
5-((3-Amidobenzyl)oxy)nicotinamides as SIRT2 Inhibitors: A Study of Constrained Analogs


Abstract
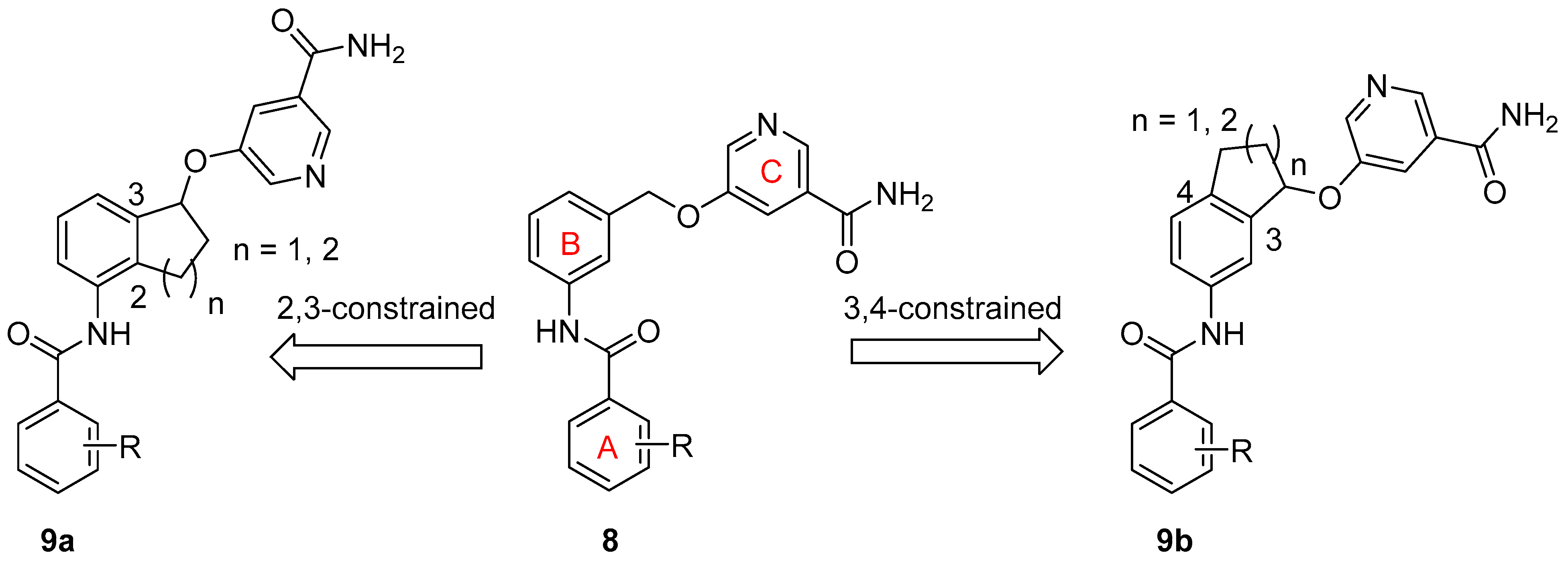
:1. Introduction
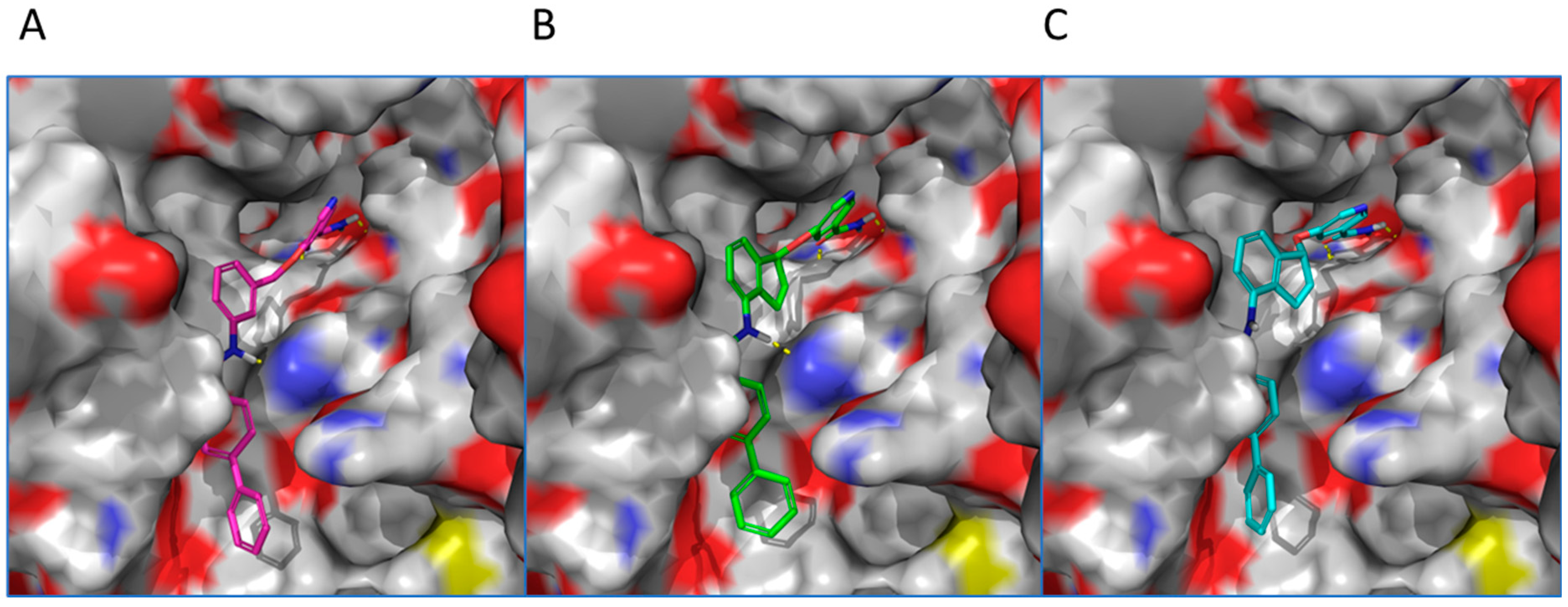
2. Results and Discussion
3. Conclusions
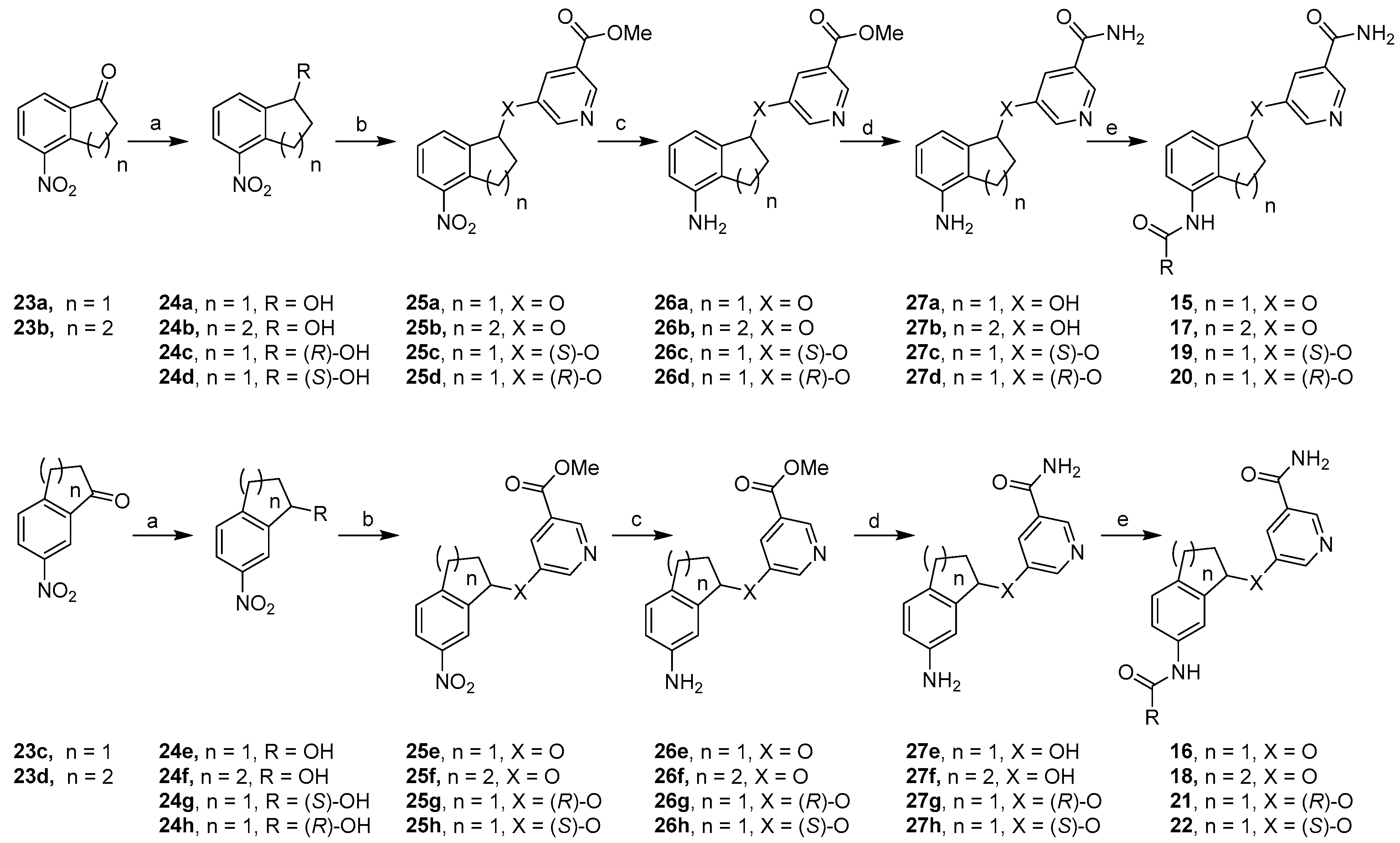
4. Experimental
- 5-((4-(4-Methyl-3-nitrobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (15a). To a solution of 27a (57 mg, 0.21 mmol) and Et3N (33 mg, 0.32 mmol) in anhydrous DMF (2 mL) at rt was added 4-methyl-3-nitrobenzoyl chloride (84 mg, 0.42 mmol) dropwise. After the mixture was allowed to stir at rt for 12 h, the reaction was quenched with saturated NH4Cl (10 mL). The resulting mixture was extracted with EtOAc, and the organic phase was washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (MeOH/CH2Cl2) to afford compound 15a as a light yellow solid (37 mg, 42%). 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.67 (s, 1H), 8.57 (s, 1H), 8.49 (d, J = 2.4 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.16 (s, 1H), 7.92 (s, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.63 (s, 1H), 7.47–7.43 (m, 1H), 7.34–7.29 (m, 2H), 6.06 (dd, J = 3.9, 5.7 Hz, 1H), 3.07–3.01 (m, 1H), 2.92–2.85 (m, 1H), 2.64–2.57 (m, 4H), 2.09–2.03 (m, 1H). HRMS (ESI+) calcd for C23H21N4O5 (M + H)+ 433.1506 found 433.1509.
- 5-((4-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (15b). White solid, 8%. 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.80 (d, J = 5.4 Hz, 2H), 8.67 (s, 1H), 8.49 (d, J = 3.0 Hz, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.88 (d, J = 4.8 Hz, 2H), 7.62 (s, 1H), 7.50–7.47 (m, 1H), 7.34–7.30 (m, 2H), 6.08–6.04 (m, 1H), 3.08–3.02 (m, 1H), 2.92–2.86 (m, 1H), 2.64–2.57 (m, 1H), 2.10–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1463.
- 5-((6-(4-Methyl-3-nitrobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (16a). Light yellow solid, 19%. 1H NMR (DMSO-d6, 600 MHz) δ 10.48 (s, 1H), 8.67 (s, 1H), 8.56 (d, J = 1.2 Hz, 1H), 8.50 (d, J = 2.4 Hz, 1H), 8.20 (d, J = 6.6 Hz, 1H), 8.16 (s, 1H), 7.90 (s, 1H), 7.85 (s, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.35 (d, J = 9.0 Hz, 1H), 6.05 (dd, J = 3.6, 3.6 Hz, 1H), 3.07–3.01 (m, 1H), 2.92–2.86 (m, 1H), 2.69–2.63 (m, 1H), 2.59 (s, 3H), 2.09–2.04 (m, 1H). HRMS (ESI+) calcd for C23H21N4O5 (M + H)+ 433.1506 found 433.1509.
- 5-((6-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (16b). White solid, 17%. 1H NMR (DMSO-d6, 600 MHz) δ 10.51 (s, 1H), 8.77 (d, J = 6.0 Hz, 2H), 8.67 (s, 1H), 8.49 (d, J = 2.4 Hz, 1H), 8.15 (s, 1H), 7.90 (s, 1H), 7.88–7.83 (m, 3H), 7.73 (d, J = 7.8 Hz, 1H), 7.62 (s, 1H), 7.35 (d, J = 7.8 Hz, 1H), 6.06–6.02 (m, 1H), 3.08–3.02 (m, 1H), 2.94–2.86 (m, 1H), 2.68–2.62 (m, 1H), 2.10–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1458.
- 5-((5-(4-Methyl-3-nitrobenzamido)-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinamide (17). White solid, 16%. 1H NMR (DMSO-d6, 600 MHz) δ 10.14 (s, 1H), 8.66 (s, 1H), 8.58 (s, 1H), 8.50 (s, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.16 (s, 1H), 7.95 (s, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.63 (s, 1H), 7.36–7.30 (m, 2H), 7.29–7.25 (m, 1H), 5.73–5.69 (m, 1H), 2.84–2.76 (m, 1H), 2.67–2.59 (m, 4H), 2.06–1.93 (m, 2H), 1.90–1.81 (m, 1H), 1.80–1.73 (m, 1H). HRMS (ESI+) calcd for C24H23N4O5 (M + H)+ 447.1663 found 447.1667.
- 5-((7-(4-Methyl-3-nitrobenzamido)-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinamide (18). White solid, 15%. 1H NMR (DMSO-d6, 600 MHz) δ 10.41 (s, 1H), 8.66 (s, 1H), 8.55 (s, 1H), 8.51 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.15 (s, 1H), 7.94 (s, 1H), 7.75–7.00 (m, 2H), 7.66 (d, J = 7.2 Hz, 1H), 7.63 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 5.68–5.64 (m, 1H), 2.86–2.79 (m, 1H), 2.76–2.68 (m, 1H), 2.58 (s, 3H), 2.07–1.95 (m, 2H), 1.92–1.83 (m, 1H), 1.82–1.74 (m, 1H). HRMS (ESI+) calcd for C23H21ClN3O3 (M + H)+ 447.1663 found 447.1671.
- (S)-5-((4-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (19a). White solid, 13%. 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.80 (d, J = 5.4 Hz, 2H), 8.67 (s, 1H), 8.49 (d, J = 2.4 Hz, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.88 (d, J = 4.8 Hz, 2H), 7.63 (s, 1H), 7.51–7.47 (m, 1H), 7.35–7.30 (m, 2H), 6.06 (dd, J = 3.6, 6.0 Hz, 1H), 3.08–3.01 (m, 1H), 2.93–2.85 (m, 1H), 2.65–2.56 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1460.
- (S)-5-((4-(4-Morpholinobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (19b). White solid, 5%. 1H NMR (CD3OD, 600 MHz) δ 8.65 (s, 1H), 8.45 (s, 1H), 7.97 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 7.2 Hz,1H), 7.34–7.26 (m, 2H), 7.03 (d, J = 8.4 Hz, 2H), 6.04–5.99 (m, 1H), 3.88–3.78 (m, 4H), 3.35–3.25 (m, 4H), 3.16–3.08 (m, 1H), 3.00–2.92 (m, 1H), 2.72–2.62 (m, 1H), 2.24–2.16 (m, 1H). HRMS (ESI+) calcd for C26H27N4O4 (M + H)+ 459.2027 found 459.2031.
- (S)-5-((4-([1,1′-Biphenyl]-4-ylcarboxamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (19c). White solid, 28%. 1H NMR (DMSO-d6, 600 MHz) δ 10.10 (s, 1H), 8.67 (s, 1H), 8.50 (d, J = 1.8 Hz, 1H), 8.17 (s, 1H), 8.08 (d, J = 8.4 Hz, 2H), 7.92 (s, 1H), 7.84 (d, J = 7.8 Hz, 2H), 7.76 (d, J = 7.8 Hz, 2H), 7.63 (s, 1H), 7.55–7.47 (m, 3H), 7.43 (dd, J = 7.2, 7.2 Hz, 1H), 7.34–7.28 (m, 2H), 6.08–6.04 (m, 1H), 3.10–3.02 (m, 1H), 2.95–2.88 (m, 1H), 2.65–2.56 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C28H24N3O3 (M + H)+ 450.1812 found 450.1812.
- (R)-5-((4-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (20a). White solid, 10%. 1H NMR (DMSO-d6, 600 MHz) δ 10.33 (s, 1H), 8.80 (d, J = 4.2 Hz, 2H), 8.67 (s, 1H), 8.49 (s, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.88 (d, J = 4.2 Hz, 2H), 7.63 (s, 1H), 7.49 (d, J = 4.8 Hz, 1H), 7.35–7.30 (m, 2H), 6.08–6.04 (m, 1H), 3.08–3.01 (m, 1H), 2.93–2.85 (m, 1H), 2.65–2.56 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1465.
- (R)-5-((4-(4-Morpholinobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (20b). White solid, 9%. 1H NMR (DMSO-d6, 600 MHz) δ 9.75 (s, 1H), 8.67 (s, 1H), 8.49 (s, 1H), 8.16 (s, 1H), 7.93–7.85 (m, 3H), 7.63 (s, 1H), 7.46 (d, J = 7.2 Hz, 1H), 7.29–7.23 (m, 2H), 7.03 (d, J = 9.0 Hz, 2H), 6.06–6.02 (m, 1H), 3.79–3.72 (m, 4H), 3.28–3.22 (m, 4H), 3.08–3.00 (m, 1H), 2.91–2.84 (m, 1H), 2.63–2.55 (m, 1H), 2.09–2.02 (m, 1H). HRMS (ESI+) calcd for C26H27N4O4 (M + H)+ 459.2027e found 459.2039.
- (R)-5-((4-([1,1′-Biphenyl]-4-carboxamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (20c). White solid, 5%. 1H NMR (CD3OD, 600 MHz) δ 8.65 (s, 1H), 8.46 (s, 1H), 8.06 (d, J = 7.8 Hz, 2H), 7.99 (s, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.52–7.46 (m, 3H), 7.42–7.39 (m, 1H), 7.38–7.32 (m, 2H), 6.06–6.02 (m, 1H), 3.20–3.13 (m, 1H), 3.03–2.96 (m, 1H), 2.74–2.66 (m, 1H), 2.26–2.19 (m, 1H). HRMS (ESI+) calcd for C28H24N3O3 (M + H)+ 450.1812 found 450.1819.
- (R)-5-((6-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (21a). White solid, 4%. 1H NMR (DMSO-d6, 600 MHz) δ 10.51 (s, 1H), 8.77 (d, J = 6.0 Hz, 2H), 8.67 (s, 1H), 8.50 (s, 1H), 8.16 (s, 1H), 7.90 (s, 1H), 7.88–7.82 (m, 3H), 7.73 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.35 (d, J = 8.4 Hz, 1H), 6.06–6.01 (m, 1H), 3.08–3.01 (m, 1H), 2.94–2.86 (m, 1H), 2.68–2.62 (m, 1H), 2.10–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1461.
- (R)-5-((6-(4-Morpholinobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (21b). White solid, 27%. 1H NMR (DMSO-d6, 600 MHz) δ 9.98 (s, 1H), 8.67 (s, 1H), 8.49 (s, 1H), 8.16 (s, 1H), 7.93–7.83 (m, 4H), 7.72 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 7.8 Hz, 2H), 6.03–5.98 (m, 1H), 3.78–3.71 (m, 4H), 3.27–3.20 (m, 4H), 3.06–2.98 (m, 1H), 2.92–2.83 (m, 1H), 2.68–2.60 (m, 1H), 2.10–2.02 (m, 1H). HRMS (ESI+) calcd for C26H27N4O4 (M + H)+ 459.2027 found 459.2034.
- (R)-5-((6-([1,1′-Biphenyl]-4-ylcarboxamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (21c). White solid, 30%. 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.68 (s, 1H), 8.51 (s, 1H), 8.16 (s, 1H), 8.05 (d, J = 7.8 Hz, 2H), 7.91 (s, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.78–7.73 (m, 3H), 7.63 (s, 1H), 7.51 (dd, J = 7.2, 7.2 Hz, 2H), 7.45–7.40 (m, 1H), 7.34 (d, J = 7.8 Hz, 1H), 6.06–6.02 (m, 1H), 3.08–3.01 (m, 1H), 2.93–2.85 (m, 1H), 2.70–2.62 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C28H24N3O3 (M + H)+ 450.1812 found 450.1820.
- (S)-5-((6-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (22a). White solid, 9%. 1H NMR (CD3OD, 600 MHz) δ 8.72 (d, J = 5.4 Hz, 2H), 8.65 (s, 1H), 8.45 (s, 1H), 7.99–7.96 (m, 1H), 7.87 (d, J = 5.4 Hz, 2H), 7.83 (s, 1H), 7.68–7.64 (m, 1H), 7.35 (d, J = 8.4 Hz, 1H), 6.01–5.97 (m, 1H), 3.17–3.10 (m, 1H), 3.00–2.93 (m, 1H), 2.74–2.67 (m, 1H), 2.26–2.19 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1465.
- (S)-5-((6-(4-Morpholinobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (22b). White solid, 7%. 1H NMR (DMSO-d6, 600 MHz) δ 9.98 (s, 1H), 8.67 (s, 1H), 8.49 (s, 1H), 8.16 (s, 1H), 7.93–7.83 (m, 4H), 7.72 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.03–5.98 (m, 1H), 3.78–3.70 (m, 4H), 3.28–3.21 (m, 4H), 3.06–2.98 (m, 1H), 2.92–2.83 (m, 1H), 2.68–2.60 (m, 1H), 2.10–2.02 (m, 1H). HRMS (ESI+) calcd for C26H27N4O4 (M + H)+ 459.2027 found 459.2037.
- (S)-5-((6-([1,1′-Biphenyl]-4-ylcarboxamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (22c). White solid, 25%. 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.67 (s, 1H), 8.51 (s, 1H), 8.17 (s, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.91 (s, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.78–7.73 (m, 3H), 7.63 (s, 1H), 7.51 (dd, J = 7.5, 7.5 Hz, 2H), 7.45–7.41 (m, 1H), 7.34 (d, J = 7.2 Hz, 1H), 6.06–6.02 (m, 1H), 3.08–3.01 (m, 1H), 2.93–2.85 (m, 1H), 2.70–2.62 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C28H24N3O3 (M + H)+ 450.1812 found 450.1820.
- 4-Nitro-2,3-dihydro-1H-inden-1-one (23a) and 6-Nitro-2,3-dihydro-1H-inden-1-one (23c) [32]. To a solution of 1-indanone (20 g, 151 mmol) in concentrated H2SO4 (210 mL) at 0 °C was added KNO3 (15.3 g, 151 mmol) in several portions. The reaction mixture was allowed to stir for 1 h and poured over ice (1 L). The mixture was extracted with EtOAc, and the organic phase was washed with brine and dried over Na2SO4. After filtration and removal of the solvent, the residue was purified by flash column chromatography (EtOAc/hexanes) to afford 23a as a yellow solid (3.00 g, 23%) and 23c as a yellow solid (4.00 g, 30%). 1H NMR data were consistent with those reported [32].
- 5-Nitro-3,4-dihydronaphthalen-1(2H)-one (23b) and 7-Nitro-3,4-dihydronaphthalen-1(2H)-one (23d). In a manner similar to that described for the preparation of compounds 23a and 23c, tetralone (5.53g, 37.8 mmol) was subjected to a nitration reaction to afford 23b as a yellow solid (1.48 g, 20%). 1H NMR (DMSO-d6, 600 MHz) δ 8.22 (d, J = 7.8 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 7.61 (dd, J = 7.5, 7.5 Hz, 1H), 3.09 (t, J = 6.3 Hz, 2H), 2.68 (t, J = 6.9 Hz, 2H), 2.06 (p, J = 6.3 Hz, 2H). HRMS (ESI−) calcd for C10H8NO3 (M − H)- 190.0510, found 190.0505 and 23d as a yellow solid (4.00 g, 55%). 1H NMR (DMSO-d6, 600 MHz) δ 8.54 (s, 1H), 8.36 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 3.08 (t, J = 5.7 Hz, 2H), 2.70 (t, J = 6.6 Hz, 2H), 2.09 (p, J = 6.3 Hz, 2H). HRMS (ESI−) calcd for C10H8NO3 (M − H)− 190.0510 found 190.0511.
- 4-Nitro-2,3-dihydro-1H-inden-1-ol (24a). To a solution of 23a (1.50 g, 8.47 mmol) in MeOH was added NaBH4 (641 mg, 16.9 mmol) in several portions. The reaction mixture was allowed to stir at rt for 2 h. After the reaction was quenched with 1 N HCl, the aqueous solution was extracted with EtOAc and the organic phase washed with brine and dried over Na2SO4. After filtration and removal of the solvent, the residue was purified by flash column chromatography (EtOAc/hexanes) to afford 24a as a light yellow solid (1.40 g, 92%). 1H NMR (CDCl3, 600 MHz) δ 8.13 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 7.2 Hz, 1H), 7.44 (dd, J = 7.5, 7.5 Hz, 1H), 5.33 (d, J = 6.6 Hz, 1H), 3.60–3.54 (m, 1H), 3.32–3.26 (m, 1H), 2.63–2.56 (m, 1H), 2.06–2.00 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0513.
- 5-Nitro-1,2,3,4-tetrahydronaphthalen-1-ol (24b). In a manner similar to that described for the preparation of compound 24a, 23b (1.35 g, 7.06 mmol) was reduced with NaBH4 to afford 24b as a light yellow solid (1.25 g, 92%). 1H NMR (DMSO-d6, 600 MHz) δ 8.26 (s, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 5.55 (d, J = 6.0 Hz, 1H), 4.66–4.61 (m, 1H), 2.88–2.75 (m, 2H), 2.01–1.94 (m, 1H), 1.93–1.85 (m, 1H), 1.76–1.62 (m, 2H). HRMS (ESI−) calcd for C10H10NO3 (M − H)− 192.0666 found 192.0659.
- (R)-4-Nitro-2,3-dihydro-1H-inden-1-ol (24c). To a solution of compound 23a (1.77 g, 10.0 mmol) and (S)-CBS (139 mg, 0.50 mmol) in CH2Cl2 (30 mL) at –20 °C was slowly added BH3.SMe2 (1.00 mL, 10.0 mmol), and the mixture was stirred at this temperature for 1 h. After the reaction was quenched with saturated NH4Cl (20 mL) and stirred at rt for 3 h, the mixture was extracted with EtOAc. The organic phase was washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/hexanes) to afford compound 24c as a white solid (1.61 g, 90%). [α]D20 = +51.4 (c = 0.43, MeOH). 1H NMR (CDCl3, 600 MHz) δ 8.12 (d, J = 8.4 Hz, 1H), 7.72 (d, J = 7.2 Hz, 1H), 7.43 (dd, J = 7.5, 7.5 Hz, 1H), 5.32 (dd, J = 6.6, 6.6 Hz, 1H), 3.60–3.52 (m, 1H), 3.32–3.24 (m, 1H), 2.63–2.56 (m, 1H), 2.06–2.00 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0512.
- (S)-4-Nitro-2,3-dihydro-1H-inden-1-ol (24d). In a manner similar to that described for the preparation of compound 24c, 23a (1.00 g, 5.65 mmol) was reduced with BH3.SMe2 (0.57 mL, 5.65 mmol) in the presence of (R)-CBS (78 mg, 0.28 mmol) to give 24d as a light yellow solid (900 mg, 89%). [α]D20 = –47.3 (c = 0.75, MeOH). 1H NMR (CDCl3, 600 MHz) δ 8.13 (d, J = 8.4 Hz, 1H), 7.73(d, J = 7.2 Hz, 1H), 7.44 (dd, J = 7.5, 7.5 Hz, 1H), 5.33 (dd, J = 6.6, 6.6 Hz, 1H), 3.60–3.52 (m, 1H), 3.32–3.24 (m, 1H), 2.63–2.56 (m, 1H), 2.06–2.00 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0513.
- 6-Nitro-2,3-dihydro-1H-inden-1-ol (24e). In a manner similar to that described for the preparation of compound 24a, 23c (2.50 g, 14.1 mmol) was reduced with NaBH4 (1.07 g, 28.2 mmol) in MeOH to give 24e as a light yellow solid (2.40 g, 95%). 1H NMR (CDCl3, 600 MHz) δ 8.26–8.22 (m, 1H), 8.14–8.10 (m, 1H), 7.39–7.35 (m, 1H), 5.34–5.28 (m, 1H), 3.16–3.09 (m, 1H), 2.94–2.86 (m, 1H), 2.63–2.56 (m, 1H), 2.09–2.01 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0512.
- 7-Nitro-1,2,3,4-tetrahydronaphthalen-1-ol (24f). In a manner similar to that described for the preparation of compound 24a, 23d (1.50 g, 7.85 mmol) was reduced with NaBH4 to give 24f as a white solid (1.40 g, 92%). 1H NMR (DMSO-d6, 600 MHz) δ 7.76 (d, J = 7.8 Hz, 2H), 7.42 (dd, J = 7.8, 7.8 Hz, 1H), 5.43 (d, J = 5.4 Hz, 1H), 4.65–4.60 (m, 1H), 2.89–2.82 (m, 1H), 2.80–2.73 (m, 1H), 1.96–1.84 (m, 2H), 1.74–1.64 (m, 2H). HRMS (ESI−) calcd for C10H10NO3 (M − H)− 192.0666 found 192.0669.
- (S)-6-Nitro-2,3-dihydro-1H-inden-1-ol (24g). In a manner similar to that described for the preparation of compound 24c, 23c (1.00 g, 5.65 mmol) was reduced with BH3.SMe2 (0.57 mL, 5.65 mmol) in the presence of (R)-CBS (78 mg, 0.28 mmol) to give 24g as a light yellow solid (900 mg, 89%). [α]D20 = +46.1 (c = 0.42, MeOH). 1H NMR (CDCl3, 600 MHz) δ 8.26–8.22 (m, 1H), 8.14 (dd, J = 8.4, 2.4 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H), 5.32 (dd, J = 6.6, 6.6 Hz, 1H), 3.16–3.09 (m, 1H), 2.94–2.86 (m, 1H), 2.64–2.58 (m, 1H), 2.09–2.01 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0513.
- (R)-6-Nitro-2,3-dihydro-1H-inden-1-ol (24h): In a manner similar to that described for the preparation of compound 24c, 23c (1.00 g, 5.65 mmol) was reduced with BH3.SMe2 (0.57 mL, 5.65 mmol) in the presence of (S)-CBS (78 mg, 0.28 mmol) in CH2Cl2 to give 24h (850 mg, 84%) as a light yellow solid. [α]D20 = –46.6 (c = 0.70, MeOH). 1H NMR (CDCl3, 600 MHz) δ 8.26–8.22 (m, 1H), 8.14 (dd, J = 8.4, 2.4 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H), 5.32 (dd, J = 6.6, 6.6 Hz, 1H), 3.16–3.09 (m, 1H), 2.94–2.86 (m, 1H), 2.64–2.58 (m, 1H), 2.09–2.01 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0514.
- Methyl 5-((4-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25a). To a solution of 24a (1.28 g, 7.16 mmol), methyl 5-hydroxynicotinate (1.32 g, 8.59 mmol), and Ph3P (2.82 g, 10.7 mmol) in anhydrous THF (45 mL) at rt was added DIAD (2.17 g, 10.7 mmol) dropwise. After the mixture was stirred for 12 h and the solvent removed, the residue was purified by flash column chromatography (EtOAc/hexanes) to afford 25a as a yellow solid (1.70 g, 76%). 1H NMR (DMSO-d6, 600 MHz) δ 8.74 (s, 1H), 8.61 (d, J = 3.0 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 7.93 (s, 1H), 7.88 (d, J = 6.6 Hz, 1H), 7.59 (dd, J = 7.8, 7.8 Hz, 1H), 6.17 (dd, J = 6.6, 4.2 Hz, 1H), 3.91 (s, 3H), 3.52–3.45 (m, 1H), 3.40–3.34 (m, 1H), 2.71–2.64 (m, 1H), 2.17–2.10 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0983.
- Methyl 5-((5-Nitro-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinate (25b). In a manner similar to that described for the preparation of compound 25a, 24b (468 mg, 2.42 mmol) and methyl 5-hydroxynicotinate (371 mg, 2.42 mmol) were treated with DIAD and Ph3P to afford 25b as a yellow solid (600 mg, 75%). 1H NMR (CDCl3, 600 MHz) δ 8.88 (s, 1H), 8.53 (d, J = 3.0 Hz, 1H), 7.91 (s, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.38 (dd, J = 8.1, 8.1 Hz, 1H), 5.50 (dd, J = 4.8, 4.8 Hz, 1H), 3.98 (s, 3H), 3.17–3.11 (m, 1H), 3.04–2.96 (m, 1H), 2.22–2.15 (m, 1H), 2.12–1.98 (m, 2H), 1.93–1.86 (m, 1H). HRMS (ESI+) calcd for C17H17N2O5 (M + H)+ 329.1132 found 329.1139.
- (S)-Methyl 5-((4-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25c). In a manner similar to that described for the preparation of compound 25a, 24c (500 mg, 2.79 mmol) and methyl 5-hydroxynicotinate (513 g, 3.35 mmol) were treated with DIAD and Ph3P to afford 25c as a yellow solid (580 g, 66%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 7.90 (s, 1H), 7.11 (dd, J = 7.8, 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.2 Hz, 1H), 5.82 (dd, J = 6.6, 3.6 Hz, 1H), 3.96 (s, 3H), 3.00–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.66–2.60 (m, 1H), 2.28–2.22 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0984.
- (R)-Methyl 5-((4-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25d). In a manner similar to that described for the preparation of compound 25a, 24d (500 mg, 2.79 mmol) and methyl 5-hydroxynicotinate (513 g, 3.35 mmol) were treated with DIAD and Ph3P to afford 25d as a yellow solid (560 g, 64%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (s, 1H), 7.90 (s, 1H), 7.11 (dd, J = 7.8, 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.2 Hz, 1H), 5.82 (dd, J = 6.6, 3.6 Hz, 1H), 3.96 (s, 3H), 3.00–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.66–2.60 (m, 1H), 2.28–2.22 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0985.
- Methyl 5-((6-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25e). In a manner similar to that described for the preparation of compound 25a, 25e was prepared from 24e (1.14 g, 6.34 mmol) as a yellow solid (1.30 g, 65%). 1H NMR (CDCl3, 600 MHz) δ 8.89 (s, 1H), 8.54 (d, J = 3.0 Hz, 1H), 8.28 (s, 1H), 8.23 (dd, J = 7.8, 1.8 Hz, 1H), 7.91 (s, 1H), 7.48 (s, 1H), 5.91–5.88 (m, 1H), 3.98 (s, 3H), 3.29–3.23 (m, 1H), 3.11–3.05 (m, 1H), 2.79–2.73 (m, 1H), 2.34–2.28 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0988.
- Methyl 5-((7-Nitro-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinate (25f). In a manner similar to that described for the preparation of compound 25a, 24f (505 mg, 3.30 mmol) and methyl 5-hydroxynicotinate (638 mg, 3.30 mmol) were treated with DIAD and Ph3P to afford 25f as a yellow solid (810 mg, 75%). 1H NMR (CDCl3, 600 MHz) δ 8.89 (s, 1H), 8.55 (d, J = 2.4 Hz, 1H), 8.24 (s, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.92 (s, 1H), 7.35 (d, J = 8.4, 1H), 5.51 (dd, J = 4.8, 4.8 Hz, 1H), 3.98 (s, 3H), 3.05–2.99 (m, 1H), 2.91–2.85 (m, 1H), 2.17–2.10 (m, 2H), 2.08–2.02 (m, 1H), 1.93–1.87 (m, 1H). HRMS (ESI+) calcd for C17H17N2O5 (M + H)+ 329.1132 found 329.1134.
- (R)-Methyl 5-((6-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25g). In a manner similar to that described for the preparation of compound 25a, 24g (550 mg, 3.07 mmol) and methyl 5-hydroxynicotinate (554 g, 3.68 mmol) were treated with DIAD and Ph3P to afford 25g as a yellow solid (629 g, 64%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 3.0 Hz, 1H), 7.89 (dd, J = 2.4, 2.4 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (dd, J = 8.1, 1.8 Hz, 1H), 5.76 (dd, J = 6.0, 4.2 Hz, 1H), 3.97 (s, 3H), 3.08–3.02 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.22–2.14 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0981.
- (S)-Methyl 5-((6-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25h). In a manner similar to that described for the preparation of compound 25a, 24h (700 mg, 3.91 mmol) and methyl 5-hydroxynicotinate (718 g, 4.69 mmol) were treated with DIAD and Ph3P to afford 25h as a yellow solid (800 g, 65%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 7.89 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (d, J = 8.4 Hz, 1H), 5.88–5.74 (m, 1H), 3.97 (s, 3H), 3.08–3.02 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.22–2.14 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0986.
- Methyl 5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26a). To a solution of compound 25a (500 mg, 1.60 mmol) and NiCl2·6H2O (760 mg, 4.48 mmol) in MeOH (800 mL) was slowly added NaBH4 (250 mg, 6.40 mmol), and the mixture was stirred at rt for 2 h. The reaction was quenched with saturated NH4Cl (50 mL) and extracted with EtOAc. The organic phase was washed with water and brine, dried over anhydrous K2CO3, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/hexanes) to afford compound 26a as a white solid (290 mg, 64%). 1H NMR (DMSO-d6, 600 MHz) δ 8.70 (s, 1H), 8.56 (d, J = 3.0 Hz, 1H), 7.88–7.86 (m, 1H), 6.93 (dd, J = 7.8, 7.8 Hz, 1H), 6.58 (d, J = 7.2 Hz, 1H), 6.56 (d, J = 8.4, 1H), 5.95 (dd, J = 6.6, 3.3 Hz, 1H), 5.02 (s, 2H), 3.90 (s, 3H), 2.82–2.77 (m, 1H), 2.69–2.64 (m, 1H), 2.57–2.51 (m, 1H), 2.05–2.00 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1241.
- Methyl 5-((5-Amino-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinate (26b). In a manner similar to that described for the preparation of compound 26a, 25b (737 mg, 2.24 mmol) was reduced to afford 26b as a light yellow solid (620 mg, 94%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.53 (s, 1H), 7.91 (s, 1H), 7.06 (dd, J = 7.8, 7.8 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 8.4 Hz, 1H), 5.45–5.40 (m, 1H), 3.96 (s, 3H), 3.68 (bs, 2H), 2.64–2.57 (m, 1H), 2.49–2.41 (m, 1H), 2.19–2.06 (m, 2H), 2.03–1.95 (m, 1H), 1.94–1.86 (m, 1H). HRMS (ESI+) calcd for C17H19N2O3 (M + H)+ 299.1390 found 299.1400.
- (S)-Methyl 5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26c). In a manner similar to that described for the preparation of compound 26a, 25c (250 mg, 0.80 mmol) was reduced to afford 26c as a light yellow solid (180 mg, 80%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 7.90 (dd, J = 1.8, 1.8 Hz, 1H), 7.11 (dd, J = 7.8, 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.8 Hz, 1H), 5.83 (dd, J = 6.0, 3.6 Hz, 1H), 3.96 (s, 3H), 3.00–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.67–2.60 (m, 1H), 2.28–2.22 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1237.
- (R)-Methyl 5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26d). In a manner similar to that described for the preparation of compound 26a, 25d (250 mg, 0.80 mmol) was reduced to afford 26d as a light yellow solid (160 mg, 70%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (s, 1H), 7.90 (s, 1H), 7.11 (dd, J = 7.8, 7.8 Hz, 1H), 6.85 (d, J = 7.2 Hz, 1H), 6.67 (d, J = 7.8 Hz, 1H), 5.84–5.80 (m, 1H), 3.96 (s, 3H), 3.00–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.67–2.59 (m, 1H), 2.29–2.22 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1241.
- Methyl 5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26e). In a manner similar to that described for the preparation of compound 26a, 25e (500 mg, 1.60 mmol) was reduced to afford 26e as a light yellow solid (270 mg, 59%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (d, J = 1.8 Hz, 1H), 8.53 (d, J = 3.0 Hz, 1H), 7.89 (dd, J = 3.0, 1.8 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (d, J = 2.4 Hz, 1H), 6.69 (dd, J = 8.4, 2.4 Hz, 1H), 5.76 (dd, J = 6.6, 4.2 Hz, 1H), 3.97 (s, 3H), 3.65 (bs, 2H), 3.08–3.02 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.02–1.95 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1243.
- Methyl 5-((7-Amino-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinate (26f). In a manner similar to that described for the preparation of compound 26a, 25f (682 mg, 2.08 mmol) was reduced to afford 26f as a white solid (580 mg, 94%). 1H NMR (CDCl3, 600 MHz) δ 8.83 (s, 1H), 8.53 (s, 1H), 7.90 (s, 1H), 6.96 (d, J = 8.4 Hz, 1H), 6.66–6.61 (m, 2H), 5.38–5.34 (m, 1H), 3.96 (s, 3H), 3.60 (bs, 2H), 2.81–2.75 (m, 1H), 2.71–2.63 (m, 1H), 2.12–2.05 (m, 1H), 2.04–1.92 (m, 2H), 1.81–1.74 (m, 1H). HRMS (ESI+) calcd for C17H19N2O3 (M + H)+ 299.1390 found 299.1390.
- (R)-Methyl 5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26g). In a manner similar to that described for the preparation of compound 26a, 25g (314 mg, 1.00 mmol) was reduced to afford 26g as a yellow solid (200 mg, 70%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 7.89 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (d, J = 8.4 Hz, 1H), 5.77–5.74 (m, 1H), 3.96 (s, 3H), 3.08–3.01 (m, 1H), 2.88–2.81 (m, 1H), 2.64–2.56 (m, 1H), 2.22–2.14 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found285.1239.
- (S)-Methyl 5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26h). In a manner similar to that described for the preparation of compound 25a, 25h (314 mg, 1.00 mmol) was reduced to afford 26h as a yellow solid (220 mg, 77%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (d, J = 1.2 Hz, 1H), 8.52 (d, J = 3.0 Hz, 1H), 7.89 (dd, J = 2.4, 2.4 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (d, J = 1.8 Hz, 1H), 6.69 (dd, J = 7.8, 1.8 Hz, 1H), 5.76 (dd, J = 6.0, 3.6 Hz, 1H), 3.97 (s, 3H), 3.07–3.02 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.21–2.14 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1239.
- 5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27a). A solution of methyl ester 26a (280 mg, 0.98 mmol) in NH3/MeOH (~7 N, 10 mL) in a seal tube was heated at 70 °C for 24 h. After the solvent was evaporated in vacuo, the residue was dissolved in EtOAc (150 mL) and the organic layer was washed with H2O (150 mL) and then with brine (100 mL). After the organic layer was dried over Na2SO4 and filtered, the filtrate was concentrated and the residue was purified by flash column chromatography (5% MeOH/CH2Cl2) to afford compound 27a as a light yellow solid (260 mg, 98%). 1H NMR (DMSO-d6, 600 MHz) δ 8.64 (s, 1H), 8.43 (d, J = 1.8 Hz, 1H), 8.14 (s, 1H), 7.86 (s, 1H), 7.61 (s, 1H), 6.93 (dd, J = 7.5, 7.5 Hz, 1H), 6.58 (d, J = 7.8, 1H), 6.50 (d, J = 7.2 Hz, 1H), 5.90 (dd, J = 6.6, 3.6 Hz, 1H), 5.02 (s, 2H), 2.82–2.77 (m, 1H), 2.68–2.63 (m, 1H), 2.58–2.52 (m, 1H), 2.04–2.00 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1247.
- 5-((5-Amino-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinamide (27b). In a manner similar to that described for the preparation of compound 27a, aminolysis of methyl ester 26b (400 mg, 1.34 mmol) in the presence of CaCl2 (149 mg, 1.34 mmol) afforded 27b as a white solid (320 mg, 84%). 1H NMR (CD3OD, 600 MHz) δ 8.62 (s, 1H), 8.40 (s, 1H), 7.95 (s, 1H), 6.95 (dd, J = 7.5, 7.5 Hz, 1H), 6.71 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.2 Hz, 1H), 5.54–5.48 (m, 1H), 2.67–2.60 (m, 1H), 2.50–2.42 (m, 1H), 2.16–2.01 (m, 2H), 2.00–1.95 (m, 1H), 1.92–1.80 (m, 1H). HRMS (ESI+) calcd for C16H18N3O2 (M + H)+ 284.1394 found 284.1397.
- (S)-5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27c). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26c (180 mg, 0.63 mmol) afforded 27c as a white solid (160 mg, 94%). 1H NMR (CDCl3, 600 MHz) δ 8.57 (d, J = 1.8 Hz, 1H), 8.50 (d, J = 3.0 Hz, 1H), 7.81 (dd, J = 2.4, 2.4 Hz, 1H), 7.11 (dd, J = 7.8 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.2 Hz, 1H), 5.84 (dd, J = 6.6, 3.6 Hz, 1H), 2.99–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.68–2.61 (m, 1H), 2.28–2.22 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1249.
- (R)-5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27d). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26d (160 mg, 0.56 mmol) afforded 27d as a white solid (140 mg, 93%). 1H NMR (CDCl3, 600 MHz) δ 8.59 (d, J = 1.8 Hz, 1H), 8.48 (d, J = 3.0 Hz, 1H), 7.81 (dd, J = 2.4, 2.4 Hz, 1H), 7.11 (dd, J = 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.67 (d, J = 7.8 Hz, 1H), 5.83 (dd, J = 7.2, 3.6 Hz, 1H), 2.98–2.91 (m, 1H), 2.78–2.72 (m, 1H), 2.66–2.60 (m, 1H), 2.27–2.21 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1235.
- 5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27e). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26e (250 mg, 0.88 mmol) afforded 27e as a light yellow solid (230 mg, 97%). 1H NMR (DMSO-d6, 600 MHz) δ 8.65 (s, 1H), 8.45 (d, J = 2.4 Hz, 1H), 8.14 (s, 1H), 7.87 (s, 1H), 7.61 (s, 1H), 6.97 (d, J = 7.8 Hz, 1H), 6.59 (s, 1H), 6.56 (d, J = 7.2, 1H), 5.86 (dd, J = 5.1, 5.1 Hz, 1H), 4.96 (s, 2H), 2.90–2.85 (m, 1H), 2.75–2.69 (m, 1H), 2.57–2.50 (m, 1H), 2.00–1.95 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1237.
- 5-((7-Amino-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinamide (27f). In a manner similar to that described for the preparation of compound 27a, aminolysis of methyl ester 26f (500 mg, 1.73 mmol) in the presence of CaCl2 (192 mg, 1.73 mmol) afforded 27f as a white solid (410 mg, 85%). 1H NMR (DMSO-d6, 600 MHz) δ 8.64 (d, J = 1.8 Hz, 1H), 8.45 (d, J = 3.0 Hz, 1H), 8.14 (s, 1H), 7.89 (dd, J = 1.8, 1.8 Hz, 1H), 7.61 (s, 1H), 6.82 (d, J = 8.4 Hz, 1H), 6.52–6.47 (m, 2H), 5.49 (t, J = 4.8 Hz, 1H), 4.86 (s, 2H), 2.68–2.62 (m, 1H), 2.60–2.53 (m, 1H), 1.96–1.91 (m, 2H), 1.86–1.79 (m, 1H), 1.73–1.66 (m, 1H). HRMS (ESI+) calcd for C16H18N3O2 (M + H)+ 284.1394 found 284.1399.
- (R)-5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27g). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26g (220 mg, 0.77 mmol) afforded 27g as a light yellow solid (180 mg, 86%). 1H NMR (CDCl3, 600 MHz) δ 8.57 (s, 1H), 8.50 (d, J = 3.0 Hz, 1H), 7.81 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (d, J = 8.4 Hz, 1H), 5.77 (dd, J = 4.8, 4.8 Hz, 1H), 3.08–3.01 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.20–2.14 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1240.
- (S)-5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27h). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26h (200 mg, 0.70 mmol) afforded 27h as a light yellow solid (150 mg, 79%). 1H NMR (CDCl3, 600 MHz) δ 8.58 (d, J = 1.2 Hz, 1H), 8.49 (d, J = 3.0 Hz, 1H), 7.80 (dd, J = 2.4, 2.4 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (dd, J = 8.4, 2.4 Hz, 1H), 5.77 (dd, J = 6.0, 4.2 Hz, 1H), 3.07–3.00 (m, 1H), 2.87–2.81 (m, 1H), 2.62–2.55 (m, 1H), 2.20–2.13 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1249.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2007, 2, e784. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, S.H.; Imbesi, M.; Nathan, W.J.; Zou, X.; Zhu, Y.; Jiang, H.; Parisiadou, L.; Gius, D. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid. Redox Signal. 2017, 26, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduino, D.M.; Silva, D.F.; Viana, S.D.; Pereira, F.C.; Cardoso, S.M. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 1440–1462. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target Ther. 2022, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci. Rep. 2016, 6, 35391. [Google Scholar] [CrossRef] [PubMed]
- Pandithage, R.; Lilischkis, R.; Harting, K.; Wolf, A.; Jedamzik, B.; Luscher-Firzlaff, J.; Vervoorts, J.; Lasonder, E.; Kremmer, E.; Knoll, B.; et al. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 2008, 180, 915–929. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Sola-Sevilla, N.; Puerta, E. SIRT2 as a potential new therapeutic target for Alzheimer’s disease. Neural Regen. Res. 2024, 19, 124–131. [Google Scholar] [CrossRef]
- Roshdy, E.; Mustafa, M.; Shaltout, A.E.; Radwan, M.O.; Ibrahim, M.A.A.; Soliman, M.E.; Fujita, M.; Otsuka, M.; Ali, T.F.S. Selective SIRT2 inhibitors as promising anticancer therapeutics: An update from 2016 to 2020. Eur. J. Med. Chem. 2021, 224, 113709. [Google Scholar] [CrossRef] [PubMed]
- Piracha, Z.Z.; Kwon, H.; Saeed, U.; Kim, J.; Jung, J.; Chwae, Y.J.; Park, S.; Shin, H.J.; Kim, K. Sirtuin 2 Isoform 1 Enhances Hepatitis B Virus RNA Transcription and DNA Synthesis through the AKT/GSK-3beta/beta-Catenin Signaling Pathway. J. Virol. 2018, 92, e00955-18. [Google Scholar] [CrossRef] [PubMed]
- Duran-Castells, C.; Llano, A.; Kawana-Tachikawa, A.; Prats, A.; Martinez-Zalacain, I.; Kobayashi-Ishihara, M.; Oriol-Tordera, B.; Pena, R.; Galvez, C.; Silva-Arrieta, S.; et al. Sirtuin-2, NAD-Dependent Deacetylase, Is a New Potential Therapeutic Target for HIV-1 Infection and HIV-Related Neurological Dysfunction. J. Virol. 2023, 97, e0165522. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.L.; Remiszewski, S.; Todd, M.J.; Kulp, J.L., 3rd; Tang, L.; Welsh, A.V.; Barry, A.P.; De, C.; Reiley, W.W.; Wahl, A.; et al. An allosteric inhibitor of sirtuin 2 deacetylase activity exhibits broad-spectrum antiviral activity. J. Clin. Investig. 2023, 133, e158978. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, W.; Zhang, J.; Li, L.; Wan, Y.; Tang, X.; Chen, X.; Liu, S.; Yao, X. Tenovin-1 inhibited dengue virus replication through SIRT2. Eur. J. Pharmacol. 2021, 907, 174264. [Google Scholar] [CrossRef]
- Zhou, Z.; Ma, T.; Zhu, Q.; Xu, Y.; Zha, X. Recent advances in inhibitors of sirtuin1/2: An update and perspective. Future Med. Chem. 2018, 10, 907–934. [Google Scholar] [CrossRef]
- Yang, W.; Chen, W.; Su, H.; Li, R.; Song, C.; Wang, Z.; Yang, L. Recent advances in the development of histone deacylase SIRT2 inhibitors. RSC Adv. 2020, 10, 37382–37390. [Google Scholar] [CrossRef]
- Penteado, A.B.; Hassanie, H.; Gomes, R.A.; Silva Emery, F.D.; Goulart Trossini, G.H. Human sirtuin 2 inhibitors, their mechanisms and binding modes. Future Med. Chem. 2023, 15, 291–311. [Google Scholar] [CrossRef]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Olah, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Olah, J.; Gerhardt, S.; Ovadi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure-Activity Relationship Study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef]
- Suzuki, T.; Khan, M.N.; Sawada, H.; Imai, E.; Itoh, Y.; Yamatsuta, K.; Tokuda, N.; Takeuchi, J.; Seko, T.; Nakagawa, H.; et al. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J. Med. Chem. 2012, 55, 5760–5773. [Google Scholar] [CrossRef] [PubMed]
- Sundriyal, S.; Moniot, S.; Mahmud, Z.; Yao, S.; Di Fruscia, P.; Reynolds, C.R.; Dexter, D.T.; Sternberg, M.J.; Lam, E.W.; Steegborn, C.; et al. Thienopyrimidinone Based Sirtuin-2 (SIRT2)-Selective Inhibitors Bind in the Ligand Induced Selectivity Pocket. J. Med. Chem. 2017, 60, 1928–1945. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Price, I.R.; Bai, J.J.; Lin, H. A Glycoconjugated SIRT2 Inhibitor with Aqueous Solubility Allows Structure-Based Design of SIRT2 Inhibitors. ACS Chem. Biol. 2019, 14, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Mellini, P.; Itoh, Y.; Tsumoto, H.; Li, Y.; Suzuki, M.; Tokuda, N.; Kakizawa, T.; Miura, Y.; Takeuchi, J.; Lahtela-Kakkonen, M.; et al. Potent mechanism-based sirtuin-2-selective inhibition by an in situ-generated occupant of the substrate-binding site, “selectivity pocket” and NAD(+)-binding site. Chem. Sci. 2017, 8, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.S.; Hong, J.Y.; Cao, J.; Lu, X.; Price, I.R.; Zhao, Q.; Kosciuk, T.; Yang, M.; Bai, J.J.; Lin, H. Novel Lysine-Based Thioureas as Mechanism-Based Inhibitors of Sirtuin 2 (SIRT2) with Anticancer Activity in a Colorectal Cancer Murine Model. J. Med. Chem. 2019, 62, 4131–4141. [Google Scholar] [CrossRef]
- Chen, S.; Zheng, Y.; Liang, B.; Yin, Y.; Yao, J.; Wang, Q.; Liu, Y.; Neamati, N. The application of PROTAC in HDAC. Eur. J. Med. Chem. 2023, 260, 115746. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Olah, J.; Ovadi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Hong, J.Y.; Jing, H.; Price, I.R.; Cao, J.; Bai, J.J.; Lin, H. Simultaneous Inhibition of SIRT2 Deacetylase and Defatty-Acylase Activities via a PROTAC Strategy. ACS Med. Chem. Lett. 2020, 11, 2305–2311. [Google Scholar] [CrossRef]
- Cui, H.; Kamal, Z.; Ai, T.; Xu, Y.; More, S.S.; Wilson, D.J.; Chen, L. Discovery of potent and selective sirtuin 2 (SIRT2) inhibitors using a fragment-based approach. J. Med. Chem. 2014, 57, 8340–8357. [Google Scholar] [CrossRef]
- Ai, T.; Wilson, D.J.; More, S.S.; Xie, J.; Chen, L. 5-((3-Amidobenzyl)oxy)nicotinamides as Sirtuin 2 Inhibitors. J. Med. Chem. 2016, 59, 2928–2941. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Pavletich, N.P. Structure of the histone deacetylase SIRT2. Nat. Struct. Biol. 2001, 8, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.L.; Cochran, F.R.; Kelley, J.L.; McLean, E.W.; Selph, J.L.; Rigdon, G.C.; Orr, G.F.; Davis, R.G.; Cooper, B.R.; Styles, V.L.; et al. Indanylidenes. 1. Design and synthesis of (E)-2-(4,6-difluoro-1-indanylidene)acetamide, a potent, centrally acting muscle relaxant with antiinflammatory and analgesic activity. J. Med. Chem. 2003, 46, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Helal, C.J. Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew. Chem. Int. Ed. Engl. 1998, 37, 1986–2012. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
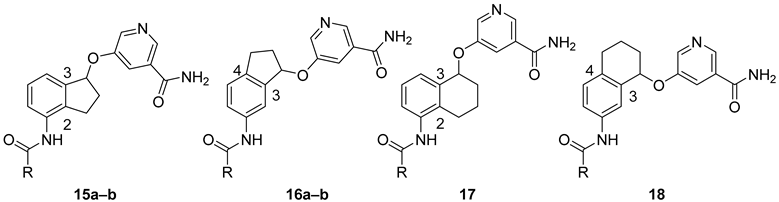 | ||||
| Compound | R | IC50 (µM) | ||
| SIRT1 | SIRT2 | SIRT3 | ||
| 7a b | (4-Me-3-NO2)-Ph | 7.14 | 0.107 | 6.39 |
| 7b b | 4-Py | 2.46 | 0.0341 | 2.40 |
| 15a | (4-Me-3-NO2)-Ph | 25.6 | 0.281 | 4.36 |
| 15b | 4-Py | 9.99 | 0.0503 | 2.86 |
| 16a | (4-Me-3-NO2)-Ph | 23.6 | 0.540 | 16.0 |
| 16b | 4-Py | 36.6 | 0.109 | 11.6 |
| 17 | (4-Me-3-NO2)-Ph | 23.9 | 3.28 | 15.0 |
| 18 | (4-Me-3-NO2)-Ph | >200 | 3.00 | 32.8 |
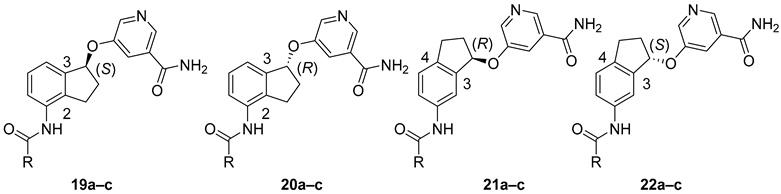 | ||||
| Compound | R | Kiapp (µM) | ||
| SIRT1 | SIRT2 | SIRT3 | ||
| 7b b | 4-Py | 3.0 ± 0.1 | 0.14 ± 0.02 | 7.2 ± 0.9 |
| 7c b | (4-morpholino)-Ph | 5.0 ± 1.2 | 0.073 ± 0.011 | 5.4 ± 1.1 |
| 7d | (4-Ph)-Ph | >100 | 0.217 ± 0.033 | >100 |
| 19a | 4-Py | 15.9 ± 1.3 | 0.147 ± 0.016 | 12.3 ± 1.5 |
| 19b | (4-morpholino)-Ph | 11.0 ± 1.3 | 0.132 ± 0.009 | 5.40 ± 0.50 |
| 19c | (4-Ph)-Ph | >100 | 0.064 ± 0.008 | >100 |
| 20a | 4-Py | 43.5 ± 2.7 | 0.717 ± 0.061 | 14.8 ± 1.1 |
| 20b | (4-morpholino)-Ph | 45.8 ± 3.0 | 0.845 ± 0.055 | 18.4 ± 1.2 |
| 20c | (4-Ph)-Ph | >100 | 0.451 ± 0.043 | >100 |
| 21a | 4-Py | >100 | 1.44 ± 0.18 | 100 ± 12 |
| 21b | (4-morpholino)-Ph | >100 | 0.863 ± 0.087 | >100 |
| 21c | (4-Ph)-Ph | >100 | 0.895 ± 0.169 | >100 |
| 22a | 4-Py | >100 | 1.11 ± 0.11 | 50.8 ± 4.2 |
| 22b | (4-morpholino)-Ph | >100 | 0.994 ± 0.114 | 88.3 ± 13.9 |
| 22c | (4-Ph)-Ph | >100 | 0.906 ± 0.183 | >100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ai, T.; Wilson, D.J.; Chen, L. 5-((3-Amidobenzyl)oxy)nicotinamides as SIRT2 Inhibitors: A Study of Constrained Analogs. Molecules 2023, 28, 7655. https://doi.org/10.3390/molecules28227655
Ai T, Wilson DJ, Chen L. 5-((3-Amidobenzyl)oxy)nicotinamides as SIRT2 Inhibitors: A Study of Constrained Analogs. Molecules. 2023; 28(22):7655. https://doi.org/10.3390/molecules28227655
Chicago/Turabian StyleAi, Teng, Daniel J. Wilson, and Liqiang Chen. 2023. "5-((3-Amidobenzyl)oxy)nicotinamides as SIRT2 Inhibitors: A Study of Constrained Analogs" Molecules 28, no. 22: 7655. https://doi.org/10.3390/molecules28227655
APA StyleAi, T., Wilson, D. J., & Chen, L. (2023). 5-((3-Amidobenzyl)oxy)nicotinamides as SIRT2 Inhibitors: A Study of Constrained Analogs. Molecules, 28(22), 7655. https://doi.org/10.3390/molecules28227655