
Relation between Halogen Bond Strength and IR and NMR Spectroscopic Markers



Abstract
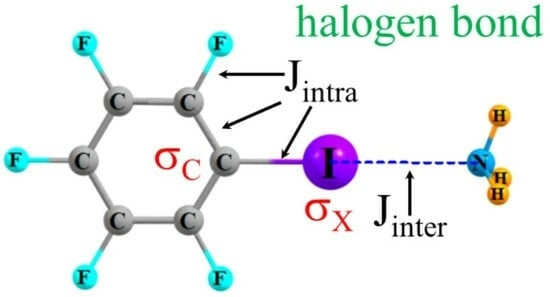
:
1. Introduction
2. Results
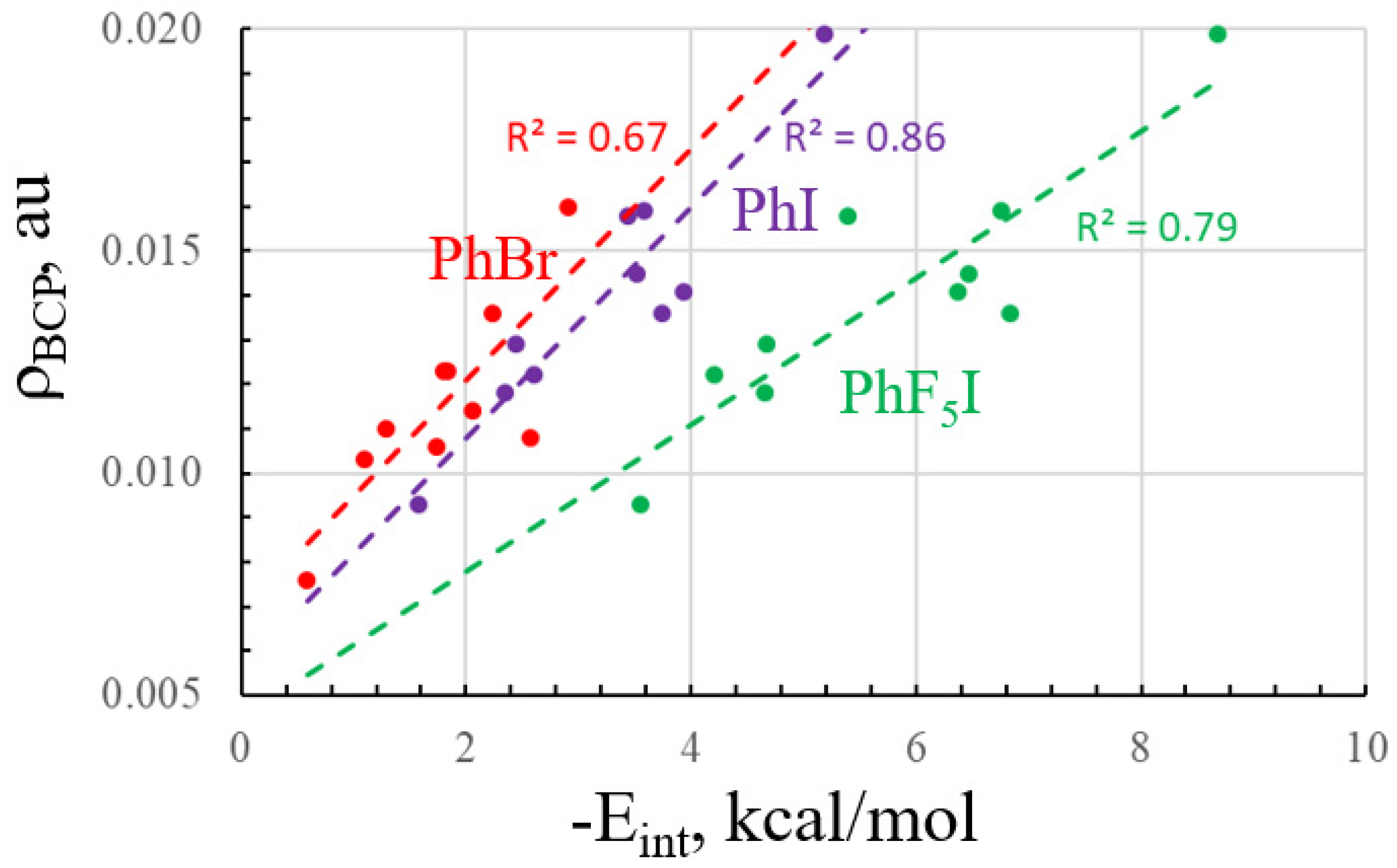
2.1. Measures of Halogen Bond Strength
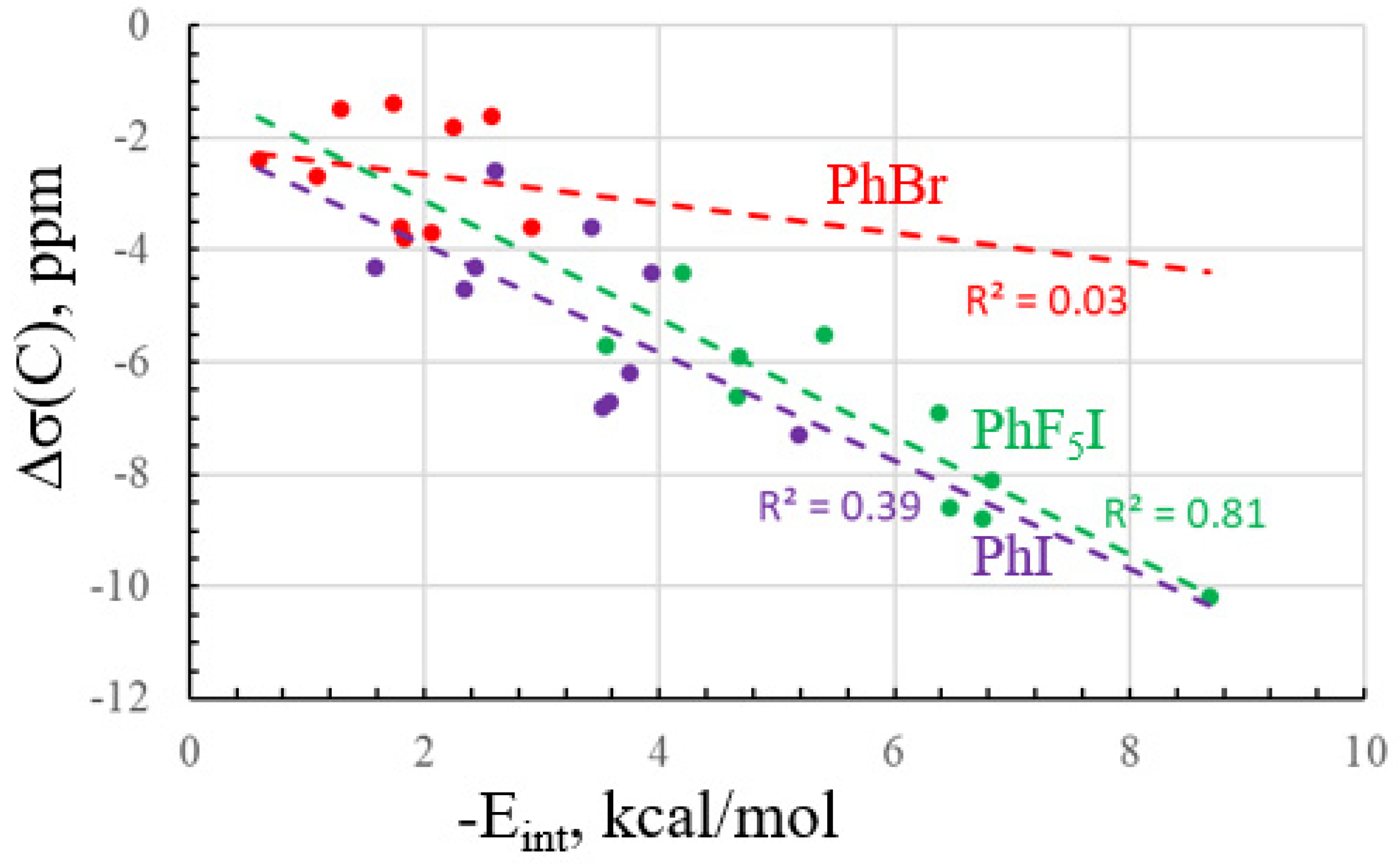
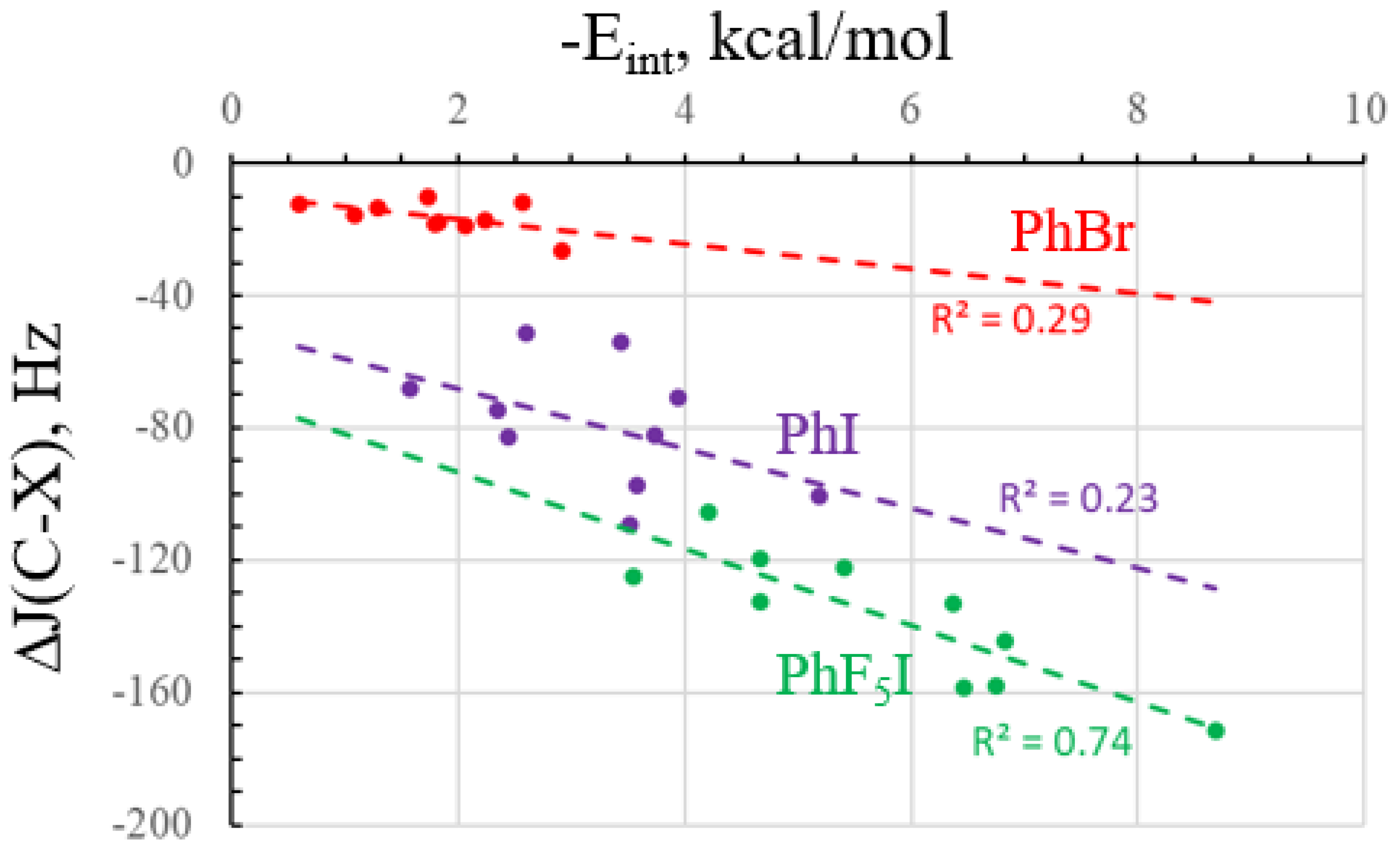
2.2. NMR Data
3. Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Hamilton, W.C.; Ibers, J.A. Hydrogen Bonding in Solids; W. A. Benjamin: New York, NY, USA, 1968; p. 284. [Google Scholar]
- Latajka, Z.; Bouteiller, Y.; Scheiner, S. Critical assessment of density functional methods for study of proton transfer processes. (FHF)−. Chem. Phys. Lett. 1995, 234, 159–164. [Google Scholar] [CrossRef]
- Joesten, M.D.; Schaad, L.J. Hydrogen Bonding; Marcel Dekker: New York, NY, USA, 1974; p. 622. [Google Scholar]
- Latajka, Z.; Scheiner, S. Ab initio comparison of H bonds and Li bonds. Complexes of LiF, LiCl, HF, and HCl with NH3. J. Chem. Phys. 1984, 81, 4014–4017. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford: New York, NY, USA, 1999; p. 507. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond; Oxford University Press: Oxford, UK, 2009; p. 313. [Google Scholar]
- Luth, K.; Scheiner, S. Excited-state energetics and proton-transfer barriers in malonaldehyde. J. Phys. Chem. 1994, 98, 3582–3587. [Google Scholar] [CrossRef]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997; p. 375. [Google Scholar]
- Gleiter, R.; Werz, D.B.; Rausch, B.J. A World Beyond Hydrogen Bonds?—Chalcogen–Chalcogen Interactions Yielding Tubular Structures. Chem. Eur. J. 2003, 9, 2676–2683. [Google Scholar] [CrossRef] [PubMed]
- Bleiholder, C.; Werz, D.B.; Koppel, H.; Gleiter, R. Theoretical investigations on chalcogen-chalcogen interactions: What makes these nonbonded interactions bonding? J. Am. Chem. Soc. 2006, 128, 2666–2674. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Halogen bonding versus chalcogen and pnicogen bonding: A combined Cambridge structural database and theoretical study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Fanfrlík, J.; Přáda, A.; Padělková, Z.; Pecina, A.; Macháček, J.; Lepšík, M.; Holub, J.; Růžička, A.; Hnyk, D.; Hobza, P. The Dominant Role of Chalcogen Bonding in the Crystal Packing of 2D/3D Aromatics. Angew. Chem. Int. Ed. 2014, 53, 10139–10142. [Google Scholar] [CrossRef]
- Alikhani, E.; Fuster, F.; Madebene, B.; Grabowski, S.J. Topological reaction sites—Very strong chalcogen bonds. Phys. Chem. Chem. Phys. 2014, 16, 2430–2442. [Google Scholar] [CrossRef]
- Trujillo, C.; Sanchez-Sanz, G.; Alkorta, I.; Elguero, J. Halogen, chalcogen and pnictogen interactions in (XNO2)2 homodimers (X = F, Cl, Br, I). New J. Chem. 2015, 39, 6791–6802. [Google Scholar] [CrossRef]
- Galmés, B.; Juan-Bals, A.; Frontera, A.; Resnati, G. Charge-Assisted Chalcogen Bonds: CSD and DFT Analyses and Biological Implication in Glucosidase Inhibitors. Chem. Eur. J. 2020, 26, 4599–4606. [Google Scholar] [CrossRef]
- Jungbauer, S.H.; Huber, S.M. Cationic Multidentate Halogen-Bond Donors in Halide Abstraction Organocatalysis: Catalyst Optimization by Preorganization. J. Am. Chem. Soc. 2015, 137, 12110–12120. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Maji, S.; Ghosh, R.; Jana, R.; Datta, A.; Ghosh, P. Aryl-platform-based tetrapodal 2-iodo-imidazolium as an excellent halogen bond receptor in aqueous medium. Chem. Commun. 2019, 55, 1506–1509. [Google Scholar] [CrossRef]
- Scheiner, S. Comparison of halide receptors based on H, halogen, chalcogen, pnicogen, and tetrel bonds. Faraday Disc. 2017, 203, 213–226. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. σ-Hole Opposite to a Lone Pair: Unconventional Pnicogen Bonding Interactions between ZF3 (Z=N, P, As, and Sb) Compounds and Several Donors. ChemPhysChem 2016, 17, 1608–1614. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Pnicogen–π complexes: Theoretical study and biological implications. Phys. Chem. Chem. Phys. 2012, 14, 14061–14066. [Google Scholar] [CrossRef]
- Grabowski, S.J. Pnicogen and tetrel bonds—Tetrahedral Lewis acid centres. Struct. Chem. 2019, 30, 1141–1152. [Google Scholar]
- Alkorta, I.; Elguero, J.; Grabowski, S.J. Pnicogen and hydrogen bonds: Complexes between PH3X+ and PH2X systems. Phys. Chem. Chem. Phys. 2015, 17, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Moaven, S.; Andrews, M.C.; Polaske, T.J.; Karl, B.M.; Unruh, D.K.; Bosch, E.; Bowling, N.P.; Cozzolino, A.F. Triple-Pnictogen Bonding as a Tool for Supramolecular Assembly. Inorg. Chem. 2019, 58, 16227–16235. [Google Scholar] [CrossRef]
- Scheiner, S. Origins and properties of the tetrel bond. Phys. Chem. Chem. Phys. 2021, 23, 5702–5717. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Effects of carbon chain substituent on the P···N noncovalent bond. Chem. Phys. Lett. 2012, 536, 30–33. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef]
- Mertsalov, D.F.; Gomila, R.M.; Zaytsev, V.P.; Grigoriev, M.S.; Nikitina, E.V.; Zubkov, F.I.; Frontera, A. On the Importance of Halogen Bonding Interactions in Two X-ray Structures Containing All Four (F, Cl, Br, I) Halogen Atoms. Crystals 2021, 11, 1406. [Google Scholar] [CrossRef]
- Palusiak, M.; Grabowski, S.J. Do intramolecular halogen bonds exist? Ab initio calculations and crystal structures’ evidences. Struct. Chem. 2008, 19, 5–11. [Google Scholar] [CrossRef]
- Kolár, M.; Hostaš, J.; Hobza, P. The strength and directionality of a halogen bond are co-determined by the magnitude and size of the σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 9987–9996. [Google Scholar] [CrossRef]
- Cunha, A.V.; Havenith, R.W.A.; van Gog, J.; De Vleeschouwer, F.; De Proft, F.; Herrebout, W. The Halogen Bond in Weakly Bonded Complexes and the Consequences for Aromaticity and Spin-Orbit Coupling. Molecules 2023, 28, 772. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Sensitivity of Noncovalent Bonds to Intermolecular Separation: Hydrogen, Halogen, Chalcogen, and Pnicogen Bonds. CrystEngComm 2013, 15, 3119–3124. [Google Scholar] [CrossRef]
- Joesten, M.D.; Drago, R.S. The Validity of Frequency Shift-Enthalpy Correlations. I. Adducts of Phenol with Nitrogen and Oxygen Donors. J. Am. Chem. Soc. 1962, 84, 3817–3821. [Google Scholar] [CrossRef]
- Drago, R.S.; Epley, T.D. Enthalpies of hydrogen bonding and changes in hydroxy frequency shifts for a series of adducts with substituted phenols. J. Am. Chem. Soc. 1969, 91, 2883–2890. [Google Scholar] [CrossRef]
- Rozenberg, M.; Loewenschuss, A.; Marcus, Y. An empirical correlation between stretching vibration redshift and hydrogen bond length. Phys. Chem. Chem. Phys. 2000, 2, 2699–2702. [Google Scholar] [CrossRef]
- Rao, C.N.R.; Dwivedi, P.C.; Ratajczak, H.; Orville-Thomas, W.J. Relation between O–H stretching frequency and hydrogen bond energy: Re-examination of the Badger–Bauer rule. J. Chem. Soc. Faraday Trans. 2 1975, 71, 955–966. [Google Scholar] [CrossRef]
- Ellington, T.L.; Reves, P.L.; Simms, B.L.; Wilson, J.L.; Watkins, D.L.; Tschumper, G.S.; Hammer, N.I. Quantifying the Effects of Halogen Bonding by Haloaromatic Donors on the Acceptor Pyrimidine. ChemPhysChem 2017, 18, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Attrell, R.J.; Widdifield, C.M.; Korobkov, I.; Bryce, D.L. Weak Halogen Bonding in Solid Haloanilinium Halides Probed Directly via Chlorine-35, Bromine-81, and Iodine-127 NMR Spectroscopy. Cryst. Growth Des. 2012, 12, 1641–1653. [Google Scholar] [CrossRef]
- Amonov, A.; Scheiner, S. Heavy pnicogen atoms as electron donors in sigma-hole bonds. Phys. Chem. Chem. Phys. 2023, 25, 23530–23537. [Google Scholar] [CrossRef]
- Amonov, A.; Scheiner, S. Competition between Binding to Various Sites of Substituted Imidazoliums. J. Phys. Chem. A 2023, 127, 6292–6299. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, S.; Elguero, J. Charge-transfer complexes between dihalogen compounds and electron donors. J. Phys. Chem. A 1998, 102, 9278–9285. [Google Scholar] [CrossRef]
- Ford, T.A. An ab initio study of some halogen-bonded complexes containing cyclic ethers. Mol. Phys. 2021, 119, e1919326. [Google Scholar] [CrossRef]
- Lu, J.; Scheiner, S. Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra. Molecules 2019, 24, 2822. [Google Scholar] [CrossRef]
- Michalczyk, M.; Zierkiewicz, W.; Wysokiński, R.; Scheiner, S. Theoretical Studies of IR and NMR Spectral Changes Induced by Sigma-Hole Hydrogen, Halogen, Chalcogen, Pnicogen, and Tetrel Bonds in a Model Protein Environment. Molecules 2019, 24, 3329. [Google Scholar] [CrossRef]
- Parra, R.D.; Grabowski, S.J. Enhancing Effects of the Cyano Group on the C-X…N Hydrogen or Halogen Bond in Complexes of X-Cyanomethanes with Trimethyl Amine: CH3−n(CN)nX…NMe3, (n = 0–3; X = H, Cl, Br, I). Int. J. Mol. Sci. 2022, 23, 11289. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Ji, B.; Tian, A. On the correlation between bond-length change and vibrational frequency shift in halogen-bonded complexes. J. Chem. Phys. 2011, 134, 224303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-J.; Liu, H.-L.; Huang, X.-R.; Li, Q.-Z.; Sun, C.-C. Effect of substitution and cooperativity on the Cl–F blue shift in single-electron halogen-bonded H3C ··· ClF complex. Mol. Phys. 2010, 108, 2021–2026. [Google Scholar] [CrossRef]
- Xu, Y.; Gabidullin, B.; Bryce, D.L. Single-Crystal NMR Characterization of Halogen Bonds. J. Phys. Chem. A 2019, 123, 6194–6209. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Yáñez, M.; Mó, O.; Montero-Campillo, M.M. Relativistic Effects on NMR Parameters of Halogen-Bonded Complexes. Molecules 2019, 24, 4399. [Google Scholar] [CrossRef]
- Karpfen, A. Charge-transfer complexes between NH3 and the halogens F2, ClF, and Cl2: An ab initio study on the intermolecular interaction. J. Phys. Chem. A 2000, 104, 6871–6879. [Google Scholar] [CrossRef]
- Lu, J.; Scheiner, S. Relationships between Bond Strength and Spectroscopic Quantities in H-Bonds and Related Halogen, Chalcogen, and Pnicogen Bonds. J. Phys. Chem. A 2020, 124, 7716–7725. [Google Scholar] [CrossRef]
- Jaźwiński, J. Chapter One—Indirect spin-spin coupling constants across noncovalent bonds. In Annual Reports on NMR Spectroscopy; Webb, G.A., Ed.; Academic Press: Cambridge, MA, USA, 2021; Volume 104, pp. 1–73. [Google Scholar]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Probing the structures, binding energies, and spin-spin coupling constants of halogen-bonded Azine:ClF complexes. Chem. Phys. Lett. 2020, 761, 137916. [Google Scholar] [CrossRef]
- Scheiner, S. Characterization of Type I and II Interactions between Halogen Atoms. Cryst. Growth Des. 2022, 22, 2692–2702. [Google Scholar] [CrossRef]
- Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Linear free energy relationships in halogen bonds. CrystEngComm 2013, 15, 3178–3186. [Google Scholar] [CrossRef]
- Tang, Q.; Li, Q. Non-additivity of F substituent in enhancing the halogen bond in C6H5I…NCH. Comput. Theor. Chem. 2015, 1070, 21–26. [Google Scholar] [CrossRef]
- Margiotta, E.; van der Lubbe, S.C.C.; de Azevedo Santos, L.; Paragi, G.; Moro, S.; Bickelhaupt, F.M.; Fonseca Guerra, C. Halogen Bonds in Ligand–Protein Systems: Molecular Orbital Theory for Drug Design. J. Chem. Infor. Model. 2020, 60, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Lambert, E.C.; Williams, A.E.; Fortenberry, R.C.; Hammer, N.I. Probing halogen bonding interactions between heptafluoro-2-iodopropane and three azabenzenes with Raman spectroscopy and density functional theory. Phys. Chem. Chem. Phys. 2022, 24, 11713–11720. [Google Scholar] [CrossRef] [PubMed]
- Côté, M.; Ovens, J.S.; Bryce, D.L. Anticooperativity and Competition in Some Cocrystals Featuring Iodine-Nitrogen Halogen Bonds. Chem. Asian J. 2023, 18, e202201221. [Google Scholar] [CrossRef]
- Toikka, Y.N.; Starova, G.L.; Suslonov, V.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y.; Bokach, N.A. Combined σ- and π-Hole Donor Properties of Perfluorinated Iodo(or bromo)benzenes: Halogen Bonding and π-Hole Interactions in Cocrystals Including Cu4I4 Clusters. Cryst. Growth Des. 2023, 23, 5194–5203. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Uchimaru, T.; Wakisaka, A.; Ono, T. Magnitude and Directionality of Halogen Bond of Benzene with C6F5X, C6H5X, and CF3X (X = I, Br, Cl, and F). J. Phys. Chem. A 2016, 120, 7020–7029. [Google Scholar] [CrossRef]
- Stoesser, J.; Rojas, G.; Bulfield, D.; Hidalgo, P.I.; Pasan, J.; Ruiz-Perez, C.; Jimenez, C.A.; Huber, S.M. Halogen bonding two-point recognition with terphenyl derivatives. New J. Chem. 2018, 42, 10476–10480. [Google Scholar] [CrossRef]
- Dang, Q.M.; Simpson, J.H.; Parish, C.A.; Leopold, M.C. Evaluating Halogen-Bond Strength as a Function of Molecular Structure Using Nuclear Magnetic Resonance Spectroscopy and Computational Analysis. J. Phys. Chem. A 2021, 125, 9377–9393. [Google Scholar] [CrossRef]
- Bramlett, T.A.; Matzger, A.J. Halogen Bonding Propensity in Solution: Direct Observation and Computational Prediction. Chem. Eur. J. 2021, 27, 15472–15478. [Google Scholar] [CrossRef]
- Grabowski, S.J. Halogen Bonds between Diiodotetrafluorobenzenes and Halide Anions: Theoretical Analysis. Cryst. Growth Des. 2023, 23, 489–500. [Google Scholar] [CrossRef]
- Vioglio, P.C.; Chierotti, M.R.; Gobetto, R. Solid-state nuclear magnetic resonance as a tool for investigating the halogen bond. CrystEngComm 2016, 18, 9173–9184. [Google Scholar] [CrossRef]
- Gao, K.; Goroff, N.S. Two New Iodine-Capped Carbon Rods. J. Am. Chem. Soc. 2000, 122, 9320–9321. [Google Scholar] [CrossRef]
- Rege, P.D.; Malkina, O.L.; Goroff, N.S. The Effect of Lewis Bases on the 13C NMR of Iodoalkynes. J. Am. Chem. Soc. 2002, 124, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.A.; Klijn, J.E.; Hill, P.A.; Bennett, J.L.; Goroff, N.S. Experimental Studies of the 13C NMR of Iodoalkynes in Lewis-Basic Solvents. J. Org. Chem. 2004, 69, 660–664. [Google Scholar] [CrossRef]
- Popa, M.M.; Man, I.C.; Draghici, C.; Shova, S.; Caira, M.R.; Dumitrascu, F.; Dumitrescu, D. Halogen bonding in 5-iodo-1-arylpyrazoles investigated in the solid state and predicted by solution 13C-NMR spectroscopy. CrystEngComm 2019, 21, 7085–7093. [Google Scholar] [CrossRef]
- Lapp, J.; Scheiner, S. Proximity Effects of Substituents on Halogen Bond Strength. J. Phys. Chem. A 2021, 125, 5069–5077. [Google Scholar] [CrossRef]
- Scheiner, S.; Hunter, S. Influence of Substituents in the Benzene Ring on the Halogen Bond of Iodobenzene with Ammonia. ChemPhysChem 2022, 23, e202200011. [Google Scholar] [CrossRef] [PubMed]
- Jimmink, B.; Sethio, D.; Turunen, L.; von der Heiden, D.; Erdélyi, M. Probing Halogen Bonds by Scalar Couplings. J. Am. Chem. Soc. 2021, 143, 10695–10699. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Kříž, K.; Řezáč, J. Non-covalent interactions atlas benchmark data sets 4: σ-hole interactions. Phys. Chem. Chem. Phys. 2022, 24, 14794–14804. [Google Scholar] [CrossRef]
- Boese, A.D. Density Functional Theory and Hydrogen Bonds: Are We There Yet? ChemPhysChem. 2015, 16, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Martin, J.M.L. Halogen bonds: Benchmarks and theoretical analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.; Harvey, A.J.A.; Sen, A.; Dessent, C.E.H. Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions. J. Phys. Chem. A 2013, 117, 12590–12600. [Google Scholar] [CrossRef]
- Thanthiriwatte, K.S.; Hohenstein, E.G.; Burns, L.A.; Sherrill, C.D. Assessment of the performance of DFT and DFT-D methods for describing distance dependence of hydrogen-bonded interactions. J. Chem. Theory Comput. 2011, 7, 88–96. [Google Scholar] [CrossRef]
- Liao, M.S.; Lu, Y.; Scheiner, S. Performance assessment of density-functional methods for study of charge-transfer complexes. J. Comput. Chem. 2003, 24, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Deible, M.J.; Tuguldur, O.; Jordan, K.D. Theoretical Study of the Binding Energy of a Methane Molecule in a (H2O)20 Dodecahedral Cage. J. Phys. Chem. B 2014, 118, 8257–8263. [Google Scholar] [CrossRef]
- Li, A.; Muddana, H.S.; Gilson, M.K. Quantum Mechanical Calculation of Noncovalent Interactions: A Large-Scale Evaluation of PMx, DFT, and SAPT Approaches. J. Chem. Theory Comput. 2014, 10, 1563–1575. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Characterizing and Understanding the Remarkably Slow Basis Set Convergence of Several Minnesota Density Functionals for Intermolecular Interaction Energies. J. Chem. Theory Comput. 2013, 9, 4453–4461. [Google Scholar] [CrossRef]
- Elm, J.; Bildeb, M.; Mikkelsena, K.V. Assessment of binding energies of atmospherically relevant clusters. Phys. Chem. Chem. Phys. 2013, 15, 16442–16445. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Bhattacherjee, A.; Shirhatti, P.R.; Wategaonkar, S. O–H···S Hydrogen Bonds Conform to the Acid–Base Formalism. J. Phys. Chem. A 2013, 117, 8238–8250. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll; TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Ditchfield, R. GIAO studies of magnetic shielding in FHF− and HF. Chem. Phys. Lett. 1976, 40, 53–56. [Google Scholar] [CrossRef]
- Hinton, J.F.; Wolinski, K. Ab initio GIAO magnetic shielding tensor for hydrogen-bonded systems. In Theoretical Treatments of Hydrogen Bonding; Hadzi, D., Ed.; John Wiley: Chichester, UK, 1997; pp. 75–93. [Google Scholar]
- Alkorta, I.; Elguero, J. Ab initio (GIAO) calculations of absolute nuclear shieldings for representative compounds containing 1(2)H, 6(7)Li, 11B, 13C, 14(15)N, 17O, 19F, 29Si, 31P, 33S, and 35Cl nuclei. Struct. Chem. 1998, 9, 187–202. [Google Scholar] [CrossRef]
- Noro, T.; Sekiya, M.; Koga, T. Segmented contracted basis sets for atoms H through Xe: Sapporo-(DK)-nZP sets (n = D, T, Q). Theor. Chem. Acc. 2012, 131, 1124. [Google Scholar] [CrossRef]
- Noro, T.; Sekiya, M.; Koga, T. Sapporo-(DKH3)-nZP (n = D, T, Q) sets for the sixth period s-, d-, and p-block atoms. Theor. Chem. Acc. 2013, 132, 1363. [Google Scholar] [CrossRef]
- Michalczyk, M.; Kizior, B.; Zierkiewicz, W.; Scheiner, S. Factors contributing to halogen bond strength and stretch or contraction of internal covalent bond. Phys. Chem. Chem. Phys. 2023, 25, 2907–2915. [Google Scholar] [CrossRef]
- Raghavendra, B.; Arunan, E. Unpaired and bond electrons as H, Cl, and Li bond acceptors: An anomalous one-electron blue-shifting chlorine. J. Phys. Chem. A 2007, 111, 9699–9706. [Google Scholar] [CrossRef]
- Otte, F.; Kleinheider, J.; Grabe, B.; Hiller, W.; Busse, F.; Wang, R.; Kreienborg, N.M.; Merten, C.; Englert, U.; Strohmann, C. Gauging the Strength of the Molecular Halogen Bond via Experimental Electron Density and Spectroscopy. ACS Omega 2023, 8, 21531–21539. [Google Scholar] [CrossRef]
- Zhang, Y.; Ji, B.; Tian, A.; Wang, W. Competition between π…π interaction and halogen bond in solution: A combined 13C NMR and density functional theory study. J. Chem. Phys. 2012, 136, 141101. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Structures, energies, bonding, and NMR properties of pnicogen complexes H2XP:NXH2 (X = H, CH3, NH2, OH, F, Cl). J. Phys. Chem. A 2011, 115, 13724–13731. [Google Scholar] [CrossRef]
- Viger-Gravel, J.; Leclerc, S.; Korobkov, I.; Bryce, D.L. Correlation between 13C chemical shifts and the halogen bonding environment in a series of solid para-diiodotetrafluorobenzene complexes. CrystEngComm 2013, 15, 3168–3177. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, Y.; Ji, B.; Tian, A.; Wang, W. Structural competition between halogen bonds and lone-pair..π interactions in solution. ChemPhysChem 2012, 13, 1411–1414. [Google Scholar] [CrossRef]
- Moss, W.N.; Goroff, N.S. Theoretical Analysis of the 13C NMR of Iodoalkynes upon Complexation with Lewis Bases. J. Org. Chem. 2005, 70, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Widdifield, C.M.; Cavallo, G.; Facey, G.A.; Pilati, T.; Lin, J.; Metrangolo, P.; Resnati, G.; Bryce, D.L. Multinuclear Solid-State Magnetic Resonance as a Sensitive Probe of Structural Changes upon the Occurrence of Halogen Bonding in Co-crystals. Chem. Eur. J. 2013, 19, 11949–11962. [Google Scholar] [CrossRef] [PubMed]
- Cerreia Vioglio, P.; Catalano, L.; Vasylyeva, V.; Nervi, C.; Chierotti, M.R.; Resnati, G.; Gobetto, R.; Metrangolo, P. Natural Abundance 15N and 13C Solid-State NMR Chemical Shifts: High Sensitivity Probes of the Halogen Bond Geometry. Chem. Eur. J. 2016, 22, 16819–16828. [Google Scholar] [CrossRef] [PubMed]
- Morin, V.M.; Szell, P.M.J.; Caron-Poulin, E.; Gabidullin, B.; Bryce, D.L. Mechanochemical Preparations of Anion Coordinated Architectures Based on 3-Iodoethynylpyridine and 3-Iodoethynylbenzoic Acid. ChemistryOpen 2019, 8, 1328–1336. [Google Scholar] [CrossRef]
- Szell, P.M.J.; Cavallo, G.; Terraneo, G.; Metrangolo, P.; Gabidullin, B.; Bryce, D.L. Comparing the Halogen Bond to the Hydrogen Bond by Solid-State NMR Spectroscopy: Anion Coordinated Dimers from 2- and 3-Iodoethynylpyridine Salts. Chem. Eur. J. 2018, 24, 11364–11376. [Google Scholar] [CrossRef]
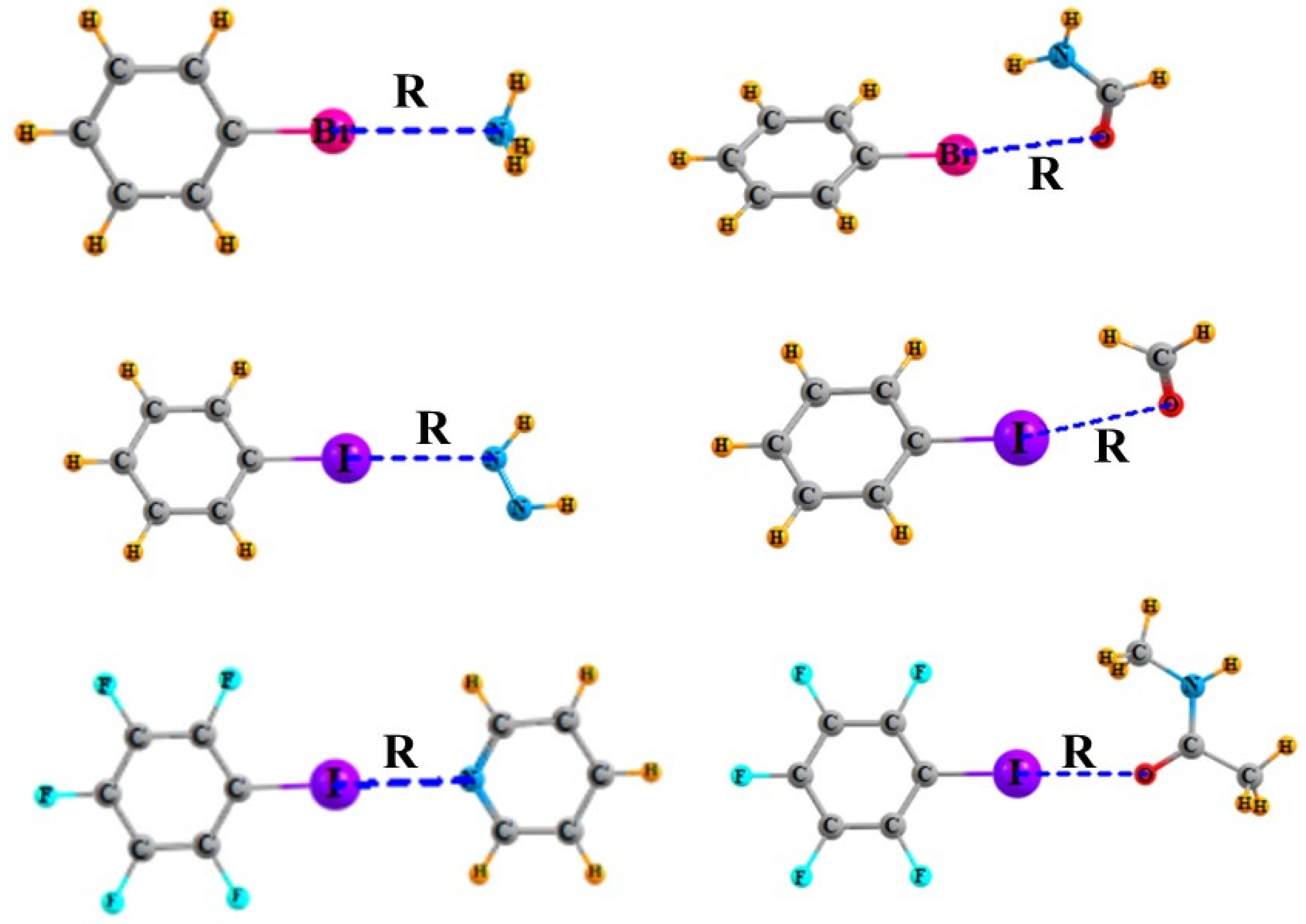


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Base | −Eint | R(X··N/O) | ρBCP a | Δr(C-X) | Δν(CX) | J(X··N/O) |
|---|---|---|---|---|---|---|
| PhBr·· | 1.8957 | 1109.7 | ||||
| N≡CH | 0.59 | 3.260 | 0.0076 | −0.0022 | 5.2 | 42.1 |
| HN=NH | 1.09 | 3.180 | 0.0103 | −0.0014 | 5.7 | 53.0 |
| PyrN | 1.80 | 3.103 | 0.0123 | 0.0001 | 1.3 | 57.3 |
| NH3 | 1.83 | 3.124 | 0.0123 | −0.0003 | 2.8 | 57.3 |
| NMe3 | 2.91 | 3.008 | 0.0160 | 0.0030 | 2.4 | 55.3 |
| OH2 | 1.29 | 3.089 | 0.0110 | −0.0001 | 0.6 | −58.3 |
| OMe2 | 2.24 | 2.982 | 0.0136 | 0.0011 | 7.7 | −69.7 |
| OCH2 | 1.73 | 3.101 | 0.0106 (0.0071) | 0.0002 | 0.8 | −43.8 |
| OCHNH2 | 2.57 | 3.091 | 0.0108 (0.0086) | 0.0016 | 0.6 | −58.5 |
| NMA | 2.06 | 3.007 | 0.0114 (0.0053) | −0.0019 | 6.1 | −103.6 |
| PhI·· | 2.0939 | 1100.5 | ||||
| N≡CH | 1.58 | 3.284 | 0.0093 | −0.0004 | 2.4 | 227.3 |
| HN=NH | 2.34 | 3.221 | 0.0118 | −0.0002 | 2.2 | 244.1 |
| PyrN | 3.58 | 3.080 | 0.0159 | 0.0026 | 3.3 | 267.1 |
| NH3 | 3.52 | 3.146 | 0.0145 | 0.0037 | 3.6 | 277.8 |
| NMe3 | 5.18 | 3.001 | 0.0199 | 0.0081 | 3.9 | 217.8 |
| OH2 | 2.44 | 3.100 | 0.0129 | 0.0005 | 3.0 | −409.2 |
| OMe2 | 3.43 | 3.013 | 0.0158 | −0.0002 | 3.0 | −343.8 |
| OCH2 | 2.60 | 3.143 | 0.0122 | −0.0011 | 4.4 | −256.7 |
| OCHNH2 | 3.94 | 3.055 | 0.0141 (0.0079) | 0.0019 | 3.4 | −357.2 |
| NMA | 3.74 | 3.022 | 0.0136 (0.0054) | −0.0005 | 4.7 | −491.2 |
| PhF5I·· | 2.0688 | 828.3 | ||||
| NCH | 3.55 | 3.101 | 0.0093 | 0.0072 | −1.0 | 334.9 |
| HN=NH | 4.66 | 3.024 | 0.0118 | 0.0109 | 4.7 | 354.6 |
| Pyr-N | 6.75 | 2.922 | 0.0159 | 0.0195 | −3.9 | 354.6 |
| NH3 | 6.47 | 2.976 | 0.0145 | 0.0186 | −3.6 | 377.9 |
| NMe3 | 8.68 | 2.823 | 0.0199 | 0.0303 | −6.4 | 308.9 |
| OH2 | 4.67 | 2.937 | 0.0129 | 0.0081 | 6.3 | −692.9 |
| OMe2 | 5.40 | 2.890 | 0.0158 | 0.0103 | 5.0 | −510.5 |
| OCH2 | 4.20 | 2.967 | 0.0122 | 0.0074 | 5.7 | −406.5 |
| OCHNH2 | 6.37 | 2.895 | 0.0141 (0.0079) | 0.0126 | −1.3 | −524.4 |
| NMA | 6.83 | 2.874 | 0.0136 (0.0054) | 0.0107 | 0.8 | −702.5 |
| Base | H | V | ∇2ρ | Ellipticity | ρrcp | Egap | E2 |
|---|---|---|---|---|---|---|---|
| PhBr·· | |||||||
| N≡CH | 0.0016 | −0.0044 | 0.0302 | 0.0017 | 0.0245 | 0.306 | 1.15 |
| HN=NH | 0.0015 | −0.0061 | 0.0364 | 0.0652 | 0.0245 | 0.277 | 1.99 |
| PyrN | 0.0015 | −0.0076 | 0.0425 | 0.0612 | 0.0268 | 0.288 | 2.70 |
| NH3 | 0.0014 | −0.0076 | 0.0414 | 0.0019 | 0.0245 | 0.305 | 3.48 |
| NMe3 | 0.0012 | −0.0103 | 0.0507 | 0.0042 | 0.0245 | 0.294 | 3.38 |
| OH2 | 0.0018 | −0.0071 | 0.0427 | 0.0569 | 0.0245 | 0.307 | 2.45 |
| OMe2 | 0.0020 | −0.0094 | 0.0532 | 0.0068 | 0.0245 | 0.307 | 2.16 |
| OCH2 | 0.0019 | −0.0068 | 0.0422 | 0.0007 | 0.0245 | 0.296 | 1.62 |
| OCHNH2 | 0.0018 | −0.0067 | 0.0416 | 0.0115 | 0.0245 | 0.308 | 2.29 |
| NMA | 0.0021 | −0.0078 | 0.0481 | 0.0208 | 0.0245 | 0.304 | 2.30 |
| PhI·· | |||||||
| N≡CH | 0.0016 | −0.0054 | 0.0342 | 0.0007 | 0.0246 | 0.295 | 1.97 |
| HN=NH | 0.0011 | −0.0111 | 0.0536 | 0.0764 | 0.0221 | 0.261 | 3.00 |
| PyrN | 0.0011 | −0.0102 | 0.0499 | 0.0569 | 0.0268 | 0.272 | 4.74 |
| NH3 | 0.0011 | −0.0090 | 0.0446 | 0.0001 | 0.0246 | 0.296 | 5.12 |
| NMe3 | 0.0004 | −0.0131 | 0.0557 | 0.0027 | 0.0246 | 0.297 | 5.43 |
| OH2 | 0.0017 | −0.0086 | 0.0479 | 0.0746 | 0.0246 | 0.296 | 3.61 |
| OMe2 | 0.0016 | −0.0110 | 0.0568 | 0.0255 | 0.0246 | 0.297 | 3.33 |
| OCH2 | 0.0017 | −0.0077 | 0.0447 | 0.0248 | 0.0246 | 0.279 | 2.47 |
| OCHNH2 | 0.0017 | −0.0094 | 0.0514 | 0.0082 | 0.0246 | 0.298 | 4.12 |
| NMA | 0.0020 | −0.0095 | 0.0538 | 0.0296 | 0.0245 | 0.294 | 3.52 |
| PhF5I·· | |||||||
| NCH | 0.0017 | −0.0084 | 0.0473 | 0.0065 | 0.0220 | 0.299 | 3.83 |
| HN=NH | 0.0011 | −0.0111 | 0.0537 | 0.0764 | 0.0221 | 0.270 | 5.87 |
| Pyr-N | 0.0006 | −0.0146 | 0.0632 | 0.0500 | 0.0222 | 0.283 | 8.46 |
| NH3 | 0.0007 | −0.0131 | 0.0578 | 0.0072 | 0.0221 | 0.300 | 9.01 |
| NMe3 | −0.0012 | −0.0197 | 0.0693 | 0.0049 | 0.0279 | 0.302 | 10.58 |
| OH2 | 0.0018 | −0.0122 | 0.0634 | 0.1057 | 0.0220 | 0.296 | 5.39 |
| OMe2 | 0.0014 | −0.0146 | 0.0692 | 0.0399 | 0.0221 | 0.300 | 5.69 |
| OCH2 | 0.0017 | −0.0117 | 0.0603 | 0.0267 | 0.0220 | 0.287 | 4.83 |
| OCHNH2 | 0.0016 | −0.0137 | 0.0675 | 0.0049 | 0.0221 | 0.300 | 7.26 |
| NMA | 0.0019 | −0.0136 | 0.0699 | 0.0344 | 0.0221 | 0.297 | 6.17 |
| Base | Δσ(X) | Δσ(C) | ΔJ(C-X) | ΔJ(C-C) | ΔJ(C-H/F) |
|---|---|---|---|---|---|
| PhBr·· | 2085.9 | 26.2 | −67.0 | 83.7 | 170.3 |
| N≡CH | −35.3 | −2.4 | −12.5 | −0.6 | −0.2 |
| HN=NH | −43.3 | −2.7 | −15.6 | −0.7 | −0.8 |
| PyrN | −44.1 | −3.6 | −18.2 | −1.1 | −1.0 |
| NH3 | −47.0 | −3.8 | −18.0 | −1.3 | −1.2 |
| NMe3 | −52.6 | −3.6 | −26.7 | −1.3 | −1.2 |
| OH2 | −20.8 | −1.5 | −13.4 | −0.7 | −0.3 |
| OMe2 | −44.8 | −1.8 | −17.2 | −0.6 | −0.4 |
| OCH2 | −31.3 | −1.4 | −10.0 | −0.3 | −0.7 |
| OCHNH2 | −34.8 | −1.6 | −11.7 | −0.5 | −0.7 |
| NMA | −61.1 | −3.7 | −19.0 | −1.1 | −1.1 |
| PhI·· | 3103.7 | 39.1 | −172.6 | 80.8 | 170.7 |
| N≡CH | −77.9 | −4.3 | −68.2 | −1.1 | −1.1 |
| HN=NH | −67.6 | −4.7 | −74.8 | −1.3 | −1.4 |
| PyrN | −80.8 | −6.7 | −97.6 | −1.6 | −1.9 |
| NH3 | −49.6 | −6.8 | −109.5 | −1.7 | −1.9 |
| NMe3 | −15.4 | −7.3 | −100.5 | −1.9 | −2.1 |
| OH2 | −64.6 | −4.3 | −83.0 | −1.2 | −1.2 |
| OMe2 | −48.5 | −3.6 | −54.2 | −1.1 | −1.4 |
| OCH2 | −77.8 | −2.6 | −51.6 | −0.7 | −1.4 |
| OCHNH2 | −118.1 | −4.4 | −71.0 | −1.1 | −1.6 |
| NMA | −120.5 | −6.2 | −82.4 | −1.3 | −2.2 |
| PhF5I·· | 3517.6 | 73.3 | −270.7 | 105.0 | −299.5 |
| NCH | −79.8 | −5.7 | −125.0 | −1.2 | 2.8 |
| HN=NH | −88.8 | −6.6 | −132.7 | −1.3 | 3.2 |
| Pyr-N | −52.2 | −8.8 | −157.9 | −1.6 | 4.8 |
| NH3 | −10.4 | −8.6 | −158.5 | −1.6 | 4.4 |
| NMe3 | −82.0 | −10.2 | −171.6 | −1.5 | 5.2 |
| OH2 | −75.9 | −5.9 | −119.7 | −1.0 | 3.0 |
| OMe2 | −56.5 | −5.5 | −122.1 | −1.2 | 3.2 |
| OCH2 a | −36.4 | −4.4 | −105.2 | −0.8 | 4.2 |
| OCHNH2 a | +0.9 | −6.9 | −132.9 | −1.0 | 7.3 |
| NMA | −77.5 | −8.1 | −144.2 | −1.2 | 6.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amonov, A.; Scheiner, S. Relation between Halogen Bond Strength and IR and NMR Spectroscopic Markers. Molecules 2023, 28, 7520. https://doi.org/10.3390/molecules28227520
Amonov A, Scheiner S. Relation between Halogen Bond Strength and IR and NMR Spectroscopic Markers. Molecules. 2023; 28(22):7520. https://doi.org/10.3390/molecules28227520
Chicago/Turabian StyleAmonov, Akhtam, and Steve Scheiner. 2023. "Relation between Halogen Bond Strength and IR and NMR Spectroscopic Markers" Molecules 28, no. 22: 7520. https://doi.org/10.3390/molecules28227520
APA StyleAmonov, A., & Scheiner, S. (2023). Relation between Halogen Bond Strength and IR and NMR Spectroscopic Markers. Molecules, 28(22), 7520. https://doi.org/10.3390/molecules28227520