

Quantitative Structure–Activity Relationship in the Series of 5-Ethyluridine, N2-Guanine, and 6-Oxopurine Derivatives with Pronounced Anti-Herpetic Activity


Abstract
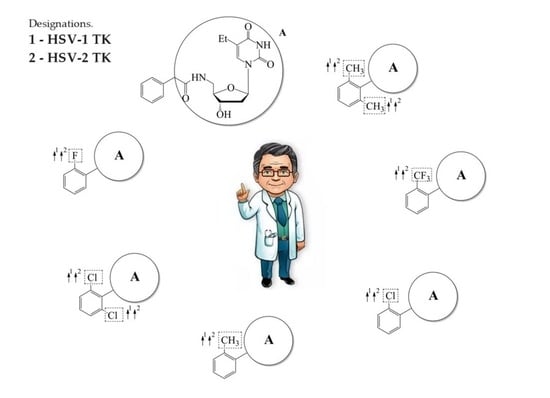
1. Introduction
2. Results
- (1)
- to show that the ideology of descriptor formation and selection implemented in the GUSAR 2019 software is applicable for modeling potential inhibitors of HSV-1 and HSV-2 TK enzymes in the series of 5-ethyluridine, N2-guanine, and 6-oxopurine derivatives;
- (2)
- to develop statistically significant QSAR models suitable for the virtual screening of HSV TK inhibitors.
- (1)
- for the full dataset in each training and test set (100% of data);
- (2)
- for 95% of the data in each training and test set (95% of the data).
- (1)
- numerical values of various coefficients of determination based on R2 (R2, R20, Q2F1, Q2F2, CCC);
- (2)
- numerical values of the MAE prediction error;
- (3)
- the scatter range of activity prediction data taking into account MAE in the mσ (or mSD) range: MAE + 3·SD. All of these parameters were computed using the XternalValidationPlus 1.2 program. In addition, this program was used to trace the systematic error that can arise in QSAR modeling.
3. Discussion
4. Computational Details
4.1. Computational Methodology
4.2. Formation of Training and Test Sets
4.3. Building QSAR Models
4.4. Assessment of Applicability
5. Evaluation of the Quality and Predictive Ability of QSAR Models
5.1. Calculating the pIC50 Values Using the Consensus Approach in the GUSAR 2019 Program
5.2. Statistical Parameters Characterizing the Predictive Ability of QSAR Models
- (1)
- based on coefficients of determination R2 (R2, R20, Q2F1, Q2F2, , CCC);
- (2)
- (1)
- different coefficients of determination, calculated by comparing the experimental data with the calculated pIC50 data contained in each of the training and test sets, respectively, were numerically similar and tended to be 1;
- (2)
- MAE values for predicted pIC50 of compounds of the training or test set, respectively, did not exceed 10% of the range of variation of the experimental pIC50 values for this set;
- (3)
- the following relation held: MAE+3·SDTrS ≤ 0.2·pIC50 TrS, where ΔpIC50 is the range of variation of pIC50 values for the TrS structures (this criterion refers to the assessment of the descriptive ability of the model);
- (4)
- the following relation held: MAE+3·SDTrS ≤ 0.2·pIC50 TrS, where ΔpIC50 is the range of variation of pIC50 values for the TrS structures (the criterion refers to the assessment of the predictive ability of the model).
- (1)
- the numerical values of different coefficients of determination, calculated by comparing the experimental data with calculated pIC50, did not exceed 0.6;
- (2)
- MAE values estimated from the results of comparing the experimental and predicted pIC50 values of compounds of the training or test set, respectively, did not exceed 20% of the range of variation of the experimental pIC50 values in the training set used to build the Mi model;
- (3)
- the following relation held: MAE + 3·SDTrS ≥ 0.25·pIC50 TrS, where ΔpIC50 is the range of variation of pIC50 values for the TrS structures (the criterion refers to the assessment of the descriptive ability of the model);
- (4)
- the following relation held: MAE + 3·SDTS ≥ 0.25·pIC50 TrS, where ΔpIC50 is the range of variation of pIC50 values for the TrS structures (the criterion refers to the assessment of the predictive ability of the model).
5.3. Evaluation of the Contribution of Atoms to the Target Activity
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sachs, S.L.; Straub, S.E.; Griffiths, P.D.; Whitley, R.J. Clinical Management of Herpes Viruses; Sachs, S.L., Straub, S.E., Griffiths, P.D., Whitley, R.J., Eds.; IOS: Washington, DC, USA, 1995; p. 398. [Google Scholar]
- Tenser, R.B. Role of herpes simplex virus thymidine kinase expression in viral pathogenesis and latency. J. Intervirol. 1991, 32, 76–92. [Google Scholar] [CrossRef]
- Jamieson, A.T.; Gentry, G.A.; Subak-Sharp, J.H. Induction of both thymidine and deoxycytidine kinase activity by herpes viruses. J. Gen. Virol. 1974, 24, 465–480. [Google Scholar] [CrossRef]
- Kukhanova, M.K.; Korovina, A.N.; Kochetkov, S.N. Virus prostogo gerpesa cheloveka: Zhiznennyy tsikl i poisk ingibitorov. J. Uspekhi Biol. Him. 2014, 54, 457–494. [Google Scholar]
- Richtin, T.; Black, M.; Mao, F.; Lewis, M.; Drake, R. Purification and Photoaffinity Labeling of Herpes Simplex Virus Type-1 Thymidine Kinase. J. Biol. Chem. 1995, 270, 7055–7060. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Crespillo, A.; Fraile-Ramos, A.; Tabares, E.; Alcina, A.; Lopez-Guerrero, J. Role of the small GTPase Rab27a during Herpes simplex virus infection of oligodendrocytic cells. BMC Microbiol. 2012, 12, 265–278. [Google Scholar] [CrossRef]
- Schuppe, H.C.; Meinhardt, A.; Allam, J.P.; Bergmann, M.; Weidner, W.; Haidl, G. Chronic orchitis: A neglected cause of male infertility? J. Androl. 2008, 40, 84–91. [Google Scholar] [CrossRef]
- Jiang, Y.-C.; Feng, H.; Lin, Y.-C.; Guo, X.-R. New strategies against drug resistance to herpes simplex virus. J. Oral Sci. 2016, 8, 1–6. [Google Scholar] [CrossRef]
- Klein, R.; Czelusniak, S. Effect of a thymidine kinase inhibitor (L-653,180) on antiviral treatment of experimental herpes simplex virus infection in mice. J. Antivir. Res. 1990, 14, 207–214. [Google Scholar] [CrossRef]
- Kim, C.U.; Luh, B.Y.; Misco, P.F.; Bisacchi, G.; Terry, B.; Mansuri, M.M. (2R, 4S, 5S)-1-(tetrahydro-4-hydroxy-5-methoxy-2-furanyl)thymine: A potent selective inhibitor of herpes simplex thymidine kinase. J. Bioorg. Med. Chem. Lett. 1993, 3, 1571–7576. [Google Scholar] [CrossRef]
- Cheng, Y.C. A rational approach to the development of antiviral chemotherapy: Alternative substrates of herpes simplex virus Type 1 (HSV-1) and Type 2 (HSV-2) thymidine kinase (TK). J. Ann. N. Y. Acad. Sci. 1977, 284, 594–598. [Google Scholar] [CrossRef]
- Watkins, A.M.; Dunford, P.J.; Moffatt, A.M.; Wong Kai-In, P.; Holland, M.J.; Pole, D.S.; Thomas, G.J.; Martin, J.A.; Roberts, N.A.; Mulqueen, M.J. Inhibition of virus-encoded thymidine kinase suppresses herpes simplex virus replication in vitro and in vivo. J. Antivir. Chem. Chemother. 1998, 9, 9–18. [Google Scholar]
- Focher, F.; Hildebrand, C.; Freese, S.; Ciarrocchi, G.; Noonan, T.; Sangalli, S.; Brown, N.; Spadari, S.; Wright, G. N2-phenyldeoxyguanosine: A novel selective inhibitor of herpes simplex thymidine kinase. J. Med. Chem. 1988, 31, 1496–1500. [Google Scholar] [CrossRef]
- Manikowski, A.; Lossani, A.; Savi, L.; Maioli, A.; Gambino, J.; Focher, F.; Spadari, S.; Wright, G.E. N2-Phenyl-9-(hydroxyalkyl)guanines and related compounds are substrates for Herpes simplex virus thymidine kinases. J. Mol. Biochem. 2012, 1, 21–25. [Google Scholar]
- Nutter, L.M.; Grill, S.P.; Dutschman, G.E.; Sharma, R.A.; Bobek, M.; Cheng, Y.C. Demonstration of viral thymidine kinase inhibitor and its effect on deoxynucleotide metabolismin cells infected with herpes simplex virus. J. Antimicrob. Agents Chemother. 1987, 31, 368–374. [Google Scholar] [CrossRef]
- Martin, J.A.; Thomas, G.J.; Merrett, J.H.; Lambert, R.W.; Bushnell, D.J.; Dunsdon, S.J.; Freeman, A.C.; Hopkins, R.A.; Johns, I.R.; Keech, E.; et al. The design, synthesis and properties of highly potent and selective inhibitors of herpes simplex virus types 1 and 2 thymidine kinase. J. Antivir. Chem. Chemother. 1998, 9, 1–8. [Google Scholar]
- Martin, J.A.; Lambert, R.W.; Thomas, G.J.; Duncan, I.D.; Hall, M.J.; Merrett, J.H. Nucleoside analogues as highly potent and selective inhibitors of herpes simplex virus thymidine kinase. J. Bioorg. Med. Chem. Lett. 2001, 11, 1655–1658. [Google Scholar] [CrossRef]
- Ferreira, M.M.C. Multivariate QSAR. J. Braz. Chem. Soc. 2002, 13, 742–753. [Google Scholar] [CrossRef]
- Aremenko, N.V.; Baskin, I.I.; Palyulin, V.A.; Zefirov, N.S. Prediction of Physical Properties of Organic Compounds Using Artificial Neural Networks within the Substructure Approach. J. Dokl. Chem. 2001, 381, 317–320. [Google Scholar] [CrossRef]
- Poroikov, V.V. Computer-aided drug design: From discovery of novel pharmaceutical agents to systems pharmacology. J. Biochem. Mosc. Suppl. Ser. B Biomed. Chem. 2020, 14, 216–227. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Rudik, A.V.; Pogodin, P.V.; Savosina, P.I.; Tarasova, O.A.; Dmitriev, A.V.; Ivanov, S.M.; Biziukova, N.Y.; Druzhilovskiy, D.S.; Filimonov, D.A.; et al. CLC-Pred 2.0: A Freely Available Web Application for In Silico Prediction of Human Cell Line Cyto-toxicity and Molecular Mechanisms of Action for Druglike Compounds. Int. J. Mol. Sci. 2023, 24, 1689. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. J. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Lampa, S.; Simeon, S.; Gleeson, M.P.; Spjuth, O.; Nantasenamat, C. Towards reproducible computational drug discovery. J. Cheminform. 2020, 2, 4–30. [Google Scholar] [CrossRef]
- Hartman, G.D.; Egbertson, M.S.; Halczenko, W.; Laswell, W.L.; Duggan, M.E.; Smith, R.L. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J. Med. Chem. 1992, 35, 4640–4642. [Google Scholar] [CrossRef]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef]
- Njoroge, F.G.; Chen, K.X.; Shih, N.Y.; Piwinski, J.J. Challenges in modern drug discovery: A case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection. Acc. Chem. Res. 2008, 41, 50–59. [Google Scholar] [CrossRef]
- McQuade, T.J.; Tomasselli, A.G.; Liu, L.; Karacostas, V.; Moss, B.; Sawyer, T.K. A synthetic HIV-1 protease inhibitor with antiviral activity arrests HIV-like particle maturation. Science 1990, 247, 454–456. [Google Scholar] [CrossRef]
- Ondetti, M.A.; Rubin, B.; Cushman, D.W. Design of specific inhibitors of angiotensin-converting enzyme: New class of orally active antihypertensive agents. Science 1977, 196, 441–444. [Google Scholar] [CrossRef]
- Cushman, D.W.; Cheung, H.S.; Sabo, E.F.; Ondetti, M.A. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 1977, 16, 5484–5491. [Google Scholar] [CrossRef]
- Cohen, N.C. Structure-based drug design and the discovery of aliskiren (Tekturna): Perseverance and creativity to overcome a R&D pipeline challenge. Chem. Biol. Drug Des. 2007, 70, 557–565. [Google Scholar] [CrossRef]
- Sokouti, B.; Hamzeh-Mivehroud, M. 6D-QSAR for predicting biological activity of human aldose reductase inhibitors using quasar receptor surface modeling. BMC Chem. 2023, 17, 1–9. [Google Scholar] [CrossRef]
- Damale, M.G.; Harke, S.N.; Kalam Khan, F.A.; Shinde, D.B.; Sangshetti, J.N. Recent advances in multidimensional QSAR (4D-6D): A critical review. Mini Rev. Med. Chem. 2014, 14, 35–55. [Google Scholar] [CrossRef]
- Hopfinger, A.J.; Wang, S.; Tokarski, J.S.; Jin, B.; Albuquerque, M.; Madhav, P.J.; Duraiswami, C. Construction of 3D-QSAR Models Using the 4D-QSAR Analysis Formalism. J. Am. Chem. Soc. 1997, 119, 10509–10524. [Google Scholar] [CrossRef]
- Giordano, D.; Biancaniello, C.; Argenio, M.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef]
- Ab, A.; Bhatt, H. 3D-QSAR (CoMFA, CoMFA-RG, CoMSIA) and molecular docking study of thienopyrimidine and thienopyridine derivatives to explore structural requirements for aurora-B kinase inhibition. Eur. J. Pharm. Sci. 2015, 79, 1–12. [Google Scholar] [CrossRef]
- Ankitkumar, P.; Hardik, B.; Bhumika, P. Structural insights on 2-phenylquinazolin-4-one derivatives as tankyrase inhibitors through CoMFA, CoMSIA, topomer CoMFA and HQSAR studies. J. Molec. Struct. 2022, 1249, 131636. [Google Scholar] [CrossRef]
- Duraiswami, C.; Madhav, P.J.; Hopfinger, A.J. Application of 4D-QSAR Analysis to a Set of Prostaglandin, PGF2α, Analogs. In Molecular Modeling and Prediction of Bioactivity; Springer: Boston, MA, USA, 2000; pp. 323–324. [Google Scholar] [CrossRef]
- Vedani, A.; Dobler, M. 5D-QSAR: The key for simulating induced fit? J. Med. Chem. 2002, 23, 2139–2149. [Google Scholar] [CrossRef]
- Vedani, A.; Dobler, M.; Lill, M.A. Combining Protein Modeling and 6D-QSAR. Simulating the Binding of Structurally Diverse Ligands to the Estrogen Receptor. J. Med. Chem. 2005, 48, 3700–3703. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Peach, M.L.; Sitzmann, M.; Nicklaus, M.C. A New Approach to Radial basis function approximation and Its application to QSAR. J. Chem. Inf. Model. 2014, 54, 713–719. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Peach, M.L.; Sitzmann, M.; Nicklaus, M.C. QSAR modeling of imbalanced high-throughput screening data in PubChem. J. Chem. Inf. Model. 2014, 54, 705–712. [Google Scholar] [CrossRef]
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. J. Mol. Inform. 2011, 30, 241–250. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Zakharov, A.V.; Lagunin, A.A.; Poroikov, V.V. QNA based “Star Track” QSAR approach. SAR QSAR En-viron. J. Resolut. 2009, 20, 679–709. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. Quantitative structure—Activity relationships of cyclin-dependent kinase 1 inhibitors. J. Biomed. Chem. 2006, 52, 3–18. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Akimov, D.V.; Poroikov, V.V. The Method of Self-Consistent Regression for the Quantitative Analysis of Relationships Between Structure and Properties of Chemicals. Pharm. Chem. J. 2004, 38, 21–24. [Google Scholar] [CrossRef]
- Ivanov, S.M.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. Relationships between the structure and severe drug-induced liver injury for low, medium, and high doses of drugs. J. Chem. Res. Texicol. 2022, 35, 402–411. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Zakharov, A.V.; Filimonov, D.A.; Poroikov, V.V. A new approach to QSAR modelling of acute toxicity. J. SAR QSAR Environ. Res. 2007, 18, 285–298. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Geronikaki, A.; Eleftheriou, P.; Pogodin, P.V.; Zakharov, A.V. Rational Use of Heterogeneous Data in Quantitative Structure–Activity Relationship (QSAR) Modeling of Cyclooxygen-ase/Lipoxygenase Inhibitors. J. Chem. Inf. Mod. 2019, 59, 713–730. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Varlamova, E.V.; Lagunin, A.A.; Dmitriev, A.V.; Muratov, E.N.; Fourches, D.; Kuz’min, V.E.; Poroikov, V.V.; Tropsha, A.; Nicklaus, M.C. QSAR Modeling and Prediction of Drug–Drug Interactions. J. Mol. Pharm. 2016, 13, 545–556. [Google Scholar] [CrossRef]
- Tarasova, O.A.; Urusova, A.F.; Filimonov, D.A.; Nicklaus, M.C.; Zakharov, A.V.; Poroikov, V.V. QSAR Modeling Using Large-Scale Databases: Case Study for HIV-1 Reverse Transcriptase Inhibitors. J. Chem. Inf. Mod. 2015, 55, 1388–1399. [Google Scholar] [CrossRef]
- Tarasova, O.A.; Rudik, A.V.; Ivanov, S.M.; Lagunin, A.A.; Poroikov, V.V.; Filimonov, D.A. Machine Learning Methods in Antiviral Drug Discovery. In Topics in Medicinal Chemistry; Tarasova, O.A., Rudik, A.V., Ivanov, S.M., Lagunin, A.A., et al., Eds.; Springer: Berlin/Heidelberg, Germany, 2021; Volume 37, pp. 245–279. [Google Scholar] [CrossRef]
- Kokurkina, G.V.; Dutov, M.D.; Shevelev, S.A.; Popkov, S.V.; Zakharov, A.V.; Poroikov, V.V. Synthesis, antifungal activity and QSAR study of 2-arylhydroxynitroindoles. Eur. J. Med. Chem. 2011, 46, 4374–4382. [Google Scholar] [CrossRef]
- Masand, V.H.; Mahajan, D.T.; Patil, K.N.; Dawale, N.E.; Hadda, T.B.; Alafeefy, A.A.; Chinchkhede, K.D. General Unrestricted Structure Activity Relationships based evaluation of quinoxaline derivatives as potential influenza NS1A protein inhibitors. Der Pharma Chem. 2011, 3, 517–525. [Google Scholar]
- Masand, V.H.; Devidas, T.; Mahajan, D.T.; Patil, K.N.; Hadda, T.B.; Youssoufi, M.H.; Jawarkar, R.D.; Shibi, I.G. Optimization of Antimalarial Activity of Synthetic Prodiginines: QSAR, GUSAR, and CoMFA analyses. J. Chem. Biol. Drug Des. 2013, 81, 527–536. [Google Scholar] [CrossRef]
- Khairullina, V.R.; Gerchikov, A.Y.; Lagunin, A.A.; Zarudii, F.S. QSAR modeling of thymidilate synthase inhibitors in a series of quinazoline derivatives. J. Pharm. Chem. 2018, 51, 884–888. [Google Scholar] [CrossRef]
- Khairullina, V.R.; Gerchikov, A.Y.; Zarudii, F.S. Analysis of the relationship “structure cyclooxygenase-2 inhibitory activity” in the series of di-tret-butylphenol, oxazolone and thiazolone. J. Vestn. Bashk. Univ. 2014, 19, 417–422. [Google Scholar]
- Khayrullina, V.R.; Gerchikov, A.Y.; Lagunin, A.A.; Zarudii, F.S. Quantitative Analysis of Structure−Activity Relationships of Tetrahydro-2H-isoindole Cyclooxygenase-2 Inhibitors. J. Biokhimiya 2015, 80, 74–86. [Google Scholar] [CrossRef]
- Khairullina, V.R.; Akbasheva, Y.Z.; Gimadieva, A.R.; Mustafin, A.G. Analysis of the relationship «structure-activity» in theseries of certain 5-ethyluridine derivatives with pronounced anti-herpetic activity. J. Vestn. Bashk. Univ. 2017, 22, 960–965. [Google Scholar]
- Martynova, Y.Z.; Khairullina, V.R.; Nasretdinova, R.N.; Garifullina, G.G.; Mitsukova, D.S.; Gerchikov, A.Y.; Mustafin, A.G. Determination of the chain termination rate constants of the radical chain oxidation of organic compounds on antioxidant molecules by the QSPR method. J. Russ. Chem. Bull. 2020, 69, 1679–1691. [Google Scholar] [CrossRef]
- Khairullina, V.; Safarova, I.; Sharipova, G.; Martynova, Y.; Gerchikov, A. QSAR Assessing the Efficiency of Antioxidants in the Termination of Radical-Chain Oxidation Processes of Organic Compounds. J. Mol. 2021, 26, 421. [Google Scholar] [CrossRef]
- Khairullina, V.; Martynova, Y.; Safarova, I.; Sharipova, G.; Gerchikov, A.; Limantseva, R.; Savchenko, R. QSPR Modeling and Experimental Determination of the Antioxidant Activity of Some Polycyclic Compounds in the Radical-Chain Oxidation Reaction of Organic Substrates. J. Mol. 2022, 27, 6511. [Google Scholar] [CrossRef]
- Martynova, Y.Z.; Khairullina, V.R.; Garifullina, G.G.; Mitsukova, D.S.; Zarudiy, F.S.; Mustafin, A.G. QSAR-modeling of the relationship “structure—Antioxidative activity” in a series of some benzopirane and benzofurane derivatives. J. Vestn. Bashk. Univ. 2019, 24, 573–580. [Google Scholar] [CrossRef]
- Martynova, Y.Z.; Khairullina, V.R.; Gerchikov, A.Y.; Zarudiy, F.S.; Mustafin, A.G. QSPR-modeling of antioxidant activity of potential and industrial used stabilizers from the class of substituted alkylphenols. J. Vestn. Bashk. Univ. 2020, 25, 723–730. [Google Scholar] [CrossRef]
- Oguri, T.; Achiwa, H.; Bessho, Y.; Muramatsu, H.; Maeda, H.; Niimi, T.; Sato, S.; Ueda, R. The role of thymidylate synthase and dihydropyrimidine dehydrogenase in resistance to 5-fluorouracil in human lung cancer cells. J. LungCan. 2005, 49, 345–351. [Google Scholar] [CrossRef]
- McGuire, J.J. Anticancer Antifolates: Current Status and Future Directions. J. Cur. Pharm. Des. 2003, 9, 2593–2613. [Google Scholar] [CrossRef]
- Roy, K.; Das, R.N.; Ambure, P.; Aher, R.B. Be aware of error measures. Further studies on validation of predictive QSAR models. J. Chemom. Intell. Lab. Syst. 2016, 152, 18–33. [Google Scholar] [CrossRef]
- Ivanov, A.S.; Veselovsky, A.V.; Dubanov, A.V.; Skvortsov, V.S.; Archakov, A.I. The integral platform “From gene to drug prototype” in silico and in vitro. J. Ross. Khim. Zh. 2006, 1, 18–35. [Google Scholar]
- Gramatica, P.; Sangion, A. A Historical Excursus on the Statistical Validation Parameters for QSAR Models: A Clarification Concerning Metrics and Terminology. J. Chem. Inform. Model. 2016, 56, 1127–1131. [Google Scholar] [CrossRef]
- Consonni, V.; Ballabio, D.; Todeschini, R. Evaluation of model predictive ability by external validation techniques. J. Chemom. 2010, 24, 194–201. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models: How to Evaluate It? Comparisonof Different Validation Criteria and Proposal of Using the Concordance Correlation Coefficient. J. Chem. Inform. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I.; Kar, S.; Ojha, P.K.; Das, R.N.; Kabir, H. Comparative Studies on Some Metrics for External Validation of QSPR Models. J. Chem. Inform. Model. 2012, 52, 396–408. [Google Scholar] [CrossRef]
- Hildebrand, C.; Sandoli, D.; Focher, F.; Gambino, J.; Ciarrocchi, G.; Spadari, S.; Wright, G. Structure-activity relationships of N2-substituted guanines as inhibitors of HSV1 and HSV2 thymidine kinases. J. Med. Chem. 1990, 33, 203–206. [Google Scholar] [CrossRef]
- Manikowski, A.; Lossani, A.; Verri, A.; Gebhardt, B.-M.; Gambino, J.; Focher, F.; Spadari, S.; Wright, G.E. Inhibition of Herpes Simplex Virus Thymidine Kinases by 2-Phenylamino-6-oxopurines and Related Compounds: Structure-Activity Relationships and Antiherpetic Activity in Vivo. J. Mol. Biochem. 2006, 48, 3919–3929. [Google Scholar] [CrossRef][Green Version]
- MarvinSketch. Available online: https://chemaxon.com/download/marvin-suite (accessed on 31 August 2023).
- DiscoveryStudioVisualiser. Available online: https://www.3ds.com (accessed on 31 August 2023).
- Dearden, J.C.; Cronin, M.T.D.; Kaiser, K.L.E. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR). J. SAR QSAR Environ. Res. 2009, 20, 241–266. [Google Scholar] [CrossRef]
- Roy, P.P.; Paul, S.; Mitra, I.; Roy, K. On Two Novel Parameters for Validation of Predictive QSAR Models. J. Mol. 2009, 14, 1660–1701. [Google Scholar] [CrossRef]
- Xternal Validation Plus. Available online: https://sites.google.com/site/dtclabxvplus (accessed on 31 August 2023).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Set | Model | N | NPM | V | ||||
|---|---|---|---|---|---|---|---|---|
| QSAR models based on the QNA descriptors | ||||||||
| TrS1 | M1 | 73 | 20 | 0.878 | 67.101 | 0.569 | 0.848 | 7 |
| TrS2 | M4 | 74 | 20 | 0.891 | 84.683 | 0.593 | 0.869 | 6 |
| TrS3 | M7 | 61 | 20 | 0.875 | 50.879 | 0.579 | 0.837 | 7 |
| TrS4 | M10 | 62 | 20 | 0.891 | 65.152 | 0.598 | 0.863 | 6 |
| QSAR models based on the MNA descriptors | ||||||||
| TrS1 | M2 | 73 | 20 | 0.878 | 63.594 | 0.568 | 0.854 | 7 |
| TrS2 | M5 | 74 | 20 | 0.906 | 79.140 | 0.552 | 0.887 | 8 |
| TrS3 | M8 | 61 | 20 | 0.882 | 51.831 | 0.565 | 0.853 | 7 |
| TrS4 | M11 | 62 | 20 | 0.894 | 70.947 | 0.589 | 0.872 | 6 |
| QSAR models based on both QNA and MNA descriptors | ||||||||
| TrS1 | M3 | 73 | 320 | 0.891 | 57.523 | 0.542 | 0.862 | 8 |
| TrS2 | M6 | 74 | 320 | 0.905 | 70.945 | 0.559 | 0.882 | 8 |
| TrS3 | M9 | 61 | 320 | 0.881 | 45.955 | 0.570 | 0.846 | 7 |
| TrS4 | M12 | 62 | 320 | 0.899 | 63.865 | 0.578 | 0.873 | 7 |
| Comments | Prediction Parameters | QSAR Model Used for Predicting pIC50 | |||||
|---|---|---|---|---|---|---|---|
| TrS1 | TrS2 | ||||||
| M1 | M2 | M3 | M7 | M8 | M9 | ||
| Classical metrics (after removing 5% of the data with high residuals) | R2 | 0.9609 | 0.9594 | 0.9653 | 0.9591 | 0.9611 | 0.9654 |
| R20 | 0.9555 | 0.9579 | 0.9614 | 0.9556 | 0.9587 | 0.9593 | |
| R2′0 | 0.8443 | 0.8804 | 0.8661 | 0.8568 | 0.8725 | 0.8483 | |
| 0.8776 | 0.9052 | 0.8952 | 0.8883 | 0.8971 | 0.8819 | ||
| ∆ | 0.0379 | 0.0355 | 0.0326 | 0.0379 | 0.0352 | 0.0342 | |
| CCC | 0.9755 | 0.9777 | 0.9790 | 0.9759 | 0.9779 | 0.9775 | |
| Mean absolute error and standard deviation for the test set (after removing 5% of the data with high residuals) | RMSE | 0.3368 | 0.3331 | 0.3193 | 0.3323 | 0.3327 | 0.3331 |
| MAE | 0.2914 | 0.2784 | 0.2673 | 0.2872 | 0.2768 | 0.2830 | |
| SD | 0.1701 | 0.1844 | 0.1758 | 0.1687 | 0.1861 | 0.1773 | |
| MAE + 3·SD | 0.8016 | 0.8314 | 0.7948 | 0.7933 | 0.8351 | 0.8149 | |
| Prediction quality | - | Good | |||||
| Presence of systematic errors | - | Absent | |||||
| Comments | Prediction Parameters | QSAR Model Used for Predicting pIC50 | |||||
|---|---|---|---|---|---|---|---|
| TrS2 | TrS4 | ||||||
| M4 | M5 | M6 | M10 | M11 | M12 | ||
| Classical metrics (after removing 5% of the data with high residuals) | R2 | 0.9714 | 0.9712 | 0.9719 | 0.9708 | 0.9676 | 0.9743 |
| R20 | 0.9687 | 0.9701 | 0.9694 | 0.9681 | 0.9664 | 0.9710 | |
| R2′0 | 0.8890 | 0.9086 | 0.8927 | 0.8889 | 0.9009 | 0.8891 | |
| 0.9137 | 0.9267 | 0.9142 | 0.9148 | 0.9216 | 0.9109 | ||
| ∆ | 0.0270 | 0.0260 | 0.0267 | 0.0273 | 0.0290 | 0.0252 | |
| CCC | 0.9830 | 0.9843 | 0.9836 | 0.9827 | 0.9823 | 0.9844 | |
| Mean absolute error and standard deviation for the test set (after removing 5% of the data with high residuals) | RMSE | 0.3278 | 0.3121 | 0.3146 | 0.3333 | 0.3328 | 0.3164 |
| MAE | 0.2712 | 0.2590 | 0.2624 | 0.2739 | 0.2822 | 0.2676 | |
| SD | 0.1856 | 0.1753 | 0.1748 | 0.1915 | 0.1781 | 0.1703 | |
| MAE + 3·SD | 0.8279 | 0.7850 | 0.7868 | 0.8484 | 0.8164 | 0.7785 | |
| Prediction quality | - | Good | |||||
| Presence of systematic errors | - | Absent | |||||
| No. | Name in ChEBIL | Structure | pIC50pred | Selectivity | |||
|---|---|---|---|---|---|---|---|
| HSV-1 | HSV-2 | ||||||
 | |||||||
| R1 | |||||||
| 1 | CHEMBL1199108 |  | 15.29 | 2.87 | 5.3359 | ||
| 2 | CHEMBL1199070 |  | 32.52 | 13.98 | 2.3267 | ||
| 3 | CHEMBL1199059 |  | 27.75 | 21.38 | 1.2980 | ||
| 4 | CHEMBL1780207 |  | 30.42 | 21.46 | 1.4176 | ||
 | |||||||
| R1 | |||||||
| 5 | CHEMBL20028 |  | 35.85 | 27.30 | 1.3131 | ||
| 6 | CHEMBL1178256 |  | 31.91 | 5.91 | 5.4029 | ||
| 7 | CHEMBL19326 |  | 14.87 | 3.82 | 3.8897 | ||
| 8 | CHEMBL1178302 |  | 13.77 | 3.27 | 4.2105 | ||
| 9 | CHEMBL19510 |  | 9.73 | 1.37 | 7.0878 | ||
| 10 | CHEMBL1178307 * |  | 13.97 | 2.63 | 5.3210 | ||
| 11 | CHEMBL19608 |  | 6.88 | 0.83 | 8.3308 | ||
| 12 | CHEMBL19725 |  | 10.33 | 2.06 | 5.0177 | ||
| 13 | CHEMBL19782 |  | 8.01 | 1.41 | 5.6706 | ||
| 14 | CHEMBL1178314 |  | 8.42 | 1.52 | 5.5286 | ||
| 15 | CHEMBL1178315 |  | 9.27 | 1.73 | 5.3491 | ||
| 16 | CHEMBL277025 |  | 12.04 | 1.51 | 7.9804 | ||
| 17 | CHEMBL1183046 |  | 11.01 | 0.99 | 11.0940 | ||
| 18 | CHEMBL277844 |  | 5.76 | 0.70 | 8.2058 | ||
| 19 | CHEMBL1183063 |  | 12.58 | 2.05 | 6.1317 | ||
| 20 | CHEMBL278626 |  | 8.87 | 0.89 | 9.9477 | ||
| 21 | CHEMBL1183081 |  | 12.39 | 2.53 | 4.9020 | ||
| 22 | CHEMBL1183082 |  | 34.36 | 7.19 | 4.7770 | ||
| 23 | CHEMBL1183089 |  | 10.95 | 1.20 | 9.1477 | ||
| 24 | CHEMBL1183095 |  | 8.78 | 0.71 | 12.4135 | ||
| 25 | CHEMBL1183096 |  | 7.31 | 1.04 | 7.0456 | ||
| 26 | CHEMBL279892 |  | 8.78 | 0.74 | 11.7868 | ||
| 27 | CHEMBL1183107 |  | 14.50 | 4.72 | 3.0716 | ||
| 28 | CHEMBL1183108 |  | 13.71 | 3.16 | 4.3415 | ||
| 29 | CHEMBL280909 |  | 5.38 | 1.07 | 5.0082 | ||
| 30 | CHEMBL1183123 |  | 8.20 | 0.83 | 9.8336 | ||
| 31 | CHEMBL1183154 |  | 15.30 | 4.26 | 3.5958 | ||
| 32 | CHEMBL1183178 |  | 11.23 | 2.77 | 4.0530 | ||
| 33 | CHEMBL1183185 |  | 8.32 | 1.57 | 5.2872 | ||
| 34 | CHEMBL1185346 |  | 8.28 | 1.06 | 7.7791 | ||
| 35 | CHEMBL1185463 |  | 32.63 | 6.28 | 5.1918 | ||
| 36 | CHEMBL1185716 |  | 8.81 | 0.93 | 9.4314 | ||
 | |||||||
| R1 | R2 | R3 | |||||
| 37 | CHEMBL217675 * | -H | -H |  | 62.83 | 26.96 | 2.3306 |
| 38 | CHEMBL238635 | -H |  |  | 36.62 | 42.98 | 0.8520 |
| 39 | CHEMBL2403290 * |  | -H | -CH3 | 26.44 | 40.28 | 0.6564 |
| 40 | CHEMBL241407 * | -H |  |  | 14.48 | 22.16 | 0.6535 |
| 41 | CHEMBL241408 * | -H |  |  | 10.05 | 6.47 | 1.5544 |
| 42 | CHEMBL1183075 * |  | 15.18 | 3.11 | 4.8817 | ||
| Designation of TrSi | Code of the Training Set | |||
|---|---|---|---|---|
| HSV-1 | HSV-2 | |||
| TrS1 | TrS3 | TrS2 | TrS4 | |
| N | 73 | 61 | 74 | 62 |
| 6.788 | 6.921 | |||
| ∆pIC50 | 5.867 | 6.250 | ||
| Thresholds used to evaluate the model’s forecast | ||||
| 0.10 × ∆pIC50 | 0.587 | 0.625 | ||
| 0.15 × ∆pIC50 | 0.880 | 0.938 | ||
| 0.20 × ∆pIC50 | 1.174 | 1.250 | ||
| 0.25 × ∆pIC50 | 1.467 | 1.563 | ||
| Designation of TSi | Code of the Test Set | |||
|---|---|---|---|---|
| HSV-1 | HSV-2 | |||
| TS1 | TS3 | TS2 | TS4 | |
| N | 15 | 12 | 15 | 12 |
| 6.788 | 6.921 | |||
| ∆pIC50 | 5.867 | 6.250 | ||
| Distribution of the observed response values of test sets TSi around the test mean | ||||
| ± 0.5, % | 26.667 | 16.667 | 20.000 | 25.000 |
| ± 1.0, % | 40.000 | 41.667 | 40.000 | 41.667 |
| ± 1.5, % | 60.000 | 58.333 | 46.667 | 50.000 |
| ± 2.0, % | 73.333 | 83.333 | 66.667 | 66.667 |
| Distribution of the observed response values of test sets TSi around the training mean | ||||
| ± 0.5, % | 13.333 | 8.333 | 26.667 | 16.667 |
| ± 1.0, % | 33.333 | 25.000 | 33.333 | 41.667 |
| ± 1.5, % | 46.667 | 50.000 | 46.667 | 50.000 |
| ± 2.0, % | 66.667 | 75.000 | 66.667 | 75.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khairullina, V.; Martynova, Y. Quantitative Structure–Activity Relationship in the Series of 5-Ethyluridine, N2-Guanine, and 6-Oxopurine Derivatives with Pronounced Anti-Herpetic Activity. Molecules 2023, 28, 7715. https://doi.org/10.3390/molecules28237715
Khairullina V, Martynova Y. Quantitative Structure–Activity Relationship in the Series of 5-Ethyluridine, N2-Guanine, and 6-Oxopurine Derivatives with Pronounced Anti-Herpetic Activity. Molecules. 2023; 28(23):7715. https://doi.org/10.3390/molecules28237715
Chicago/Turabian StyleKhairullina, Veronika, and Yuliya Martynova. 2023. "Quantitative Structure–Activity Relationship in the Series of 5-Ethyluridine, N2-Guanine, and 6-Oxopurine Derivatives with Pronounced Anti-Herpetic Activity" Molecules 28, no. 23: 7715. https://doi.org/10.3390/molecules28237715
APA StyleKhairullina, V., & Martynova, Y. (2023). Quantitative Structure–Activity Relationship in the Series of 5-Ethyluridine, N2-Guanine, and 6-Oxopurine Derivatives with Pronounced Anti-Herpetic Activity. Molecules, 28(23), 7715. https://doi.org/10.3390/molecules28237715