

Determination and Kinetic Characterization of a New Potential Inhibitor for AmsI Protein Tyrosine Phosphatase from the Apple Pathogen Erwinia amylovora
 , ,
, ,  and
and
Abstract
:
1. Introduction
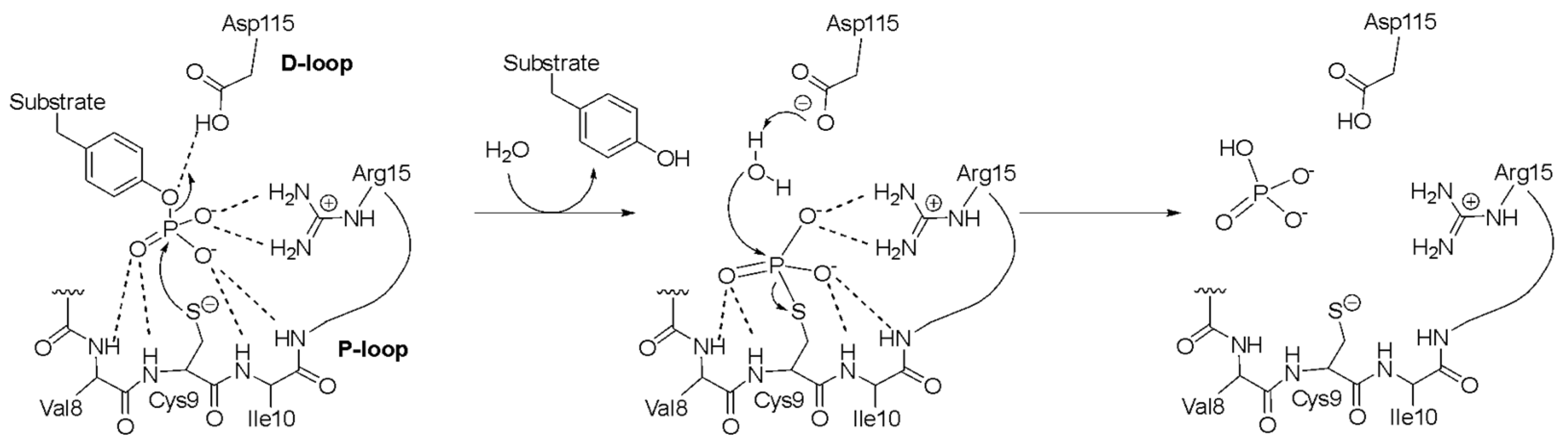
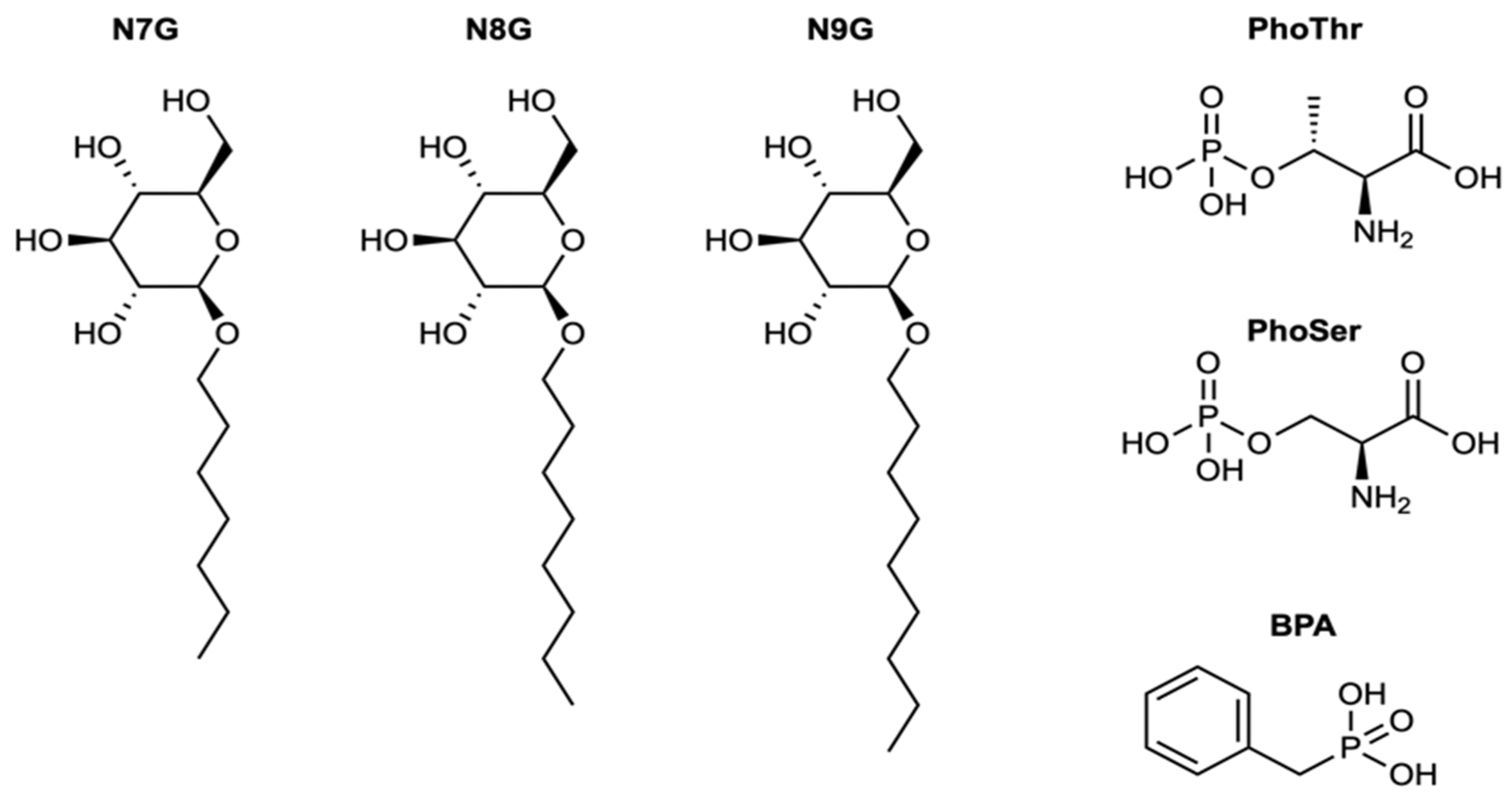
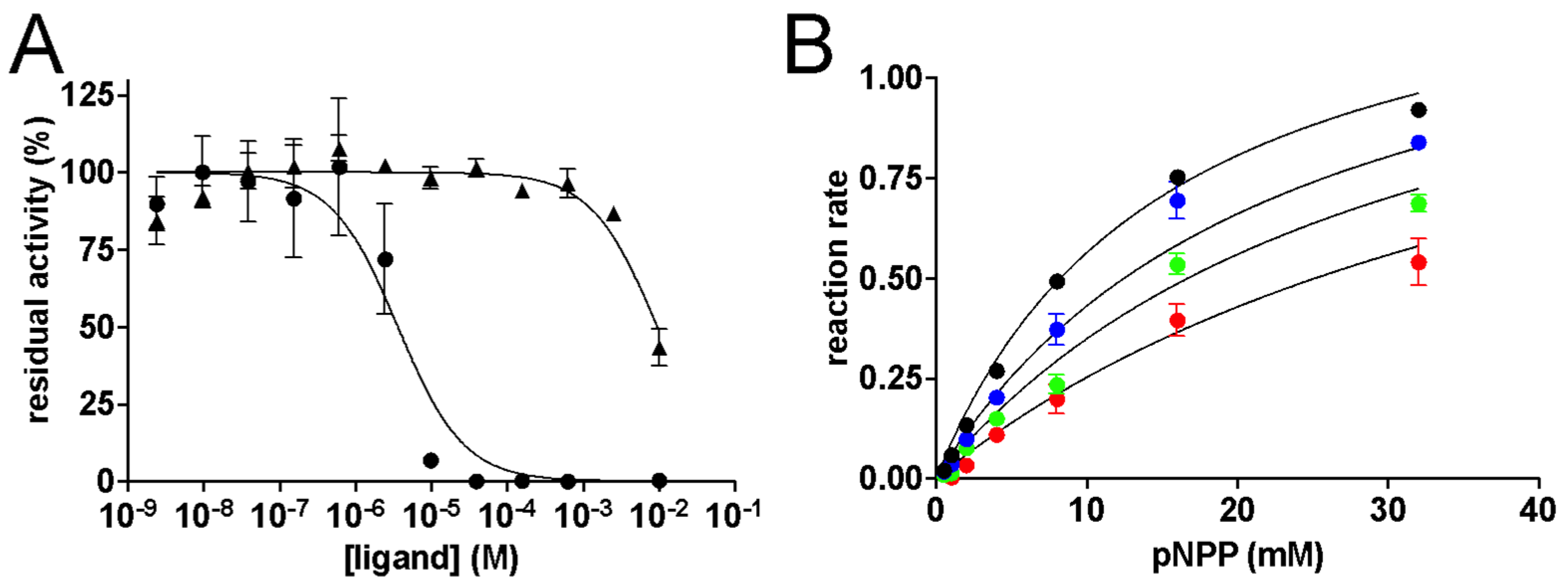
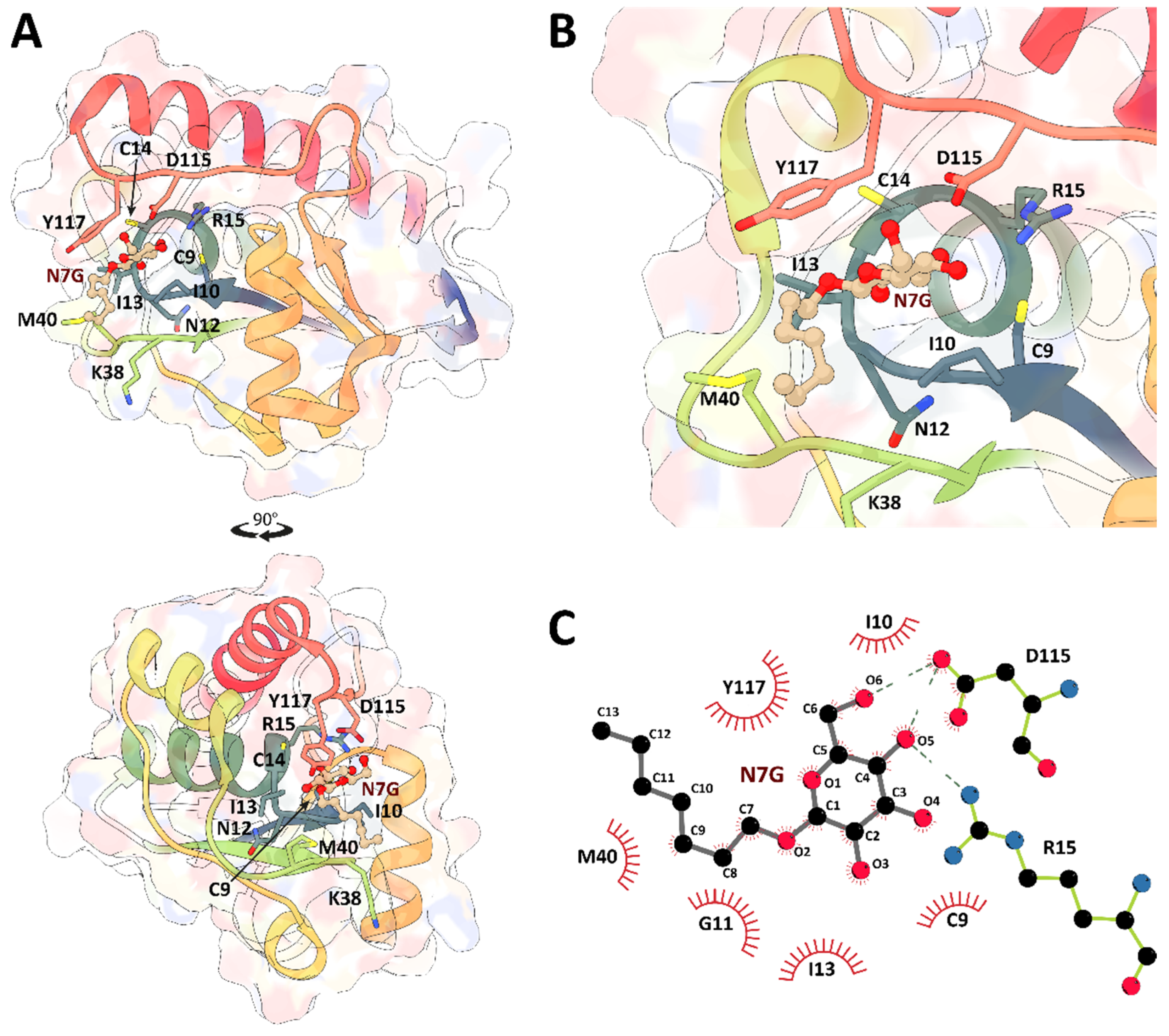
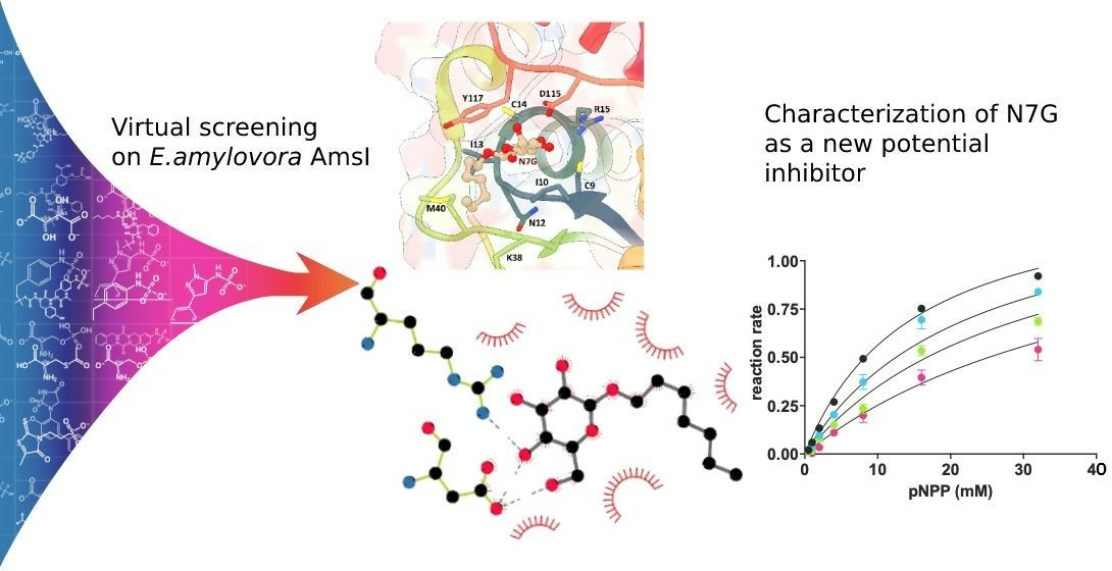
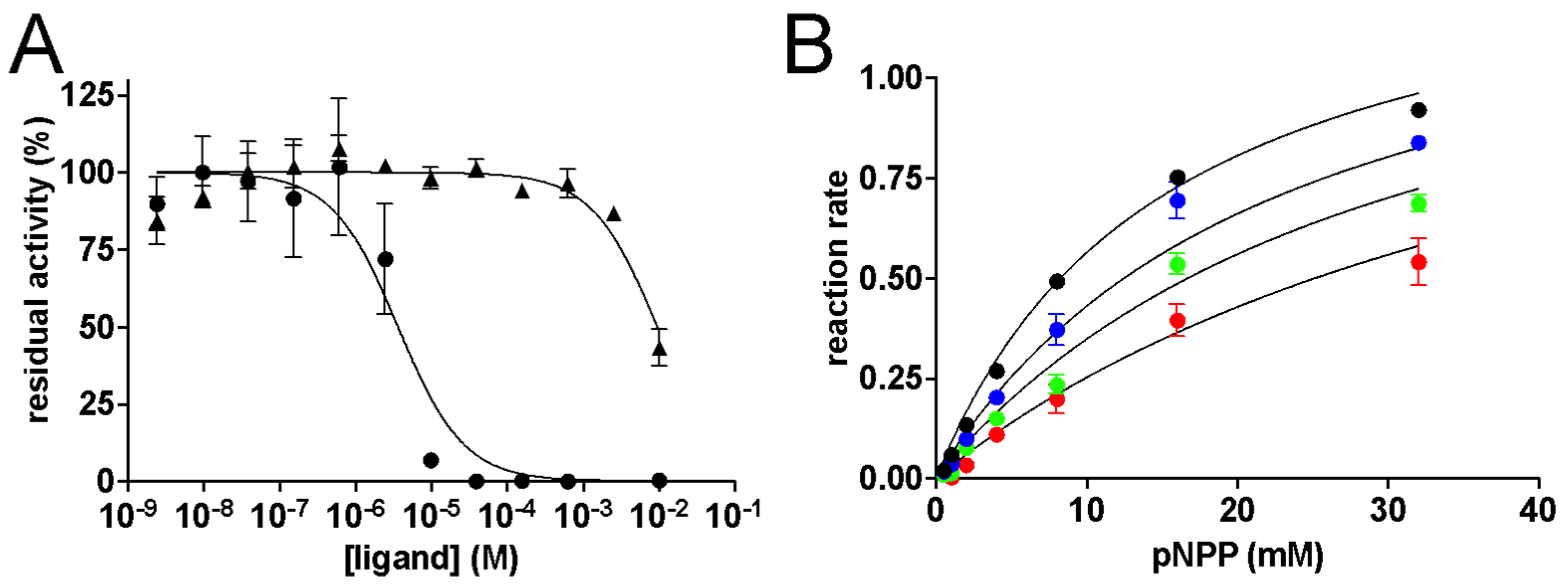
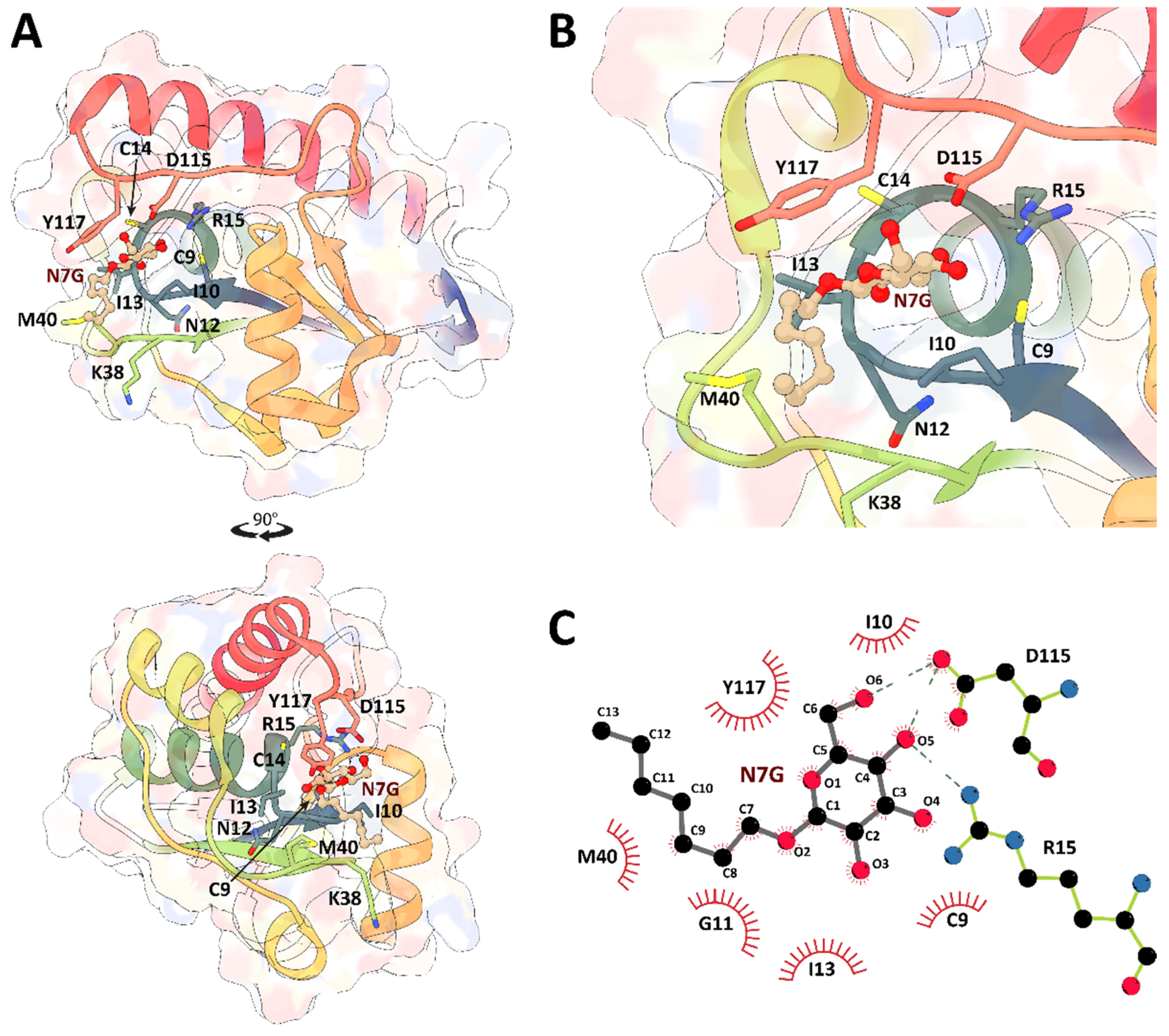
2. Results and Discussion
3. Materials and Methods
3.1. Selection of the Docking Software
3.2. OpenEye Fred Docking
3.3. DOCK6 Docking
3.4. Autodock
3.5. AutoDock Vina
3.6. Production of Recombinant EaAmsI
3.7. Kinetic Measurements
3.8. Software for Schemes and Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 Plant Pathogenic Bacteria in Molecular Plant Pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.L. Fire Blight: The Disease and Its Causative Agent, Erwinia Amylovora; CABI: Wallingford, UK, 2000; ISBN 978-1-84593-298-5. [Google Scholar]
- Zeng, Q.; Cui, Z.; Wang, J.; Childs, K.L.; Sundin, G.W.; Cooley, D.R.; Yang, C.; Garofalo, E.; Eaton, A.; Huntley, R.B.; et al. Comparative Genomics of Spiraeoideae-infecting Erwinia amylovora Strains Provides Novel Insight to Genetic Diversity and Identifies the Genetic Basis of a Low-virulence Strain. Mol. Plant Pathol. 2018, 19, 1652–1666. [Google Scholar] [CrossRef] [PubMed]
- Myung, I.-S.; Lee, J.-Y.; Yun, M.-J.; Lee, Y.-H.; Lee, Y.-K.; Park, D.-H.; Oh, C.-S. Fire Blight of Apple, Caused by Erwinia Amylovora, a New Disease in Korea. Plant Dis. 2016, 100, 1774. [Google Scholar] [CrossRef]
- Rhouma, A.; Helali, F.; Chettaoui, M.; Hajjouji, M.; Hajlaoui, M.R. First Report of Fire Blight Caused by Erwinia Amylovora on Pear in Tunisia. Plant Dis. 2014, 98, 158. [Google Scholar] [CrossRef] [PubMed]
- Soukainen, M.; Santala, J.; Tegel, J. First Report of Erwinia Amylovora, the Causal Agent of Fire Blight, on Pear in Finland. Plant Dis. 2015, 99, 1033. [Google Scholar] [CrossRef]
- Laala, S.; Manceau, C.; Valentini, F.; Kerkoud, M.; Kheddam, M. Fireblight Survey and First Characterization of Erwinia Amylovora Isolates from Algeria. J. Plant Pathol. 2012, 94, 693–696. [Google Scholar]
- Gusberti, M.; Klemm, U.; Meier, M.S.; Maurhofer, M.; Hunger-Glaser, I. Fire Blight Control: The Struggle Goes On. A Comparison of Different Fire Blight Control Methods in Switzerland with Respect to Biosafety, Efficacy and Durability. Int. J. Environ. Res. Public. Health 2015, 12, 11422–11447. [Google Scholar] [CrossRef]
- Bugert, P.; Geider, K. Molecular Analysis of the Ams Operon Required for Exopolysaccharide Synthesis of Erwinia Amylovora. Mol. Microbiol. 1995, 15, 917–933. [Google Scholar] [CrossRef]
- Borruso, L.; Salomone-Stagni, M.; Polsinelli, I.; Schmitt, A.O.; Benini, S. Conservation of Erwinia Amylovora Pathogenicity-Relevant Genes among Erwinia Genomes. Arch. Microbiol. 2017, 199, 1335–1344. [Google Scholar] [CrossRef]
- Wuerges, J.; Caputi, L.; Cianci, M.; Boivin, S.; Meijers, R.; Benini, S. The Crystal Structure of Erwinia Amylovora Levansucrase Provides a Snapshot of the Products of Sucrose Hydrolysis Trapped into the Active Site. J. Struct. Biol. 2015, 191, 290–298. [Google Scholar] [CrossRef]
- Caputi, L.; Cianci, M.; Benini, S. Cloning, Expression, Purification, Crystallization and Preliminary X-Ray Analysis of EaLsc, a Levansucrase from Erwinia Amylovora. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Caputi, L.; Nepogodiev, S.A.; Malnoy, M.; Rejzek, M.; Field, R.A.; Benini, S. Biomolecular Characterization of the Levansucrase of Erwinia Amylovora, a Promising Biocatalyst for the Synthesis of Fructooligosaccharides. J. Agric. Food Chem. 2013, 61, 12265–12273. [Google Scholar] [CrossRef] [PubMed]
- Geier, G.; Geider, K. Characterization and Influence on Virulence of the Levansucrase Gene from the Fireblight Pathogen Erwinia Amylovora. Physiol. Mol. Plant Pathol. 1993, 42, 387–404. [Google Scholar] [CrossRef]
- Nimtz, M.; Mort, A.; Domke, T.; Wray, V.; Zhang, Y.; Qiu, F.; Coplin, D.; Geider, K. Structure of Amylovoran, the Capsular Exopolysaccharide from the Fire Blight Pathogen Erwinia Amylovora. Carbohydr. Res. 1996, 287, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, F.; Coplin, D.L.; Geider, K. A Gene Cluster for Amylovoran Synthesis in Erwinia Amylovora: Characterization and Relationship to Cps Genes in Erwinia Stewartii. Mol. Gen. Genet. MGG 1993, 239, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Orye, K.; Bobev, S.; Devreese, B.; Van Beeumen, J.; De Bruyn, A.; Busson, R.; Herdewijn, P.; Morreel, K.; Messens, E. Influence of Amylovoran Production on Virulence of Erwinia Amylovora and a Different Amylovoran Structure in E. Amylovora Isolates from Rubus. Eur. J. Plant Pathol. 2001, 107, 839–844. [Google Scholar] [CrossRef]
- Raugei, G.; Ramponi, G.; Chiarugi, P. Low Molecular Weight Protein Tyrosine Phosphatases: Small, but Smart. Cell. Mol. Life Sci. CMLS 2002, 59, 941–949. [Google Scholar] [CrossRef]
- Salomone-Stagni, M.; Musiani, F.; Benini, S. Characterization and 1.57 Å Resolution Structure of the Key Fire Blight Phosphatase AmsI from Erwinia Amylovora. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 903–910. [Google Scholar] [CrossRef]
- Fauman, E.B.; Saper, M.A. Structure and Function of the Protein Tyrosine Phosphatases. Trends Biochem. Sci. 1996, 21, 413–417. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R. Low Molecular Weight Phosphotyrosine Protein Phosphatases as Emerging Targets for the Design of Novel Therapeutic Agents. J. Med. Chem. 2012, 55, 2–22. [Google Scholar] [CrossRef]
- Tautz, L.; Critton, D.A.; Grotegut, S. Protein Tyrosine Phosphatases: Structure, Function, and Implication in Human Disease. In Phosphatase Modulators; Millán, J.L., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; pp. 179–221. ISBN 978-1-62703-562-0. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Banerjee, R.; Sen, U. Atomic Resolution Crystal Structure of VcLMWPTP-1 from Vibrio Cholerae O395: Insights into a Novel Mode of Dimerization in the Low Molecular Weight Protein Tyrosine Phosphatase Family. Biochem. Biophys. Res. Commun. 2014, 450, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Stanford, S.M.; Aleshin, A.E.; Zhang, V.; Ardecky, R.J.; Hedrick, M.P.; Zou, J.; Ganji, S.R.; Bliss, M.R.; Yamamoto, F.; Bobkov, A.A.; et al. Diabetes Reversal by Inhibition of the Low-Molecular-Weight Tyrosine Phosphatase. Nat. Chem. Biol. 2017, 13, 624–632. [Google Scholar] [CrossRef]
- PubChem 3[N-Morpholino]Propane Sulfonic Acid. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/70807 (accessed on 9 August 2023).
- PubChem 2-(N-Morpholino)-Ethanesulfonic Acid. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/78165 (accessed on 9 August 2023).
- PubChem 4-(2-Hydroxyethyl)-1-Piperazine Ethanesulfonic Acid. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/23831 (accessed on 9 August 2023).
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK Prediction and the Preparation of Biomolecular Structures for Atomistic Molecular Modeling and Simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- He, R.; Wang, J.; Yu, Z.-H.; Zhang, R.-Y.; Liu, S.; Wu, L.; Zhang, Z.-Y. Inhibition of Low Molecular Weight Protein Tyrosine Phosphatase by an Induced-Fit Mechanism. J. Med. Chem. 2016, 59, 9094–9106. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Madhurantakam, C.; Rajakumara, E.; Mazumdar, P.A.; Saha, B.; Mitra, D.; Wiker, H.G.; Sankaranarayanan, R.; Das, A.K. Crystal Structure of Low-Molecular-Weight Protein Tyrosine Phosphatase from Mycobacterium Tuberculosis at 1.9-Å Resolution. J. Bacteriol. 2005, 187, 2175–2181. [Google Scholar] [CrossRef]
- Ku, B.; Keum, C.W.; Lee, H.S.; Yun, H.-Y.; Shin, H.-C.; Kim, B.Y.; Kim, S.J. Crystal Structure of SP-PTP, a Low Molecular Weight Protein Tyrosine Phosphatase from Streptococcus Pyogenes. Biochem. Biophys. Res. Commun. 2016, 478, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Prlić, A.; Bliven, S.; Rose, P.W.; Bluhm, W.F.; Bizon, C.; Godzik, A.; Bourne, P.E. Pre-Calculated Protein Structure Alignments at the RCSB PDB Website. Bioinformatics 2010, 26, 2983–2985. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Benini, S.; Caputi, L.; Cianci, M. Cloning, Purification, Crystallization and 1.57 Å Resolution X-Ray Data Analysis of AmsI, the Tyrosine Phosphatase Controlling Amylovoran Biosynthesis in the Plant Pathogen Erwinia Amylovora. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 1693–1696. [Google Scholar] [CrossRef] [PubMed]
- Dümmler, A.; Lawrence, A.-M.; de Marco, A. Simplified Screening for the Detection of Soluble Fusion Constructs Expressed in E. Coli Using a Modular Set of Vectors. Microb. Cell Factories 2005, 4, 34. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. Available online: https://www.embopress.org/doi/full/10.1038/msb.2011.75 (accessed on 6 July 2023). [CrossRef]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
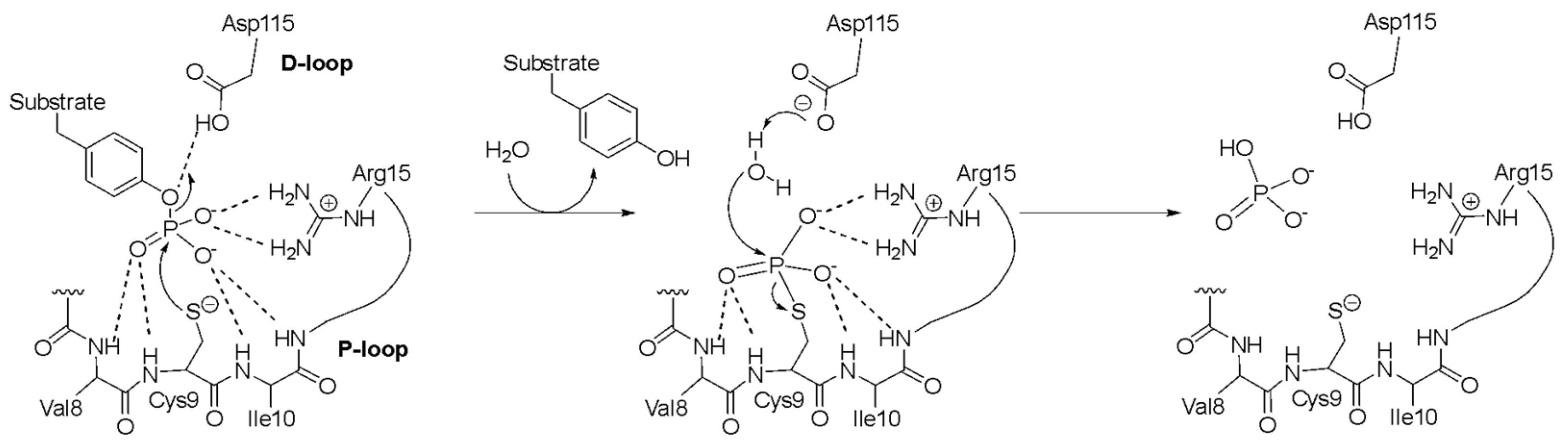{kind=link}
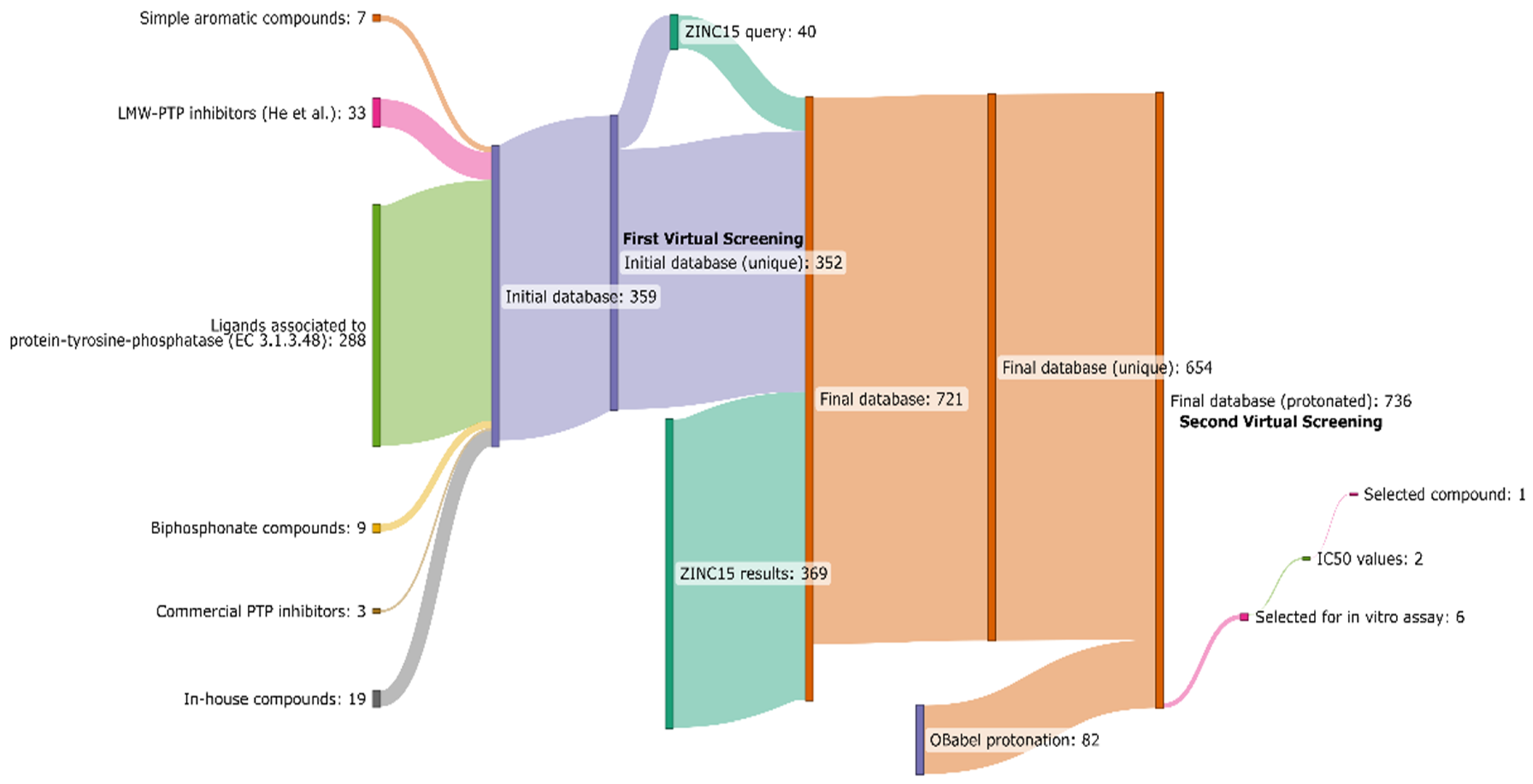{kind=link}
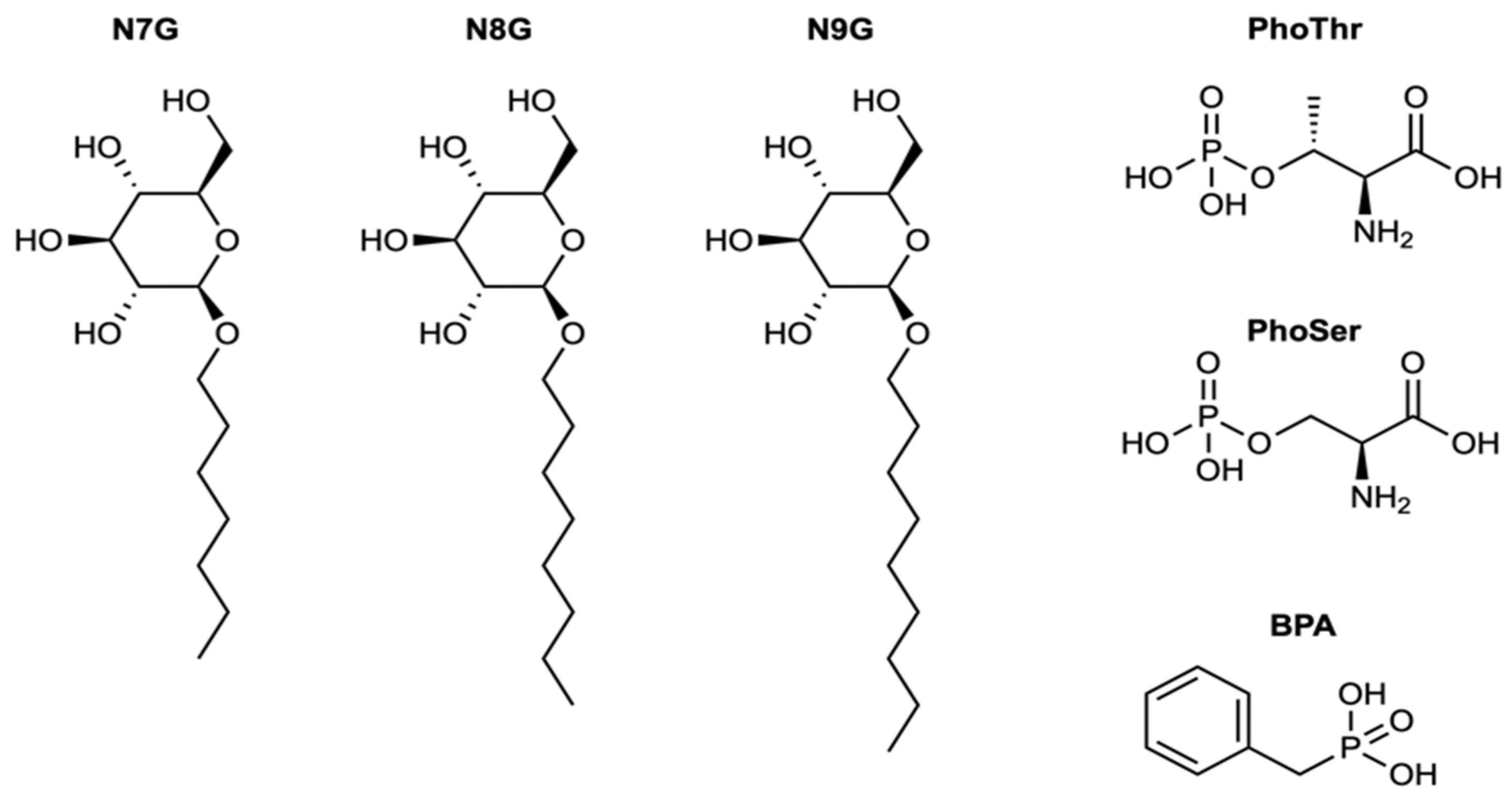
| Name | IUPAC Name | DOCK6 Score | FRED Score | LogP |
|---|---|---|---|---|
| BPA | benzylphosphonic acid | −26.72 | −10.11 | 0.55 |
| PhoThr | (2S,3R)-2-amino-3-phosphonooxybutanoic acid | −26.42 | −10.97 | −1.33 |
| PhoSer | (2S)-2-amino-3-(phosphonooxy)propanoic acid | −27.38 | −9.29 | −1.75 |
| N7G | (2R,3R,4S,5S,6R)-2-(heptyloxy)-6- (hydroxymethyl)oxane-3,4,5-triol | −33.50 | −4.66 | 0.37 |
| N8G | (2R,3S,4S,5R,6R)-2-(hydroxymethyl)-6- (octyloxy)oxane-3,4,5-triol | −33.22 | −4.26 | 0.81 |
| N9G | (2R,3S,4S,5R,6R)-2-(hydroxymethyl)-6- (nonyloxy)oxane-3,4,5-triol | −32.81 | −4.31 | 1.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albani, S.; Polsinelli, I.; Mazzei, L.; Musiani, F.; Benini, S. Determination and Kinetic Characterization of a New Potential Inhibitor for AmsI Protein Tyrosine Phosphatase from the Apple Pathogen Erwinia amylovora. Molecules 2023, 28, 7774. https://doi.org/10.3390/molecules28237774
Albani S, Polsinelli I, Mazzei L, Musiani F, Benini S. Determination and Kinetic Characterization of a New Potential Inhibitor for AmsI Protein Tyrosine Phosphatase from the Apple Pathogen Erwinia amylovora. Molecules. 2023; 28(23):7774. https://doi.org/10.3390/molecules28237774
Chicago/Turabian StyleAlbani, Simone, Ivan Polsinelli, Luca Mazzei, Francesco Musiani, and Stefano Benini. 2023. "Determination and Kinetic Characterization of a New Potential Inhibitor for AmsI Protein Tyrosine Phosphatase from the Apple Pathogen Erwinia amylovora" Molecules 28, no. 23: 7774. https://doi.org/10.3390/molecules28237774
APA StyleAlbani, S., Polsinelli, I., Mazzei, L., Musiani, F., & Benini, S. (2023). Determination and Kinetic Characterization of a New Potential Inhibitor for AmsI Protein Tyrosine Phosphatase from the Apple Pathogen Erwinia amylovora. Molecules, 28(23), 7774. https://doi.org/10.3390/molecules28237774