
Synthesis of Novel Arginine Building Blocks with Increased Lipophilicity Compatible with Solid-Phase Peptide Synthesis

 , , , , , ,
, , , , , ,
Abstract
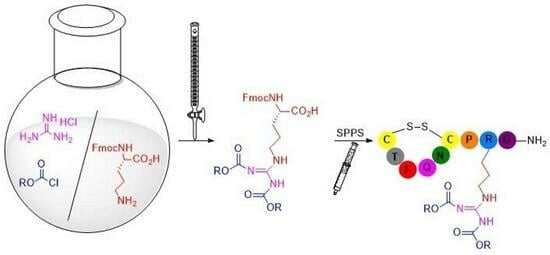
:
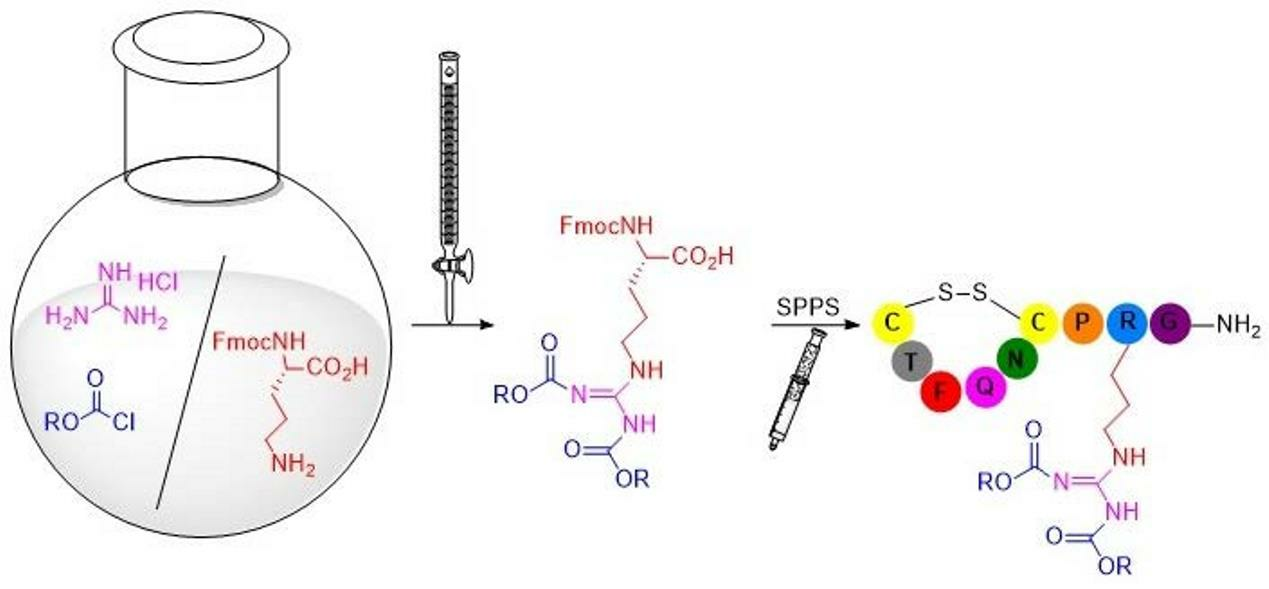
1. Introduction
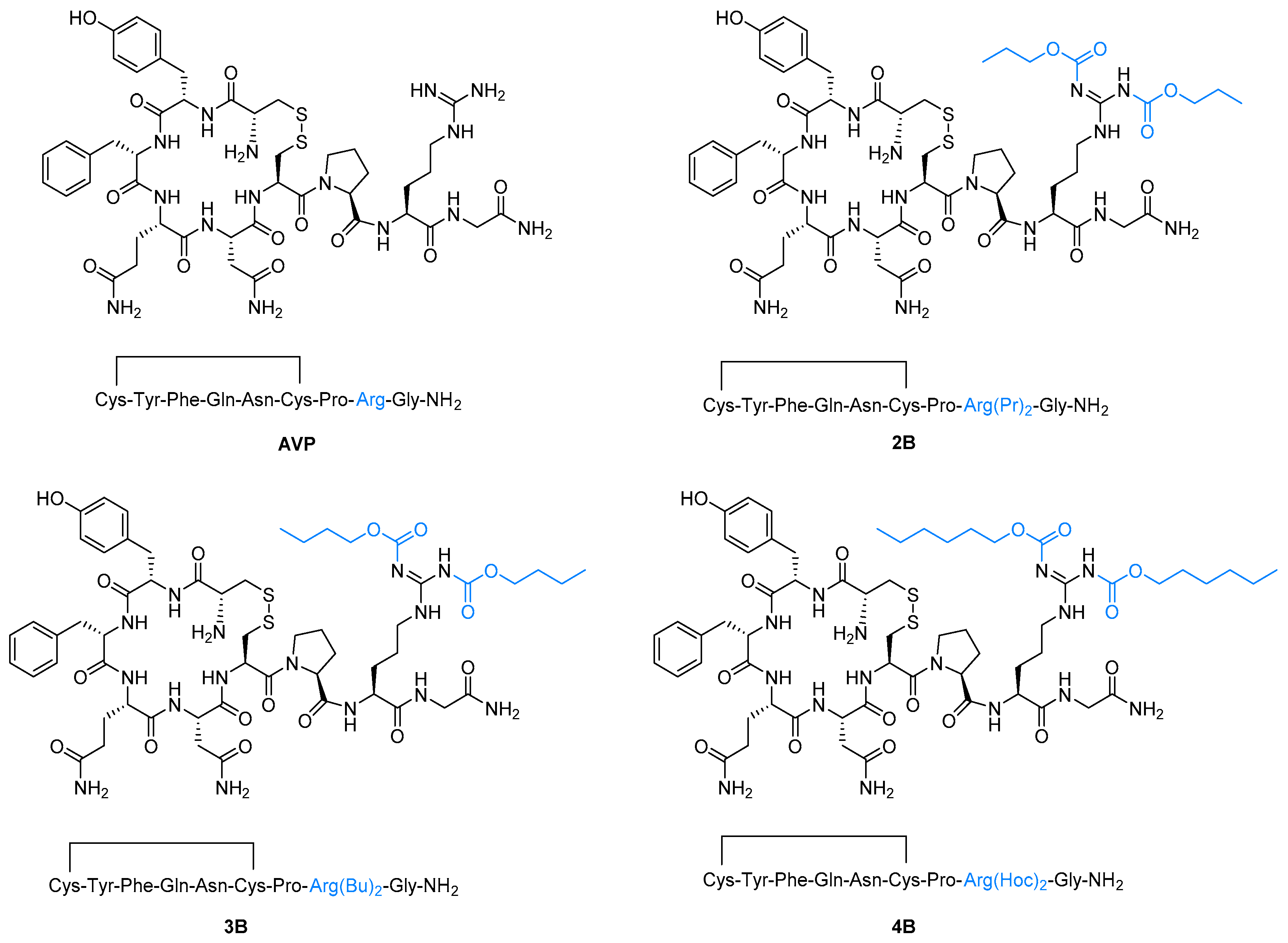
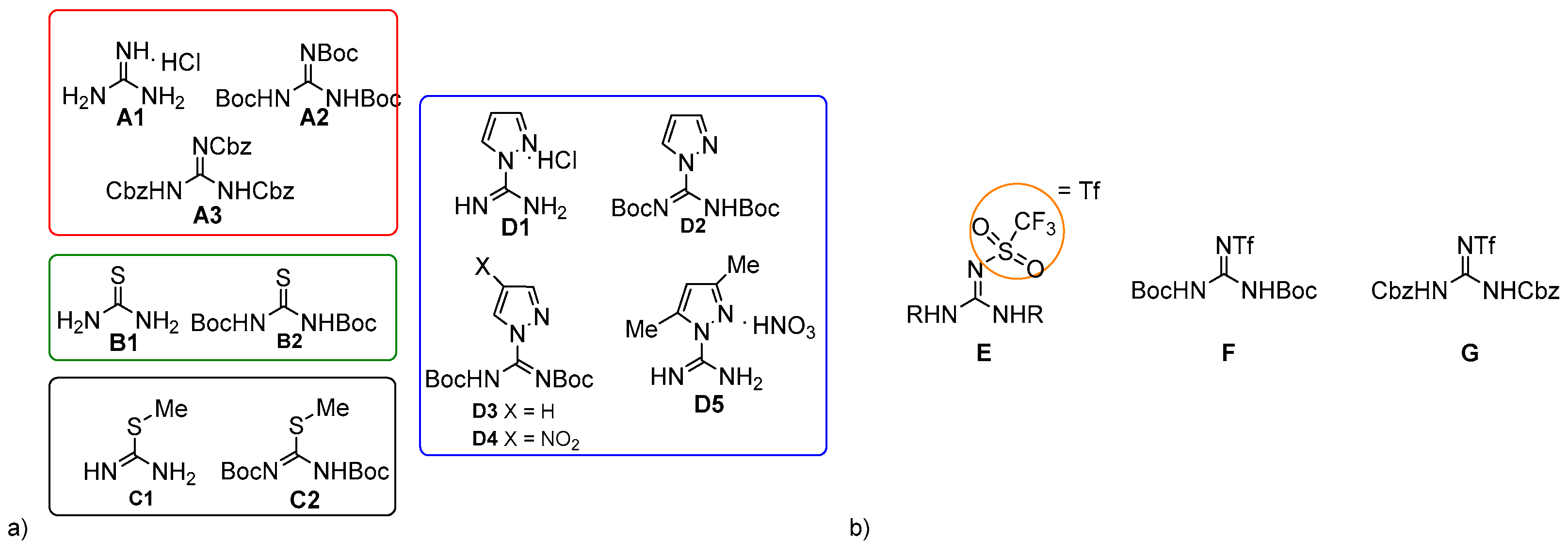
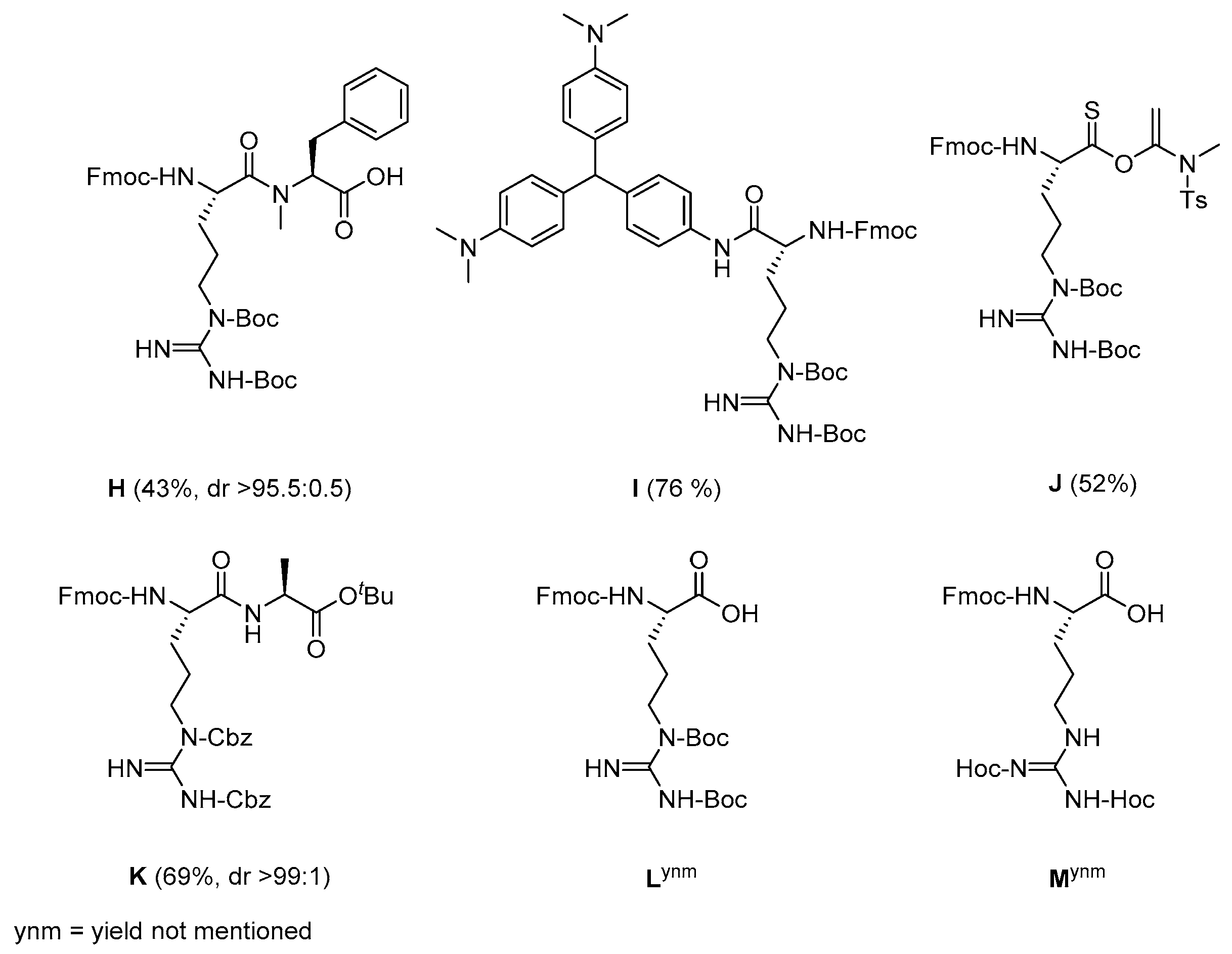
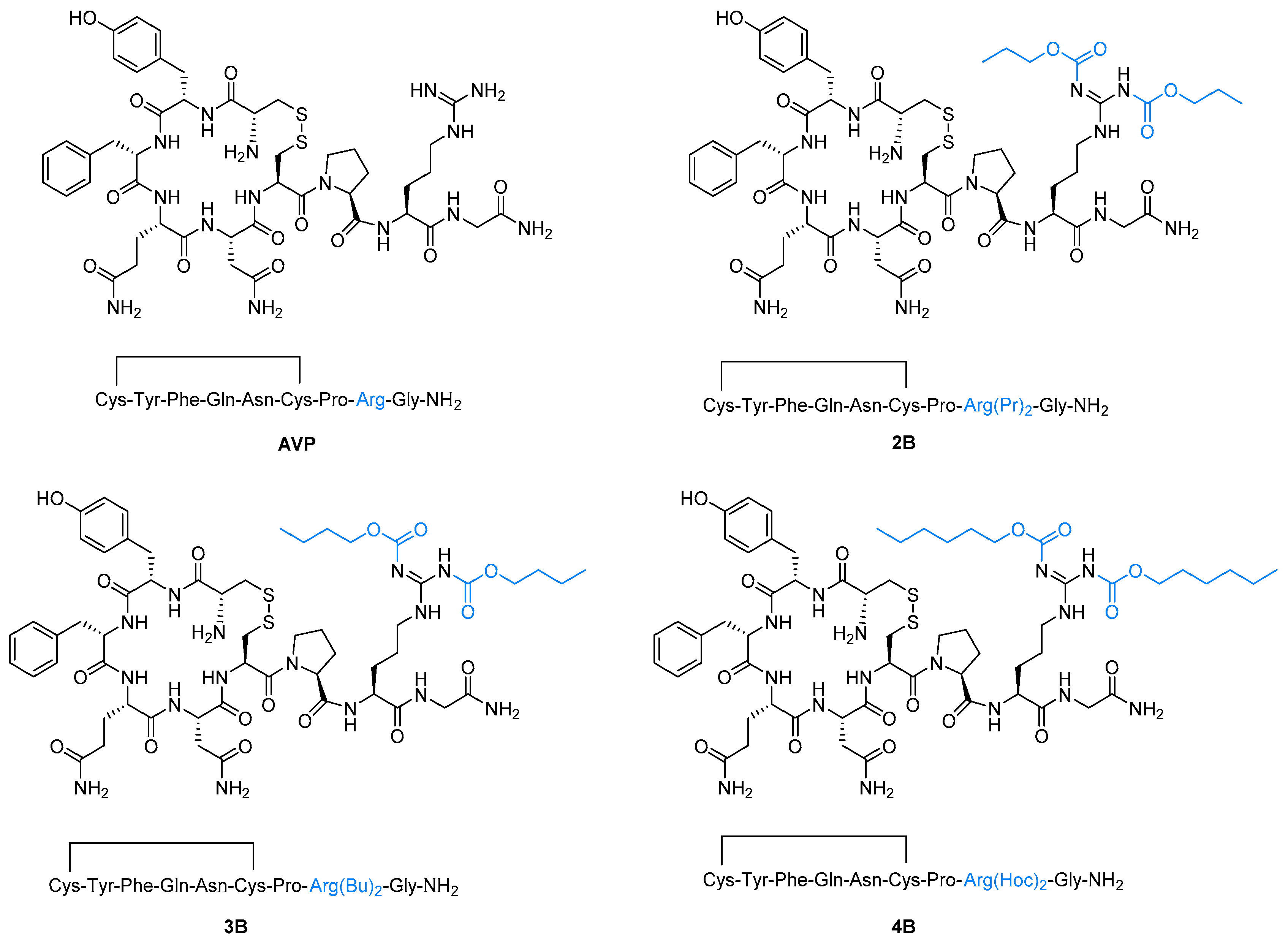
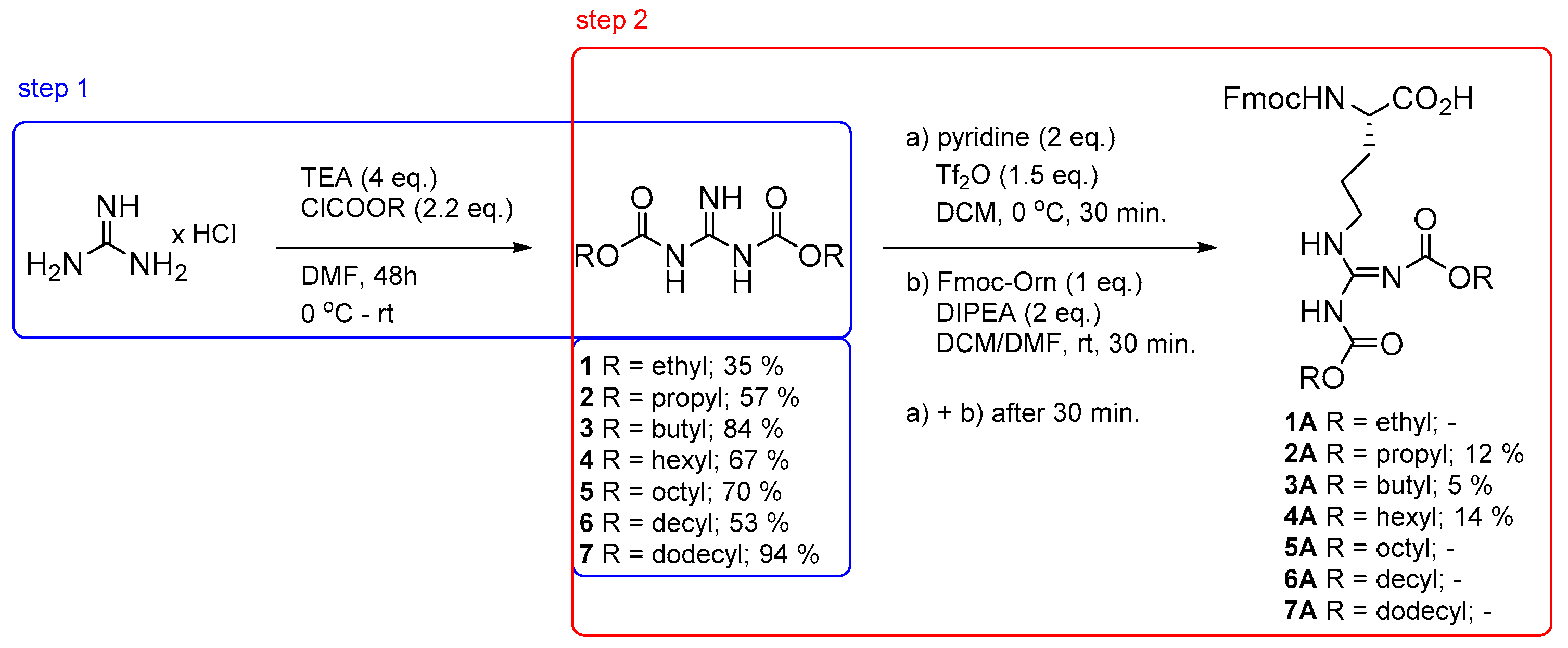
2. Results and Discussion
3. Materials and Methods
3.1. General
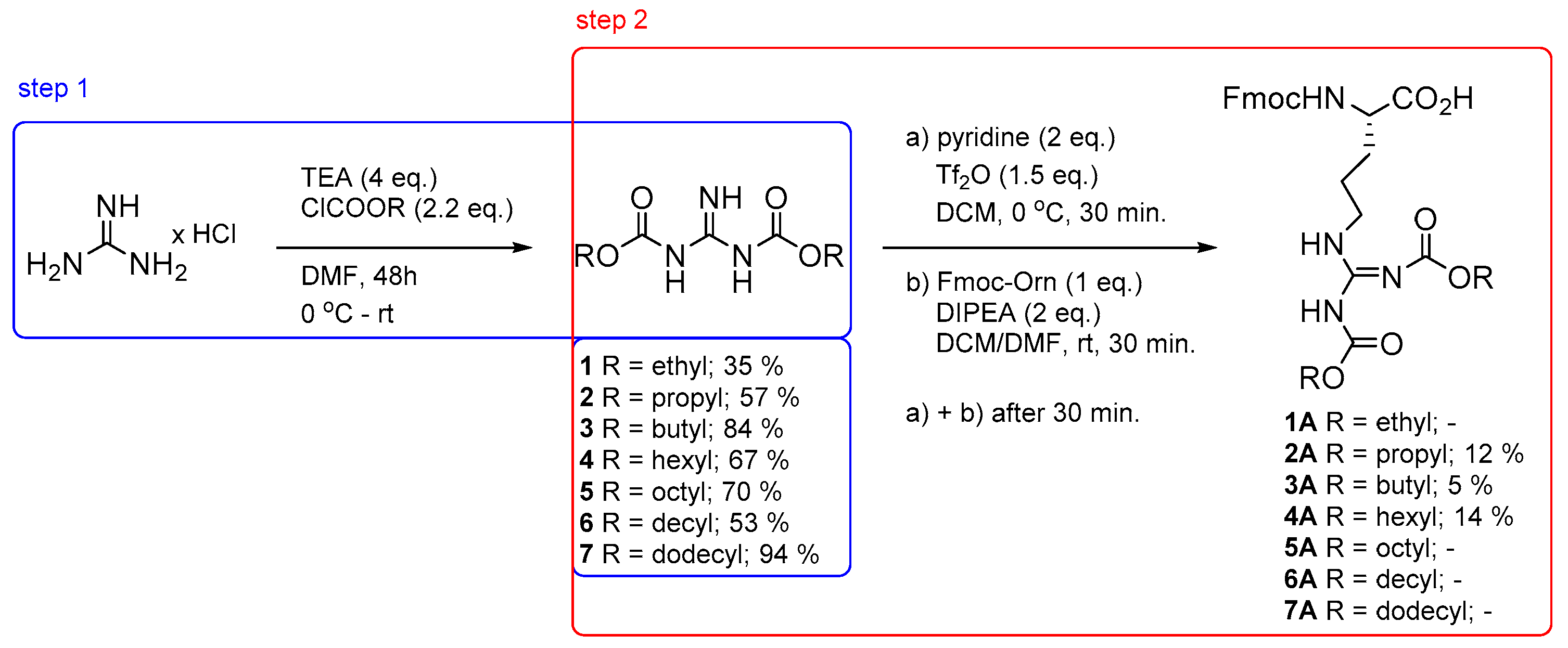
3.2. General Procedure for the Synthesis of Dicarbamates 1–7
3.3. Synthesis of Arginine Derivatives 2A–4A
3.3.1. (S,E)-N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-Nω,Nω′-bis(propoxycarbonyl)-l-arginine (2A)
3.3.2. (S,E)-N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-Nω,Nω′-bis(butoxycarbonyl)-l-arginine (3A)
3.3.3. (S,E)-N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-Nω,Nω′-bis((hexyloxy)carbonyl)-l-arginine (4A)
3.4. General Procedure for the Synthesis of Arginine Vasopressin Analogues 2B–4B
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Witkowska, E.; Kubik, K.; Krosnicka, J.; Grabowska, K.; Niescioruk, A.; Wilenska, B.; Misicka, A. Microwave-assisted guanidinylation in solid phase peptide synthesis: Comparison of various reagents. Tetrahedron Lett. 2014, 55, 6198–6203. [Google Scholar] [CrossRef]
- Feichtinger, K.; Sings, H.L.; Baker, T.J.; Matthews, K.; Goodman, M. Triurethane-protected guanidines and triflyldiurethane-protected guanidines: New reagents for guanidinylation reactions. J. Org. Chem. 1998, 63, 8432–8439. [Google Scholar] [CrossRef]
- Schug, K.A.; Lindner, W. Noncovalent binding between guanidinium and anionic groups: Focus on biological- and synthetic-based arginine/guanidinium interactions with phosph[on]ate and sulf[on]ate residues. Chem. Rev. 2005, 105, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Fenselau, C. Proton affinity of arginine measured by the kinetic approach. Rapid Commun. Mass Spectrom. 1992, 6, 403–405. [Google Scholar] [CrossRef]
- Burley, S.K.; Petsko, G.A. Amino-aromatic interactions in proteins. FEBS Lett. 1986, 203, 139–143. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S. Natural guanidine derivatives. Nat. Prod. Rep. 1999, 16, 339–365. [Google Scholar] [CrossRef]
- Feichtinger, K.; Zapf, C.; Sings, H.L.; Goodman, M. Diprotected triflylguanidines: A new class of guanidinylation reagents. J. Org. Chem. 1998, 63, 3804–3805. [Google Scholar] [CrossRef]
- Giltrap, M.; Dowman, L.J.; Nagalingam, G.; Ochoa, J.L.; Linington, R.G.; Britton, W.J.; Payne, R.J. Total synthesis of teixobactin. Org. Lett. 2016, 18, 2788–2791. [Google Scholar] [CrossRef]
- Yong, Y.F.; Kowalski, J.A.; Lipton, M.A. Facile and efficient guanylation of amines using thioureas and mukaiyama’s reagent. J. Org. Chem. 1997, 62, 1540–1542. [Google Scholar] [CrossRef]
- Bergeron, R.J.; McManis, J.S. Total synthesis of (+/−)-15-deoxyspergualin. J. Org. Chem. 1987, 52, 1700–1703. [Google Scholar] [CrossRef]
- Bernatowicz, M.S.; Wu, Y.; Matsueda, G.R. Urethane protected derivatives of 1-guanylpyrazole for the mild and efficient preparation of guanidines. Tetrahedron Lett. 1993, 34, 3389–3392. [Google Scholar] [CrossRef]
- Yong, Y.F.; Kowalski, J.A.; Thoen, J.C.; Lipton, M.A. A new reagent for solid and solution phase synthesis of protected guanidines from amines. Tetrahedron Lett. 1999, 40, 53–56. [Google Scholar] [CrossRef]
- Fotsch, C.H.; Wong, C.-H. Synthesis of guanidino-sugar as a glycosyl cation mimic. Tetrahedron Lett. 1994, 35, 3481–3484. [Google Scholar] [CrossRef]
- Levallet, C.; Lerpinier, J.; Ko, S.Y. The HgCl2-promoted guanylation reaction: The scope and limitations. Tetrahedron 1997, 53, 5291–5304. [Google Scholar] [CrossRef]
- Guo, Z.-X.; Cammidge, A.N.; Horwell, D.C. A convenient and versatile method for the synthesis of protected guanidines. Synth. Comm. 2000, 30, 2933–2943. [Google Scholar] [CrossRef]
- Bionda, N.; Cudic, P. Solid-Phase Guanidinylation of Peptidyl Amines Compatible with Standard Fmoc-Chemistry: Formation of Monosubstituted Guanidines. In Peptide Modifications to Increase Metabolic Stability and Activity; Methods in Molecular Biology; Springer: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Rogovoy, B.V. Recent developments in guanylatin reagents. ARKIVOC 2005, 4, 49–87. [Google Scholar] [CrossRef]
- Robinson, S.; Roskamp, E.J. Solid phase synthesis of guanidines. Tetrahedron 1997, 53, 6697–6705. [Google Scholar] [CrossRef]
- Ohara, K.; Vasseur, J.-J.; Smietana, M. NIS-promoted guanylation of amines. Tetrahedron Lett. 2009, 50, 1463–1465. [Google Scholar] [CrossRef]
- Schneider, S.E.; Bishop, P.A.; Salazar, M.A.; Bishop, O.A.; Anslyn, E.V. Solid phase synthesis of oligomeric guanidiniums. Tetrahedron 1998, 54, 15063–15086. [Google Scholar] [CrossRef]
- Santana, A.G.; Francisco, C.G.; Suárez, E.; González, C.C. Synthesis of guanidines from azides: A general and straightforward methodology in carbohydrate chemistry. J. Org. Chem. 2010, 75, 5371–5374. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bandyopadhyay, A. Synthesis and characterization of two potential impurities (des-ethyl-Ganirelix) generated in the Ganirelix manufacturing process. J. Pept. Sci. 2023, 29, e3489. [Google Scholar] [CrossRef] [PubMed]
- Shainyan, B.A.; Tolstikova, L.L.; Schilde, U. Simple methods for the preparation of N-trifly guanidines and the structure of compounds with the CF3SO2N=C-N fragment. J. Fluorine Chem. 2012, 135, 261–264. [Google Scholar] [CrossRef]
- Petrik, V.N.; Kondratenko, N.V.; Yagupolskii, L.M. N-Bis(mehylthio)methylene-trifluoromethanesulfonylamide CF3SO2N=C(SCH3)2: New reagent for the preparation of N-trifluoromethylenesulfonylimino carbonic and thiocarbonic acids derivatives. J. Fluor. Chem. 2003, 124, 151–158. [Google Scholar] [CrossRef]
- Yagupolskii, L.M.; Shelyazhenko, S.V.; Maletina, I.I.; Petrik, V.N.; Rusanov, E.B.; Chernega, A.N. The aza Curtis rearrangement. Eur. J. Org. Chem. 2001, 2001, 1225–1233. [Google Scholar] [CrossRef]
- Yagupolskii, L.M.; Yagupolskii, L.M.; Shelyazhenko, S.V.; Maletina, I.I.; Sokolenko, L.V.; Chernega, A.N.; Rusanov, E.B.; Tsymbal, Y.F. Trifluoromethanesulfonylimides of arenehydroxamic acids and their aza Lossen rearrangement. J. Fluor. Chem. 2007, 128, 515–523. [Google Scholar] [CrossRef]
- Yagupolskii, L.M.; Maletina, I.I.; Sokolenko, L.V.; Vlasenko, Y.G.; Buth, S.A. N-Perfluoroalkylsulfonylimido derivatives of arenecarboxylic acid amides and their oxidative aza Hofmann rearrangement. J. Fluor. Chem. 2008, 129, 486–492. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Y. One-pot synthesis of sulfamoylguanidines and sulfonylguanidines. Tetrahedron Lett. 2000, 41, 8075–8078. [Google Scholar] [CrossRef]
- Kurasaki, H.; Nagaya, A.; Kobayashi, Y.; Matsuda, A.; Matsumoto, M.; Morimoto, K.; Taguri, T.; Takeuchi, H.; Handa, M.; Cary, D.R.; et al. Isostearyl Mixed Anhydrides for the Preparation of N-Methylated Peptides Using C-Terminally Unprotected N-Methylamino Acids. Org. Lett. 2020, 22, 8039–8043. [Google Scholar] [CrossRef]
- Ayele, T.M.; Knutson, S.D.; Ellipilli, S.; Hwang, H.; Heemstra, J.M. Fluorogenic Photoaffinity Labeling of Proteins in Living Cells. Bioconjugate Chem. 2019, 30, 1309–1313. [Google Scholar] [CrossRef]
- Yang, J.; Wang, C.; Yao, C.; Chen, C.; Hu, Y.; He, G.; Zhao, J. Site-Specific Incorporation of Multiple Thioamide Substitutions into a Peptide Backbone via Solid Phase Peptide Synthesis. J. Org. Chem. 2020, 85, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Kamioka, S.; Shimazu, S.; Doi, T.; Takahashi, T. Combinatorial Synthesis of RGD Model Cyclic Peptides Utilizing a Palladium-Catalyzed Carbonylative Macrolactamization on a Polymer Support. J. Comb. Chem. 2008, 10, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, W.; Manthena, C.; Nakashima, E.; Yamamoto, H. Peptide Bond-Forming Reaction via Amino Acid Silyl Esters:New Catalytic Reactivity of an Aminosilane. ACS Catal. 2020, 10, 9594–9603. [Google Scholar] [CrossRef]
- Weinmüller, M.; Rechenmacher, F.; Marelli, U.K.; Reichart, F.; Kapp, T.G.; Räder, A.F.B.; Di Leva, F.S.; Marinelli, L.; Novellino, E.; Muñoz-Felix, J.M.; et al. Overcoming the Lack of Oral Availability of Cyclic Hexapeptides: Design of a Selective and Orally Available Ligand for the Intergrin αvβ3. Angew. Chem. Int Ed. Engl. 2017, 56, 16405–16409. [Google Scholar] [CrossRef] [PubMed]
- Schumacher-Klinger, A.; Fanous, J.; Merzbach, S.; Weinmueller, M.; Reichart, F.; Räder, A.F.B.; Gitlin-Domagalska, A.; Gilon, C.; Kessler, H.; Hoffman, A. Enhancing, oral bioavailability of cyclce RGD hexa-peptides by the lipophilic prodrug charge masking approach: Redirection of peptide intestinal permeability from paracellular to transcellular pathway. Mol. Pharm. 2018, 15, 3468–3477. [Google Scholar] [CrossRef]
- Räder, A.F.B.; Weinmüller, M.; Reichart, F.; Schumacher-Kilinger, A.; Merzbach, S.; Gilon, C.; Hoffman, A.; Kessler, H. Orally active peptides—Is there a magic bullet. Angew. Chem. Int. Ed. Engl. 2018, 57, 14414–14438. [Google Scholar] [CrossRef] [PubMed]
- Koopmanschap, G.; Ruijter, E.; Orru, R.V.A. Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–598. [Google Scholar] [CrossRef]
- Sharma, K.; Sharma, K.K.; Sharma, A.; Jain, R. Peptide-based drug discovery:Current status and recent advances. Drug Discov. Today 2023, 28, 103464. [Google Scholar] [CrossRef]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef]
- Wang, D.; Zou, L.; Jin, Q.; Hou, J.; Ge, G.; Yang, L. Human carboxylesterases: A comprehensive review. Acta Pharma. Sin. B 2018, 8, 699–712. [Google Scholar] [CrossRef]
- Siddiqui, F.M.; Qureshi, A.I. Dabigatran etexilate, a new oral direct thrombin inhibitor, fore stroke prevention in patients with atrial fibrilattion. Expert Opin. Pharmacother. 2010, 11, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [PubMed]
- Blech, S.; Ebner, T.; Ludwig-Schwellinger, E.; Stangier, J.; Roth, W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug. Metab. Dispos. 2008, 36, 386–399. [Google Scholar] [CrossRef]
- Glavaš, M.; Gitlin-Domagalska, A.; Dębowski, D.; Ptaszyńska, N.; Łęgowska, A.; Rolka, K. Vasopressin and Its Analogues: From Natural Hormones to Multitasking Peptides. Int. J. Mol. Sci. 2022, 23, 3068. [Google Scholar] [CrossRef] [PubMed]
- Peterlin-Mašič, L.; Kikelj, D. Arginine mimetics. Tetrahedron 2001, 57, 7073–7105. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Vojkovsky, T. Detection of secondary amines on solid phase. Peptide Res. 1995, 8, 236–237. [Google Scholar]
- Pearson, D.A.; Blanchette, M.; Baker, M.L.; Guindon, C.A. Trialkylsilanes as scavangers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989, 30, 2739–2742. [Google Scholar] [CrossRef]
- Yang, Y.; Sweeney, W.V.; Schneider, K.; Chait, B.T.; Tam, J.P. Two-step selective formation of three disulfide bridge in the synthesis of the C-terminal epidermal growth factor-like domain in human blood coagulation factor IX. Protein Sci. 1994, 3, 1267–1275. [Google Scholar] [CrossRef]
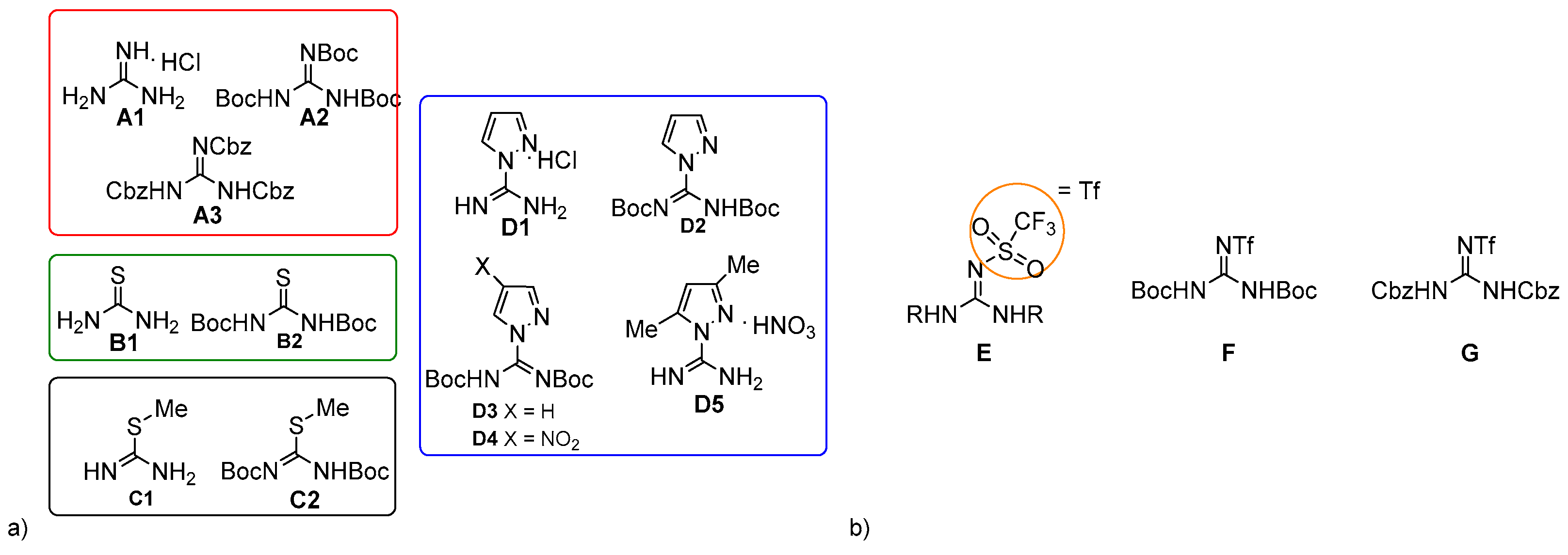


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | m (Guanidine Hydrochloride)/g | Base/eq. | Solvent/mL | t/°C | Chloroformate/2.2 eq. | ƞ/% |
|---|---|---|---|---|---|---|
| 1 | 0.25 | TEA/4 | ACN/H2O 3/1 | 0→rt | butyl | 13 |
| 2 a | 0.1 | TEA/4 | ACN/H2O 1.2/1.2 | 0→rt | butyl | - |
| 3 | 0.1 | TEA/4 | ACN/H2O 1.2/1.2 | 40 | butyl | - |
| 4 | 0.1 | 1M NaOH | ACN/H2O 1.2/1.2 | 0→rt | butyl | - |
| 5 | 0.1 | DIPEA/4 | ACN/H2O 1.2/1.2 | 0→rt | butyl | 44 |
| 6 | 0.1 | NaH/4 | ACN/H2O 1.2/1.2 | 0→rt | butyl | 70 |
| 7 | 0.1 | TEA/4 | DMF/2 | 0→rt | butyl | 76 |
| 8 | 1 | TEA/4 | DMF/20 | 0→rt | butyl | 84 |
| 9 | 2 | TEA/4 | DMF/40 | 0→rt | ethyl | 23 |
| 10 | 1 | TEA/4 | DMF/80 | 0→rt | ethyl | 35 |
| 11 | 1 | TEA/4 | DMF/20 | 0→rt | propyl | 57 |
| 12 | 1 | TEA/4 | DMF/20 | 0→rt | hexyl | 67 |
| 13 | 1 | TEA/4 | DMF/20 | 0→rt | octyl | 70 |
| 14 | 1 | TEA/4 | DMF/20 | 0→rt | decyl | 53 |
| 15 | 1 | TEA/4 | DMF/20 | 0→rt | dodecyl | 94 |
| Mixture A | Mixture B | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry [Compound (1–7)/mmol] a | Tf2O/eq. | Base/eq. | Solvent/mL | t/°C | Fmoc-Orn/eq. | Base/eq. | Solvent/mL | t/°C; t/h | ƞ/% |
| 1 [1/2.461] b | 1.5 | Py/2 | DCM/23.5 | 0 | 1 | DIPEA/2 | DCM/DMF/23.5/11 | rt; 24 | - |
| 2 [1/0.492] b | 1.5 | Py/2 | DCM/4.7 | 0 | 1 | DIPEA/2 | DCM/DMF/4.7/2.2 | rt; 24 | - |
| 3 [1/1.058] c | 1.5 | Py/2 | DCM/10 | 0 | 1 | DIPEA/2 | DCM/DMF/10/4.7 | rt; 24 | - |
| 4 [1/0.246] b | 1.5 | Py/2 | DCM/2.4 | 0 | 1 | DIPEA/2 | DCM/DMF/2.4/1.1 | rt; 24 | - |
| 5 [1/1.826] | 1.5 | Py/1.2 | dioxane/7 | 0 | 0.2 | TEA/1.2 | dioxane/5 | rt; 24 | - |
| 6 [3/0.157] | 1.5 | Py/2 | DCM/1.4 | rt | 1 | DIPEA/1.5 | DCM/DMF/1.4/0.7 | rt; 24 | - |
| 7 [3/0.104] d | 1 | NaH/2 | H2O/ACN/0.1/0.1 | −45 | - | - | - | rt; 24 | - |
| 8 [3/0.096] | 1.5 | Py/2 | DCM/1.3 | 0 | 1 | DIPEA/1.5 | DCM/DMF/1.3/0.5 | rt; 3 | - |
| 9 [3/0.096] | 1.5 | Py/2 | DCM/1.3 | 0 | 1 | DIPEA/1.5 | DCM/DMF/1.3/0.5 | 40; 2 | - |
| 10 [3/0.386] c,e | 1.5 | Py/2 | DCM/4.7 | 0 | 1 | DIPEA/2 | DCM/DMF/4.7/2.2 | rt/; 8 | 8 |
| 11 [3/0.386] c,e | 1.5 | Py/2 | DCM/4.7 | −78 | 1 | DIPEA/2 | DCM/DMF/4.7/2.2 | 0→rt; 96 | 3 |
| 12 [3/0.386] b,e | 1.5 | Py/2 | DCM/4.7 | 0 | 1 | DIPEA/2 | DCM/DMF/4.7/2.2 | 40; 24 | 6 |
| 13 [3/0.386] b,e | 1.5 | Py/2 | DCM/4.7 | 0 | 1 | DIPEA/2 | DCM/DMF/3.7/3.2 | 40; 24 | 8 |
| 14 [3/0.386] b,e | 1.5 | Py/2 | dioxane/4.7 | 0 | 1 | DIPEA/2 | dioxane/DMF/4.7/2.2 | 65; 24 | - |
| 15 [3/0.386] a,c | 1.5 | Py/2 | toluene/4.7 | 0 | 1 | DIPEA/2 | toluene/DMF/4.7/1 | 90; 5 | 1 |
| 16 [3/1.928] b,e | 1.5 | Py/2 | DCM/23.5 | 0 | 1 | DIPEA/2 | DCM/DMF/18.5/16 | rt; 5 | 5 |
| 17 [3/1.928] b,e | 1.5 | Py/2 | DCM/23.5 | 0 | 1 | DIPEA/2 | DCM/DMF/18.5/16 | rt; 5 | - |
| 18 [4/0.317] | 1.5 | Py/2 | DCM/4.5 | 0 | 1 | DIPEA/2 | DCM/DMF/4.5/2.1 | rt; 24 | 4 |
| 19 [4/0.793] b | 1.5 | Py/2 | DCM/12 | 0 | 1 | DIPEA/2 | DCM/DMF/12/5.4 | rt; 24 | 4 |
| 20 [4/1.585] b,e | 1.5 | Py/2 | DCM/23.5 | 0 | 1 | DIPEA/2 | DCM/DMF/23.5/11 | rt; 24 | 14 |
| 21 [4/0.317] d,f | TEA/2.2 | DMF/0.50 | rt; 24 | - | |||||
| 22 [4/0.317] d,f | TEA/2.2 | DCM/3.5 | rt; 24 | - | |||||
| 23 [4/0.159] d,f | DIPEA/2.2 | DCM/DMF0.3/0.1 | rt; 24 | - | |||||
| 24 [5/0.269] e | 1.5 | Py/2 | DCM/4.7 | 0 | 1 | DIPEA/2 | DCM/DMF/4.7/3.2 | rt; 24 | - |
| 25 [5/0.269] e | 1 | NaH/3.2 | DCM/3 | 0 | - | - | - | - | - |
| 26 [6/0.234] e | 1 | Py/2 | DCM/5 | 0 | 1 | DIPEA/2 | DCM/DMF/5/3 | rt; 24 | - |
| 27 [7/0.207] e | 1.5 | Py/2 | DCM/10 | 0 | 1 | DIPEA/2 | DCM/DMF/10/4 | rt; 24 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glavaš, M.; Gitlin-Domagalska, A.; Ptaszyńska, N.; Starego, D.; Freza, S.; Dębowski, D.; Helbik-Maciejewska, A.; Łęgowska, A.; Gilon, C.; Rolka, K. Synthesis of Novel Arginine Building Blocks with Increased Lipophilicity Compatible with Solid-Phase Peptide Synthesis. Molecules 2023, 28, 7780. https://doi.org/10.3390/molecules28237780
Glavaš M, Gitlin-Domagalska A, Ptaszyńska N, Starego D, Freza S, Dębowski D, Helbik-Maciejewska A, Łęgowska A, Gilon C, Rolka K. Synthesis of Novel Arginine Building Blocks with Increased Lipophilicity Compatible with Solid-Phase Peptide Synthesis. Molecules. 2023; 28(23):7780. https://doi.org/10.3390/molecules28237780
Chicago/Turabian StyleGlavaš, Mladena, Agata Gitlin-Domagalska, Natalia Ptaszyńska, Dominika Starego, Sylwia Freza, Dawid Dębowski, Aleksandra Helbik-Maciejewska, Anna Łęgowska, Chaim Gilon, and Krzysztof Rolka. 2023. "Synthesis of Novel Arginine Building Blocks with Increased Lipophilicity Compatible with Solid-Phase Peptide Synthesis" Molecules 28, no. 23: 7780. https://doi.org/10.3390/molecules28237780
APA StyleGlavaš, M., Gitlin-Domagalska, A., Ptaszyńska, N., Starego, D., Freza, S., Dębowski, D., Helbik-Maciejewska, A., Łęgowska, A., Gilon, C., & Rolka, K. (2023). Synthesis of Novel Arginine Building Blocks with Increased Lipophilicity Compatible with Solid-Phase Peptide Synthesis. Molecules, 28(23), 7780. https://doi.org/10.3390/molecules28237780