologs. Mbio 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Ertl, G.; Huber, M.; Lee, S.B.; Paal, Z.; Weiss, M. Interactions of Nitrogen and Hydrogen on Iron Surfaces. Appl. Surf. Sci. 1981, 8, 373–386. [Google Scholar] [CrossRef]
- Davis, L.C.; Henzl, M.T.; Burris, R.H.; Orme-Johnson, W.H. Iron-sulfur clusters in the molybdenum-iron protein component of nitrogenase. Electron paramagnetic resonance of the carbon monoxide inhibited state. Biochemistry 1979, 18, 4860–4869. [Google Scholar] [CrossRef]
- Lee, H.I.; Cameron, L.M.; Hales, B.J.; Hoffman, B.M. CO binding to the FeMo cofactor of CO-inhibited nitrogenase: (CO)-13C and 1H Q-band ENDOR investigation. J. Am. Chem. Soc. 1997, 119, 10121–10126. [Google Scholar] [CrossRef]
- Pollock, R.C.; Lee, H.I.; Cameron, L.M.; Derose, V.J.; Hales, B.J.; Orme-Johnson, W.H.; Hoffman, B.M. Investigation of CO Bound to Inhibited Forms of Nitrogenase MoFe Protein by 13C ENDOR. J. Am. Chem. Soc. 1995, 117, 8686–8687. [Google Scholar] [CrossRef]
- George, S.J.; Ashby, G.A.; Wharton, C.W.; Thorneley, R.N.F. Time-resolved binding of carbon monoxide to nitrogenase monitored by stopped-flow infrared spectroscopy. J. Am. Chem. Soc. 1997, 119, 6450–6451. [Google Scholar] [CrossRef]
- Cameron, L.M.; Hales, B.J. Investigation of CO binding and release from Mo-nitrogenase during catalytic turnover. Biochemistry 1998, 37, 9449–9456. [Google Scholar] [CrossRef] [PubMed]
- Maskos, Z.; Hales, B.J. Photo-lability of CO bound to mo-nitrogenase from Azotobacter vinelandii. J. Inorg. Biochem. 2003, 93, 11–17. [Google Scholar] [CrossRef]
- Yan, L.F.; Pelmenschikov, V.; Dapper, C.H.; Scott, A.D.; Newton, W.E.; Cramer, S.P. IR-Monitored Photolysis of CO-Inhibited Nitrogenase: A Major EPR-Silent Species with Coupled Terminal CO Ligands. Chem.-Eur. J. 2012, 18, 16349–16357. [Google Scholar] [CrossRef]
- Spatzal, T.; Perez, K.A.; Einsle, O.; Howard, J.B.; Rees, D.C. Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase. Science 2014, 345, 1620–1623. [Google Scholar] [CrossRef] [PubMed]
- Rohde, M.; Grunau, K.; Einsle, O. CO Binding to the FeV Cofactor of CO-Reducing Vanadium Nitrogenase at Atomic Resolution. Angew. Chem. Int. Edit. 2020, 59, 23626–23630. [Google Scholar] [CrossRef]
- Buscagan, T.M.; Perez, K.A.; Maggiolo, A.O.; Rees, D.C.; Spatzal, T. Structural Characterization of Two CO Molecules Bound to the Nitrogenase Active Site. Angew. Chem. Int. Edit. 2021, 60, 5704–5707. [Google Scholar] [CrossRef]
- Rohde, M.; Laun, K.; Zebger, I.; Stripp, S.T.; Einsle, O. Two ligand-binding sites in CO-reducing V nitrogenase reveal a general mechanistic principle. Sci. Adv. 2021, 7, eabg4474. [Google Scholar] [CrossRef]
- Einsle, O. Catalysis and structure of nitrogenase. Curr. Opin. Struct. Biol. 2023, 83, 102719. [Google Scholar] [CrossRef]
- Van Stappen, C.; Thorhallsson, A.T.; Decamps, L.; Björnsson, R.; DeBeer, S. Resolving the structure of the E1 state of Mo nitrogenase through Mo and Fe K-edge EXAFS and QM/MM calculations. Chem. Sci. 2019, 10, 9807–9821. [Google Scholar] [CrossRef]
- Smith, J.M.; Lachicotte, R.J.; Holland, P.L. N=N bond cleavage by a low-coordinate iron(II) hydride complex. J. Am. Chem. Soc. 2003, 125, 15752–15753. [Google Scholar] [CrossRef] [PubMed]
- Rittle, J.; McCrory, C.C.L.; Peters, J.C. A 106-Fold Enhancement in N2-Binding Affinity of an Fe2(µ-H)2 Core upon Reduction to a Mixed-Valence FeIIFeI State. J. Am. Chem. Soc. 2014, 136, 13853–13862. [Google Scholar] [CrossRef] [PubMed]
- Van Stappen, C.; Davydov, R.; Yang, Z.Y.; Fan, R.; Guo, Y.; Bill, E.; Seefeldt, L.C.; Hoffman, B.M.; DeBeer, S. Spectroscopic Description of the E1 State of Mo Nitrogenase Based on Mo and Fe X-ray Absorption and Mössbauer Studies. Inorg. Chem. 2019, 58, 12365–12376. [Google Scholar] [CrossRef]
- Van Stappen, C.; Decamps, L.; Cutsail, G.E.; Bjornsson, R.; Henthorn, J.T.; Birrell, J.A.; DeBeer, S. The Spectroscopy of Nitrogenases. Chem. Rev. 2020, 120, 5005–5081. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.K.; Ugalde, R.A.; Imperial, J.; Brill, W.J. Molybdenum in Nitrogenase. Annu. Rev. Biochem. 1984, 53, 231–257. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.S.; Costas, A.M.G.; Hamilton, T.L.; Mus, F.; Peters, J.W. Evolution of Molybdenum Nitrogenase during the Transition from Anaerobic to Aerobic Metabolism. J. Bacteriol. 2015, 197, 1690–1699. [Google Scholar] [CrossRef]
- Pi, H.W.; Lin, J.J.; Chen, C.A.; Wang, P.H.; Chiang, Y.R.; Huang, C.C.; Young, C.C.; Li, W.H. Origin and Evolution of Nitrogen Fixation in Prokaryotes. Mol. Biol. Evol. 2022, 39, 9. [Google Scholar] [CrossRef]
- Rebelein, J.G.; Lee, C.C.; Hu, Y.L.; Ribbe, M.W. The in vivo hydrocarbon formation by vanadium nitrogenase follows a secondary metabolic pathway. Nat. Commun. 2016, 7, 13641. [Google Scholar] [CrossRef]
- Durrant, M.C. A molybdenum-centred model for nitrogenase catalysis. Inorg. Chem. Commun. 2001, 4, 60–62. [Google Scholar] [CrossRef]
- Coucouvanis, D.; Mosier, P.E.; Demadis, K.D.; Patton, S.; Malinak, S.M.; Kim, C.G.; Tyson, M.A. The Catalytic Reduction of Hydrazine to Ammonia by the MoFe3S4 Cubanes and Implications Regarding the Function of Nitrogenase—Evidence for Direct Involvement of the Molybdenum Atom in Substrate Reduction. J. Am. Chem. Soc. 1993, 115, 12193–12194. [Google Scholar] [CrossRef]
- Lovell, T.; Li, J.; Case, D.A.; Noodleman, L. FeMo cofactor of nitrogenase: Energetics and local interactions in the protein environment. J. Biol. Inorg. Chem. 2002, 7, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, P.C.; Igarashi, R.Y.; Lee, H.I.; Hoffman, B.M.; Seefeldt, L.C.; Dean, D.R. Substrate interactions with the nitrogenase active site. Acc. Chem. Res. 2005, 38, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Lukoyanov, D.A.; Yang, Z.Y.; Perez-Gonzalez, A.; Raugei, S.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. 13C-ENDOR Characterization of the Central Carbon within the Nitrogenase Catalytic Cofactor Indicates That the CFe6 Core Is a Stabilizing “Heart of Steel”. J. Am. Chem. Soc. 2022, 2022, 6149. [Google Scholar]
- Lukoyanov, D.A.; Harris, D.F.; Yang, Z.Y.; Pérez-González, A.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. The One-Electron Reduced Active-Site FeFe-Cofactor of Fe-Nitrogenase Contains a Hydride Bound to a Formally Oxidized Metal-Ion Core. Inorg. Chem. 2022, 61, 5459–5464. [Google Scholar] [CrossRef]
- Bröcker, M.J.; Virus, S.; Ganskow, S.; Heathcote, P.; Heinz, D.W.; Schubert, W.D.; Jahn, D.; Moser, J. ATP-driven reduction by dark-operative protochlorophyllide oxidoreductase from Chlorobium tepidum mechanistically resembles nitrogenase catalysis. J. Biol. Chem. 2008, 283, 10559–10567. [Google Scholar] [CrossRef]
- Moore, S.J.; Sowa, S.T.; Schuchardt, C.; Deery, E.; Lawrence, A.D.; Ramos, J.V.; Billig, S.; Birkemeyer, C.; Chivers, P.T.; Howard, M.J.; et al. Elucidation of the biosynthesis of the methane catalyst coenzyme F430. Nature 2017, 543, 78–82. [Google Scholar] [CrossRef]
- Zheng, K.Y.; Ngo, P.D.; Owens, V.L.; Yang, X.P.; Mansoorabadi, S.O. The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science 2016, 354, 339–342. [Google Scholar] [CrossRef]
- Garcia, A.K.; Harris, D.F.; Rivier, A.J.; Carruthers, B.M.; Pinochet-Barros, A.; Seefeldt, L.C.; Kacar, B. Nitrogenase resurrection and the evolution of a singular enzymatic mechanism. Elife 2023, 12, 1384. [Google Scholar] [CrossRef]
- Buckel, W.; Hetzel, M.; Kim, J. ATP-driven electron transfer in enzymatic radical reactions. Curr. Opin. Chem. Biol. 2004, 8, 462–467. [Google Scholar] [CrossRef]
- Seefeldt, L.C.; Peters, J.W.; Beratan, D.N.; Bothner, B.; Minteer, S.D.; Raugei, S.; Hoffman, B.M. Control of electron transfer in nitrogenase. Curr. Opin. Chem. Biol. 2018, 47, 54–59. [Google Scholar] [CrossRef]
- Rutledge, H.L.; Tezcan, F.A. Electron Transfer in Nitrogenase. Chem. Rev. 2020, 120, 5158–5193. [Google Scholar] [CrossRef] [PubMed]
- Burén, S.; Jimenez-Vicente, E.; Echavarri-Erasun, C.; Rubio, L.M. Biosynthesis of Nitrogenase Cofactors. Chem. Rev. 2020, 120, 4921–4968. [Google Scholar] [CrossRef]
- Wang, L.Y.; Zhang, L.H.; Liu, Z.Z.; Zhao, D.H.; Liu, X.M.; Zhang, B.; Xie, J.B.; Hong, Y.Y.; Li, P.F.; Chen, S.F.; et al. A Minimal Nitrogen Fixation Gene Cluster from Paenibacillus sp. WLY78 Enables Expression of Active Nitrogenase in Escherichia coli. PLoS Genet. 2013, 9, e1003865. [Google Scholar] [CrossRef]
- Yang, J.G.; Xie, X.Q.; Wang, X.; Dixon, R.; Wang, Y.P. Reconstruction and minimal gene requirements for the alternative iron-only nitrogenase in Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, E3718–E3725. [Google Scholar] [CrossRef] [PubMed]
- Wiig, J.A.; Lee, C.C.; Rebelein, J.G.; Sickerman, N.S.; Tanifuji, K.; Stiebritz, M.T.; Hu, Y.; Ribbe, M.W. Biosynthesis of the M-Cluster of Mo-Nitrogenase. Molybdenum Tungsten Enzym. Biochem. 2017, 5, 297–312. [Google Scholar]
- Dos Santos, P.C. Genomic manipulations of the diazotroph Azotobacter vinelandii. In Metalloproteins. Methods in Molecular Biology, 1st ed.; Hu, Y., Ed.; Humana Press: New York, NY, USA, 2019; Volume 1876, pp. 91–109. [Google Scholar]
- Brigle, K.E.; Setterquist, R.A.; Dean, D.R.; Cantwell, J.S.; Weiss, M.C.; Newton, W.E. Site-Directed Mutagenesis of the Nitrogenase MoFe Protein of Azotobacter vinelandii. Proc. Natl. Acad. Sci. USA 1987, 84, 7066–7069. [Google Scholar] [CrossRef]
- Hoffman, B.M.; Dean, D.R.; Seefeldt, L.C. Climbing Nitrogenase: Toward a Mechanism of Enzymatic Nitrogen Fixation. Acc. Chem. Res. 2009, 42, 609–619. [Google Scholar] [CrossRef]
- Smil, V. Detonator of the population explosion. Nature 1999, 400, 415. [Google Scholar] [CrossRef]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Gu, B.J.; Zhang, X.M.; Lam, S.K.; Yu, Y.L.; van Grinsven, H.J.M.; Zhang, S.H.; Wang, X.X.; Bodirsky, B.L.; Wang, S.T.; Duan, J.K.; et al. Cost-effective mitigation of nitrogen pollution from global croplands. Nature 2023, 613, 77–84. [Google Scholar] [CrossRef]
- Buren, S.; Rubio, L.M. State of the art in eukaryotic nitrogenase engineering. FEMS Microbiol. Lett. 2018, 365, 2. [Google Scholar] [CrossRef] [PubMed]
- Johnston, E.; Okada, S.; Gregg, C.M.; Warden, A.C.; Rolland, V.; Gillespie, V.; Byrne, K.; Colgrave, M.L.; Eamens, A.L.; Allen, R.S.; et al. The structural components of the iron-only nitrogenase, AnfDKG, form a protein complex within the plant mitochondrial matrix. Plant Mol. Biol. 2023, 112, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Gregg, C.M.; Allen, R.S.; Menon, A.; Hussain, D.; Gillespie, V.; Johnston, E.; Byrne, K.; Colgrave, M.L.; Wood, C.C. A Synthetic Biology Workflow Reveals Variation in Processing and Solubility of Nitrogenase Proteins Targeted to Plant Mitochondria, and Differing Tolerance of Targeting Sequences in a Bacterial Nitrogenase Assay. Front. Plant Sci. 2020, 11, 552160. [Google Scholar] [CrossRef] [PubMed]
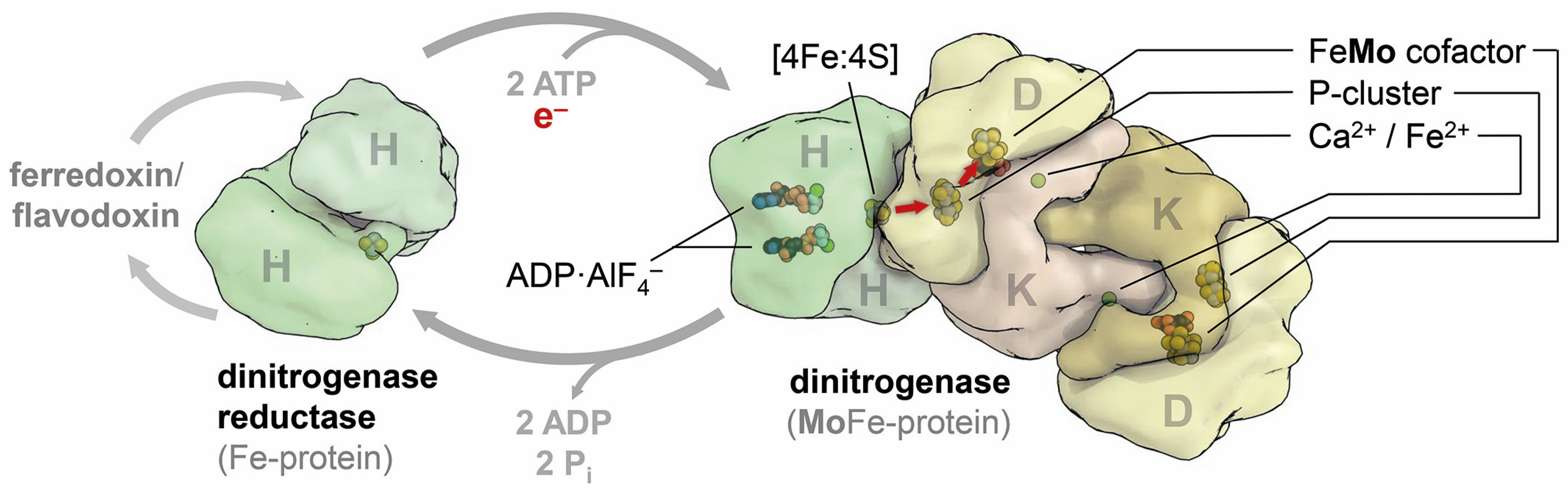

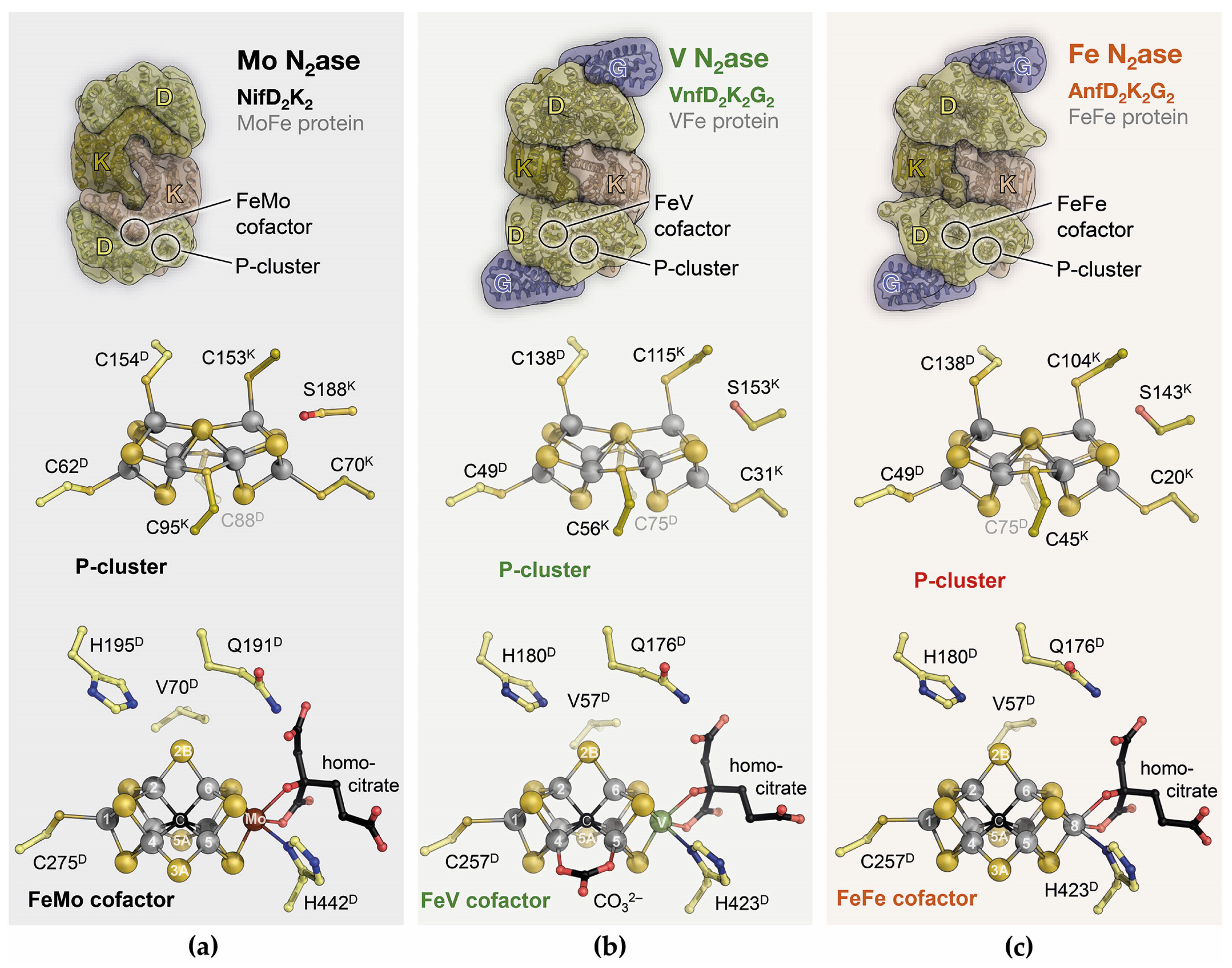
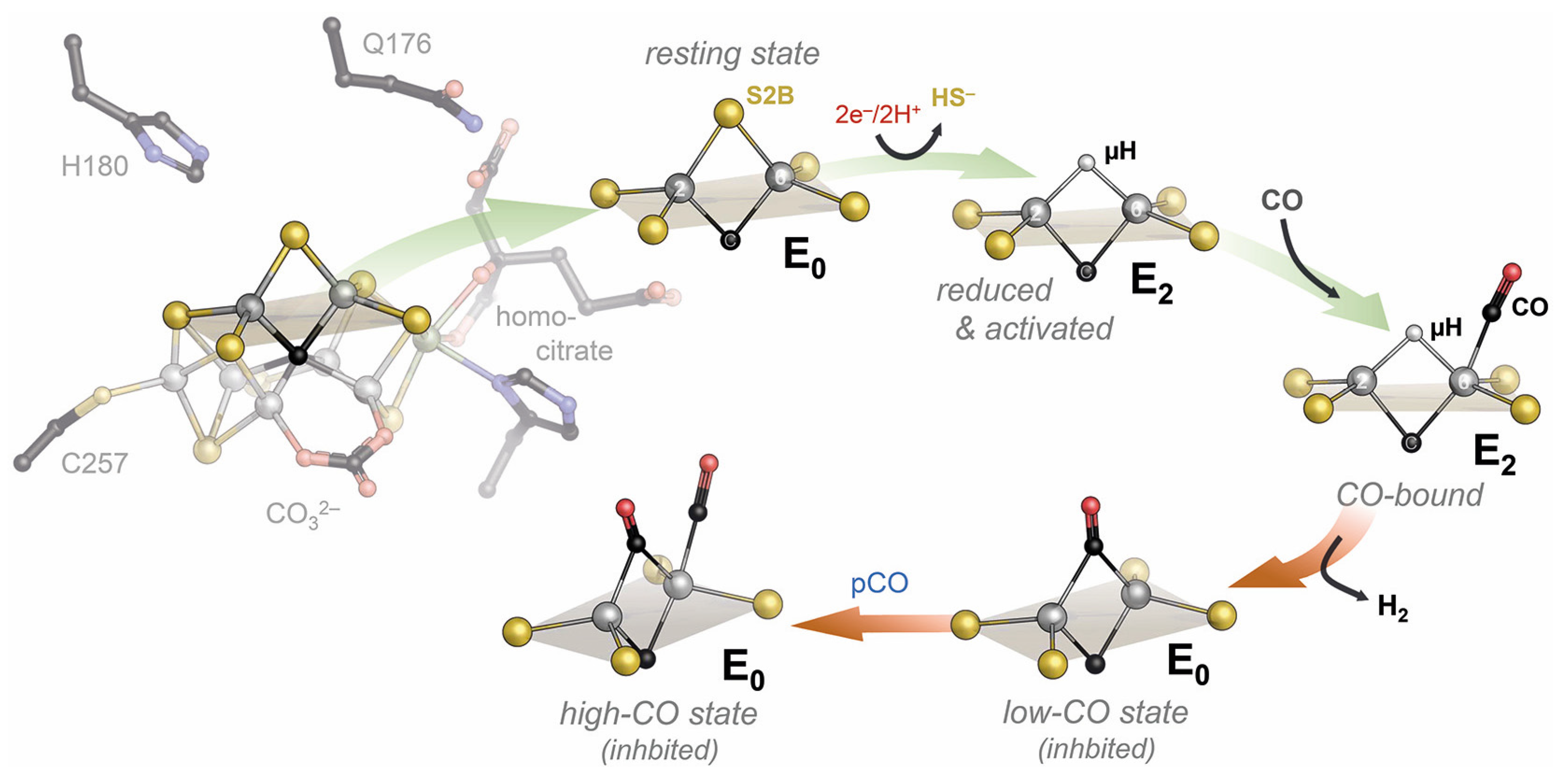
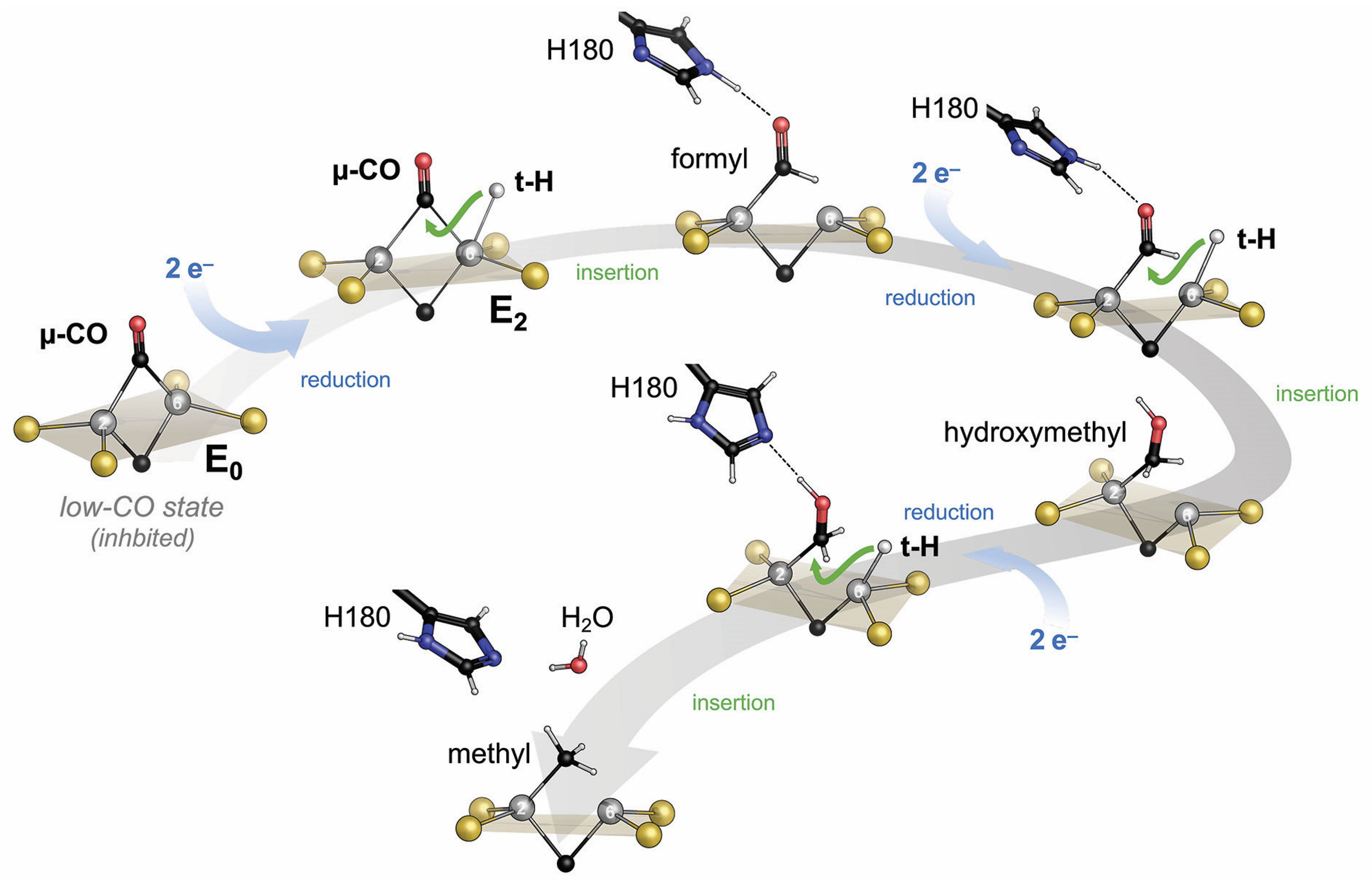

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Einsle, O. On the Shoulders of Giants—Reaching for Nitrogenase. Molecules 2023, 28, 7959. https://doi.org/10.3390/molecules28247959
Einsle O. On the Shoulders of Giants—Reaching for Nitrogenase. Molecules. 2023; 28(24):7959. https://doi.org/10.3390/molecules28247959
Chicago/Turabian StyleEinsle, Oliver. 2023. "On the Shoulders of Giants—Reaching for Nitrogenase" Molecules 28, no. 24: 7959. https://doi.org/10.3390/molecules28247959
APA StyleEinsle, O. (2023). On the Shoulders of Giants—Reaching for Nitrogenase. Molecules, 28(24), 7959. https://doi.org/10.3390/molecules28247959