
Study of Amiloride Binding to Human Serum Albumin: Insights from Thermodynamic, Spectroscopic, and Molecular Docking Investigations

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Investigating the Interaction between HSA and AML
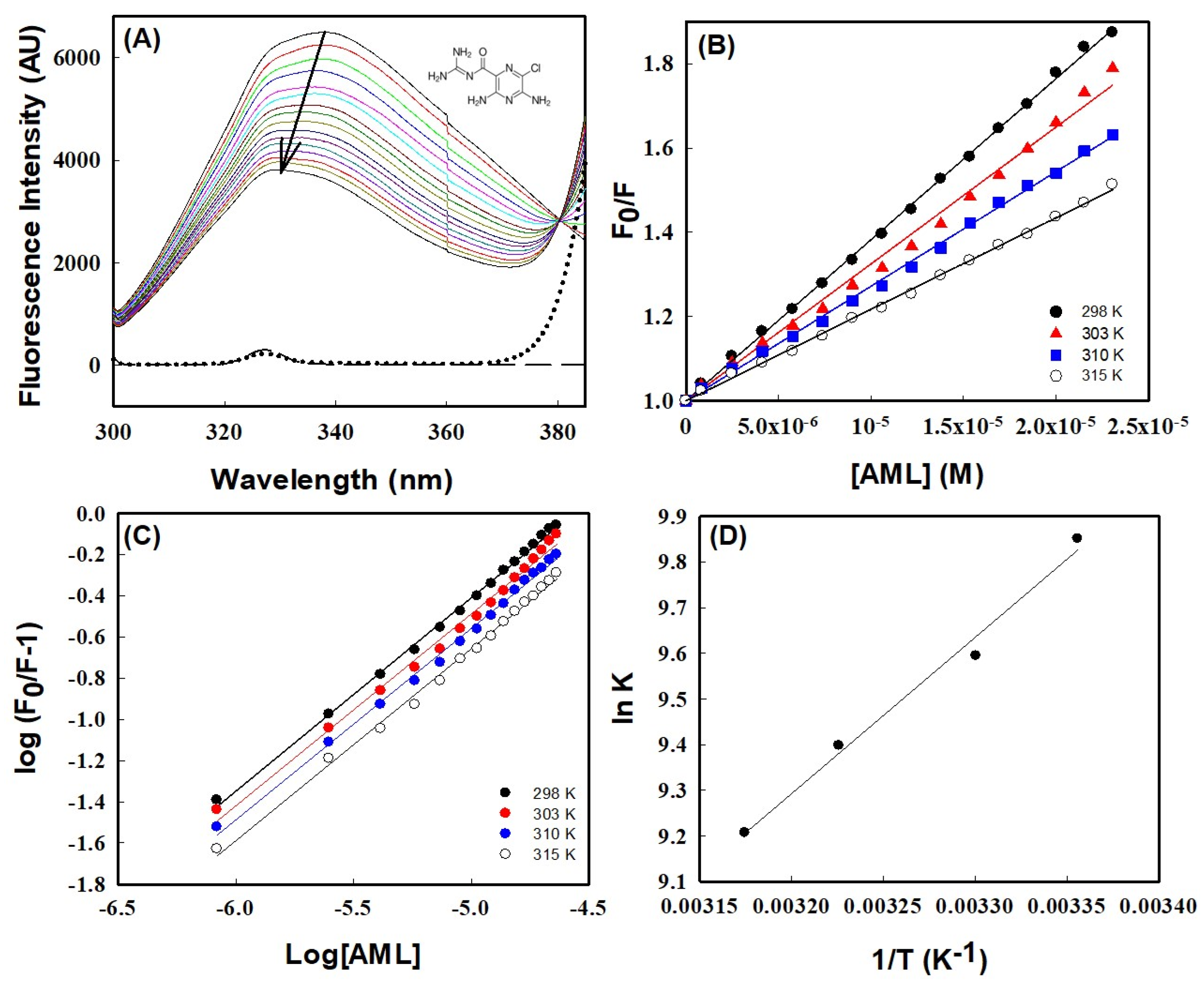
2.1.1. Fluorescence-Quenching Analysis
2.1.2. Evaluations of Thermodynamic and Binding Parameters
2.1.3. Förster Resonance Energy Transfer (FRET) between HSA and AML
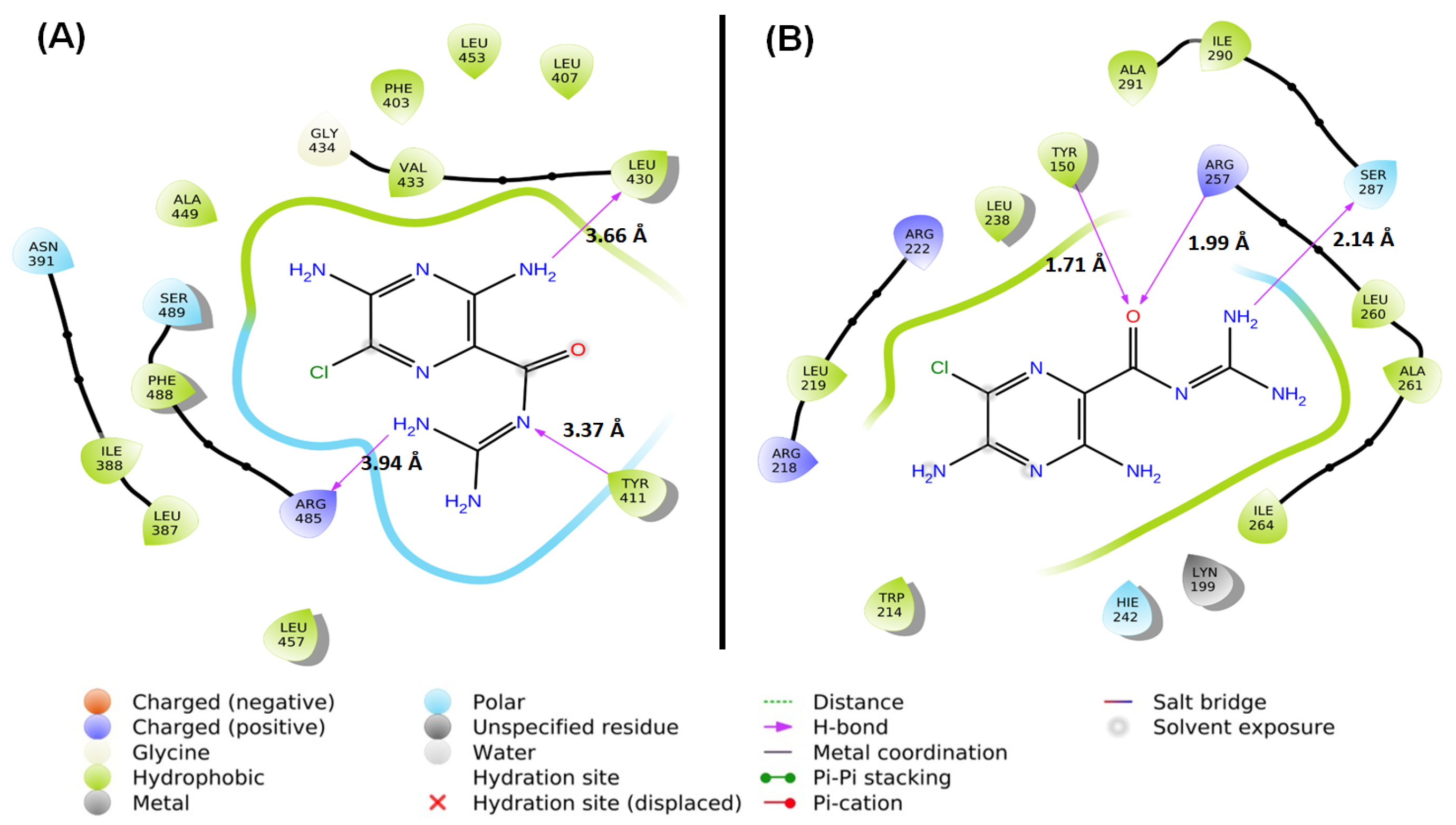
2.1.4. Location of the AML Binding Site in HSA
2.2. Effect of AML on the Conformation of HSA
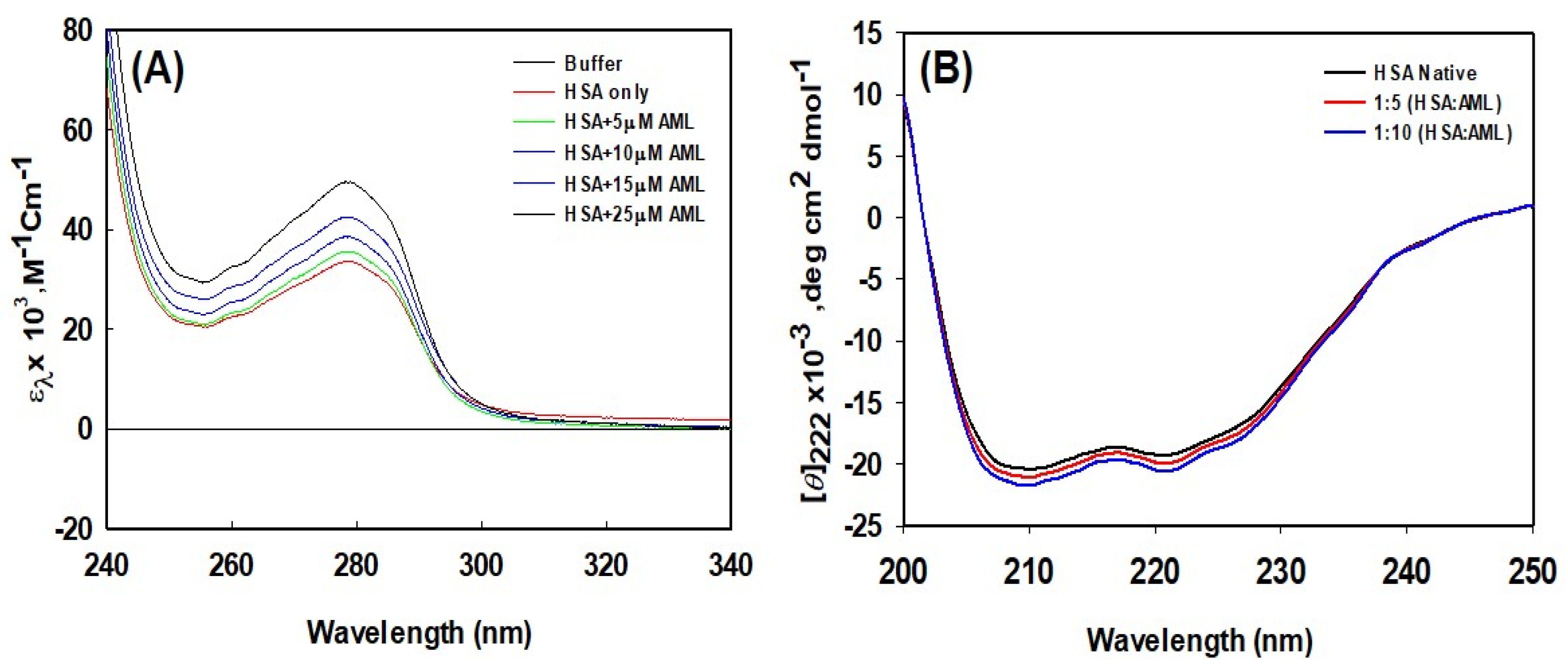
2.2.1. UV-Vis Spectroscopy
2.2.2. Analysis of Secondary Structural Changes Using Circular Dichroism (CD) in the Far-UV
2.2.3. Analysis of Tertiary Structure Changes Using Synchronous Fluorescence
2.2.4. Three-Dimensional Fluorescence Analysis
2.3. Molecular Dynamics Simulation
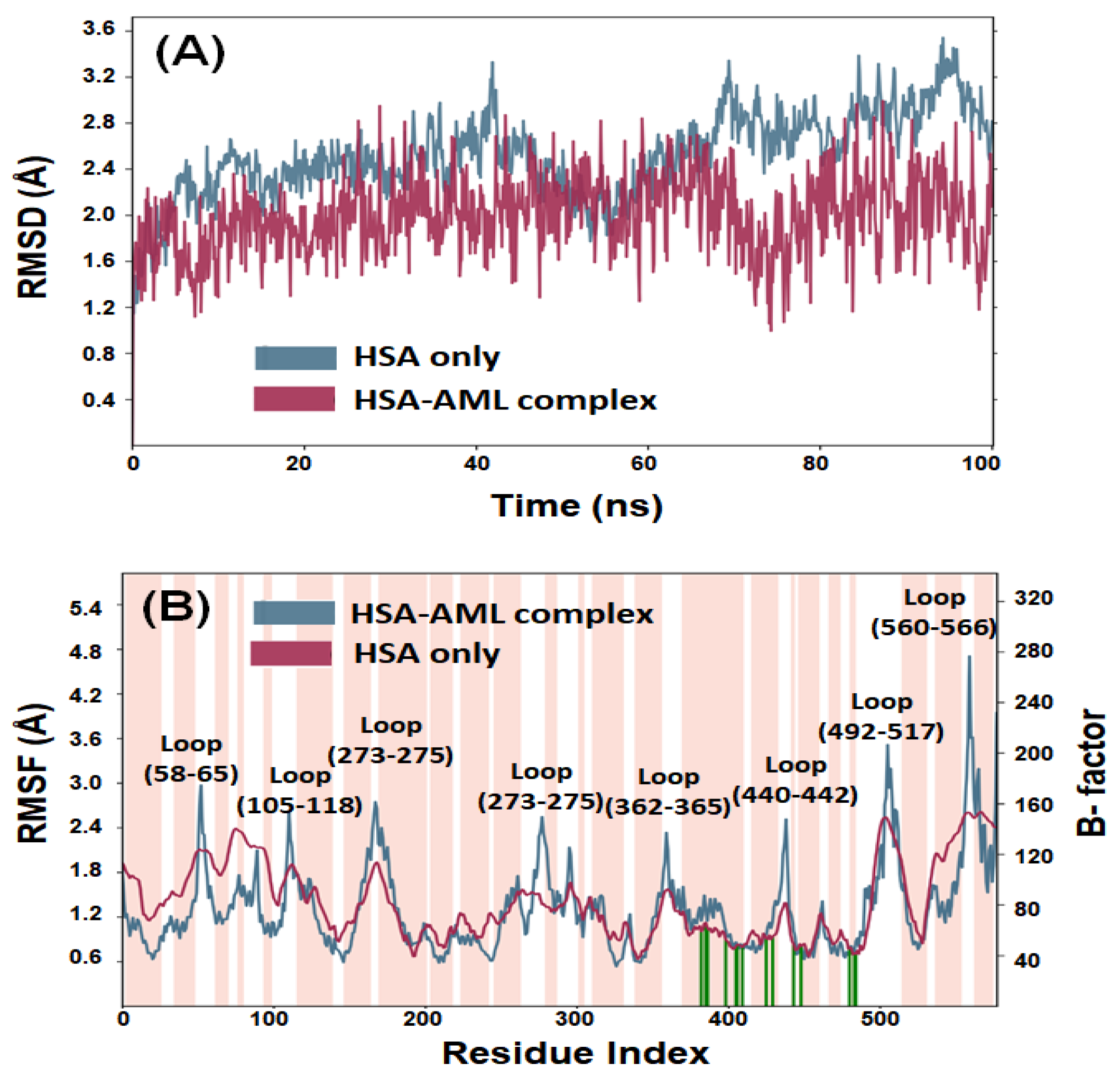
2.3.1. Root-Mean-Square Deviation (RMSD)
2.3.2. Root-Mean-Square Fluctuation (RMSF)
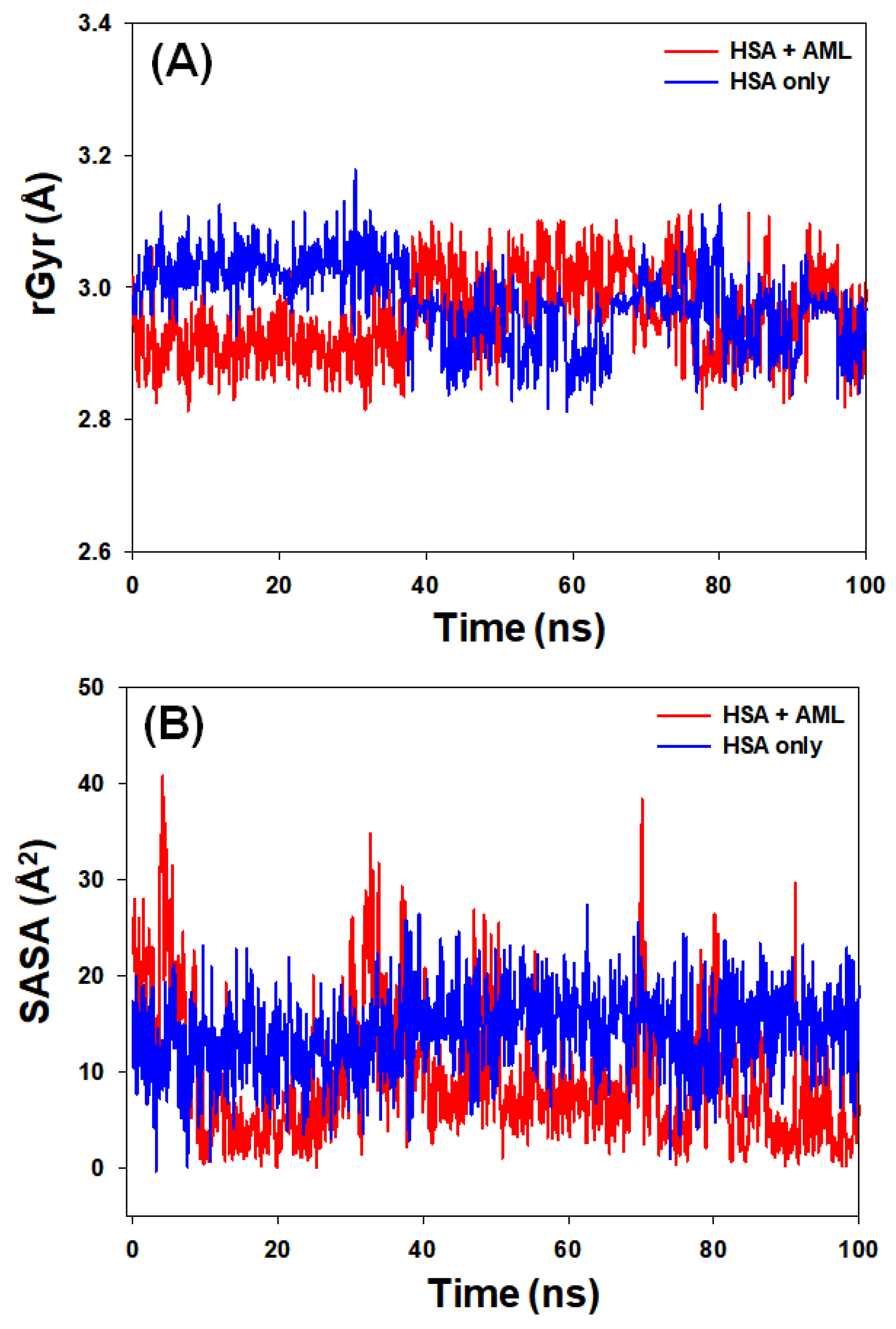
2.3.3. Radius of Gyration (rGyr) and Solvent-Accessible Surface Area (SASA)
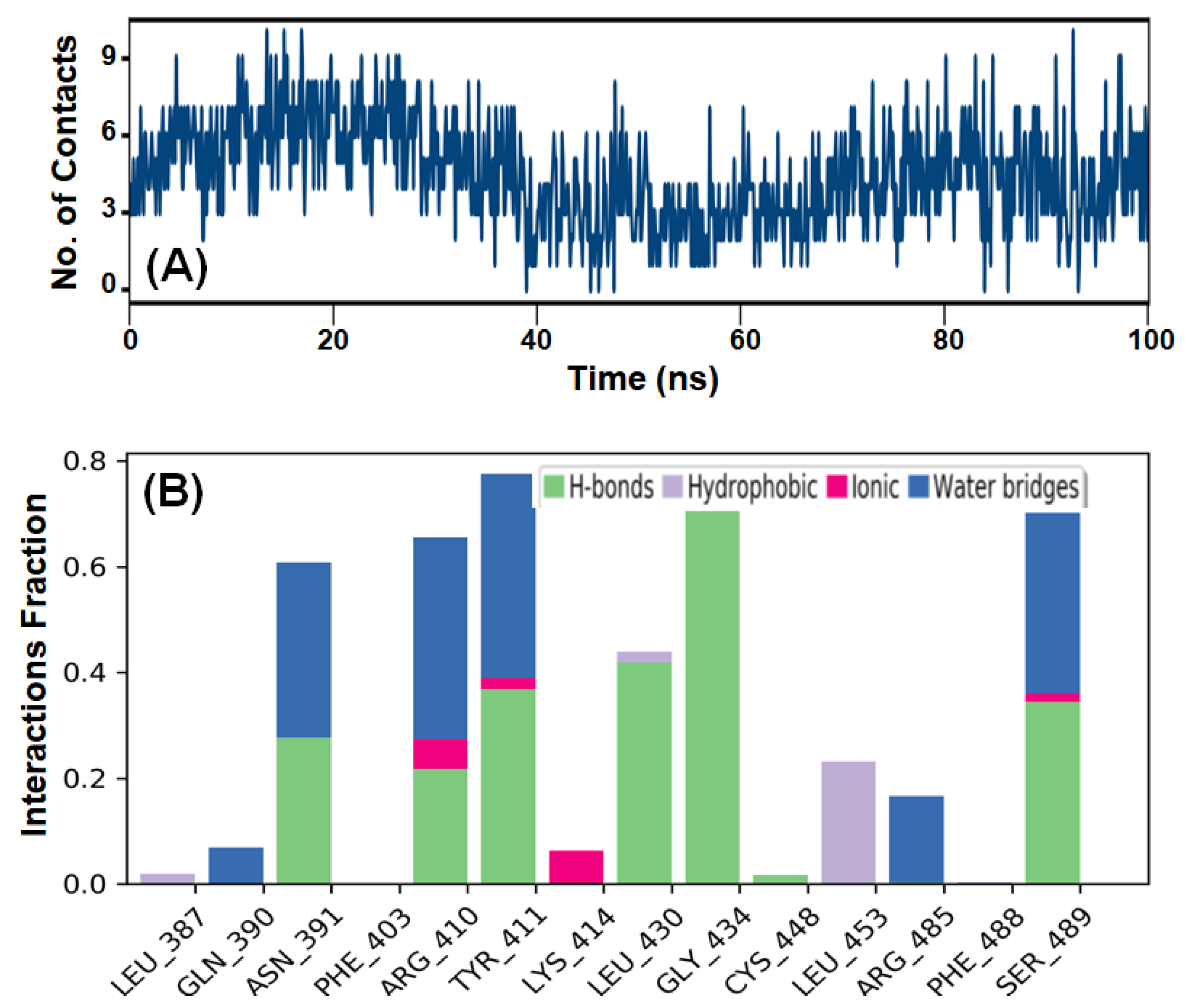
2.3.4. Total Number of Contacts and Interacting Residues
3. Materials and Methods
3.1. Materials
3.2. Sample Preparation
3.3. Fluorescence-Quenching Measurements
3.4. Synchronous Fluorescence Measurements
3.5. Three-Dimensional Fluorescence Spectroscopy
3.6. UV-Vis Spectra Measurements
3.7. Far-UV Circular Dichroism (far-UV CD) Measurements
3.8. Molecular Docking (MD)
3.9. Molecular Dynamics Simulation (MDS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef]
- Rabbani, G.; Ahn, S.N. Structure, enzymatic activities, glycation and therapeutic potential of human serum albumin: A natural cargo. Int. J. Biol. Macromol. 2019, 123, 979–990. [Google Scholar] [CrossRef]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. Des. Sel. 1999, 12, 439–446. [Google Scholar] [CrossRef]
- Rabbani, N.; Tabrez, S.; Islam, B.U.; Rehman, M.T.; Alsenaidy, A.M.; AlAjmi, M.F.; Khan, R.A.; Alsenaidy, M.A.; Khan, M.S. Characterization of colchicine binding with normal and glycated albumin: In vitro and molecular docking analysis. J. Biomol. Struct. Dyn. 2018, 36, 3453–3462. [Google Scholar] [CrossRef]
- Rehman, M.T.; Faheem, M.; Khan, A.U. Insignificant beta-lactamase activity of human serum albumin: No panic to nonmicrobial-based drug resistance. Lett. Appl. Microbiol. 2013, 57, 325–329. [Google Scholar] [CrossRef]
- Hu, Y.J.; Li, W.; Liu, Y.; Dong, J.X.; Qu, S.S. Fluorometric investigation of the interaction between methylene blue and human serum albumin. J. Pharm. Biomed. Anal. 2005, 39, 740–745. [Google Scholar] [CrossRef]
- Śliwińska-Hill, U.; Wiglusz, K. Multispectroscopic studies of the interaction of folic acid with glycated human serum albumin. J. Biomol. Struct. Dyn. 2019, 37, 3731–3739. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Evoli, S.; Mobley, D.L.; Guzzi, R.; Rizzuti, B. Multiple binding modes of ibuprofen in human serum albumin identified by absolute binding free energy calculations. Phys. Chem. Chem. Phys. 2016, 18, 32358–32368. [Google Scholar] [CrossRef]
- Rabbani, G.; Baig, M.H.; Jan, A.T.; Ju Lee, E.; Khan, M.V.; Zaman, M.; Farouk, A.E.; Khan, R.H.; Choi, I. Binding of erucic acid with human serum albumin using a spectroscopic and molecular docking study. Int. J. Biol. Macromol. 2017, 105, 1572–1580. [Google Scholar] [CrossRef]
- Khan, F.I.; Rehman, M.T.; Fathima, S.; Hussain, T.; AlAjmi, M.F.; Lai, D.; Khan, M.K.A. Investigating the binding mechanism of topiramate with bovine serum albumin using spectroscopic and computational methods. J. Mol. Recognit. 2022, 35, e2958. [Google Scholar] [CrossRef]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal structure analysis of warfarin binding to human serum albumin: Anatomy of drug site I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef]
- Matthews, H.; Ranson, M.; Kelso, M.J. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: An orally active anti-cancer drug waiting for its call-of-duty? Int. J. Cancer 2011, 129, 2051–2061. [Google Scholar] [CrossRef]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef]
- Antolini, M.; Trevisi, L.; Debetto, P.; Luciani, S. Effect of amiloride on sodium-calcium exchange activity in rat cardiac myocytes. Pharmacol. Res. 1993, 27, 227–231. [Google Scholar] [CrossRef]
- Frelin, C.; Barbry, P.; Vigne, P.; Chassande, O.; Cragoe, E.J., Jr.; Lazdunski, M. Amiloride and its analogs as tools to inhibit Na+ transport via the Na+ channel, the Na+/H+ antiport and the Na+/Ca2+ exchanger. Biochimie 1988, 70, 1285–1290. [Google Scholar] [CrossRef]
- Kleyman, T.R.; Cragoe, E.J., Jr. Amiloride and its analogs as tools in the study of ion transport. J. Membr. Biol. 1988, 105, 1–21. [Google Scholar] [CrossRef]
- Sparks, R.L.; Pool, T.B.; Smith, N.K.; Cameron, I.L. Effects of amiloride on tumor growth and intracellular element content of tumor cells in vivo. Cancer Res. 1983, 43, 73–77. [Google Scholar]
- Tatsuta, M.; Iishi, H.; Baba, M.; Yano, H.; Iseki, K.; Uehara, H.; Nakaizumi, A. Inhibition by amiloride of experimental carcinogenesis induced by azaserine in rat pancreas. Cancer Lett. 1996, 106, 23–28. [Google Scholar] [CrossRef]
- Oka, N.; Okumura, Y.; Kanayama, H.O.; Izaki, H.; Okamoto, M.; Kido, H.; Kagawa, S. Amiloride and urinary trypsin inhibitor inhibit urothelial cancer invasion. Eur. Urol. 2003, 44, 737–741. [Google Scholar] [CrossRef]
- Ray, P.; Bhatti, R.; Gadarowski, J.; Bell, N.; Nasruddin, S. Inhibitory effect of amiloride on the urokinase plasminogen activators in prostatic cancer. Tumour Biol. 1998, 19, 60–64. [Google Scholar] [CrossRef]
- Reshkin, S.J.; Bellizzi, A.; Cardone, R.A.; Tommasino, M.; Casavola, V.; Paradiso, A. Paclitaxel induces apoptosis via protein kinase A- and p38 mitogen-activated protein-dependent inhibition of the Na+/H+ exchanger (NHE) NHE isoform 1 in human breast cancer cells. Clin. Cancer Res. 2003, 9, 2366–2373. [Google Scholar]
- Izzo, J.L., Jr.; Hong, M.; Hussain, T.; Osmond, P.J. Long-term BP control and vascular health in patients with hyperaldosteronism treated with low-dose, amiloride-based therapy. J. Clin. Hypertens. 2019, 21, 922–928. [Google Scholar] [CrossRef]
- AlAjmi, M.F.; Hussain, A.; Rehman, M.T.; Khan, A.A.; Shaikh, P.A.; Khan, R.A. Design, Synthesis, and Biological Evaluation of Benzimidazole-Derived Biocompatible Copper(II) and Zinc(II) Complexes as Anticancer Chemotherapeutics. Int. J. Mol. Sci. 2018, 19, 1492. [Google Scholar] [CrossRef]
- Baig, M.H.; Rahman, S.; Rabbani, G.; Imran, M.; Ahmad, K.; Choi, I. Multi-Spectroscopic Characterization of Human Serum Albumin Binding with Cyclobenzaprine Hydrochloride: Insights from Biophysical and In Silico Approaches. Int. J. Mol. Sci. 2019, 20, 662. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2004; p. 2006. [Google Scholar]
- Chaves, O.A.; Amorim, A.P.; Castro, L.H.; Sant’Anna, C.M.; de Oliveira, M.C.; Cesarin-Sobrinho, D.; Netto-Ferreira, J.C.; Ferreira, A.B. Fluorescence and Docking Studies of the Interaction between Human Serum Albumin and Pheophytin. Molecules 2015, 20, 19526–19539. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Jiao, Y.; Dong, C. Spectroscopic analysis and molecular modeling on the interaction of jatrorrhizine with human serum albumin (HSA). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 118, 48–54. [Google Scholar] [CrossRef]
- Bian, Q.; Liu, J.; Tian, J.; Hu, Z. Binding of genistein to human serum albumin demonstrated using tryptophan fluorescence quenching. Int. J. Biol. Macromol. 2004, 34, 275–279. [Google Scholar] [CrossRef]
- Kamtekar, N.; Pandey, A.; Agrawal, N.; Pissurlenkar, R.R.; Borana, M.; Ahmad, B. Interaction of multimicrobial synthetic inhibitor 1,2-bis(2-benzimidazolyl)-1,2-ethanediol with serum albumin: Spectroscopic and computational studies. PLoS ONE 2013, 8, e53499. [Google Scholar] [CrossRef]
- Yeggoni, D.P.; Gokara, M.; Manidhar, D.M.; Rachamallu, A.; Nakka, S.; Reddy, C.S.; Subramanyam, R. Binding and molecular dynamics studies of 7-hydroxycoumarin derivatives with human serum albumin and its pharmacological importance. Mol. Pharm. 2014, 11, 1117–1131. [Google Scholar] [CrossRef]
- Zaidi, N.; Ajmal, M.R.; Rabbani, G.; Ahmad, E.; Khan, R.H. A comprehensive insight into binding of hippuric acid to human serum albumin: A study to uncover its impaired elimination through hemodialysis. PLoS ONE 2013, 8, e71422. [Google Scholar] [CrossRef]
- Rehman, M.T.; Shamsi, H.; Khan, A.U. Insight into the binding mechanism of imipenem to human serum albumin by spectroscopic and computational approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef]
- Stryer, L.; Haugland, R.P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719–726. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef]
- Rahman, S.; Rehman, M.T.; Rabbani, G.; Khan, P.; AlAjmi, M.F.; Hassan, M.I.; Muteeb, G.; Kim, J. Insight of the Interaction between 2,4-thiazolidinedione and Human Serum Albumin: A Spectroscopic, Thermodynamic and Molecular Docking Study. Int. J. Mol. Sci. 2019, 20, 2727. [Google Scholar] [CrossRef]
- Rabbani, G.; Baig, M.H.; Lee, E.J.; Cho, W.K.; Ma, J.Y.; Choi, I. Biophysical Study on the Interaction between Eperisone Hydrochloride and Human Serum Albumin Using Spectroscopic, Calorimetric, and Molecular Docking Analyses. Mol. Pharm. 2017, 14, 1656–1665. [Google Scholar] [CrossRef]
- Painter, L.; Harding, M.M.; Beeby, P.J. Synthesis and interaction with human serum albumin of the first 3,18-disubstituted derivative of bilirubin. J. Chem. Soc. Perkin Trans. 1 1998, 3041–3044. [Google Scholar] [CrossRef]
- Rehman, M.T.; Ahmed, S.; Khan, A.U. Interaction of meropenem with ‘N’ and ‘B’ isoforms of human serum albumin: A spectroscopic and molecular docking study. J. Biomol. Struct. Dyn. 2016, 34, 1849–1864. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Toukmaji, A.Y.; Board, J.A., Jr. Ewald summation techniques in perspective: A survey. Comput. Phys. Commun. 1996, 95, 73–92. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters/Temperature (K) | 298 | 303 | 310 | 315 |
|---|---|---|---|---|
| KSV (M−1 ) | 3.82 × 104 | 3.25 × 104 | 2.72 × 104 | 2.17 × 104 |
| kq (M−1 s−1) | 6.60 × 1012 | 5.62 × 1012 | 4.70 × 1012 | 3.75 × 1012 |
| Kb (M−1) | 1.89 × 104 | 1.46 × 104 | 1.20 × 104 | 0.99 × 104 |
| N | 0.93 | 0.93 | 0.92 | 0.93 |
| ∆H° (kcal mol−1) | −6.80 | |||
| T∆S° (kcal mol−1) | −0.98 | −1.00 | −1.02 | −1.04 |
| ∆G° (kcal mol−1) | −5.81 | −5.80 | −5.77 | −5.76 |
| J (M−1 cm3) | Ro (nm) | r (nm) | EFRET | |
|---|---|---|---|---|
| HSA–AML complex | 5.99 × 10−15 | 2.26 | 2.33 | 0.45 |
| Peak No. | Peak Position [λex/λem (nm/nm)] | Peak Intensity | |
|---|---|---|---|
| HSA only | 1 | 280/335 | 3736 |
| 2 | 230/330 | 1097 | |
| HSA + AML (1:5 molar ratio) | 1 | 280/325 | 3325 |
| 2 | 230/315 | 976 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.; Iram, S.; Rehman, M.T.; Hussain, A.; Jan, A.T.; Kim, J. Study of Amiloride Binding to Human Serum Albumin: Insights from Thermodynamic, Spectroscopic, and Molecular Docking Investigations. Molecules 2023, 28, 7688. https://doi.org/10.3390/molecules28237688
Rahman S, Iram S, Rehman MT, Hussain A, Jan AT, Kim J. Study of Amiloride Binding to Human Serum Albumin: Insights from Thermodynamic, Spectroscopic, and Molecular Docking Investigations. Molecules. 2023; 28(23):7688. https://doi.org/10.3390/molecules28237688
Chicago/Turabian StyleRahman, Safikur, Sana Iram, Md Tabish Rehman, Afzal Hussain, Arif Tasleem Jan, and Jihoe Kim. 2023. "Study of Amiloride Binding to Human Serum Albumin: Insights from Thermodynamic, Spectroscopic, and Molecular Docking Investigations" Molecules 28, no. 23: 7688. https://doi.org/10.3390/molecules28237688
APA StyleRahman, S., Iram, S., Rehman, M. T., Hussain, A., Jan, A. T., & Kim, J. (2023). Study of Amiloride Binding to Human Serum Albumin: Insights from Thermodynamic, Spectroscopic, and Molecular Docking Investigations. Molecules, 28(23), 7688. https://doi.org/10.3390/molecules28237688