Abstract
D-Glucuronic acid is a fundamental building block of many biologically important polysaccharides, either in its non-substituted form or bearing a variety of substituents, among them sulfates. We have previously performed a study of the effects of exhaustive sulfation on the conformational behavior of β-gluronopyranosides. Herein, we report an investigation comparing α- and β-derivatives of this monosaccharide within the title disaccharides using NMR and quantum chemistry approaches. It was found that for α-linked disaccharides, the introduction of sulfates did not greatly affect their conformational behavior. However, for β-derivatives, considerable conformational changes were observed. In general, they resemble those that took place for the monosaccharides, except that NOESY experiments and calculations of intra-ring spin–spin coupling constants suggest the presence of a 1S5 conformer along with 3S1 in the fully sulfated disaccharide. During the synthesis of model compounds, hydrogen bond-mediated aglycone delivery was used as an α-directing stereocontrol approach in the glucuronidation reaction.
1. Introduction
d-Glucuronic acid is an important building block which can be found in a great number of natural compounds [1,2,3], some of them with promising biological activity profiles [4,5]. In these compounds, GlcA can be found in both α- and β-form. Glycosaminoglycans (chondroitin sulfates [6], heparan sulfates [7], hyaluronic acid [8], fucosylated chondroitin sulfates [9,10]) are one of the most promising and intensively investigated classes bearing β-d-GlcA residues. Previously, we encountered an interesting unit in fucosylated chondroitin sulfate, isolated from the sea cucumber Eupentacta fraudatrix [11]. Its structure was determined as →4)-β-d-GlcpA2S3S-(1→3)-β-d-GalpNAc6S-(1→ (Figure 1), and its GlcA residue’s chemical shifts and coupling constants were quite unusual. They differed from the spectral data of similar polysaccharides derived from Cucumaria frondosa [12], C. djakonovi [13], and C. japonica [14], which contained the same fragment but were either without sulfates or monosulfated at O-3. In order to gain insight into the biological importance of these unusual structures? we performed a thorough investigation of the conformation of a fully sulfated β-d-GlcA residue employing a series of synthetic monosaccharides [15]. The obtained data clearly indicated that two skew-boat conformers, OS2 and 3S1, provided a significant contribution to the conformational equilibrium and could, in turn, affect intramolecular interactions.
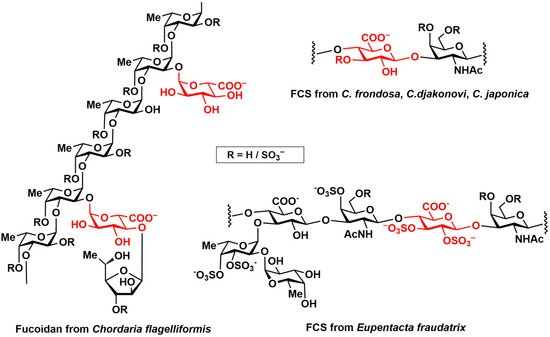
Figure 1.
Fragments of polysaccharides bearing GlcA units.
Although the α-d-GlcA unit is more rarely encountered in natural compounds than the β-isomer, some of its compounds have prospects for practical application. In particular, fucoidans (polysaccharides with a backbone built of α-L-fucose residues) are already being actively investigated as potential medical agents [16]. There are a few known examples of fucoidans bearing α-d-GlcA as a side chain [17,18,19,20]. The structure of one of them, fucoidan isolated from the seaweed Chordaria flagelliformis, is shown in Figure 1. In natural fucoidans, the GlcA units, unlike fucose ones, do not bear any sulfate groups. It is noteworthy that chemical sulfation significantly changes the effect of modified fucoidans on blood coagulation and platelet aggregation [21]. This is probably related to the increased charge density, but conformational changes should not be discarded as an explanation at once.
In order to investigate this possibility, in the present study the scope of objects has been expanded to include disaccharides with glucuronic acid as the glycosylating residue. Both α- and β-configurations are investigated. The model compounds 1–6 used in this work are shown in Figure 2. During the synthesis, we also tried to address the problem of α-directing stereocontrol in the glucuronidation reaction. We performed a series of model glycosylations varying the substituent at O-4 in the glucuronic donors to examine the application of the hydrogen bond-mediated aglycone delivery (HAD) stereocontrol approach using the picoloyl protecting group suggested by Yasomanee and Demchenko in 2012 [22].
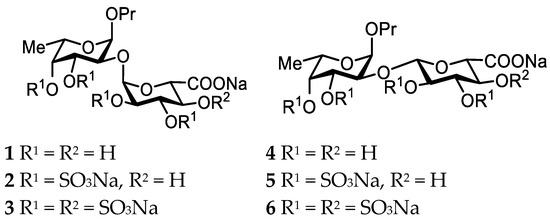
Figure 2.
Model compounds studied in this work.
2. Results and Discussion
In order to carry out NMR and conformational studies, we synthesized six model compounds—α-linked disaccharides 1–3 and β-linked disaccharides 4–6. For both types of linkages, we obtained non-sulfated, fully sulfated, and partially sulfated disaccharides. As a model acceptor, allyl α-L-fucopyranoside 7 bearing 3,4-O-isopropylidene protection was chosen. For the synthesis of glucuronic acid donors, we used commercially available β-d-glucose pentaacetate 8. In the first step, 5-tert-butyl-2-methylbenzenethiol (MTBTP) was introduced in the anomeric position and the product was treated with MeONa in methanol followed by the installation of 4,6-para-methoxybenzylidene protection to give diol 9 with good yield (72%). The latter was benzylated with NaH and BnBr in DMF, the diol protection was cleaved in acidic conditions, and the primary hydroxyl group was oxidized with TEMPO and BAIB in a DCM/water mixture. The uronic acid intermediate product was treated with MeI and K2CO3 in DMF to give methyl ester 10 with an overall yield of 46%. The remaining hydroxyl group was substituted with TBS, picoloyl, or benzoyl protection. Then, the anomeric thiophenol was cleaved employing NBS in acetone/water, followed by treatment with TCAN and cesium carbonate in DCM to give trichloroacetimidate donors 11–13 with moderate yields of 52–69% (Scheme 1).
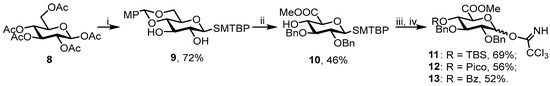
Scheme 1.
Synthesis of glucuronic acid donors 11–13. Reagents and conditions: i: 1. MTBTP, BF3·Et2O, DCM, 2. MeONa, MeOH, 3. 4-anisaldehyde, HC(OMe)3, CSA, CH3CN; ii: 1. BnBr, NaH, DMF, 2. TFA, 3. BAIB, TEMPO, DCM/H2O 5:1, 0 °C, 4. MeI, K2CO3, DMF; iii: a. TBSOTf, NEt3, DCM; b. PicoA, DCC, DMAP, DCM; c. BzCl, NEt3, DCM iv: 1. NBS, H2O/acetone, 2. TCAN, Cs2CO3, DCM.
Efficient α-directing stereocontrol in glucuronidation reactions is still a challenging problem [23,24]. Early on in the synthesis of fucoidan-related oligosaccharides bearing α-d-GlcA units [25], we used 2,3-di-O-benzyl-4-O-tert-butyldimethylsilyl 11 and 2,3,4-tri-O-benzyl 15 trichloroacetimidate donors and achieved α:β ratios of 4:1 and 5:1, respectively (Scheme 2a). In this model glycosylation, we decided to employ the HAD concept, based on a preliminary coordination between donor and acceptor due to hydrogen bond formation [22]. This is the second attempt to elaborate this approach on uronic donors. Previously, Demchenko employed it to obtain a β-(1→3)-linked mannuronic tetrasaccharide [26].
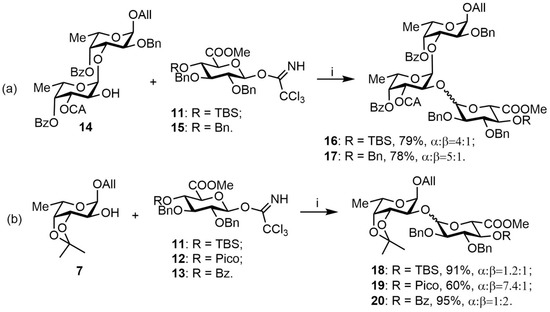
Scheme 2.
Model glycosylations with glycosyl acceptors 14 (a) and 7 (b). Reagents and conditions: i: TMOSTf, MS-4Å, DCM.
Out of our three trichloroacetimidate donors bearing 4-O-TBS-, 4-O-Pico-, and 4-O-Bz-protecting groups (11, 12, 13, respectively), the first one was chosen to connect our new dataset with the results obtained earlier, while the third one was used as a non-coordinating analogue of the picoloyl group. All new glycosylation reactions were performed at the same conditions with 2 h preliminary stirring, which is required for coordination between donor and acceptor. The obtained results clearly indicate that the HAD approach is a prospective method for α-directing glycosylation stereocontrol for our GlcA-donors—the α:β ratio in the case of 4-O-picoloyl donor 12 was determined to be 7.4:1 compared to 1.2:1 for 4-O-TBS donor 11 and 1:2 for 4-O-Bz donor 13 (Scheme 2b). However, the yield in case of the HAD approach was significantly lower (60%). We anticipate that in the case of oligosaccharides and more hindered acceptors, the ratio would be even more favorable towards α-glycosylation, based on the comparison of acceptors 14 and 7 as glycosylated by donor 11.
For the subsequent synthesis of model compounds 1–6, only disaccharide 20 was used (Scheme 3). After cleavage of 3,4-O-isopropylidene protection in acidic conditions followed by hydrogenolysis, α- and β-isomers were separated to give tetraols 21a and 21b with an overall yield of 80% for both isomers. Final saponification of 21a and 21b with LiOH 1 M gave disaccharides 1 (74%) and 4 (70%), respectively. Treatment of 1 and 4 with Py·SO3 in DMF gave fully sulfated compounds 3 (72%) and 6 (78%). To obtain the last couple of disaccharides, 21a and 21b were on the first step treated with Py·SO3 in DMF, followed by saponification with LiOH to give partially sulfated disaccharides 2 (64%) and 4 (57%).
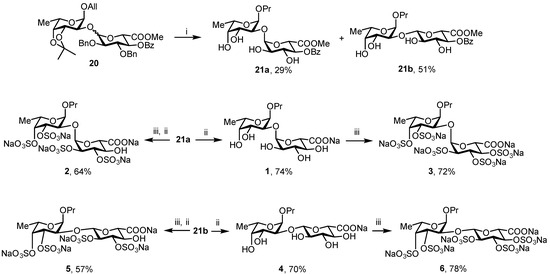
Scheme 3.
Synthesis of model compounds 1–6. Reagents and conditions: i: 1. TFA, DCM, 2. H2, Pd/C, EtOAc/MeOH. ii: LiOH 1 M, H2O; iii: 1. Py·SO3, DMF, 2. NaHCO3, MeOH.
The measured 1H NMR chemical shifts for the studied compounds 1–6 are given in Table 1. Inter-proton coupling constants for glucuronic acid residues (GlcA) are shown in Table 2 (coupling constants for fucose residues can be found in Section 3).

Table 1.
1H chemical shifts (ppm) observed for compounds 1–6.
Table 2.
Intra-ring coupling constants (Hz) and peak multiplicity in the glucuronic acid residues of compounds 1–6.
Preliminary analysis of chemical shifts for GlcA residues does not demonstrate any unexpected changes upon the introduction of sulfate groups in the α-series (structures 1–3). However, some decrease in coupling constants can be observed for compound 3 (Table 2). For the β-series, the situation is different, resembling that which was observed in the case of GlcA monosaccharides. When comparing structures 4 and 6, it can be seen that drastic changes both in chemical shifts and coupling constants occur to protons H-3, H-4, and H-5. Besides the sulfation effect caused by the introduction of the electronegative sulfate group, they may arise from conformational changes. To explain the observed effects and in continuation of our ongoing computer studies of carbohydrate conformations [27,28], a DFT analysis of the studied glucuronides was performed.
First, relaxed potential energy scans of ψ torsions of the glycosidic linkages were performed for both disaccharides at B3LYP/6-31G(d,p) level of theory. The scanned range was (−60°…+60°) with a 20° step. The φ angles were allowed to change during the constrained optimizations. This was in order to save computation time. This approach is justified since the behavior of the φ angles is mostly governed by the exo-anomeric effect and they have a more restricted range to change within. Since in our previous investigations the non-sulfated glucuronic acid residue was shown to exist exclusively in the 4C1 conformation, it was the only one considered for compounds 1 and 4.
The resulting graphs are presented in Figure 3. They demonstrate that in the α-series, the 4C1 conformer always retains the lowest energy (structures 1–3). However, in structure 5, the 1S5 conformer has energy comparable to that of 4C1, and in structure 6 it seems to dominate along with 3S1. The minima found during these scans for the sulfated structures were reoptimized using M062X/def2-tzvp approximation. The resulting relative energies are shown in Table 3.
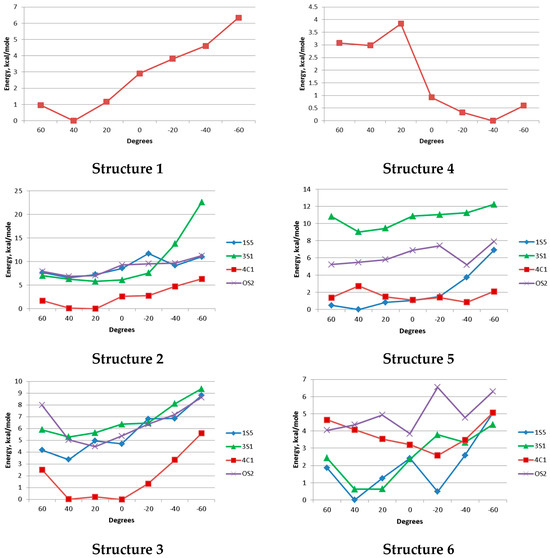
Figure 3.
Scan graphs of the glycosidic linkages in structures 1–6 obtained at B3LYP/6-31G(d,p) level.
Table 3.
Relative energies for sulfated structures 2, 3, 5, and 6 obtained after M062X/def2-tzvp reoptimization.
The values shown in Table 3 confirm the tendency described above. While for disaccharides from the α-series the 4C1 conformer always has much lower energy than any skew-boat, for compound 5 and especially 6, 3S1 and 1S5 become more preferred. This observation is additionally supported by 2D NOESY spectroscopy (Figure 4A, see full range NOESY spectra in Supplementary Materials). While in the case of non-sulfated β-disaccharide 4 cross-peaks corresponding to H-1/H-5 and H-1/H-3 intra-ring interactions in GlcA residue can be observed, they disappear in the spectrum of the fully sulfated compound 6. Interestingly, an H-1/H-5 interaction was present in the case of monosaccharides. Here, its absence might be explained by the contribution from the 1S5 conformer where the H-1 and H-5 protons have a distance of about 3.6 Å between them due to the equatorial orientation of H-1 (Figure 4B).
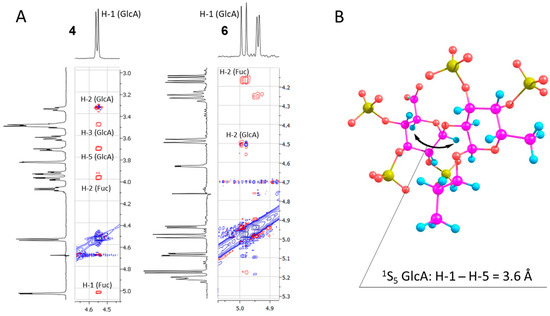
Figure 4.
Fragments of 2D NOESY spectra of model disaccharides 4 and 6 (A). 1S5 conformer of compound 6, atoms representation: hydrogen—cyan, carbon—magenta, oxygen—red, and sulfur—yellow (B).
Chemical shieldings were calculated for all conformers of structure 6 (Table 4). We used this work as an opportunity to test a recent DLPNO-MP2 method developed by the ORCA team [29] in its application to calculate NMR shielding constants for such highly sulfated systems. It was found that in the 3S1 conformer deshielding of H-3, H-4 and H-5 is present, while in 1S5 deshielding of H-4 occurs. This can serve as an explanation to the fact that lower field chemical shifts are observed in the NMR spectra of these compounds.
Table 4.
Chemical shielding constants (ppm) calculated for different conformers of molecule 6.
Additionally, intra-ring 1H-1H spin–spin coupling constants (J) were calculated for the revealed conformers of molecule 6. The calculation was performed at the B3LYP/pcJ-2 level of theory. The results are presented in Table 5.
Table 5.
Intra-ring 1H-1H spin–spin coupling constants (J, Hz) calculated for different conformers of molecule 6.
In the experimental spectra (Table 2), those J-values that could be definitely determined demonstrated a tendency towards a decrease in H-1/H-2, H-2/H-3, and H-4/H-5 interactions. From Table 5, it can be seen that in all the skew-boat conformers some constants also become lower as compared to the 4C1 conformer. Obviously, the experimentally observed J-values pattern does not correspond to a single conformer, as was also the case in the glucuronic acid monosaccharide. Thus, in the studied β-disaccharides, glucuronic acid retains conformational flexibility upon the introduction of sulfates and exhibits a prevalence of skew-boat conformers.
3. Materials and Methods
3.1. General Information
In this study, commercially purchased chemicals were used without purification unless noted. Standard procedures were used for the distilling and drying of solvents when necessary (DCM, MeOH, acetone). Some solvents were purchased as dry (DMF, THF, CH3CN, Sigma-Aldrich, Darmstadt, Germany) and used without additional manipulations. Dry solvents under argon atmosphere were used for chemical transformations involving air- or moisture-sensitive reagents. Aluminum sheets coated with silica gel 60 F254 (Merck) were used for thin-layer chromatography (TLC). The TLC plates were analyzed by UV light (λ = 254 nm) and developed with a mixture of 15% H3PO4 and orcinol (1.8 g/L) in EtOH/H2O (95:5, v/v) followed by heating (200 °C). Column chromatography was performed using Silica Gel 60 (40–63 μm, E. Merck). Solvents for column and thin layer chromatography are listed in volume-to-volume ratios. Sephadex G-15 column (400 × 16 mm) by elution with water at a flow rate of 0.5 mL/min was used for size-exclusion chromatography.
NMR spectra were measured on Bruker AM300 (300 MHz) or AV600 (600 MHz) spectrometers equipped with 5 mm pulsed-field-gradient (PFG) probes at temperatures denoted in the spectra in supplementary. Microtubes (Shigemi, Inc., Tokyo, Japan) were applied for sensitivity enhancement of small concentration probes. The resonance assignment in 1H and 13C NMR spectra was performed using various 2D experiments (e.g., COSY, NOESY, HSQC). Chemical shifts are reported in ppm referenced either to the solvent residual peaks as standard for chloroform (δ 7.26 1H, δ 77.16 13C) or methanol as internal standard for D2O (δ 3.34 1H, δ 49.50 13C). The ratio between isomers in glycosylation reactions was measured based on the ratio of integral intensities between GlcA residues’ anomeric correlations in HSQC spectrum.
Optical rotations were measured using a JASCO P-2000 polarimeter at ambient temperature (22–25 °C).
Bruker micrOTOF II instrument was used to record high-resolution mass spectra (HR MS) with electrospray ionization (ESI). The positive ion mode (interface capillary voltage −4500 V) or the negative ion mode (3200 V) were used; mass range was measured from m/z 50 to m/z 3000 Da; calibration was performed with Electrospray Calibrant Solution (Fluka). A syringe sample loading was apply for solutions in a mixture of acetonitrile and water (50:50, v/v, flow rate 3 μL/min). Nitrogen was used as a dry gas; interface temperature was set at 180 °C.
Geometry optimizations were performed using ORCA 5.0.2 program. RKS approximation with 6-31G(d,p) [30] basis set and D3 dispersion correction was employed for initial scans. Sulfates in the studied structures were treated as anions alone. CPCM model was applied with the built-in parameters for water. Geometry optimizations were performed until the RMS gradient reached a value less than 10−4. Further optimizations were conducted using M062X functional with def2-tzvp [31,32,33] basis set. Chemical shifts were calculated using DLPNO-MP2 approximation and gauge-independent atomic orbital approximation with PCSSEG-2 basis set, as described in work [34]. Spin–spin couplings were calculated using DFT approach with B3LYP functional and pcJ-2 [35] basis set. Only Fermi contact terms were considered.
3.2. Standard Procedures
3.2.1. Substitution Thiophenyl with Trichloroacetimidate (Standard Procedure A)
To a solution of thioglycoside [0.2 mmol] in a mixture of acetone [1.8 mL] and H2O [0.2 mL] at 0 °C, NBS [0.6 mmol, 3 eq.] was added. Reaction mixture was stirred at 0 °C for 2 h. After, TLC showed full consumption of the 5 eq.
Starting material of the reaction mixture was diluted with EtOAc [50 mL] and washed with Na2S2O3 1 M [50 mL] and H2O [50 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc) to give hemiacetal. The latter was dissolved in DCM [2 mL], and Cs2CO3 [0.04 mmol, 0.2 eq.] and CCl3CN [0.24 mmol, 1.2 eq.] were added. Reaction mixture was stirred for 20 min. After TLC showed full consumption of the starting material, the mixture was purified by chromatography (silica gel neutralized with NEt3, eluent: hexane/EtOAc) to give a trichloroacetimidate product.
3.2.2. Glycosylation (Standard Procedure B)
A mixture of a glycosyl acceptor [0.16 mmol] and a glycosyl donor [0.16 mmol, 1 eq.] was co-evaporated with toluene twice and dried for 1 h in vacuo, and then the flask was filled with Ar. Freshly activated molecular sieves 4 Å [100 mg] and anhydrous DCM [2 mL] were added and the mixture was stirred for 2 h at rt. Then, the solution of TMSOTf in DCM [10% v/v, 60 μL, 0.2 eq.] was added at −50 °C under argon protection. The mixture was stirred under argon protection until TLC showed full consumption of the starting materials, and then neutralized with Et3N and the mixture was purified by chromatography (silica gel, eluent: hexane/EtOAc) to give a glycosylation product as a mixture of α- and β-isomers.
3.2.3. Saponification (Standard Procedure C)
To a solution of disaccharide [0.02 mmol] in H2O [0.5 mL], 1 M solution of LiOH in H2O [0.3 mL] was added at 0 °C. The reaction mixture was kept at 0 °C until TLC showed formation of the final product and then quenched with 0.5 M AcOH and evaporated to dryness. Deprotected disaccharide was isolated from the reaction mixture by gel chromatography on a Sephadex G-15 column with water elution followed by lyophilization to give the desired product.
3.2.4. Sulfation (Standard Procedure D)
To a solution of a deprotected disaccharide [0.02 mmol] in DMF [1 mL], complex Py·SO3 [16–20 eq., 4 eq.×-OH] was added. The reaction mixture was kept at rt overnight, and then quenched with excess of NaHCO3 [5 eq.×-OH] and MeOH [0.4 mL]. The mixture was stirred for 3 h and evaporated to dryness. The product was purified either by chromatography (silica gel: DCM/MeOH) or by gel chromatography on a Sephadex G-15 column to give the desired product.
3.3. Synthesis of Compounds 9, 10, 11, 12, 13, 18, 19, 20, 21a, 21b, 1, 2, 3, 4, 5, and 6
3.3.1. (2-Methyl-5-tert-butylphenyl) 4,6-O-(4-methoxybenzylidene)-1-thio-β-d-glucopyranoside (9)
To a solution of glucose β-d-pentaacetate 8 [2 g, 5.1 mmol] in DCM [20 mL], MTBTP [1.32 mL, 1.4 eq.] was added. The mixture was cooled to 0 °C and BF3·Et2O [1.26 mL, 2 eq.] was added dropwise. The reaction mixture was allowed to warm to rt and stirred for 2 h. After 2 h, TLC [hexane/EtOAc = 1:1] showed full consumption of the starting material, and the reaction mixture was neutralized with Et3N, diluted with EtOAc [150 mL], and washed with H2O [2 × 100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The crude thioglycoside was dissolved in MeOH [10 mL] and 1 M solution of MeONa in MeOH was added [1 mL]. After 20 min, TLC [EtOAc] showed formation of the final product and the reaction mixture was neutralized with ion exchange resin IR-120 in H+ form. The resin was filtered off and the filtrate was concentrated in vacuo. The crude residue was dissolved in CH3CN [15 mL] and CSA was added [150 mg, 10 mg/mL] followed by HC(OMe)3 [0.84 mL, 1.5 eq.] and 4-anisaldehyde [0.81 mL, 1.3 eq.]. After 30 min, TLC showed full consumption of the staring material, and the reaction mixture was diluted with EtOAc [150 mL] and washed with NaHCO3 [100 mL] and H2O [100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: toluene/EtOA c = 10:1→3:1) to give 9 (1.68 g, 72%) as white crystals. Rf = 0.4 (toluene/EtOAc = 2:1). [α]D = −41.9° (c = 1, EtOAc). 1H NMR (300 MHz, CDCl3): δ 7.61 (d, 1H, J = 2.0 Hz, SMTBP), 7.41 (d, 2H, J = 8.7 Hz, o-MPh), 7.28–7.20 (m, 1H, SMTBP), 7.15 (d, 1H, J = 8.0 Hz, SMTBP), 6.88 (d, 2H, J = 8.8 Hz, m-MPh), 5.48 (s, 1H, CHMPh), 4.62 (d, 1H, J1–2 = 9.8 Hz, H-1), 4.38–4.27 (m, 1H, H-6), 3.87–3.69 (m, 5H, H-3. H-6′, OCH3), 3.58–3.43 (m, 3H, H-2, H-4, H-5), 3.08 (br s, 1H, 3-OH), 2.84 (s, 1H, 2-OH), 2.42 (s, 3H, Me (SMTBP)), 1.31 (s, 9H, tert-Bu (SMTBP)). 13C NMR (75 MHz, CDCl3): δ 160.39 (SMTBP), 149.75 (i-MBn), 137.15 (SMTBP), 130.90 (SMTBP), 130.38 (SMTBP), 130.23 (SMTBP), 129.46 (p-MPh), 127.72 (o-MPh), 125.56 (SMTBP), 113.84 (m-MPh), 101.99 (MPhCH), 88.83 (C-1), 80.31 (C-4), 74.79 (C-3), 73.02 (C-2), 70.52 (C-5), 68.71 (C-6), 55.42 (OMe), 31.42 (tert-Bu(SMTBP)), 20.59 (Me(SMTBP)). HRMS (ESI): Calcd. m/z for [M + H]+ C25H32O6S 461.1992 found 461.1991.
3.3.2. Methyl [(2-methyl-5-tert-butylphenyl) 2,3-di-O-benzyl-1-thio-β-d-glucopyranosyl] Uronate (10)
A solution of 9 [700 mg, 1.58 mmol] in DMF [7 mL] was cooled to 0 °C and NaH [250 mg, 4 eq.] was added portionwise. The reaction mixture was allowed to warm to rt and stirred for 30 min. Then, the mixture was cooled to 0 °C again and BnBr [0.57 mL, 3 eq.] was added dropwise. After 2 h, TLC [hexane/EtOAc = 2:1] showed formation of the final product and the reaction mixture was cooled to 0 °C and TFA [0.5 mL, 90% aq] was added. Reaction mixture was stirred at rt until TLC [hexane/EtOAc = 2:1] showed full consumption of the starting material. Then, the mixture was diluted with EtOAc [100 mL] and washed with NaHCO3 [100 mL] and H2O [100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The crude residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 10:1→3:1) to give diol. The latter was dissolved in DCM [5 mL] and H2O [1 mL] and TEMPO [50 mg, 0.2 eq.] were added. The mixture was cooled to 0 °C and BAIB [1g, 2.5 eq.] was added portionwise. The reaction mixture was vigorously stirred for 3 h until TLC [hexane/EtOAc = 2:1] showed full consumption of the starting material. Then, the mixture was diluted with EtOAc [100 mL] and washed with Na2S2O3 [100 mL] and H2O [100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The crude residue was dissolved in DMF [5 mL] and K2CO3 [200 mg, 1.5 eq.] and MeI [175 μL, 3 eq.] were added. The mixture was vigorously stirred for 3 h until TLC [hexane/EtOAc = 2:1] showed full consumption of the starting material. Then, the mixture was diluted with EtOAc [100 mL] and washed with H2O [2 × 100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 15:1→6:1) to give 10 (400 mg, 46%) as colorless oil. Rf = 0.55 (hexane/EtOAc = 2:1). [α]D = 7.2° (c = 1, EtOAc). 1H NMR (300 MHz, CDCl3): δ 7.63 (d, 1H, J = 1.9 Hz, SMTBP), 7.43–7.13 (m, 11H, SMTBP, 2xBn), 7.08 (d, 1H, J = 8.0 Hz, SMTBP), 4.90 (d, 1H, J = 10.3 Hz, CHH’Ph), 4.84 (s, 2H, CH2Ph), 4.75 (d, 1H, J = 10.3 Hz, CHH’Ph), 4.61 (d, 1H, J1–2 = 9.4 Hz, H-1), 3.97–3.86 (m, 1H, H-4), 3.79–3.75 (m, 4H, H-5, OMe), 3.61–3.44 (m, 2H, H-2, H-3), 2.97 (br s, 1H, 4-OH), 2.35 (s, 3H, Me(SMTBP)), 1.25 (s, 9H, tert-Bu(SMTBP)). 13C NMR (75 MHz, CDCl3): δ 169.67 (C-6), 149.76 (Bn), 138.01 (SMTBP), 136.43 (Bn), 132.90 (SMTBP), 129.99 (SMTBP), 129.61 (SMTBP), 128.64 (Bn), 128.51 (Bn), 128.35 (Bn), 128.11 (Bn), 127.99 (Bn), 124.97 (SMTBP), 89.03 (C-1), 85.38 (C-3), 80.20 (C-2), 77.45 (C-5), 75.83 (CH2Ph), 75.77 (CH2Ph), 71.99 (C-4), 52.78 (OMe), 31.35 (tert-Bu(SMTBP)), 20.45 (Me(SMTBP). HRMS (ESI): Calcd. m/z for [M + Na]+ C32H38O6S 573.2281 found 573.2287.
3.3.3. Methyl (2,3-di-O-benzyl-4-O-tert-butyldimethylsilyl-α,β-d-glucopyranosyl) Uronate Trichloroacetimidate (11)
NEt3 [65 μL, 2 eq.] and TBSOTf [80 μL, 1.5 eq.] were added under Ar protection to a solution of 10 [125 mg, 0.23 mmol] in DCM [2 mL] and the resulting mixture was stirred at rt for 2 h. After TLC showed full consumption of the starting material, the mixture was diluted with EtOAc [50 mL] and washed with H2O [2 × 50 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The crude product was treated according to the standard procedure A and after purification by chromatography (silica gel neutralized with NEt3, eluent: hexane/EtOAc = 30:1→10:1) gave 11 (102 mg, 69%) as a mixture of α/β-isomers = 1:1.4. Rf = 0.9 (hexane/EtOAc = 2:1). Analytical data for 11 were in agreement with those previously reported [25].
3.3.4. Methyl (2,3-di-O-benzyl-4-O-picoloyl-α,β-d-glucopyranosyl) Uronate Trichloroacetimidate (12)
PicoA [47 mg, 1.5 eq.], DCC [70 mg, 1.5 eq.], and DMAP [6 mg, 0.2 eq.] were added under Ar protection to a solution of 10 [124 mg, 0.23 mmol] in DCM [2 mL] and the resulting mixture was stirred at rt for 1 h. After TLC showed full consumption of the starting material, the mixture was diluted with EtOAc [50 mL] and washed with H2O [2 × 50 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 8:1→2:1) to give fully protected thioglycoside. The latter was treated according to the standard procedure A and after purification by chromatography (silica gel neutralized with NEt3, eluent: hexane/EtOAc = 6:1→1:1) gave 12 (80 mg, 56%) as a mixture of α/β-isomers 1:7. Rf = 0.3 and 0.15 (hexane/EtOAc = 2:1). 1H NMR (400 MHz, CDCl3): δ 8.75 (d, 1H, J = 3.8 Hz, Pico), 8.66 (s, 1H, NH), 7.99 (d, J = 7.8 Hz, Pico), 7.79 (td, 1H, J = 7.7, J = 1.8 Hz, Pico), 7.46 (ddd, 1, J = 7.6 Hz, J = 4.8 Hz, J = 1.2 Hz, Pico), 7.34–6.96 (m, 10H, 2xBn), 6.58 (d, 1H, J1–2 = 3.5 Hz, H-1 (β)), 6.55 (d, 1H, J1–2 = 2.5 Hz, H-4 (α)) 6.19 (d, 1H, J4–5 = 2.8 Hz, H-4 (α)), 5.49–5.33 (m, 1H, H-4 (β)), 4.81 (d, 1H, J = 11.3 Hz, CHH’Ph), 4.76–4.64 (m, 3H, CHH’Ph, CH2Ph), 4.59 (d, 1H, J4–5 = 10.3 Hz, H-5 (β)), 4.43 (dd, 1H, J2–3 = 7.8 Hz, J3-4 = 2.8 Hz, H-3 (α)), 4.25 (t, 1H, J2–3, J3–4 = 9.5 Hz, H-3 (β)), 3.93 (dd, J2–3 = 7.7 Hz, J1–2 = 2.5 Hz, H-2 (α)), 3.85 (dd, 1H, J2–3 = 9.5 Hz, J1–2 = 3.6 Hz, H-2 (β)), 3.59 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3): δ 167.91 (C-6), 164.21 (CO(Pico)), 161.01 (CCl3), 147.37 (Pico), 137.99 (i-Bn), 137.65 (i-Bn), 137.14 (Pico), 128.56-127.28 (Ar), 125.74 (Pico), 111.19 (C-1 (α)), 93.90 (C-1 (β)), 78.44 (C-2 (β)), 77.65 (C-3 (β)), 75.57 (CH2Ph), 73.42 (CH2Ph), 72.04 (C-4 (β)), 70.89 (C-5 (β)), 52.98 (OMe). HRMS (ESI): Calcd. m/z for [M + Na]+ C29H27Cl3N2O8 659.0725, found 659.0715.
3.3.5. Methyl (2,3-di-O-benzyl-4-O-benzoyl-α,β-d-glucopyranosyl) Uronate Trichloroacetimidate (13)
NEt3 [100 μL, 3 eq.] and BzCl [40 μL, 1.5 eq.] were added to a solution of 10 [129 mg, 0.23 mmol] in DCM [2 mL] and the resulting mixture was stirred overnight. After TLC showed full consumption of the starting material, an excess of MeOH was added and the mixture was kept for 30 min and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 30:1→12:1) to give fully protected thioglycoside. The latter was treated according to the standard procedure A and after purification by chromatography (silica gel neutralized with NEt3, eluent: hexane/EtOAc = 30:1→4:1) gave 13 (77 mg, 52%) as a mixture of α/β-isomers 1:2.1. Rf = 0.8 (hexane/EtOAc = 2:1). 1H NMR (400 MHz, CDCl3): δ 8.73 (s, 1H, NH(β)), 8.66 (s, 1H, NH(α)), 7.97–7.87 (m, 4H, o-Bz(α), o-Bz(β)), 7.56–7.52 (m, 2H, p-Bz(α), p-Bz(β)), 7.43–7.35 (m, 4H, m-Bz(α), m-Bz(β)), 7.29–7.06 (m, 20H, 2xBn(α), 2xBn(β)), 6.55 (d, 1H, J1–2 = 3.4 Hz, H-1 (α)), 5.91 (d, 1H, J1–2 = 7.3 Hz, H-1 (β)), 5.48 (t, 1H, J3–4, J4–5 = 9.2 Hz, H-4 (β)), 5.37 (t, 1H, J3–4, J4–5 = 9.8 Hz, H-4 (α)), 4.90 (d, 1H, J = 10.9 Hz, CHH’Ph(β)), 4.77–4.63 (m, 7H, CHH’Ph(β), CH2Ph(β), 2xCH2Ph(α)), 4.46 (d, 1H, J4–5 = 10.3 Hz, H-5 (α)), 4.22 (d, 1H, J4–5 = 9.6 Hz, H-5 (β)), 4.14 (t, 1H, J2–3, J3–4 = 9.4 Hz, H-3 (α)), 3.94–3.84 (m, 3H, H-2 (α), H-2 (β), H-3 (β)), 3.58 (s, 3H, OMe (β)), 3.56 (s, 3H, OMe (α)). 13C NMR (100 MHz, CDCl3): δ 168.04 (C-6 (α)), 167.57 (C-6 (β)), 165.47 (COPh (α)), 165.37 (COPh (β)), 161.15 (CCl3 (β)), 160.85 (CCl3 (α)), 137.67 (i-Bn), 133.51 (p-Bz (α), p-Bz (β)), 129.95 (o-Bz(α)), 129.90 (o-Bz(β)), 128.60–127.81 (Ar), 97.72 (C-1 (β)), 93.86 (C-1(α)), 80.73 (C-3 (β)), 80.15 (C-2 (β)), 78.48 (C-2 (α)), 77.49 (C-3 (α)), 75.47 (CH2Ph (α)), 75.24 (CH2Ph (β)), 75.10 (CH2Ph (β)), 73.45 (C-5 (β)), 73.40 (CH2Ph (α)), 71.34 (C-4 (β)), 71.14 (C-4 (α), C-5 (α)), 52.96 (OMe (α)), 52.92 (OMe (β)). HRMS(ESI): Calcd. m/z for [M + Na]+ C30H28Cl3NO8 658.0773 found 658.0769.
3.3.6. Allyl 3,4-O-isopropylidene-2-O-(methyl 2,3-di-O-benzyl-4-O-tert-butyldimethylsilyl-α,β-d-glucopyranosyluronate)-α-L-fucopyranoside (18)
The glycosylation was performed according to the standard procedure B with 7 [40 mg, 0.16 mmol] and 11 [102 mg, 0.16 mmol]. Purification by chromatography (silica gel, eluent: hexane/EtOAc = 20:1→8:1) gave 18 (109 mg, 91%) as a mixture of α/β-isomers 1.2:1. Rf = 0.8 (hexane/EtOAc = 2:1). 1H NMR (600 MHz, CDCl3): δ 7.36–7.21 (m, 20H, 2xBn (α), 2xBn (β)), 6.01 (ddd, 1H, J = 22.7 Hz, J = 10.8 Hz, J = 5.6 Hz, CH2CH=CH2 (α)), 5.80 (ddd, 1H, J = 22.5 Hz, J = 10.8 Hz, J = 5.6 Hz, CH2CH=CH2 (β)), 5.41 (dd, 1H, J = 17.2, J = 1.6 Hz, CH2CH=CHH’ (α)), 5.28–5.22 (m, 3H, H-1 (GlcA, α) CH2CH=CHH’ (α), CH2CH=CHH’ (β)), 5.13–5.08 (m, 2H, CH2CH=CHH’ (β), CHH’Ph (α)), 5.04–5.02 (m, 2H, 2xCHH’Ph (β)), 4.97 (d, 1H, J1–2 = 3.3 Hz, H-1 (Fuc, β)), 4.96 (d, 1H, J1–2 = 3.6 Hz, H-1(Fuc, α)), 4.76 (d, 1H, J = 11.3 Hz, CHH’Ph (α)), 4.72–4.68 (m, 2H, CHH’Ph (α), CHH’Ph (β)), 4.63–4.61 (m, 2H, H-1 (GlcA (β), CHH’Ph (β)), 4.58 (d, 1H, J = 11.3 Hz, CHH’Ph (α)), 4.45 (dd, 1H, J2–3 = 8.1 Hz, J3–4 = 5.5 Hz, H-3 (Fuc, α)), 4.40 (dd, 1H, J2–3 = 7.8 Hz, J3–4 = 5.5 Hz, H-3 (Fuc, β)), 4.28 (d, 1H, J4–5 = 9.5 Hz, H-5 (GlcA, α)), 4.26–4.08 (m, 7H, H-4 (Fuc, α), H-4 (Fuc, β), H-5 (Fuc, α), H-5 (Fuc, β), OCH2 (α), OCHH’ (β)), 4.04–3.98 (m, 2H, H-2 (Fuc, β), H-4 (GlcA, β), 3.94 (dd, 1H, J = 12.9 Hz, J = 6.0 Hz, CHH’ (β)), 3.87 (t, 1H, J4–5 = 9.1 Hz, H-4 (GlcA, α)), 3.83–3.80 (m, 2H, H-3 (GlcA, α), H-5 (GlcA, β)), 3.77–3.72 (m, 7H, H-2 (Fuc, α), OMe (α), OMe (β)), 3.64–3.59 (m, 2H, H-2 (GlcA, α), H-2 (GlcA, β)), 3.48 (t, 1H, J2–3, J3–4 = 8.7 Hz, H-3 (Glc, β)), 1.55 (s, 3H, CMe (α)), 1.50 (s, 3H, CMe (β)), 1.40–1.34 (m, 12H, H-6 (Fuc, α), H-6 (Fuc, β), CMe (α), CMe (β)), 0.85 (s, 9H, tert-BuSi (α)), 0.83 (s, 9H, tert-BuSi (β)), 0.01 (s, 3H, SiMe (α)), −0.02 (s, 3H, SiMe (β)), −0.02 (s, 3H, SiMe (α)), −0.03 (s, 3H, SiMe (β)). 13C NMR (150 MHz, CDCl3): δ 170.08 (C-6 (GlcA, β)), 168.63 (C-6 (GlcA, α)), 139.26 (i-Bn (α)), 138.89 (i-Bn (β)), 138.48 (i-Bn (β)), 138.06 (i-Bn (α)), 133.94 (CH2CH=CH2 (α)), 133.87 (CH2CH=CH2 (β)), 128.50–127.04 (Ar), 117.72 (CH2CH=CH2 (α)), 117.38 (CH2CH=CH2 (β)), 109.05 (CMe3 (β)), 108.92 (CMe3 (α)), 102.13 (C-1 (GlcA, β)), 99.17 (C-1 (GlcA, α)), 97.59 (C-1 (Fuc, α)), 95.45 (C-1 (Fuc, β)), 84.10 (C-3 (Glc, β)), 81.67 (C-2 (GlcA, β)), 80.83 (C-3 (GlcA, α)), 79.51 (C-2 (GlcA, α)), 78.78 (C-2 (Fuc, α)), 76.58 (C-5 (Glc, β)), 76.44 (C-4 (Fuc, α)), 76.34 (C-4 (Fuc, β)), 75.38 (C-2 (Fuc, β)), 75.21 (C-3 (Fuc, α)), 75.00 (CH2Ph, α), 74.84 (CH2Ph, β), 74.27 (C-3 (Fuc, β), CH2Ph, β), 72.61 (C-5 (GlcA, α)), 72.27 (C-4 (GlcA, β)), 72.07 (C-4 (GlcA, β)), 71.98 (OCH2Ph, α), 68.77 (OCH2, α), 68.62 (OCH2, β), 64.07 (C-5 (Fuc, α)), 63.25 (C-5 (Fuc, α)), 52.30 (OMe, α), 52.16 (OMe, β), 28.65 (CMe, α), 27.82 (CMe, β), 26.62 (CMe, α), 26.43 (CMe, β), 25.86 (tert-BuSi, β), 25.81 (tert-BuSi, α), 16.41 (C-6 (Fuc, α), C-6 (Fuc, β)), −3.83 (SiMe, α), −3.93 (SiMe, β), −5.20 (SiMe, β), −5.23 (SiMe, α). HRMS (ESI): Calcd. m/z for [M + NH4]+ C39H56O11Si 746.3930, found 746.3933.
3.3.7. Allyl 3,4-O-isopropylidene-2-O-(methyl 2,3-di-O-benzyl-4-O-picoloyl-α,β-d-glucopyranosyluronate)-α-L-fucopyranoside (19)
The glycosylation was performed according to the standard procedure B with 7 [30 mg, 0.12 mmol] and 12 [80 mg, 0.12 mmol]. Purification by chromatography (silica gel, eluent: hexane/EtOAc = 4:1→1:1) gave 19 (53 mg, 60%) as a mixture of α/β-isomers 7.4:1. Rf = 0.2 (hexane/EtOAc = 2:1). 1H NMR (600 MHz, CDCl3): δ 8.77 (d, 1H, J = 4.2 Hz, 1-Pico, α), 8.74 (d, 1H, J = 4.1 Hz, 1-Pico, β), 8.02 (d, 1H, J = 7.9 Hz, 4-Pico, β), 8.00 (d, 1H, J = 7.8 Hz, 4-Pico, α), 7.82–7.79 (m, 2H, 3-Pico (α), 3-Pico (β)), 7.50–7.44 (m, 2H, 2-Pico (α), 2-Pico (β)), 7.41–7.06 (m, 20H, 2xBn (α), 2xBn (β)), 5.93 (ddd, 1H, J = 22.8 Hz, J = 10.8 Hz, J = 5.7 Hz, CH2CH=CH2 (α)), 5.82 (ddd, 1H, J = 22.4 Hz, J = 10.7 Hz, J = 5.6 Hz, CH2CH=CH2 (β)), 5.52 (t, 1H, J3–4, J4–5 = 9.7 Hz, H-4 (GlcA, β)), 5.37 (t, 1H, J3–4, J4–5 = 9.8 Hz, H-4 (GlcA, α), 5.31 (dd, 1H, J = 17.2 Hz, J = 1.6 Hz, CH2CH=CHH’ (α)), 5.28–5.24 (m, 2H, H-1 (GlcA, α), CH2CH=CHH’ (β)), 5.16 (dd, 1H, J = 10.4 Hz, J = 1.2 Hz, CH2CH=CHH’ (α)), 5.12 (dd, 1H, J = 10.4 Hz, J = 1.3 Hz, CH2CH=CHH’ (β)), 5.03 (d, 1H, J = 11.4 Hz, CHH’Ph, β), 4.98 (d, 1H, J1–2 = 3.3 Hz, H-1 (Fuc, β)), 4.91 (d, 1H, J1–2 = 3.5 Hz, H-1 (Fuc, α)), 4.87 (d, 1H, J = 11.3 Hz, CHH’Ph, α), 4.78–4.71 (m, 6H, CHH’Ph (α), CHH’Ph (β), CH2Ph (α), CH2Ph (β)), 4.66–4.63 (m, 2H, H-1 (GlcA, β), H-5 (GlcA, α)), 4.42–4.40 (m, 2H, H-3 (Fuc, α), H-3 (Fuc, β)), 4.27 (t, 1H, J2–3, J3–4 = 9.4 Hz, H-3 (GlcA, α)), 4.18–4.08 (m, 7H, H-5 (GlcA, β), H-5 (Fuc, α), H-5 (Fuc, β), H-4 (Fuc, α), H-4 (Fuc, β), OCHH’ (α), OCHH’ (β)), 4.02 (dd, 1H, J2–3 = 7.9 Hz, J1–2 = 3.3 Hz, H-2 (Fuc, β)), 3.98–3.93 (m, 2H,OCHH’ (α), OCHH’ (β)), 3.90 (t, 1H, J2–3, J3–4 = 9.2 Hz, H-3 (GlcA, β)), 3.75 (dd, 1H, J2–3 = 8.0 Hz, J1–2 = 3.6 Hz, H-2 (Fuc, α)), 3.70–3.67 (m, 2H, H-2 (GlcA, α), H-2 (GlcA, β)), 3.63 (s, 3H, OMe, β), 3.61 (s, 3H, OMe, α), 1.55 (s, 3H, CMe, α), 1.52 (s, 3H, CMe, β), 1.38 (s, 3H, CMe’, α), 1.35–1.33 (m, 9H, H-6 (Fuc, α), H-6 (Fuc, β), CMe’ (β)). 13C NMR (150 MHz, CDCl3): δ 168.84 (C-6 (GlcA, α), 164.16 (CO (Pico, α), 150.06 (1-Pico, α), 147.70 (1-Pico, β), 138.51 (i-Bn), 138.19 (i-Bn), 137.00 (3-Pico α, β), 133.97 (CH2CH=CHH’, α, β), 128.48–127.06 (2xBn α, 2xBn β, 2 × 2-Pico α, β) 125.66 (4-Pico, β), 125.50 (4-Pico, α), 117.56 (CH2CH=CH2, α, β), 101.91 (C-1 (GlcA, β)), 98.96 (C-1 (GlcA, α)), 97.37 (C-1 (Fuc, α)), 95.39 (C-1 (Fuc, β)), 81.24 (C-3 (GlcA, β)), 81.10 (C-2 (GlcA, β)), 79.19 (C-2 (Fuc, α)), 78.66 (C-2 (GlcA, α)), 78.43 (C-3 (GlcA, α)), 76.36 (C-4 (Fuc, α)), 76.28 (C-4 (Fuc, β)), 75.95 (C-2 (Fuc, β)), 75.58 (CH2Ph, α), 75.39 (CH2Ph, β), 74.98 (C-3 (Fuc, α)), 74.69 (C-3 (Fuc, β), 74.28 (CH2Ph, β)), 72.71 (C-4 (GlcA, α), CH2Ph, α), 72.64 (C-5 (GlcA, β)), 72.26 (C-4 (GlcA, β)), 68.98 (C-5 (GlcA, α)), 68.75 (OCH2, α), 68.64 (OCH2, β), 64.08 (C-5 (Fuc, β)), 63.37 (C-5 (Fuc, α)), 52.68 (OMe, α, β), 28.60 (CMe, α), 28.00 (CMe, β), 26.52 (CMe’, α), 26.40 (CMe’, β), 16.41 (C-6 (Fuc, β), 16.35 (C-6 (Fuc, α). HRMS (ESI): Calcd. m/z for [M + Na]+ C39H45NO12 742.2834 found 742.2839.
3.3.8. Allyl 3,4-O-isopropylidene-2-O-(methyl 2,3-di-O-benzyl-4-O-benzoyl-α,β-d-glucopyranosyluronate)-α-L-fucopyranoside (20)
The glycosylation was performed according to the standard procedure B with 7 [30 mg, 0.12 mmol] and 13 [77 mg, 0.12 mmol]. Purification by chromatography (silica gel, eluent: toluene/EtOAc = 20:1→10:1) gave 20 (84 mg, 95%) as a mixture of α/β-isomers 1:2. Rf = 0.7 (hexane/EtOAc = 2:1). 1H NMR (600 MHz, CDCl3): δ 7.97 (d, 2H, J = 7.1 Hz, o-Bz, α), 7.94 (d, 2H, J = 7.1 Hz, o-Bz, β), 7.60–7.54 (m, 2H, p-Bz, α, β), 7.46–7.39 (m, 8H, m-Bz, α, β, Bn, β), 7.36–7.26 (m, 13H, Bn, α, β), 7.18–7.07 (m, 15H, Bn, α, β), 5.93 (ddd, 1H, J = 22.7 Hz, J = 10.8 Hz, J = 5.6 Hz, CH2CH=CH2, α), 5.81 (ddd, 1H, J = 22.2 Hz, J = 10.8, J = 5.6 Hz, CH2CH=CH2, β), 5.44 (t, 1H, J3–4, J4–5 = 9.6 Hz, H-4 (GlcA, β)), 5.36–5.25 (m, 3H, H-4 (GlcA, α), CH2CH=CHH’, α, CH2CH=CHH’, β), 5.22 (d, 1H, J1–2 = 3.7 Hz, H-1 (GlcA, α)), 5.18 (dd, 1H, J = 10.4 Hz, J = 1.4 Hz, CH2CH=CHH’, α), 5.13 (dd, 1H, J = 10.4 Hz, J = 1.4 Hz, CH2CH=CHH’, β), 5.05 (d, 1H, J = 11.3 Hz, CHH’Ph, β), 4.99 (d, 1H, J1–2 = 3.3 Hz, H-1 (Fuc, β)), 4.92 (d, 1H, J1–2 = 3.6 Hz, H-1 (Fuc, α)), 4.88 (d, 1H, J = 11.2 Hz, CHH’, α), 4.79–4.73 (m, 4H, CH2Ph, α, CHH’Ph, β, CHH’Ph, β), 4.69 (d, 1H, J = 11.2 Hz, CHH’Ph, α), 4.66–4.63 (m, 2H, H-1 (GlcA, β), CHH’Ph, β), 4.51 (d, 1H, J4–5 = 10.2 Hz, H-5 (GlcA, α)), 4.45–4.42 (m, 2H, H-3 (Fuc, α), H-3 (Fuc, β)), 4.22–4.12 (m, 5H, H-3 (GlcA, α), H-5 (Fuc, α), H-5 (Fuc, β), OCHH’, α, OCHH’, β), 4.12–4.09 (m, 2H, H-4 (Fuc, α), H-4 (Fuc, β)), 4.03–3.99 (m, 2H, H-2 (Fuc, β), H-5 (GlcA, β)), 3.98–3.91 (m, 2H, OCHH’, α, OCHH’, β), 3.81 (t, 1H, J2–3, J3–4 = 9.2 Hz, H-3 (GlcA, β)), 3.74 (dd, 1H, J2–3 = 8.0 Hz, J1–2 = 3.6 Hz, H-2 (Fuc, α)), 3.71–3.68 (m, 2H, H-2 (GlcA, α), H-2 (GlcA, β)), 3.62 (s, 3H, OMe, β), 3.58 (s, 3H, OMe, α), 1.57 (s, 3H, CMe, α), 1.52 (s, 3H, CMe, β), 1.41 (s, 3H, CMe’, α), 1.37–1.33 (m, 9H, H-6 (Fuc, α, H-6 (Fuc, β, CMe’, β). 13C NMR (150 MHz, CDCl3): δ 168.93 (C-6 (GlcA, α)), 167.66 (C-6 (GlcA, β)), 165.51 (CO (Bz, α)), 165.34 (CO, (Bz, β)), 138.35 (i-Bn), 137.83 (i-Bn), 133.76 (CH2CH=CH2, β), 133.72 (CH2CH=CH2, α), 133.37 (p-Bz, α, β), 129.84 (o-Bz, β), 129.82 (o-Bz, α), 128.54–127.65 (m-Bz, α, β, Bn, α, β), 117.64 (CH2CH=CH2, α), 117.51 (CH2CH=CH2, β), 109.16 (CMe2, α, β), 101.79 (C-1 (GlcA, β)), 99.05 (C-1 (GlcA, α)), 97.19 (C-1 (Fuc, α)), 95.30 (C-1 (Fuc, β)), 81.03 (C-2 (GlcA, β)), 80.95 (C-3 (GlcA, β)), 79.17 (C-2 (Fuc, α)), 78.53 (C-2 (GlcA, α)), 78.38 (C-3 (GlcA, α)), 76.30 (C-4 (Fuc, α)), 76.24 (C-4 (Fuc, β)), 75.95 (C-2 (Fuc, β)), 75.22 (CH2Ph, β), 74.84 (CH2Ph, α), 74.70 (CH2Ph, β), 74.18 (C-3 (Fuc, α), C-3 (Fuc, β)), 72.87 (CH2Ph, α), 72.82 (C-5 (GlcA, β)), 71.79 (C-4 (GlcA, α)), 71.48 (C-4 (GlcA, β)), 69.03 (C-5 (GlcA, α)), 68.58 (OCH2, β), 68.56 (OCH2, α), 63.98 (C-5 (Fuc, β)), 63.28 (C-5 (Fuc, α)), 52.73 (OMe, α), 52.65 (OMe, β), 28.63 (CMe, α), 28.01 (CMe, β), 26.54 (CMe’, α), 26.42 (CMe’, β), 16.41 (C-6 (Fuc, β)), 16.36 (C-6 (Fuc, α)). HRMS (ESI): Calcd. m/z for [M + NH4]+ C40H46O12 736.3328 found 736.3326.
3.3.9. Propyl 2-O-(methyl 4-O-benzoyl-α-d-glucopyranosyluronate)-α-L-fucopyranoside (21a) and propyl 2-O-(methyl 4-O-benzoyl-β-d-glucopyranosyluronate)-α-L-fucopyranoside (21b)
TFA [150 μL, 90% aq.] was added to a solution of 20 [84 mg, 0.11 mmol] in DCM [1.5 mL]. The reaction mixture was kept at 40 °C for 30 min. After TLC [hexane/EtOAc = 1:1] showed full consumption of the starting material, the mixture was neutralized with NEt3 and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 3:1→1:2) to give diol. A mixture of the resulting product and the catalyst 10% Pd/C [25 mg] in EtOAc–MeOH (1:1) [2 mL] was stirred under H2 at rt for 2 h. After TLC (toluene:EtOAc = 1:1) showed formation of the final product, the reaction mixture was filtered through a nylon membrane syringe filter (0.45 μm). The filtrate was concentrated in vacuo and the residue was purified by chromatography (silica gel, eluent: DCM/MeOH = 20:1→10:1) to give 21a (17 mg, 29%) and 21b (30 mg, 51%). Rf = 0.25 and 0.1 (toluene/EtOAc = 1:1).
For 21a: [α]D = −98.5° (c = 1, MeOH). 1H NMR (600 MHz, CD3OD3): 8.05 (d, 2H, J = 7.1 Hz, o-Bz)), 7.66 (t, 1H, J = 7.4 Hz, p-Bz), 7.52 (t, 2H, J = 7.8 Hz, m-Bz), 5.13–5.12 (m, 2H, H-1 (GlcA), H-4 (GlcA)), 4.91 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 4.66 (d, 1H, J4–5 = 10.3 Hz, H-5 (GlcA)), 4.07 (t, 1H, J2–3, J3–4= 9.5 Hz, H-3 (GlcA)), 4.02 (dd, 1H, J2–3 = 10.0 Hz, J3–4 = 3.3 Hz, H-3 (Fuc)), 3.95 (q, 1H, J5–6 = 6.5 Hz, H-5 (Fuc)), 3.81 (dd, 1H, J2–3 = 10.0 Hz, J1–2 = 3.7 Hz, H-2 (Fuc)), 3.75 (d, 1H, J3–4 = 3.4 Hz, H-4 (Fuc)), 3.67–3.60 (m, 2H, H-2 (GlcA), OCHH’), 3.22 (dt, 1H, J = 9.3 Hz, J = 6.6 Hz, OCHH’), 1.66 (dq, 2H, J = 14.2 Hz, J = 7.1 Hz, OCH2CH2CH3), 1.23 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.99 (t, 3H, J = 7.4 Hz, OCH2CH2CH3). 13C NMR (150 MHz, CD3OD): δ 13C NMR (151 MHz, MeOD) δ 170.84 (C-6 (GlcA)), 167.20 (COPh), 134.53 (p-Bz), 130.66 (o-Bz), 129.59 (m-Bz), 103.26 (C-1 (GlcA)), 99.35 (C-1 (Fuc)), 81.39 (C-2 (Fuc)), 74.02 (C-4 (GlcA)), 73.43 (C-2 (GlcA), C-4 (Fuc)), 72.14 (C-3 (GlcA)), 70.39 (C-5 (GlcA)), 70.10 (C-3 (Fuc), OCH2), 67.19 (C-5 (Fuc)), 53.04 (OMe), 23.87 (OCH2CH2CH3), 16.50 (C-6 (Fuc)), 11.25 (OCH2CH2CH3). HRMS (ESI): Calcd. m/z for [M + Na]+ C23H32O12 523.1786 found 523.1786.
For 21b: [α]D = −17.6° (c = 1, MeOH). 1H NMR (600 MHz, CD3OD3): δ 8.07 (d, 2H, J = 7.9 Hz, o-Bz), 7.65 (t, 1H, J = 7.4 Hz, p-Bz), 7.51 (t, 2H, J = 7.8 Hz, m-Bz), 5.12 (t, 1H, J3–4, J4–5 = 9.7 Hz, H-4 (GlcA)), 4.98 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 4.66 (d, 1H, J1–2 = 7.9 Hz, H-1 (GlcA)), 4.30 (d, 1H, J4–5 = 10.0 Hz, H-5 (GlcA)), 4.04 (dd, 1H, J2–3 = 10.2 Hz, J1–2 = 3.7 Hz, H-2 (Fuc)), 4.00 (q, 1H, J5–6 = 6.6 Hz, H-5 (Fuc)), 3.94 (dd, 1H, J2–3 = 10.2 Hz, J3–4 = 3.3 Hz, H-3 (Fuc)), 3.87 (t, 1H, J2–3, J3–4 = 9.3 Hz, H-3 (GlcA)), 3.76 (d, 1H, J3–4 = 3.3 Hz, H-4 (Fuc)), 3.66–3.60 (m, 1H, OCHH’), 3.57 (s, 3H, OMe), 3.54–3.50 (m, 1H, H-2 (GlcA)), 3.49–3.46 (m, 1H, OCHH’), 1.71–1.63 (m,2H, CH2CH2CH3), 1.25 (d, 3H, J5–6 = 5.6 Hz, H-6 (Fuc)), 0.97 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, CD3OD): δ 170.09 (C-6 (GlcA)), 167.32 (COPh), 134.46 (p-Bz), 130.69 (o-Bz), 129.52 (m-Bz), 102.78 (C-1 (GlcA)), 98.24 (C-1 (Fuc)), 77.11 (C-2 (Fuc)), 74.71 (C-3 (GlcA)), 74.08 (C-2 (GlcA)), 73.64 (C-4 (GlcA)), 73.39 (C-5 (GlcA)), 73.14 (C-4 (Fuc)), 70.85 (OCH2), 69.74 (C-3 (Fuc)), 67.22 (C-5 (Fuc)), 53.10 (OMe), 23.70 (CH2CH2CH3), 16.53 (C-6 (Fuc)), 11.03 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M + Na]+ C23H32O12 523.1786 found 523.1786.
3.3.10. Propyl 2-O-(α-d-glucopyranosyluronic acid)-α-L-fucopyranoside Sodium Salt (1)
Final deprotection of 21a [11.5 mg, 0.024 eq.] was performed according to the standard procedure C. Purification by gel chromatography followed by lyophilization gave 1 (7 mg, 74%). Analytical data for 1 were in agreement with those previously reported [36]. [α]D = −21° (c = 1, H2O). 1H NMR (600 MHz, D2O): δ 5.10 (d, 1H, J1–2 = 3.9 Hz, H-1 (GlcA)), 5.02 (d, 1H, J1–2 = 3.8 Hz, H-1 (Fuc)), 4.07 (q, 1H, J5–6 = 6.5 Hz, H-5 (Fuc)), 4.03–3.98 (m, 2H, H-5 (GlcA), H-3 (Fuc)), 3.84 (d, 1H, J3–4 = 3.0 Hz, H-4 (Fuc)), 3.78 (dd, 1H, J2–3 = 10.3 Hz, J1–2 = 3.8 Hz, H-2 (Fuc)), 3.74 (t, 1H, J2–3, J3–4 = 9.5 Hz, H-3 (GlcA)), 3.65–3.60 (m, 1H, OCHH’), 3.58 (dd, 1H, J2–3 = 10.0 Hz, J1–2 = 3.9 Hz, H-2 (GlcA)), 3.48 (t, 1H, J3–4, J4–5 = 9.6 Hz, H-4 (GlcA)), 3.43–3.39 (m, 1H, OCHH’), 1.66–1.58 (m, 2H, CH2CH2CH3), 1.21 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.91 (t, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, D2O): δ 101.66 (C-1 (GlcA)), 98.39 (C-1 (Fuc)), 78.64 (C-2 (Fuc)), 73.27 (C-3 (GlcA)), 72.42 (C-4 (Fuc)), 72.37 (C-4 (GlcA)), 72.24 (C-5 (GlcA)), 72.02 (C-2 (GlcA)), 70.76 (OCH2), 69.02 (C-3 (Fuc)), 66.80 (C-5 (Fuc)), 22.66 (CH2CH2CH3), 15.68 (C-6 (Fuc)), 10.51 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − H]− C15H26O11 381.1402 found 381.1393.
3.3.11. Propyl 2-O-(2,3-di-O-sulfo-α-d-glucopyranosyluronic acid)-3,4-di-O-sulfo-α-L-fucopyranoside Sodium Salt (2)
According to the standard procedure D, 21a [5.5 mg, 0.011 eq.] was treated followed by chromatography (silica gel, eluent: DCM/MeOH = 5:1→2:1). Product of sulfation was treated according to standard procedure C. Purification by gel chromatography followed by lyophilization gave 2 (3.5 mg, 64%). [α]D =−44° (c = 1, H2O). 1H NMR (600 MHz, D2O): δ 5.39 (d, 1H, J1–2 = 3.7 Hz, H-1 (GlcA)), 5.06 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 4.97 (d, 1H, J3–4 = 2.9 Hz, H-4 (Fuc)), 4.72 (dd, 1H, J2–3 = 10.6 Hz, J3–4 = 3.0 Hz, H-3 (Fuc)), 4.62 (t, 1H, J2–3, J3–4 = 9.5 Hz, H-3 (GlcA)), 4.29 (dd, 1H, J2–3 = 10.0 Hz, J1–2 = 3.8 Hz, H-2 (GlcA)), 4.22 (q, 1H, J5–6 = 6.5 Hz, H-5 (Fuc)), 4.11 (d, 1H, J4–5 = 10.2 Hz, H-5 (GlcA)), 4.01 (dd, 1H, J2–3 = 10.6 Hz, J1–2 = 3.7 Hz, H-2 (Fuc)), 3.71 (dd, 1H, J4–5 = 10.0 Hz, J3–4 = 9.0 Hz, H-4 (GlcA)), 3.66–3.61 (m, 1H, OCHH’), 3.47 (dt, 1H, J = 9.5 Hz, J = 6.6 Hz, OCHH’), 1.70–1.61 (m, 2H, CH2CH2CH3), 1.27 (d, 3H, J5–6 = 6.5 Hz, H-6 (Fuc)), 0.93 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, D2O): δ 176.27 (C-6 (GlcA)), 99.35 (C-1 (GlcA)), 98.29 (C-1 (Fuc)), 79.74 (C-4 (Fuc)), 78.87 (C-3 (GlcA)), 76.18 (C-2 (Fuc)), 75.26 (C-2 (GlcA)), 73.94 (C-3 (Fuc)), 72.78 (C-5 (GlcA)), 71.79 (C-4 (GlcA)), 66.41 (C-5 (Fuc)), 22.80 (CH2CH2CH3), 16.36 (C-6 (Fuc)), 10.64 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − 2Na + H]− C15H21Na5O23S4 766.9133 found 766.9118.
3.3.12. Propyl 2-O-(2,3,4-tri-O-sulfo-α-d-glucopyranosyluronic acid)-3,4-di-O-sulfo-α-L-fucopyranoside Sodium Salt (3)
Exhaustive sulfation of 1 [3.2 mg, 0.008 eq.] was performed according to the standard procedure D. Purification by gel chromatography followed by lyophilization gave 3 (5 mg, 72%). Analytical data for 3 were in agreement with those previously reported [37]. [α]D = −13° (c = 1, H2O). 1H NMR (600 MHz, D2O): δ 5.44 (d, 1H, J1–2 = 2.6 Hz, H-1 (GlcA)), 5.07 (d, 1H, J1–2 = 3.8 Hz, H-1 (Fuc)), 4.95–4.93 (m, 2H, H-3 (GlcA), H-4 (Fuc)), 4.76–4.71 (m, 2H, H-3 (Fuc), H-4 (GlcA)), 4.52 (dd, 1H, J2–3 = 6.4 Hz, J1–2 = 2.6 Hz, H-2 (GlcA)), 4.39 (d, 1H, J4–5 = 5.4 Hz, H-5 (GlcA)), 4.24–4.19 (m, 2H, H-2 (Fuc), H-5 (Fuc)), 3.66–3.57 (m, 2H, OCH2), 1.73–1.61 (m, 2H, CH2CH2CH3), 1.28 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.93 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, D2O): δ 98.53 (C-1 (Fuc)), 96.94 (C-1 (GlcA)), 79.75 (C-4 (Fuc)), 75.61 (C-5 (GlcA)), 74.86 (C-2 (Fuc)), 74.81 (C-4 (GlcA)), 74.55 (C-3 (GlcA)), 74.44 (C-3 (Fuc)), 73.35 (C-2 (GlcA)), 71.13 (OCH2), 66.42 (C-5 (Fuc)), 22.67 (CH2CH2CH3), 16.39 (C-6 (Fuc)), 10.56 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − 2Na + H]− C15H20Na6O26S5 868.8515 found 868.8501.
3.3.13. Propyl 2-O-(β-d-glucopyranosyluronic acid)-α-L-fucopyranoside Sodium Salt (4)
Final deprotection of 21b [8.5 mg, 0.016 eq.] was performed according to the standard procedure C. Purification by gel chromatography followed by lyophilization gave 4 (4.8 mg, 70%). [α]D = −83° (c = 1, H2O). 1H NMR (600 MHz, D2O): δ 5.05 (d, 1H, J1–2 = 3.4 Hz, H-1 (Fuc)), 4.56 (d, 1H, J1–2 = 7.9 Hz, H-1 (GlcA)), 4.09 (q, 1H, J5–6 = 6.6 Hz, H-5 (Fuc)), 4.00 (dd, 1H, J2–3 = 10.4 Hz, J1–2 = 3.6 Hz, H-2 (Fuc)), 3.95 (dd, 1H, J2–3 = 10.3 Hz, J1–2 = 3.2 Hz, H-3 (Fuc)), 3.84 (d, 1H, J3–4 = 2.9 Hz, H-4 (Fuc)), 3.76–3.71 (m, 1H, H-5 (GlcA)), 3.63 (dt, 1H, J = 14.4 Hz, J = 7.2 Hz, OCHH’), 3.54–3.48 (m, 3H, H-3 (GlcA), H-4 (GlcA), OCHH’), 3.36 (t, 1H, J1–2, J2–3 = 8.0 Hz, H-2 (GlcA)), 1.62 (dd, 2H, J = 14.3 Hz, J = 7.1 Hz, CH2CH2CH3), 1.22 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.91 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, D2O): δ 101.56 (C-1 (GlcA)), 96.73 (C-1 (Fuc)), 76.38 (C-5 (GlcA)), 76.14 (C-2 (Fuc)), 76.08 (C-3 (GlcA)), 73.39 (C-2 (GlcA)), 72.32 (C-4 (GlcA)), 72.25 (C-4 (Fuc)), 70.72 (OCH2), 68.76 (C-3 (Fuc)), 66.96 (C-5 (Fuc)), 22.59 (CH2CH2CH3), 15.78 (C-6 (Fuc)), 10.49 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − H]− C15H26O11 381.1402 found 381.1405.
3.3.14. Propyl 2-O-(2,3-di-O-sulfo-β-d-glucopyranosyluronic acid)-3,4-di-O-sulfo- α-L-fucopyranoside Sodium Salt (5)
According to the standard procedure D, 21b [10.5 mg, 0.021 eq.] was treated followed by chromatography (silica gel, eluent: DCM/MeOH = 4:1→1:1). Product of sulfation was treated according to standard procedure C. Purification by gel chromatography followed by lyophilization gave 5 (6 mg, 57%). [α]D = −23° (c = 1, H2O). 1H NMR (600 MHz, D2O): δ 5.23 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 4.95 (d, 1H, J3–4 = 2.6 Hz, H-4 (Fuc)), 4.92 (d, 1H, J1–2 = 7.1 Hz, H-1 (GlcA)), 4.75 (dd, 1H, J2–3 = 6.3 Hz, J3–4 = 2.5 Hz, H-3 (Fuc)), 4.48 (t, 1H, J2–3, J3–4 = 7.8 Hz, H-3 (GlcA)), 4.28 (t, 1H, J1–2, J2–3 = 7.3 Hz, H-2 (Fuc)), 4.24 (q, 1H, J5–6 = 6.4 Hz, H-5 (Fuc)), 4.16 (dd, 1H, J2–3 = 10.5 Hz, J1–2 = 3.7 Hz, H-2 (Fuc)), 3.94–3.88 (m, 2H, H-4 (GlcA), H-5 (GlcA)), 3.64–3.57 (m, 2H, OCH2), 1.64 (dq, 2H, J = 14.5 Hz, J = 7.3 Hz, CH2CH2CH3), 1.27 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.91 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, D2O): δ 101.41 (C-1 (GlcA)), 97.24 (C-1 (Fuc)), 81.63 (C-3 (GlcA)), 79.70 (C-4 (Fuc)), 77.99 (C-2 (GlcA)), 77.12 (C-5 (GlcA)), 75.44 (C-2 (Fuc)), 74.86 (C-3 (Fuc)), 71.33 (C-4 (GlcA)), 71.11 (OCH2), 66.52 (C-5 (Fuc)), 22.67 (CH2CH2CH3), 16.33 (C-6 (Fuc)), 10.48 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − 3Na + 2H]− C15H21Na5O23S4 744.9314 found 744.9217.
3.3.15. Propyl 2-O-(2,3,4-tri-O-sulfo-β-d-glucopyranosyluronic acid)-3,4-di-O-sulfo- α-L-fucopyranoside Sodium Salt (6)
Exhaustive sulfation of 4 [3.8 mg, 0.09 eq.] was performed according to the standard procedure D. Purification by gel chromatography followed by lyophilization gave 6 (6.8 mg, 78%). [α]D = −47° (c = 1, H2O). 1H NMR (400 MHz, D2O): δ 5.21 (dt, 1H, J3–4 = 4.1 Hz, J4–5 = 1.3 Hz, H-4 (GlcA)), 5.17 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 5.08 (dd, 1H, J3–4 = 4.1 Hz, J2–3 = 1.2 Hz, H-3 (GlcA)), 4.98 (d, 1H, J1–2 = 6.8 Hz, H-1 (GlcA)), 4.94 (d, 1H, J3–4 = 3.0 Hz, H-4 (Fuc)), 4.74 (dd, 1H, J2–3 = 10.6 Hz, J3–4 = 3.0 Hz, H-3 (Fuc)), 4.62 (d, 1H, J4–5 = 1.6 Hz, H-5 (GlcA)), 4.48 (d, 1H, J1–2 = 6.7 Hz, H-2 (GlcA)), 4.25 (q, 1H, J5–6 = 6.5 Hz, H-5 (Fuc)), 4.16 (dd, 1H, J2–3 = 10.6 Hz, J2–3 = 3.7 Hz, H-2 (Fuc)), 3.62 (t, 2H, J = 6.9 Hz, OCH2), 1.65 (h, 2H, J = 7.2 Hz, CH2CH2CH3), 1.27 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.91 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (100 MHz, D2O): δ 99.91 (C-1 (GlcA)), 97.02 (C-1 (Fuc)), 79.70 (C-4 (Fuc)), 78.90 (C-5 (GlcA)), 78.60 (C-2 (GlcA)), 75.89 (C-3 (GlcA)), 75.28 (C-4 (GlcA)), 74.83 (C-2 (Fuc)), 74.33 (C-3 (Fuc)), 71.42 (OCH2), 66.56 (C-5 (Fuc)), 22.69 (CH2CH2CH3), 16.37 (C-6 (Fuc)), 10.51 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − 3Na + 2H]− C15H20Na6O26S5 846.8701 found 846.8489.
All spectra data obtained are presented in Supplementary Materials.
4. Conclusions
Model glycosylations clearly indicate that the picoloyl-assisted HAD method is a prospective approach to α-directing stereocontrol of glycosylation with certain GlcA donors. The α:β ratio when employing HAD was 7.4:1 compared to 1.2:1 and 1:2 for non-HAD-mediated cases. The stereoselectivity for larger oligosaccharides and more hindered acceptors could be even higher.
Six disaccharide models were synthesized and studied by means of NMR and DFT methods of different levels. Analysis of 1H chemical shifts along with the intra-ring coupling constants in the case of the β-linked saccharides suggested conformational changes occurring in the glucuronic acid pyranoside ring upon the exhaustive sulfation. Calculations of NMR shielding constants by means of the DLPNO-MP2 approach confirmed that the observed changes in chemical shifts corresponded to the change from 4C1 to 3S1 and 1S5 conformation. Analysis of NOE spectra for fully sulfated and non-sulfated β-disaccharides provided additional support for this result. In substances where glucuronic acid residue had α-configuration, no changes occurred. Absence of the sulfate group at O-4 in the β-glucuronic acid residue resulted in a less pronounced but similar effect.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/molecules28227571/s1, Copies of the NMR spectra of compounds 9, 10, 11, 12, 13, 18, 19, 20, 21a, 21b, 1, 2, 3, 4, 5, and 6.
Author Contributions
A.G.G., D.Z.V. and N.E.N. conceived the project and designed the experiments; A.G.G., D.Z.V., A.I.T. and A.S.D. performed the experiments. A.G.G., D.Z.V., A.I.T., V.B.K., N.E.U. and N.E.N. interpreted the data and wrote the manuscript. V.B.K. acquired funding. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (theme No. FFZZ-2022-0010).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are available from the corresponding author.
Acknowledgments
The authors thank D.A. Argunov for recording NMR spectra.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
TEMPO—(2,2,6,6-Tetramethylpiperidin-1-yl)oxyl; DMF—Dimethylformamide; DCM—Dichloromethane; TBS—tert-butyldimethylsilyl; NBS—N-bromosuccinimide; TCAN—Trichloroacetonitrile; DCC—N,NCCdicyclohexylcarbodiimide; DMAP—4-Dimethylaminopyridine; TBSOTf—tert-Butyldimethylsilyl trifluoromethanesulfonate; MP—4-Methoxyphenyl; Pico—Picoloyl; TFA—Trifluoroacetic acid.
References
- Vinnitskiy, D.Z.; Ustyuzhanina, N.E.; Nifantiev, N.E. Natural Bacterial and Plant Biomolecules Bearing α-D-Glucuronic Acid Residues. Russ. Chem. Bull. 2015, 64, 1273–1301. [Google Scholar] [CrossRef]
- Kjellén, L.; Lindahl, U. Proteoglycans: Structures and Interactions. Annu. Rev. Biochem. 1991, 60, 443–475. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Kulshrestha, S.; Bhardwaj, K.; Jangir, R. A Review on Properties and Applications of Xanthan Gum. In Microbial Polymers: Applications and Ecological Perspectives; Springer: Singapore, 2021; pp. 87–107. [Google Scholar] [CrossRef]
- Petri, D.F. Xanthan Gum: A Versatile Biopolymer for Biomedical and Technological Applications. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Wijesinghe, W.; Jeon, Y.-J. Biological Activities and Potential Industrial Applications of Fucose Rich Sulfated Polysaccharides and Fucoidans Isolated from Brown Seaweeds: A Review. Carbohydr. Polym. 2012, 88, 13–20. [Google Scholar] [CrossRef]
- Mikami, T.; Kitagawa, H. Biosynthesis and Function of Chondroitin Sulfate. Biochim. Biophys. Acta 2013, 1830, 4719–4733. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, D.L. Heparin and Heparan Sulfate: Structure and Function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic Acid in the Third Millennium. Polymers 2018, 10, 701. [Google Scholar] [CrossRef]
- Merry, C.L.R.; Lindahl, U.; Couchman, J.; Esko, J.D. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; ISBN 978-1-62182-421-3. [Google Scholar]
- Pomin, V.H. Holothurian Fucosylated Chondroitin Sulfate. Mar. Drugs 2014, 12, 232–254. [Google Scholar] [CrossRef]
- Ustyuzhanina, N.E.; Bilan, M.I.; Dmitrenok, A.S.; Nifantiev, N.E.; Usov, A.I. Two Fucosylated Chondroitin Sulfates from the Sea Cucumber Eupentacta fraudatrix. Carbohydr. Polym. 2017, 164, 8–12. [Google Scholar] [CrossRef]
- Ustyuzhanina, N.E.; Bilan, M.I.; Dmitrenok, A.S.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. The Structure of a Fucosylated Chondroitin Sulfate from the Sea Cucumber Cucumaria frondosa. Carbohydr. Polym. 2017, 165, 7–12. [Google Scholar] [CrossRef]
- Ustyuzhanina, N.E.; Bilan, M.I.; Panina, E.G.; Sanamyan, N.P.; Dmitrenok, A.S.; Tsvetkova, E.A.; Ushakova, N.A.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. Structure and Anti-Inflammatory Activity of a New Unusual Fucosylated Chondroitin Sulfate from Cucumaria djakonovi. Mar. Drugs 2018, 16, 389. [Google Scholar] [CrossRef] [PubMed]
- Ustyuzhanina, N.E.; Bilan, M.I.; Dmitrenok, A.S.; Shashkov, A.S.; Kusaykin, M.I.; Stonik, V.A.; Nifantiev, N.E.; Usov, A.I. Structure and Biological Activity of a Fucosylated Chondroitin Sulfate from the Sea Cucumber Cucumaria japonica. Glycobiology 2016, 26, 449–459. [Google Scholar] [CrossRef]
- Gerbst, A.G.; Vinnitsky, D.Z.; Dmitrenok, A.S.; Ustyuzhanina, N.E.; Nifantiev, N.E. Conformational Study of Persulfated Propyl Glucuronide. Carbohydr. Res. 2018, 455, 81–85. [Google Scholar] [CrossRef]
- Kiselevskiy, M.V.; Anisimova, N.Y.; Ustyuzhanina, N.E.; Vinnitskiy, D.Z.; Tokatly, A.I.; Reshetnikova, V.V.; Chikileva, I.O.; Shubina, I.Z.; Kirgizov, K.I.; Nifantiev, N.E. Perspectives for the Use of Fucoidans in Clinical Oncology. Int. J. Mol. Sci. 2022, 23, 11821. [Google Scholar] [CrossRef]
- Nagaoka, M.; Shibata, H.; Kimura-Takagi, I.; Hashimoto, S.; Kimura, K.; Makino, T.; Aiyama, R.; Ueyama, S.; Yokokura, T. Structural Study of Fucoidan from Cladosiphon okamuranus Tokida. Glycoconj. J. 1999, 16, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Tako, M.; Yoza, E.; Tohma, S. Chemical Characterization of Acetyl Fucoidan and Alginate from Commercially Cultured Cladosiphon okamuranus. Bot. Mar. 2000, 43, 393–398. [Google Scholar] [CrossRef]
- Sakai, T.; Ishizuka, K.; Shimanaka, K.; Ikai, K.; Kato, I. Structures of Oligosaccharides Derived from Cladosiphon okamuranus Fucoidan by Digestion with Marine Bacterial Enzymes. Mar. Biotechnol. 2003, 5, 536–544. [Google Scholar] [CrossRef]
- Bilan, M.I.; Vinogradova, E.V.; Tsvetkova, E.A.; Grachev, A.A.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. A Sulfated Glucuronofucan Containing Both Fucofuranose and Fucopyranose Residues from the Brown Alga Chordaria flagelliformis. Carbohydr. Res. 2008, 343, 2605–2612. [Google Scholar] [CrossRef]
- Ustyuzhanina, N.E.; Ushakova, N.A.; Zyuzina, K.A.; Bilan, M.I.; Elizarova, A.L.; Somonova, O.V.; Madzhuga, A.V.; Krylov, V.B.; Preobrazhenskaya, M.E.; Usov, A.I.; et al. Influence of Fucoidans on Hemostatic System. Mar. Drugs 2013, 11, 2444–2458. [Google Scholar] [CrossRef]
- Yasomanee, J.P.; Demchenko, A.V. Effect of Remote Picolinyl and Picoloyl Substituents on the Stereoselectivity of Chemical Glycosylation. J. Am. Chem. Soc. 2012, 134, 20097–20102. [Google Scholar] [CrossRef]
- Takahashi, D.; Toshima, K. 1, 2-Cis O-Glycosylation Methods. In Comprehensive Glycoscience, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 365–412. [Google Scholar]
- Wadouachi, A.; Kovensky, J. Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: An Overview. Molecules 2011, 16, 3933–3968. [Google Scholar] [CrossRef]
- Vinnitskiy, D.Z.; Krylov, V.B.; Ustyuzhanina, N.E.; Dmitrenok, A.S.; Nifantiev, N.E. The Synthesis of Heterosaccharides Related to the Fucoidan from Chordaria flagelliformis Bearing an α-L-Fucofuranosyl Unit. Org. Biomol. Chem. 2016, 14, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Alex, C.; Visansirikul, S.; Demchenko, A.V. A Versatile Approach to the Synthesis of Glycans Containing Mannuronic Acid Residues. Org. Biomol. Chem. 2021, 19, 2731–2743. [Google Scholar] [CrossRef] [PubMed]
- Gerbst, A.G.; Krylov, V.B.; Nifantiev, N.E. Application of Computational Methods for the Studies of Carbohydrate Reactivity. Carbohydr. Chem. 2021, 44, 151–169. [Google Scholar] [CrossRef]
- Komarova, B.S.; Novikova, N.S.; Gerbst, A.G.; Sinitsyna, O.A.; Rubtsova, E.A.; Kondratyeva, E.G.; Sinitsyn, A.P.; Nifantiev, N.E. Combination of 3-O-Levulinoyl and 6-O-Trifluorobenzoyl Groups Ensures α-Selectivity in Glucosylations: Synthesis of the Oligosaccharides Related to Aspergillus fumigatus α-(1→3)-Glucan. J. Org. Chem. 2023, 88, 12542–12564. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Hartree–Fock Exchange Fitting Basis Sets for H to Rn. J. Comput. Chem. 2008, 29, 167–175. [Google Scholar] [CrossRef]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized Accurate Auxiliary Basis Sets for RI-MP2 and RI-CC2 Calculations for the Atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Stoychev, G.L.; Auer, A.A.; Gauss, J.; Neese, F. DLPNO-MP2 Second Derivatives for the Computation of Polarizabilities and NMR Shieldings. J. Chem. Phys. 2021, 154, 164110. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. The Optimum Contraction of Basis Sets for Calculating Spin–Spin Coupling Constants. Theor. Chem. Acc. 2010, 126, 371–382. [Google Scholar] [CrossRef]
- Zlotina, N.S.; Ustyuzhanina, N.E.; Grachev, A.A.; Gerbst, A.G.; Nifantiev, N.E. Synthesis, NMR and Conformational Studies of Fucoidan Fragments. X. Stereoselective Synthesis of Di- and Trisaccharide Fucoidan Fragments Bearing α-D-Glucuronic Acid Residue. J. Carbohydr. Chem. 2008, 27, 429–445. [Google Scholar] [CrossRef]
- Krylov, V.B.; Kaskova, Z.M.; Vinnitskiy, D.Z.; Ustyuzhanina, N.E.; Grachev, A.A.; Chizhov, A.O.; Nifantiev, N.E. Acid-Promoted Synthesis of per-O-Sulfated Fucooligosaccharides Related to Fucoidan Fragments. Carbohydr. Res. 2011, 346, 540–550. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).