2. Results
The bulk amount of salts of cations C
3H
5+ and C
4H
7+ can be easily obtained by adding to the acid H(Cl
11) powder such a small amount of 1,2-dichloropropane or 1,2-dichloro-2-methylpropane, respectively, with the sample remaining powdery. The quality of the resulting salts can be controlled by IR spectroscopy (their IR spectra should coincide with the reference spectra of salts [
15,
16], indicating the absence of impurities). These salts are soluble in pentafluorobenzene only in the presence of small amounts of water. Storage of such solutions for 1 day under ambient conditions led to a release of colorless crystals from it. The solubility of the salts and the yield of crystals increased with an increase in the content of water. X-ray diffraction analysis of crystals isolated from C
4H
7+(Cl
11−) solutions showed that they are a salt of proton disolvate L−H
+−L formed by two alcohol molecules L, representing 1-hydroxy-2-methylpropene (
Figure 2).
The O⋅⋅⋅O distance in the OH
+O moiety (2.420 Å) is typical for proton disolvates with very strong H-bonds [
19,
20]. The position of the bridging proton was determined by means of an electron density difference map (short O-H distance of 1.14 Å). Bond lengths and bond angles of the cation, which we will designate as I
a (
Figure 3), are given in
Table 1 and
Figure 2. Four carbon atoms C1-C4 and an oxygen atom of the alcohol molecule are in the same plane, and their CCC and CCO angles are close to 120°, i.e., C1 and C2 atoms have sp
2 hybridization and, therefore, are double bonded. The C-O∙∙∙O angle is 118(3)°, which means that the O atom also has sp
2 hybridization and belongs to the alcohol OH group. The C=C double bond length of 1.286 Å was determined as the average of two isomeric alcohol molecules (
Figure S1 in Supplementary Materials). It is shorter than that in molecular C=C(–OH) fragments of enol tautomers (1.362 Å) [
22]. The C−O distance of 1.252 Å is also slightly shortened compared to that in a single C–O bond in the same (C=)C–OH fragment (1.333 Å) [
22].
The IR spectrum of the crystals is characteristic of proton disolvates: it contains an intense absorption pattern of the OH
+O group, consisting of three broad bands at 905, 1297 and 1552 cm
−1 (
Figure 4, red). The band at 905 cm
−1 belongs to ν
as(OH
+O), and bands 1297 and 1552 cm
−1 to mixed stretching and bending vibrations of the OH
+O group [
20]. The shape of the band at 905 cm
−1 is distorted by the resonance effect leading to so-called transparent windows (Evans holes) [
23], which appear as dips in the spectrum at 855 and 640 cm
−1.
The bands of CH stretching vibrations of CH
3 groups of cation I
a (
Figure 4, inset) are easily interpreted because they are similar to those of CH
3 groups of acetone in the previously studied disolvate Acetone−H
+−Acetone [
24], (
Table 2). A weak band at 3048 cm
−1 may belong to the stretching vibration of the =C-H bond, the C atom of which is adjacent to the OH
+O group.
There are more uncertainties in the interpretation of the C-O and C=C stretching vibrations. It has been found that absorption bands of C−O stretching vibrations of alcohol molecules directly bound to H
+ in proton disolvates are so strongly broadened and weakened in intensity that they are not detectable in IR spectra [
24]. Two bands at 1560 and 1683 cm
−1 (
Figure 4) are in the expected frequency range of C=C stretch vibrations [
15,
16,
17] and can be attributed to them. The fact that the position of the bridging proton is determined by X-ray diffraction means that its two-well potential has a high enough potential barrier for the proton to be at the bottom of one of the wells for a sufficiently long time (at the time scale of IR spectroscopy) in order to demonstrate (in an IR spectrum) the non-equivalence of two alcohol molecules in I as two C=C stretching frequencies. One of them at 1560 cm
−1 is close to the frequency (1555 cm
−1) of the cation in contact ion pair (CH
3)
2C=C
+−H…(Cl
11−). Therefore, it can be assumed that the frequency 1560 cm
−1 belongs to the isobutylene alcohol molecule, which has a shorter O
+−H bond (1.14 Å) and imitates a protonated alcohol molecule solvated by the second L alcohol molecule (CH
3)
2C=CH−OH−
+H…L, which is less influenced by the positive charge and has an increased C=C stretch frequency: 1683 cm
−1. The CC and CH stretch frequencies are summarized in
Table 2.
It follows from the obtained results that disolvate Ia is generated by the interaction of the C
4H
7+ carbocation with water molecules, as shown in
Scheme 1. The weak spectrum of the H
3O
+ cation [
25], which should arise simultaneously, actually manifests itself in the IR spectrum of the viscous phase, which precipitates concurrently with the crystalline phase.
X-ray diffraction analysis of crystals—grown from a solution of the C
3H
5+(Cl
11−) salt under the same conditions under which crystals of the salt of Ia were obtained—showed partially disordered structure. This property does not allow to see in detail the entire structure of the cation but enables us to determine its similarity with cation Ia. The similarity is confirmed by the identity of the IR spectrum of the crystals to that of the salt of disolvate Ia (
Figure 4), which means that the crystals growing from solutions of C
3H
5+(Cl
11−) are proton disolvates L
2-H
+-L
2, where L
2 is CH
3CH=CHOH. Hereafter, they will be referred to as cations Ib (
Figure 2).
The solubility of salts of cations C
3H
5+ and C
4H
7+ in carefully dehydrated pentafluorobenzene is very low, and crystals cannot be grown from them. To increase the solubility of the salts, 1 vol% acetone was added to pentafluorobenzene containing trace amounts of water. This approach helped to obtain a solution with a heightened salt content and a reduced H
2O/cation
+ molar ratio. Keeping this solution over hexane vapor for 1 to 2 weeks led to the appearance of crystals. The X-ray diffraction analysis of the crystals obtained from solutions of the C
4H
7+(Cl
11−) salts revealed that they contain the C
4H
7+ cation solvated by one molecule of 1-hydroxy-2-methylpropane (
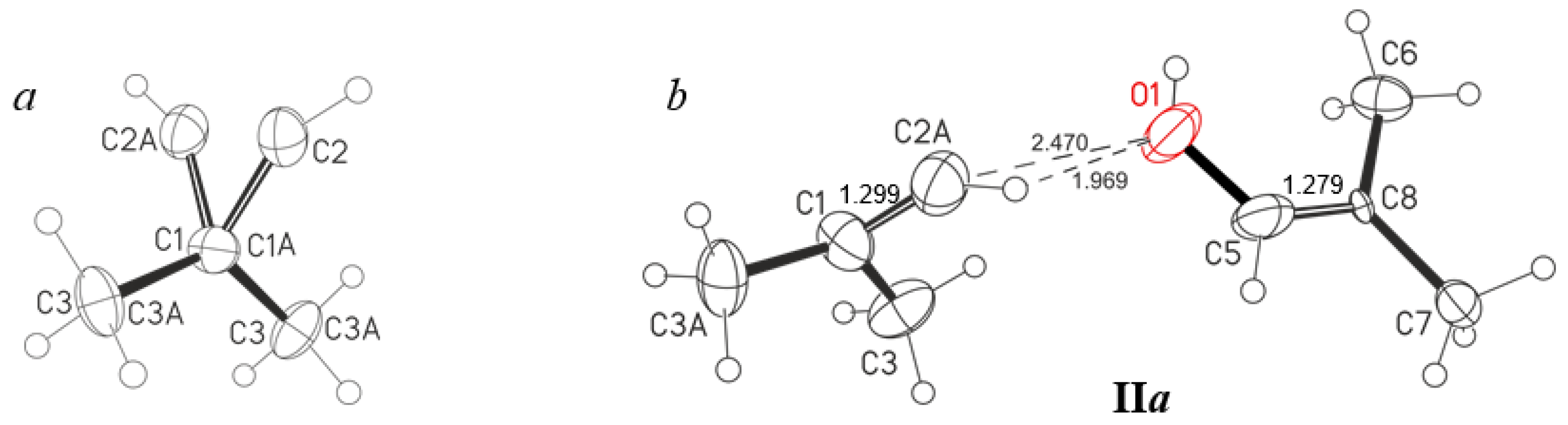
Figure 5). The crystal lattice does not have an acetone molecule but contains one solvent molecule, C
6HF
5, per two salt molecules. The C
4H
7+ cation is disordered over two positions differing in the location of the C2A atom slightly above or below the plane of three atoms: C1, C3, and C3A (
Figure 5a and
Figure S1 in Supplementary Materials). The C1-C2A∙∙∙O1 and C5-O1∙∙∙C2A angles are 137° and 121°, respectively (
Figure 5b,
Table 3), which means sp
2 hybridization of C2A and O1 atoms, i.e., an H atom is attached to each of them. The C∙∙∙O distance is 2.470 Å, which matches the maximum allowable distance between the O atoms of the symmetric O∙∙∙H
+∙∙∙O moiety in proton disolvates (2.47 Å for the most unstable proton disolvates obtained: H
+(nitrobenzene)
2) [
19]. Therefore, with a high probability, a bridging proton is located between the C2A and O1 atoms, forming a short, strong, and low-barrier double-well H-bond, although compounds containing asymmetric moiety (C)C−H
+∙∙∙O with a strong H-bond are currently unknown. The short distance (C2A)H∙∙∙O1 of 1.97 Å, also points to the presence of a strong H- bond. The significant difference of the C2A-H∙∙∙O1 angle at 111° from the optimal one at 180° may be partly due to the inaccuracy of determining the localization of the H atom by the calculation method. The question of the presence of a strong H-bond with double-well proton potential is discussed below when the IR spectra of these crystals are examined. The C
4H
7+·OHC
4H
7 cation under consideration is hereafter denoted as IIa (
Figure 2).
X-ray diffraction analysis of crystals obtained from a solution of C
3H
5+(Cl
11−) in C
6HF
5 + 1% acetone indicated that they contain the hydrocarbon cations and C
6HF
5 inclusion molecules of the solvents with significantly disordered C and F-atoms. We failed to localize the highly disordered structure of the cation. For this reason, we do not present or discuss these X-ray diffraction data. Nonetheless, they provide some useful information. For instance, in the crystal lattice, alcohol molecules C
3H
5OH with reliably fixed C and O atoms were identified, possibly indicating the solvation of the cation with the C
3H
5OH molecule in the same way as the C
4H
7OH molecule solvates the C
4H
7+ cation in adduct II
a. That is, we can assume the emergence of cationic adduct C
3H
5+·C
3H
5OH, similar to IIa, and designate it as IIb (
Figure 2). The IR spectra of crystals containing IIa and IIb adducts are identical (
Figure 6), which confirms that IIb is C
3H
5+·C
3H
5OH.
In the IR spectra of cationic adducts IIa and IIb, specific absorption of a strong C–H
+∙∙∙O H-bond should be present; however, this type of strong H-bond has not yet been found. Well-studied strong hydrogen bonds are symmetric O∙∙∙H
+∙∙∙O or N∙∙∙H
+∙∙∙N H-bonds in proton disolvates L
2H
+ with double- well proton potential separated by a low potential barrier that transforms it into flat-bottom potential for vibrational transitions. In these cases, the proton vibrations appear in the IR spectra as an intense and broad absorption pattern with a maximum at 850–1000 cm
−1 [
25], as observed in the spectra of cations Ia and Ib (
Figure 4). In proton disolvates with an asymmetric moiety, for example, N−H
+∙∙∙O, the bottom of the double-well potential is asymmetric and the maximum of the broad and intense absorption shifts to 1400–1700 cm
−1 [
25]. In the spectra of the analyzed adducts IIa and IIb, a broad band at 1630 cm
−1 is observed, as is absorption in the region of 1200–1500 cm
−1, which is not described by a single Gaussian (
Figure 6, inset). They correspond fairly well to the asymmetric X
1−H
+∙∙∙X
2 fragment, in our case =C−H
+∙∙∙O.
The C=C bond of cations C
3H
5+ and C
4H
7+ is affected by the bridging proton, and this effect should specifically influence the absorption of its stretch vibrations. For example, it has been found [
24] that if a single C-O bond is attached to a bridged proton its absorption band broadens and decreases in intensity so much that it is undetectable in the IR spectrum. If the C=O bond is attached to H
+, as in proton disolvate Acetone-H
+-Acetone (discussed below), it still appears in the spectrum as a weak-to-moderately intense broadened band [
24]. Therefore, absorption of the C=C stretch vibration of C
3H
5+ and C
4H
7+ may look like a weak band at 1633 cm
−1 (
Figure 6). It is higher in frequency than νC=C at ~1560–1590 cm
−1 in the contact ion pairs (CIPs) formed with the (Cl
11−) anion in solutions and in a solid phase [
15,
16,
17]. This means that the interaction of C
3H
5+ and C
4H
7+ cations with an alcohol molecule in II is stronger than the interaction with the (Cl
11−) anion in the CIP.
The C=C band of the alcohol molecule in adducts IIa and IIb is subject to a weaker influence of the bridging proton. Its C=C stretching vibration is observed at 1704 cm
−1 (
Figure 6). It is a single band for adduct IIb, whereas in the spectrum of IIa it is split into two components at 1715 and 1694 cm
−1 of equal intensity (
Figure 6, inset). Obviously, in the crystal lattice of the II
a adduct salt, there are two weakly nonequivalent C
4H
7OH molecules.
In the CH stretch frequency region, the three bands at 2961, 2932 and 2874 cm
−1 can be unambiguously interpreted as vibrations of the CH
3 group (
Table 2). The IR spectra of the crystals also contain absorption bands of the captured pentafluorobenzene molecule. They are easily identified because of the finding that when a crystal is crushed on an ATR accessory and its crystal lattice is destroyed, pentafluorobenzene is released and slowly evaporates when the sample is kept in a glove box atmosphere. Therefore, the intensity of its absorption decreases over time. The most intense C
6F
5H bands in
Figure 6 are marked with asterisks.
The band at 3370 cm−1 may belong to OH groups of the C4H7OH alcohol molecule, which are engaged in weak H bonds with the (Cl11−) anion.
The solution from which crystals of the salt of II
b grew was kept in a sealed ampoule, and after a few days a small number of tiny crystals grew from it. It was possible to find one crystal of sufficient size for X-ray analysis. It turned out that this was a salt of the monohydrate of the propylene cation (C
3H
5+∙OH
2)(Cl
11−) (
Figure 7). The C
3H
5+∙OH
2 species has two localizations in the unit cell with slightly different positions of C and O atoms, but they can be distinguished (
Figure 7a). Similarly, an anion can be disordered over two positions, as indicated by the presence of electron density peaks in the difference map in the region of the anion. Nonetheless, we failed to localize the second position of the anion. This may be the reason for substantial deterioration of the R
f factor, but does not interfere with the determination of coordinates of the C and O atoms of the cation with accuracy sufficient to establish the topology of the cation qualitatively and its main geometric parameters (
Table 4).
The main feature of the structure of the C
3H
5+∙OH
2 cationic adduct, which is denoted below as III
b (
Figure 2), is as follows. The H
2O molecule is attached to the C
3H
5+ cation through the O atom in the direction perpendicular to the C=C double bond at a distance of 2.32 Å, which is very close to the average Na···O(H
2) distance of 2.333 Å in the first hydration shell of hexahydrate Na(OH
2)
6 as determined by X-ray diffraction analysis for 13 structures (retrieval was made according to the Cambridge structural data base, ConQuest 2021.3.0) [
26]. Therefore, the nature of the interaction of the H
2O molecule with the C=C
+ bond of the vinyl cation is similar to that of the interaction of water molecules with the alkali metal cation in its first hydration shell.
Unfortunately, the insufficient amount of the obtained crystals of (C
3H
5+∙OH
2)(Cl
11−) did not allow us to register their IR spectrum. Nevertheless, the spectra of these compounds have been obtained by us earlier, when we characterized crystalline salts C
3H
5+(Cl
11−) and C
4H
7+(Cl
11−) by X-ray diffraction and IR spectroscopy [
15,
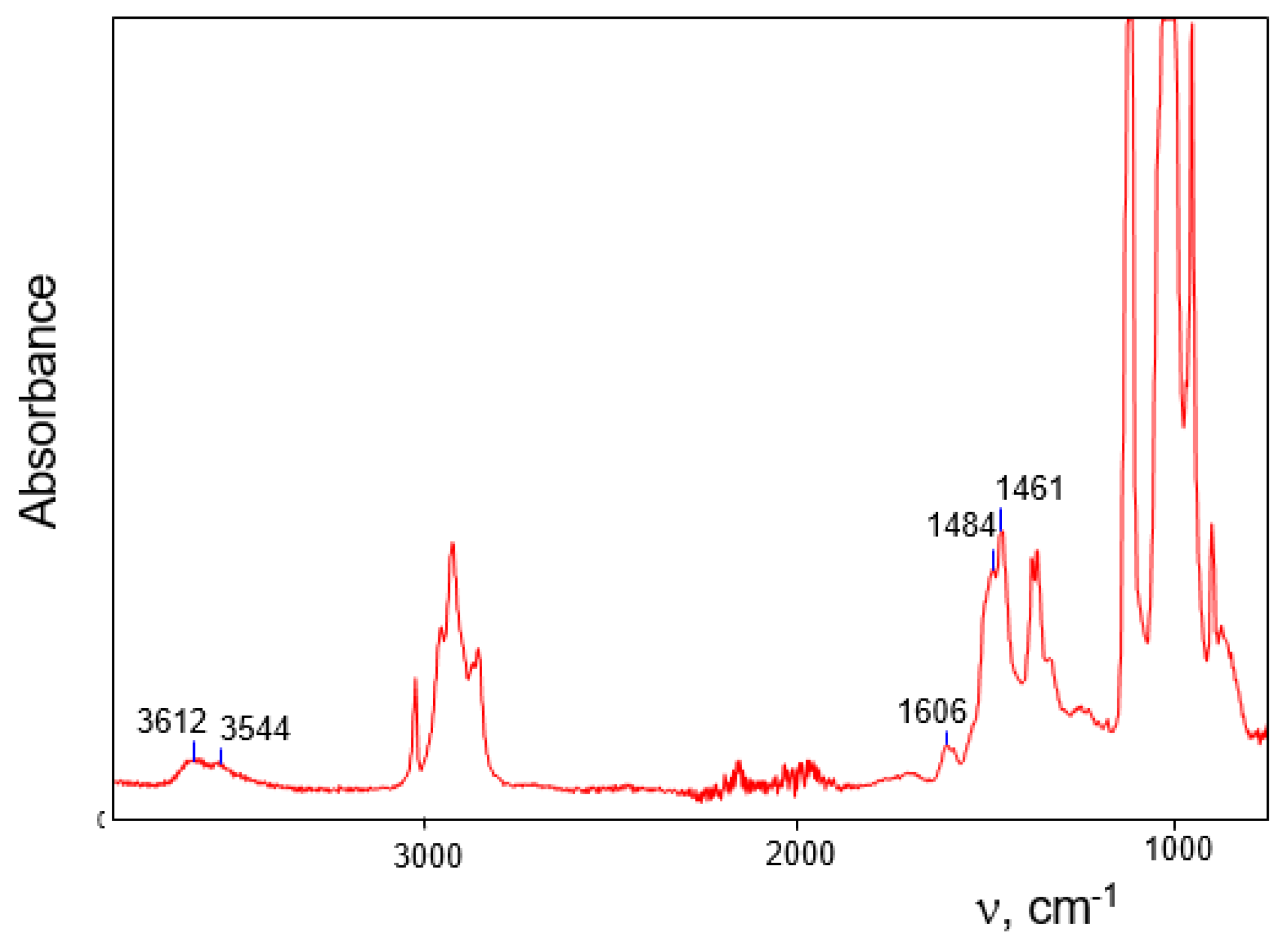
16]. In the IR spectra of individually selected single crystals of these salts subjected to X-ray analysis, no traces of water absorption were found. On the other hand, during the recording of IR spectra of an aggregate of small crystals that arose simultaneously with larger ones, a weak spectrum of water molecules was observed (
Figure 8). Its frequencies ν
as, ν
s and δ at 3612, 3544, and 1606 cm
−1, respectively, are similar to those of monomeric molecules dissolved in organic solvents or hydrated alkali metal cations (
Table 5). This result was surprising and could not be explained. Now it is clear that the observed bands belong to non–H-bonded water molecules of monohydrates (C
4H
7+∙OH
2)(Cl
11−) and (C
3H
5+∙OH
2)(Cl
11−) designated subsequently as IIIa and IIIb (
Figure 2), which are formed and co-crystallize with salts of anhydrous vinyl cations. They are typical of the H
2O molecules that hydrate the H
3O
+ cation in H
3O
+(H
2O)
3(Cl
11−) crystals [
27], or alkali metal cations (
Table 5). Such mostly ionic interaction Cat
+···OH
2 leads to a slight decrease in the frequencies of OH stretching, compared to those of the dissolved monomer molecule. In adducts III
a/b, OH stretching frequencies are somewhat lower than those in hydrates of alkali metal cations and H
3O
+; hence the strength of bond Cat
+···OH
2 in III
a/b is higher.
As mentioned above, crystals of compounds IIa and IIb were obtained from saturated solutions of vinyl cation salts in C
6HF
5 containing 1 vol% acetone. It could be expected that after an increase in the acetone content, crystals containing acetone could be obtained. Incubation of saturated solutions of the C
3H
5+(Cl
11−) or C
4H
7+(Cl
11−) salts in C
6HF
5 + 5% acetone over hexane vapor gave rise to needle-like crystals. X-ray diffraction analysis indicated that this was proton disolvate salt Ac-H
+-Ac (IV), where Ac is the acetone (
Figure 2 and
Figure 9a). The same salt was obtained in a different way by reacting the H(Cl
11) acid with acetone vapor and its subsequent dissolution in dichloromethane. Slow evaporation of the solution in the glove box led to the formation of crystals. Their X-ray diffraction analysis showed that this was also proton disolvate salt (Ac)
2H
+(Cl
11−) but with other crystal lattice parameters, that is, it is a different polymorph (
Figure 9b). The O⋅⋅⋅O distance in cations for both polymorphs is almost the same, 2.429 and 2.423 Å. Selected geometric parameters of IV and IV′ are given in
Table 6.
3. Discussion
The results make it possible to establish the sequence of the interaction of vinyl cations with water molecules. Initially, an H2O molecule is attached to the C=C+ bond of cation RCH=C+H or R2C=C+H (where R is CH3) via the O- atom (Equation (1), in it and in subsequent equations, the R2C=C+H cation is used).
The nature of this bond is close to that of bonds formed by water molecules with alkali metal cations during their hydration or with H3O+ in crystal salts of H3O+·3H2O, i.e., the bond is strongly ionic.
With an increase in the concentration of the salt of cationic adduct R
2C=C
+H···H
2O in solutions, the self-association of ion pairs increases (which is typical for salts of carbocations in solutions [
29]). This enhances the contact interaction of H
2O with the cation and promotes the transfer of a proton to a water molecule with the formation of H
3O
+ and cationic adduct II:
With a further increase in the contents of water and of the carbocation salt, the concentration of the resulting adducts II and of the H3O+ cation increases. The latter can protonate an alcohol molecule that is more basic than H2O, thereby producing a proton disolvate and regenerating cation R2C=C+ H (Equation (3)),

which next interacts with water, closing the cycle. The alcohol molecule of adduct II can also be protonated directly by cation III, thus by passing the stage of H
3O
+ formation:

The studied cationic adducts enable one to trace the change in the nature of the interaction of vinyl cations with basic molecules of H
2O, alcohol, and acetone as their basicity increases. The H
2O molecule is attached to the C=C bond of the vinyl cation (
Scheme 2, III) with a strength similar to that of an H
2O molecule attached to an alkali metal cation or to the H
3O
+ cation in H
3O
+(H
2O)
3. As the basicity of the O atom increases from H
2O to alcohol molecule, its interaction with the cation shifts to the H atom, thereby producing a strong H-bond, with a partially covalent character [
30], and an increase in the C⋅ ⋅ ⋅ ⋅ ⋅O distance to 2.47 Å (
Scheme 2, II). In this case, the asymmetric double-well potential of the bridging proton has a deeper minimum at the C atom. A further increase in the basicity of the oxygen atom (acetone molecule) causes a shift of the minimum of the double-well potential to the O atom with a high probability of proton transfer to the acetone molecule. The loss of a proton by the vinyl cation results in an extremely reactive carbene molecule (with C=C: as the active site), which then reacts with the components of the mixture, forming non-crystallizing products (wax phase). The released protonated acetone adds a second acetone molecule thereby generating proton disolvate IV.
Thus, the interaction of the vinyl cation with nucleophile L proceeds: (1) through its addition to a charged double bond (if L = H
2O) or H-bonded to the =C-H group (if L = alcohol molecule), and (2) through the protonation of L with the transition of the vinyl cation to the neutral carbene molecule. The second mechanism of interaction was proposed in ref. [
31] and proved for the benzyl carbocation [
32].
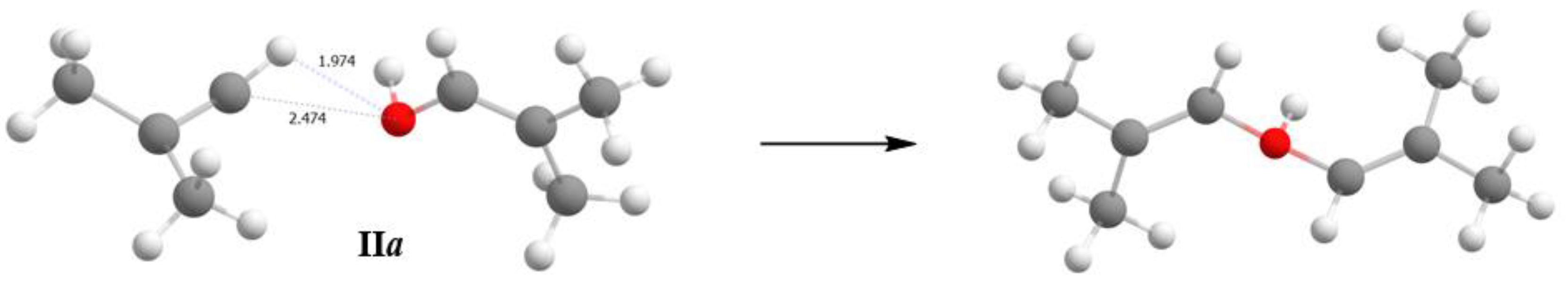
The finding that adducts II and III with strongly ionic Cat
+−L interaction exist is surprising. They can exist if the basicity of L slightly exceeds that of counterion (Cl
11−). This means that vinyl cations behave like rather chemically inert particles, contradicting predictions of quantum chemical calculations. For example, if the crystallographic structures of adducts II and III are optimized, then the covalent interaction of the cation with the H
2O or alcohol molecule results in the emergence (in the case of C
4H
7+) of molecules of protonated isobutenyl alcohol and diisobutenyl ether, respectively (
Figure 10), with a significant gain in energy.
For example, if we compare energies of the structures calculated at the UB3LYP/6- 311++G(d,p) level of theory: (1) energy with optimized H atomic coordinates and fixed coordinates of C and O that are equal to the coordinates that follow from the X-Ray data for IIa, and (2) energy with fully optimized structure (including the C, O and H atomic coordinates), energies (1) and (2) are related as 69.71 and 0 kcal/mol with the transformation of structure IIa into protonated diisobutenyl ether. The environment cannot have a stronger stabilizing effect on the IIa structure because the purely ionic interaction of C and O atoms with Cl atoms of the anionic environment is weak (C···Cl and O···Cl distances exceed the sum of van der Waals atomic radii). Thus, it follows from quantum chemical calculations that adducts II and III should not exist, which contradicts the experimental findings. Therefore, the use of quantum chemical calculations requires caution in studies on mechanisms of the interaction of vinyl cations with neutral molecules.
5. Conclusions
Vinyl cations C3H5+ and C4H7+ (Cat+) in solutions of their salts in dichloromethane and C6HF5 interact with O-containing nucleophiles as follows.
An H2O molecule attaches to the C=C bond of Cat+ in a similar manner to the hydration of alkali metal cations, thereby yielding Cat+·H2O adducts with a strongly ionic bond. Its strength only slightly exceeds the strength of the interaction of the (Cl11−) anion with the vinyl cation in contact ion pairs Cat+(Cl11−).
With an increase in the content of Cat+·H2O adducts in solutions, the adduct self-associates and interacts with a transfer of a proton to one water molecule and attachment of the second one to the C=C bond, thus forming H3O+ and an alcohol molecule, respectively.
The alcohol molecule interacts predominantly with the H atom of the C=C+−H moiety of the vinyl cation, thereby producing a proton disolvate with strong asymmetric H-bond =C−+H···O having double-well proton potential with a deeper minimum near the C atom.
A further increase in the water content in the solutions leads to complete conversion of vinyl cations into alcohol molecules with the formation of symmetric proton disolvates LH
+L containing strong and partially covalent O−H
+−O hydrogen bonds [
30] and H
3O
+ cations.
The interaction of the vinyl cations with acetone molecules, which are more basic than H2O or alcohol molecules, causes the formation of only symmetrical proton disolvates, LH+L, in the absence of acetone-containing cationic adducts. The vinyl cation is converted into carbene containing a highly reactive C=C: moiety.
Summing up, we can say that the interaction of the vinyl cation with base L proceeds through two mechanisms: via the formation of adducts (SN1 reaction), and via the mechanism where vinyl cation acts as a protonating agent. When the basicity of L is close to that of a single water molecule, L attaches to the double C=C bond thereby producing an adduct. As the basicity of L increases, the interaction with the C=C+−H moiety of the vinyl cation strengthens and is shifted to the H atom, thus forming a solvate having a strong asymmetrical =C−+H···O hydrogen bond. Further strengthening of the basicity of L leads to the transfer of a proton to L and to the emergence of the eventually symmetric LH+L cation. The loss of a proton by the vinyl cation converts it into a neutral reactive carbene molecule containing a C=C: moiety.
The formation of adducts with water and alcohol molecules by vinyl cations is unexpected, because according to quantum chemical calculations, they are energetically unfavorable and should not exist.
The very existence of these adducts means that the alleged high reactivity of vinyl carbocations is an overestimation.








 which next interacts with water, closing the cycle. The alcohol molecule of adduct II can also be protonated directly by cation III, thus by passing the stage of H3O+ formation:
which next interacts with water, closing the cycle. The alcohol molecule of adduct II can also be protonated directly by cation III, thus by passing the stage of H3O+ formation:













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}