

Preparation of Thin Films Containing Modified Hydroxyapatite Particles and Phospholipids (DPPC) for Improved Properties of Biomaterials
 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of HA
2.2. HA Characterization
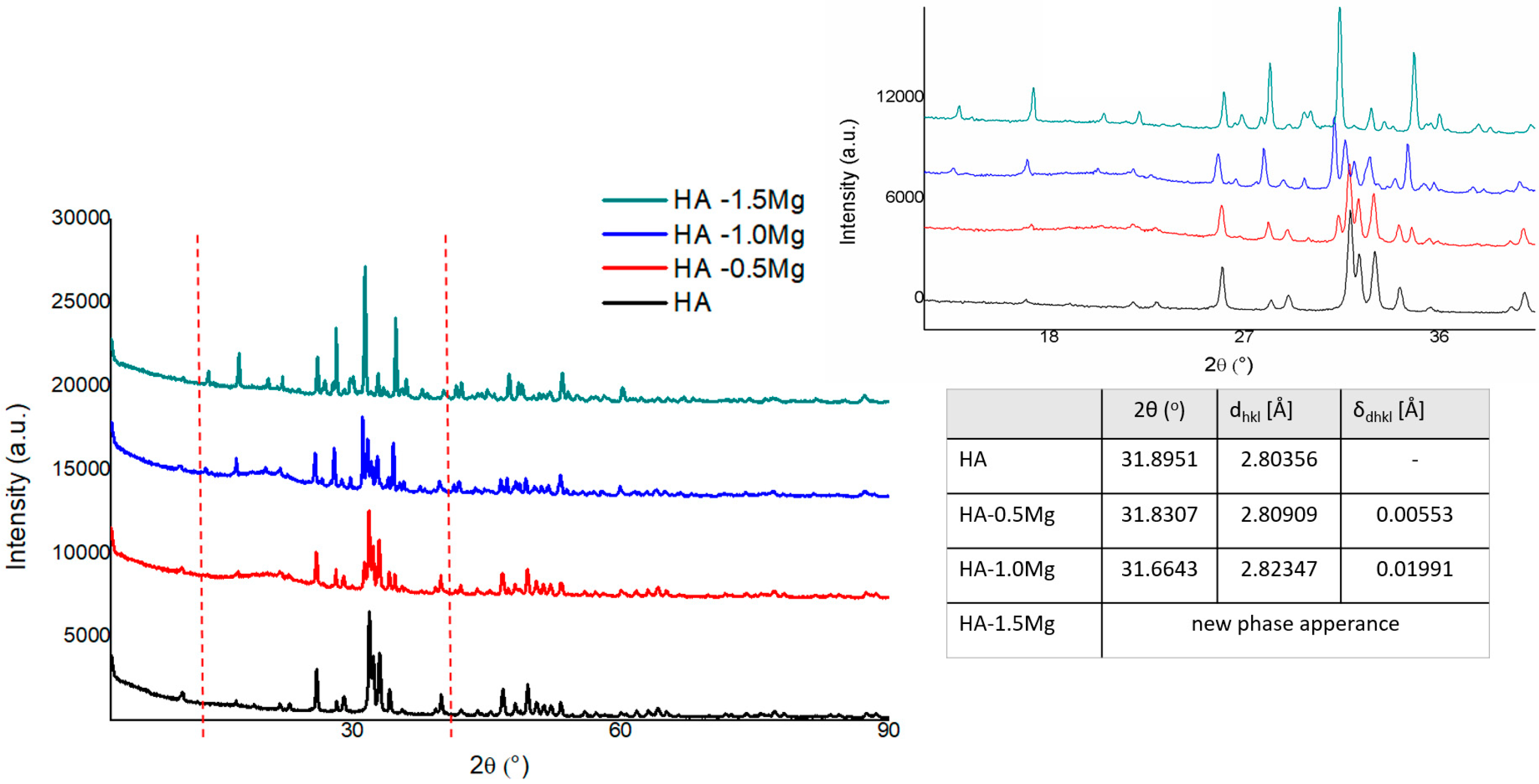
2.2.1. XRD Analysis
2.2.2. FT-IR Analysis
2.2.3. SEM Analysis
2.3. The π–A Isotherm
3. Materials and Methods
3.1. Substances
3.2. Sample Characterization
3.2.1. XRD Analysis
3.2.2. Fourier-Transform Infrared Spectroscopy
3.2.3. Scanning Electron Microscopy/Energy-Dispersive Spectroscopy
3.2.4. Dynamic Light Scattering Method (DLS)
3.3. Registration of π–A Isotherms
3.3.1. π–A Isotherm of Modified HA Microparticles
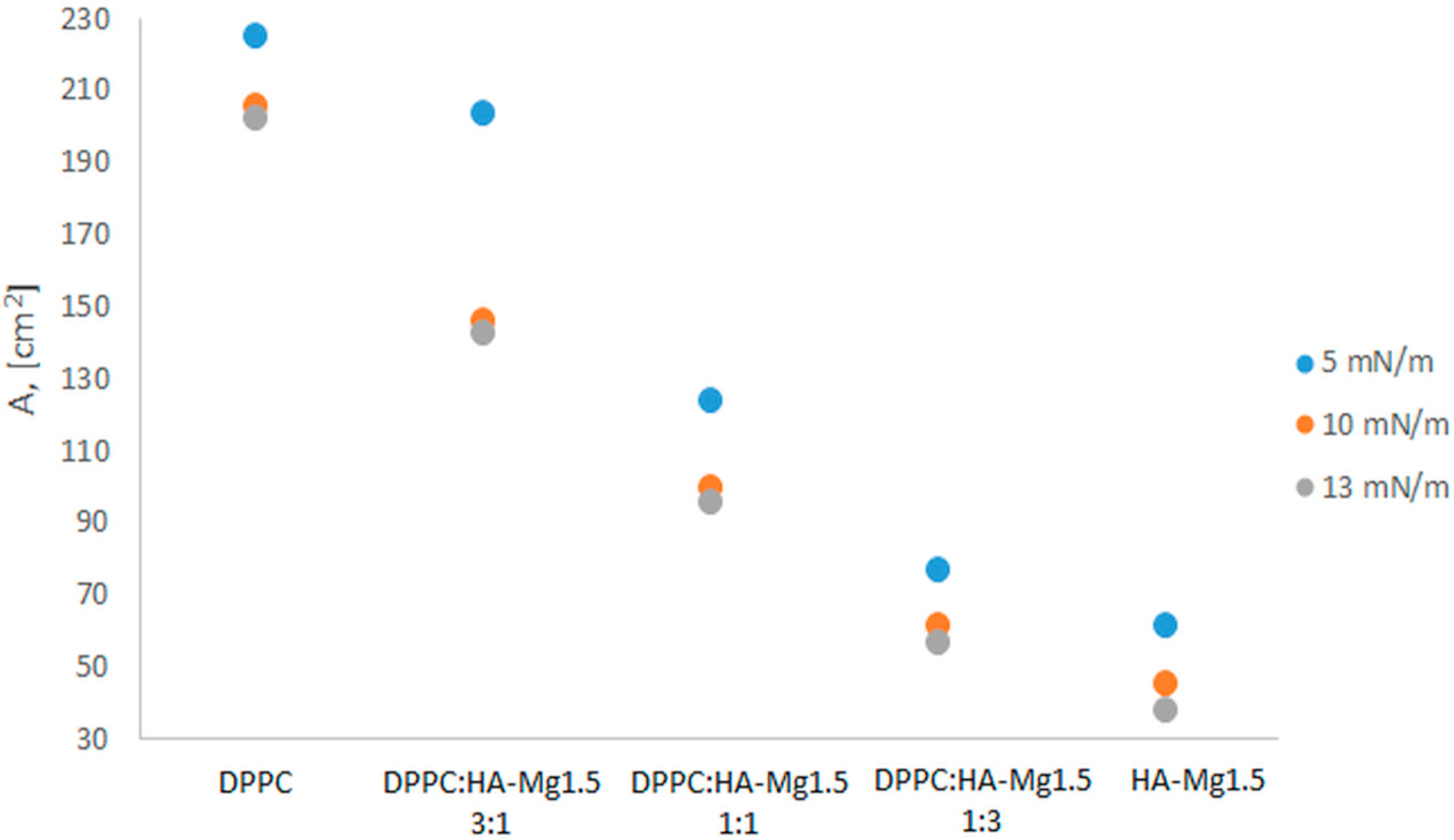
3.3.2. π–A Isotherm of Films Containing HA Microparticles and DPPC
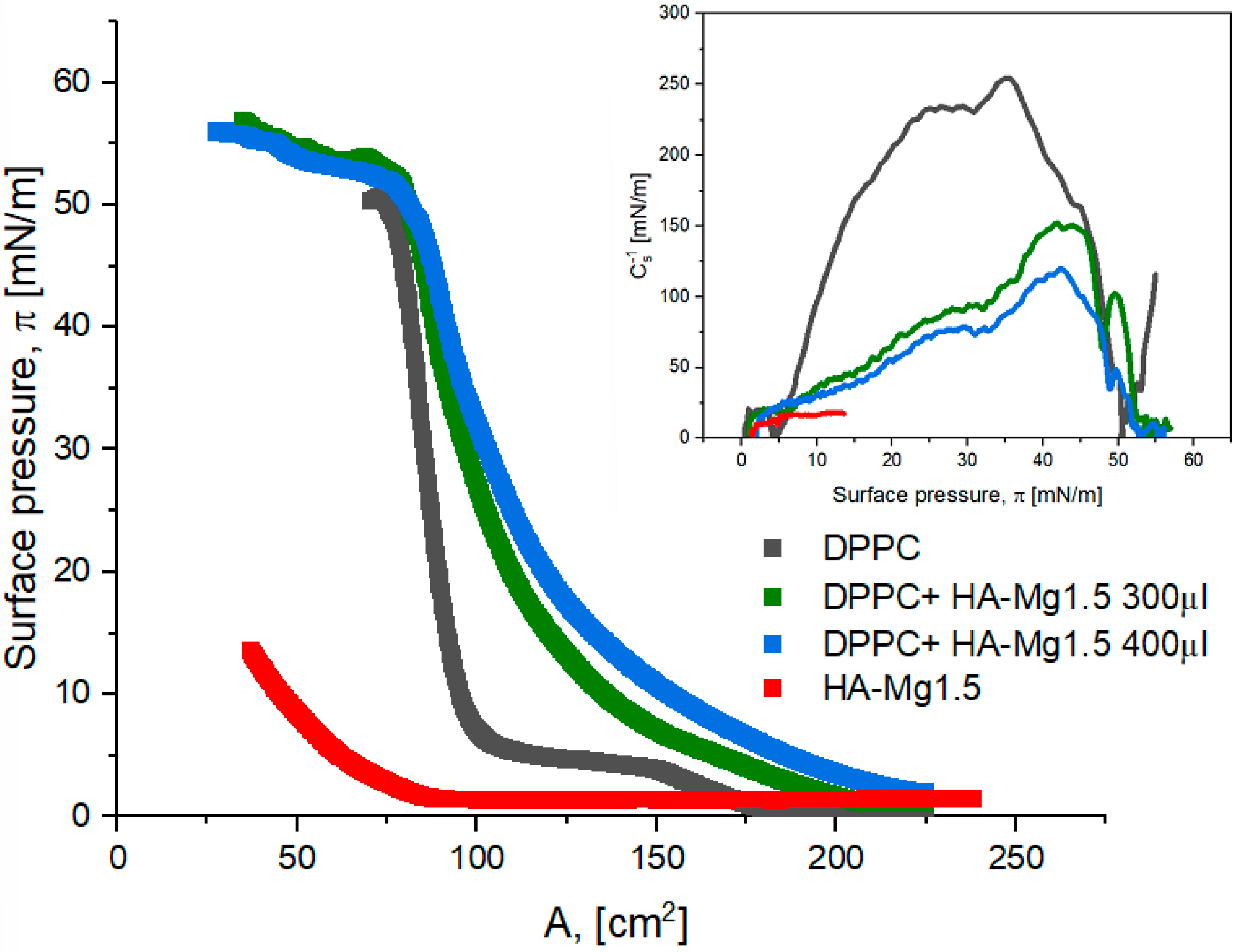
3.3.3. π–A Isotherm of DPPC Monolayer and Deposited HA Dispersion
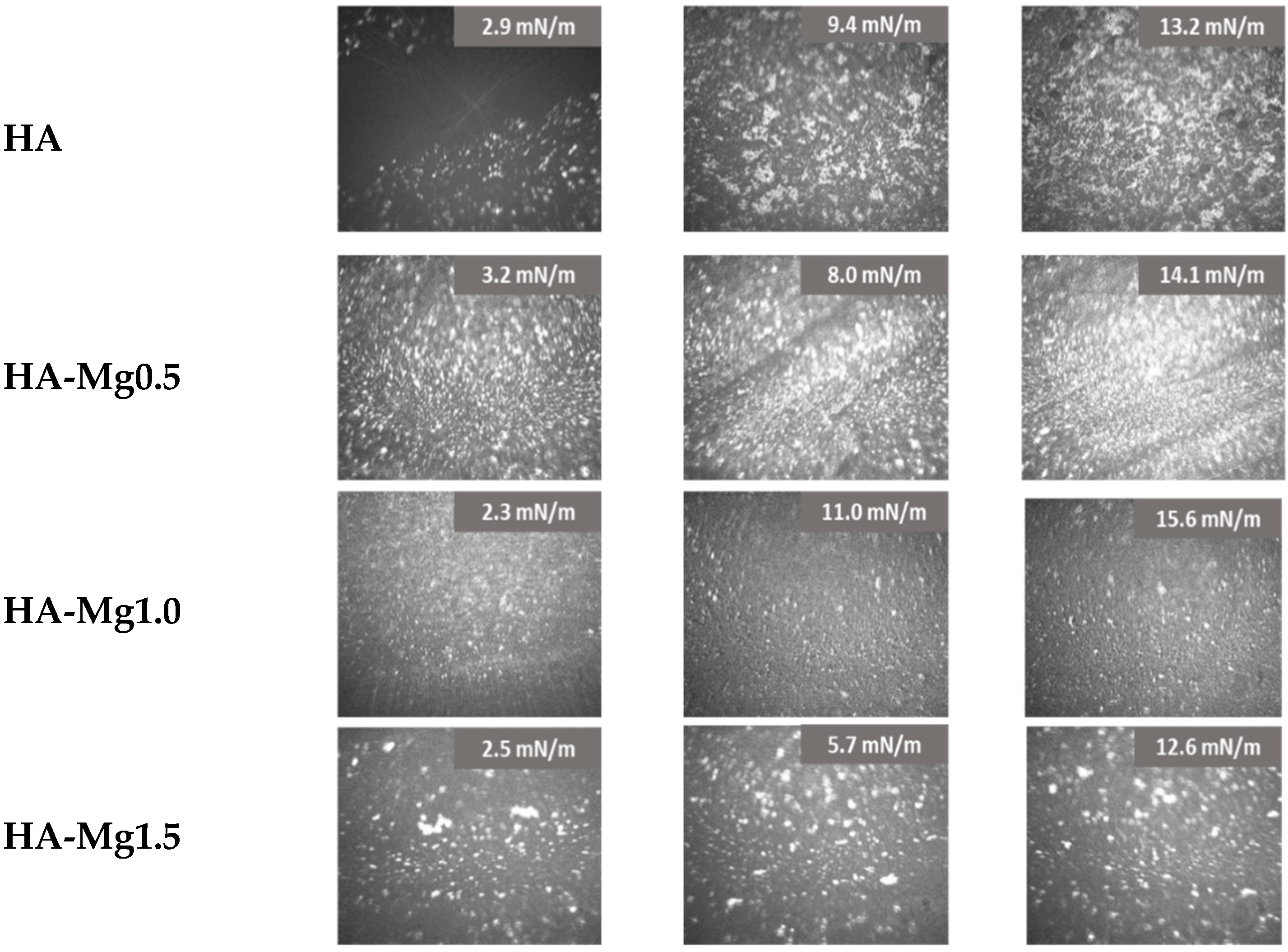
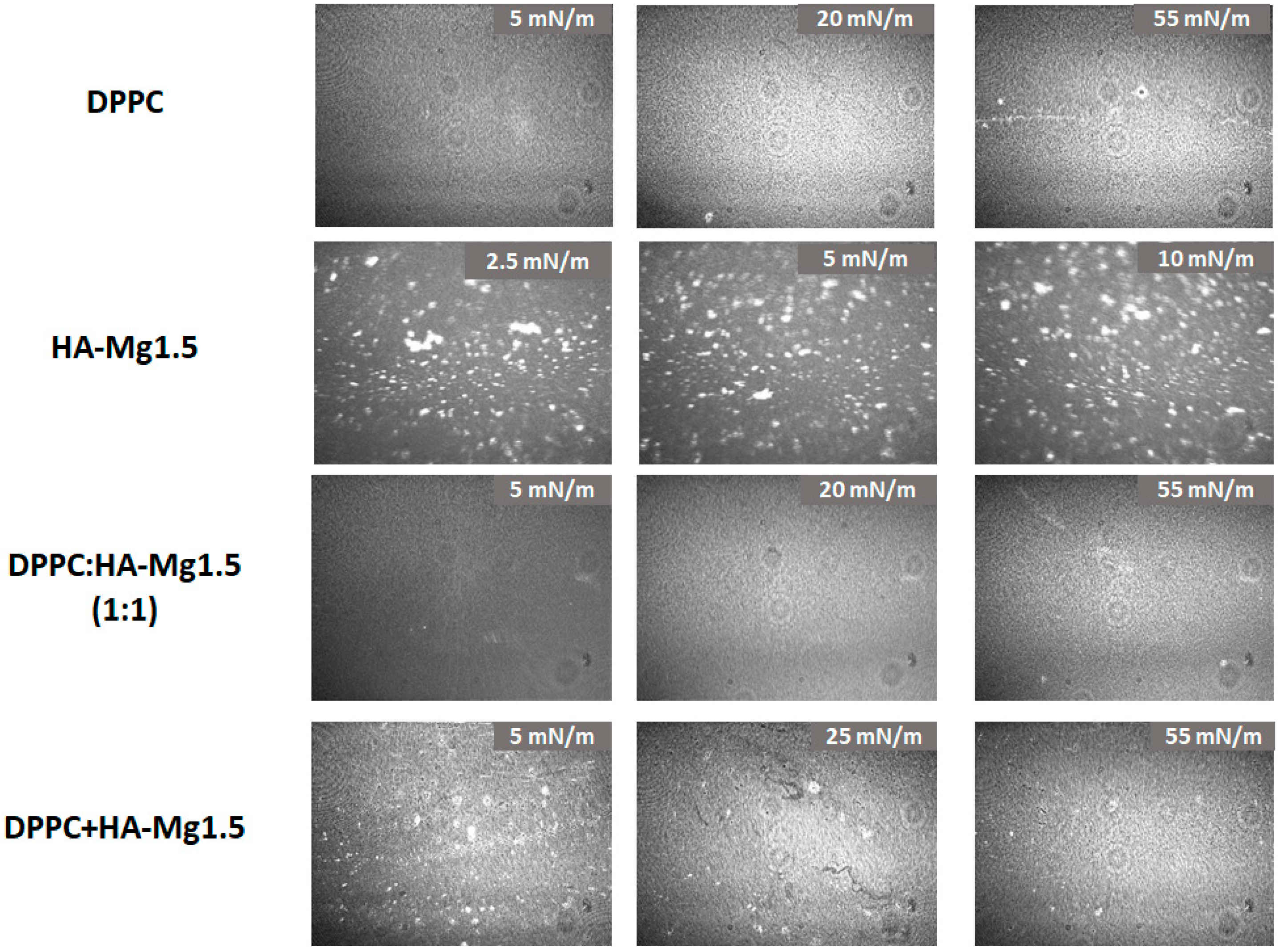
3.4. Brewster Angle Microscopy (BAM)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- He, M.; Zhu, L.; Yang, N.; Li, H.; Yang, Q. Recent advances of oral film as platform for drug delivery. Int. J. Pharm. 2021, 604, 120759. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Liu, X.; Jiang, X.; Lian, R.; Du, B.; Rogachev, A.V. Studies of magnesium—Hydroxyapatite micro/nano film for drug sustained release. Appl. Surf. Sci. 2021, 565, 150598. [Google Scholar] [CrossRef]
- Meyer, F.; Enax, J. Hydroxyapatite in Oral Biofilm Management. Eur. J. Dent. 2019, 13, 287–290. [Google Scholar] [CrossRef]
- Izzetti, R.; Gennai, S.; Nisi, M.; Gulia, F.; Miceli, M.; Gucia, M.R. Clinical Applications of Nano-Hydroxyapatite in Dentistry. Appl. Sci. 2022, 12, 10762. [Google Scholar] [CrossRef]
- Balhuc, S.; Campian, R.; Labunet, A.; Negucioiu, M.; Buduru, S.; Kui, A. Dental Applications of Systems Based on Hydroxyapatite Nanoparticles—An Evidence-Based Update. Crystals 2021, 11, 674. [Google Scholar] [CrossRef]
- Ielo, I.; Calabrese, G.; De Luca, G.; Conoci, S. Recent Advances in Hydroxyapatite-Based Biocomposites for Bone Tissue Regeneration in Orthopedics. Int. J. Mol. Sci. 2022, 23, 9721. [Google Scholar] [CrossRef] [PubMed]
- Farzadi, A.; Solati-Hashjin, M.; Bakhshi, F.; Aminian, A. Synthesis and characterization of hydroxyapatite/β-tricalcium phosphate nanocompo- sites using microwave irradiation. Ceram. Int. 2011, 37, 65–71. [Google Scholar] [CrossRef]
- Rabiei, A.; Blalock, T.; Thomas, B.; Cuomo, J.; Yang, Y.; Ong, J. Microstructure, mechanical properties, and biological response to functionally graded HA coatings. Mater. Sci. Eng. C 2007, 27, 529–533. [Google Scholar] [CrossRef]
- Du, M.; Chen, J.; Liu, K.; Xing, H.; Song, C. Recent advances in biomedical engineering of nano-hydroxyapatite including dentistry, cancer treatment and bone repair. Compos. Part B Eng. 2021, 215, 108790. [Google Scholar] [CrossRef]
- Pelin, I.M.; Maier, S.S.; Chitanu, G.C.; Bulacovschi, V. Preparation and characterization of a hydroxyapatite–collagen composite as component for injectable bone substitute. Mater. Sci. Eng. C 2009, 29, 2188–2194. [Google Scholar] [CrossRef]
- O’Hare, P.; Meenan, B.J.; Burke, G.A.; Byrne, G.; Dowling, D.; Hunt, J.A. Biological responses to hydroxyapatite surfaces deposited via a co-incident microblasting technique. Biomaterials 2010, 31, 515–522. [Google Scholar] [CrossRef]
- Syazwan, M.N.M.; Marliana, B.I.Y. The influence of simultaneous divalent cations (Mg2+, Co2+ and Sr2+) substitution on the physico-chemical properties of carbonated hydroxyapatite. Ceram. Int. 2019, 45, 14783–14788. [Google Scholar] [CrossRef]
- Bodhak, S.; Bose, S.; Bandyopadhyay, A. Bone Cell-Materials Interactions on Metal-Ion Doped Polarized Hydroxyapatite. Mater. Sci. Eng. C 2011, 31, 755–761. [Google Scholar] [CrossRef]
- Choi, S.; Kim, K.-J.; Cheon, S.; Kim, E.-M.; Kim, Y.-A.; Park, C.; Kim, K.K. Biochemical activity of magnesium ions on human osteoblast migration. Biochem. Bioph. Res. Commun. 2020, 531, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rath, B.; Tingart, M.; Eschweiler, J. Role of implants surface modification in osseointegration: A systematic review. J. Biomed. Mater Res. A 2020, 108, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, G.; Li, J.J. Advances in implant surface modifications to improve osseointegration. Mater. Adv. 2021, 2, 6901–6927. [Google Scholar] [CrossRef]
- Nobles, K.P.; Janorkar, A.V.; Williamson, R.S. Surface modifications to enhance osseointegration-Resulting material properties and biological responses. J. Biomed. Mater Res. B Appl. Biomater. 2021, 109, 1909–1923. [Google Scholar] [CrossRef]
- Hu, C.; Ashok, D.; Nisbet, D.R.; Gautam, V. Bioinspired surface modification of orthopedic implants for bone tissue engineering. Biomaterials 2019, 219, 119366. [Google Scholar] [CrossRef]
- Rupp, F.; Liang, L.; Geis-Gerstorfer, J.; Scheideler, L.; Hüttig, F. Surface characteristics of dental implants: A review. Dent. Mater. 2018, 34, 40–57. [Google Scholar] [CrossRef]
- Gittens, R.A.; McLachlan, T.; Olivares-Navarrete, R.; Cai, Y.; Berner, S.; Tannenbaum, R.; Schwartz, Z.; Sandhage, K.H.; Boyan, B.D. The effects of combined micron-/submicron-scale surface roughness and nanoscale features on cell proliferation and differentiation. Biomaterials 2011, 32, 3395–3403. [Google Scholar] [CrossRef]
- Lee, E.M.; Smith, K.; Gall, K.; Boyan, B.D.; Schwartz, Z. Change in surface roughness by dynamic shape-memory acrylate networks enhances osteoblast differentiation. Biomaterials 2016, 110, 34–44. [Google Scholar] [CrossRef]
- Joshi, R.; Akram, W.; Chauhan, R.; Garud, N. Thin Films: A Promising Approach for Drug Delivery System. In Drug Carriers; Villarreal-Gómez, L.J., Ed.; Intech Open: London, UK, 2022. [Google Scholar] [CrossRef]
- Russo, E.; Selmin, F.; Baldassari, S.; Gennari, C.G.M.; Caviglioli, G.; Cilurzo, F.; Minghetti, P.; Parodi, B. A focus on mucoadhesive polymers and their application in buccal dosage forms. J. Drug Deliv. Sci. Technol. 2015, 32, 113–125. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, N.; Sharma, P.K. Buccal film: An advance technology for oral drug delivery. Adv. Biol. Res. 2014, 8, 260–267. [Google Scholar]
- Crowley, M.M.; Zhang, F. Pharmaceutical applications of hot-melt extrusion: Part I. Drug Dev. Ind. Pharm. 2007, 33, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Preis, M.; Breitkreutz, J.; Sandler, N. Perspective: Concepts of printing technologies for oral film formulations. Int. J. Pharm. 2015, 494, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Dey, B.; Bhattacharjee, D.; Mehta, N. Unique supramolecular assembly through Langmuir Blodgett (LB) technique. Heliyon 2018, 4, e01038. [Google Scholar] [CrossRef]
- Bodik, M.; Jergel, M.; Majkova, E.; Siffalovic, P. Langmuir films of low-dimensional nanomaterials. Adv. Colloid Interface Sci. 2020, 283, 102239. [Google Scholar] [CrossRef] [PubMed]
- Quan, P.; Bu, W.; Wang, L.; Chen, C.; Wu, X.; Heffern, C.; Lee, K.Y.C.; Meron, M.; Lin, B. Structure of polymer-capped gold nanorods binding to model phospholipid monolayers. J. Phys. Mater. 2021, 4, 034004. [Google Scholar] [CrossRef]
- Schöne, A.-C.; Roch, T.; Schulz, B.; Lendlein, A. Evaluating polymeric biomaterial–environment interfaces by Langmuir monolayer techniques. J. R. Soc. Interface 2017, 14, 20161028. [Google Scholar] [CrossRef]
- Dopierała, K.; Krajewska, M.; Prochaska, K. Study on PH-Dependent Interactions of Linoleic Acid with α-Lactalbumin. Food Hydrocoll. 2021, 111, 106217. [Google Scholar] [CrossRef]
- Wydro, P.; Krajewska, B.; Hac-Wydro, K. Chitosan as a Lipid Binder: A Langmuir Monolayer Study of Chitosan-Lipid Interactions. Biomacromolecules 2007, 8, 2611–2617. [Google Scholar] [CrossRef]
- Ruiz, G.C.M.; Cruz, M.A.E.; Faria, A.N.; Zancanela, D.C.; Ciancaglini, P.; Ramos, A.P. Biomimetic Collagen/Phospholipid Coatings Improve Formation of Hydroxyapatite Nanoparticles on Titanium. Mater. Sci. Eng. C 2017, 77, 102–110. [Google Scholar] [CrossRef]
- Broniatowski, M.; Bojarski, J.; Wydro, P. Langmuir Monolayers as Models of the Lipid Matrix of Cyanobacterial Thylakoid Membranes. J. Mol. Liq. 2022, 368, 120727. [Google Scholar] [CrossRef]
- Oliveira, O.N.O., Jr.; Caseli, L.; Ariga, K. The Past and the Future of Langmuir and Langmuir–Blodgett Films. Chem. Rev. 2022, 122, 6459–6513. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Yoon, I.; Hubble, D.; Tran, T.; Kostecki, R.; Liu, G. Recent Applications of Langmuir-Blodgett Technique in Battery Research. ACS Appl. Mater. Interfaces 2002, 14, 2431–2439. [Google Scholar] [CrossRef] [PubMed]
- Corrales-Ureña, Y.R.; Souza-Schiaber, Z.; Noronha, P.; Filho, L.; Marquenet, F.; Noeske, P.M.; Gätjen, L.; Rischka, K. Functionalization of hydrophobic surfaces with antimicrobial peptides immobilized on a bio-interfactant layer. RSC Adv. 2019, 10, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, M.; Tim, B.; Prochaska, K. Interactions between silica particles and model phospholipid monolayers. J. Mol. Liquids 2022, 345, 116999. [Google Scholar] [CrossRef]
- Tim, B.; Rojewska, M.; Prochaska, K. Effect of Silica Microparticles on Interactions in Mono- and Multicomponent Membranes. Int. J. Mol. Sci. 2022, 23, 12822. [Google Scholar] [CrossRef]
- Sheridan, A.J.; Thompson, K.C.; Slater, J.M. Interaction of negatively and positively capped gold nanoparticle with different lipid model membranes. Biophys. Chem. 2022, 290, 106896. [Google Scholar] [CrossRef] [PubMed]
- Alioui, H.; Bouras, O.; Bollinger, J.C. Toward an efficient antibacterial agent: Zn- and Mg-doped hydroxyapatite nanopowders. J. Environ. Sci. Health Part A 2019, 54, 315–327. [Google Scholar] [CrossRef]
- Dubey, A.; Jaiswal, S.; Garg, A.; Jain, V.; Lahiri, D. Synthesis and evaluation of magnesium/co-precipitated hydroxyapatite based composite for biomedical application. J. Mech. Behav. Biomed. Mater. 2021, 118, 104460. [Google Scholar] [CrossRef]
- Farzadi, A.; Bakhshi, F.; Solati-Hashjin, M.; Asadi-Eydivand, M.; Osman, N.A.A. Magnesium incorporated hydroxyapatite: Synthesis and structural properties characterization. Ceram. Int. 2014, 40, 6021–6029. [Google Scholar] [CrossRef]
- Trusca, R.; Predoi, D. Influence of annealing treatment of nano-hydroxyapatite bioceramics on the vibrational properties. Dig. J. Nanomater. Biostructures 2011, 6, 609–624. [Google Scholar]
- Costescu, A.; Pasuk, I.; Ungureanu, F.; Dinischiotu, A.; Huneau, F.; Galaup, S. Physico-chemical properties of nano-sized hexagonal hydroxyapatite powder synthesized by sol-gel. Dig. J. Nanomater. Biostructures 2010, 5, 989–1000. [Google Scholar]
- Panda, R.N.; Hsieh, M.F.; Chung, R.J.; Chin, T.S. FTIR, XRD, SEM and solid state NMR investigations of synthesized by hydroxide-gel technique. J. Phys. Chem. Solids. 2003, 64, 193–199. [Google Scholar] [CrossRef]
- Gopi, D.; Nithiya, S.; Shinyjoy, E.; Kavitha, L. Spectroscopic investigation on formation and growth of mineralized nanohydroxyapatite for bone tissue engineering applications. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 92, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Wojciech, L.; Suchanek, L.; Byrappa, K.; Shuk, P.; Riman, R.E.; Janas, V.F.; TenHuisen, K.S. Preparation of magnesium-substituted hydroxyapatite powders by the mechanochemical–hydrothermal method. Biomaterials 2004, 25, 4647–4657. [Google Scholar] [CrossRef]
- Mróz, W.; Bombalska, A.; Burdyńska, S.; Jedyński, M.; Prokopiuk, A.; Budner, B.; Ślósarczyk, A.; Zima, A.; Menaszek, E.; Ścisłowska-Czarnecka, A.; et al. Structural studies of magnesium doped hydroxyapatite coatings after osteoblast culture. J. Mol. Struct. 2010, 977, 145–152. [Google Scholar] [CrossRef]
- Cox, S.C.; Jamshidi, P.; Grover, L.M.; Mallick, K.K. Preparation and characterisation of nanophase Sr, Mg, and Zn substituted hydroxyapatite by aqueous precipitation. Mater. Sci. Eng. C Mater Biol. Appl. 2014, 35, 106–114. [Google Scholar] [CrossRef]
- Jurak, M.; Szafran, K.; Cea, P.; Martin, S. Analysis of Molecular Interactions between Components in Phospholipid-Immunosuppressant-Antioxidant Mixed Langmuir Films. Langmuir 2021, 37, 5601–5616. [Google Scholar] [CrossRef]
- Davies, J.T.; Rideal, E.K. Interfacial Phenomena, 2nd ed.; Academic Press: New York, NY, USA, 1963; p. 265. [Google Scholar]
- Guzmán, E.; Liggieri, L.; Santini, E.; Ferrari, M.; Ravera, F. Influence of silica nanoparticles on phase behavior and structural properties of DPPC-Palmitic acid Langmuir monolayers. Colloid. Surf. A 2012, 413, 280–287. [Google Scholar] [CrossRef]
- Pereira, A.R.; de Oliveira Junior, O.N. Recent advances in the use of Langmuir monolayers as cell membrane models. Eclét. Quím. 2021, 46, 18–29. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ca n (mmol) | P n (mmol) | Mg n (mmol) | Final Chemical Formula |
|---|---|---|---|---|
| HA | 100 | 60 | 0 | |
| HA-Mg0.5 | 95 | 60 | 5 | |
| HA-Mg1.0 | 90 | 60 | 10 | |
| HA-Mg1.5 | 85 | 60 | 15 |
| Sample | Ca | P | Mg (Ex) * | Mg (T) * | (Ca + Mg)/P |
|---|---|---|---|---|---|
| HA-Mg0.5 | 37.6 ± 2.5 | 18.8 ± 1.1 | 1.0 ± 0.1 | 1.2 | 2.05 |
| HA-Mg1.0 | 36.4 ± 1.7 | 20.5 ± 1.3 | 2.4 ± 0.16 | 2.4 | 1.89 |
| HA-Mg1.5 | 33.9 ± 0.8 | 22.7 ± 1.1 | 4.3 ± 0.6 | 3.6 | 1.68 |
| System | (mN/m) | (mN/m) | (cm2) | (cm2) | max. Cs−1 (mN/m) |
|---|---|---|---|---|---|
| HA | 1.6 | 13.6 | 26.6 | 44.7 | 26.4 |
| HA-Mg0.5 | 1.2 | 20.1 | 26.0 | 75.8 | 21.0 |
| HA-Mg1.0 | 0.6 | 16.8 | 44.5 | 125.4 | 22.0 |
| HA-Mg1.5 | 1.4 | 13.5 | 39.3 | 85.8 | 18.2 |
| System (50 µL) | (mN/m) | (cm2) | Alift off (cm2) | max. Cs−1 (mN/m) |
|---|---|---|---|---|
| DPPC | 56.8 | 161.7 | 225.0 | 292.9 |
| DPPC: HA-Mg1.5 1:1 | 50.3 | 61.5 | 174.7 | 212.9 |
| DPPC: HA-Mg1.5 1:3 | 49.2 | 29.1 | 225.0 | 128.6 |
| DPPC: HA-Mg1.5 3:1 | 54.6 | 116.4 | 107.6 | 299.4 |
| HA-Mg1.5 (400 µL) | ) 13.5 | (Amax) 39.3 | 85.8 | 18.2 |
| System | (mN/m) | (cm2) | Alift off (cm2) | Cs−1 (mN/m) |
|---|---|---|---|---|
| DPPC 25 (μL) | 57.3 | 59.0 | 174.3 | 254.6 |
| DPPC + HA-Mg1.5 300 (μL) | 56.9 | 34.7 | 225.0 | 152.0 |
| DPPC + HA-Mg1.5 400 (μL) | 56.0 | 27.4 | 230.0 | 119.5 |
| HA-Mg1.5 400 (μL) | ) 13.5 | (Amax) 39.3 | 85.8 | 18.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojewska, M.; Adamska, K.; Kurnatowska, J.; Miklaszewski, A.; Bartkowska, A.; Prochaska, K. Preparation of Thin Films Containing Modified Hydroxyapatite Particles and Phospholipids (DPPC) for Improved Properties of Biomaterials. Molecules 2023, 28, 7843. https://doi.org/10.3390/molecules28237843
Rojewska M, Adamska K, Kurnatowska J, Miklaszewski A, Bartkowska A, Prochaska K. Preparation of Thin Films Containing Modified Hydroxyapatite Particles and Phospholipids (DPPC) for Improved Properties of Biomaterials. Molecules. 2023; 28(23):7843. https://doi.org/10.3390/molecules28237843
Chicago/Turabian StyleRojewska, Monika, Katarzyna Adamska, Justyna Kurnatowska, Andrzej Miklaszewski, Aneta Bartkowska, and Krystyna Prochaska. 2023. "Preparation of Thin Films Containing Modified Hydroxyapatite Particles and Phospholipids (DPPC) for Improved Properties of Biomaterials" Molecules 28, no. 23: 7843. https://doi.org/10.3390/molecules28237843
APA StyleRojewska, M., Adamska, K., Kurnatowska, J., Miklaszewski, A., Bartkowska, A., & Prochaska, K. (2023). Preparation of Thin Films Containing Modified Hydroxyapatite Particles and Phospholipids (DPPC) for Improved Properties of Biomaterials. Molecules, 28(23), 7843. https://doi.org/10.3390/molecules28237843