4.2. Synthesis of Scutellarein Derivatives
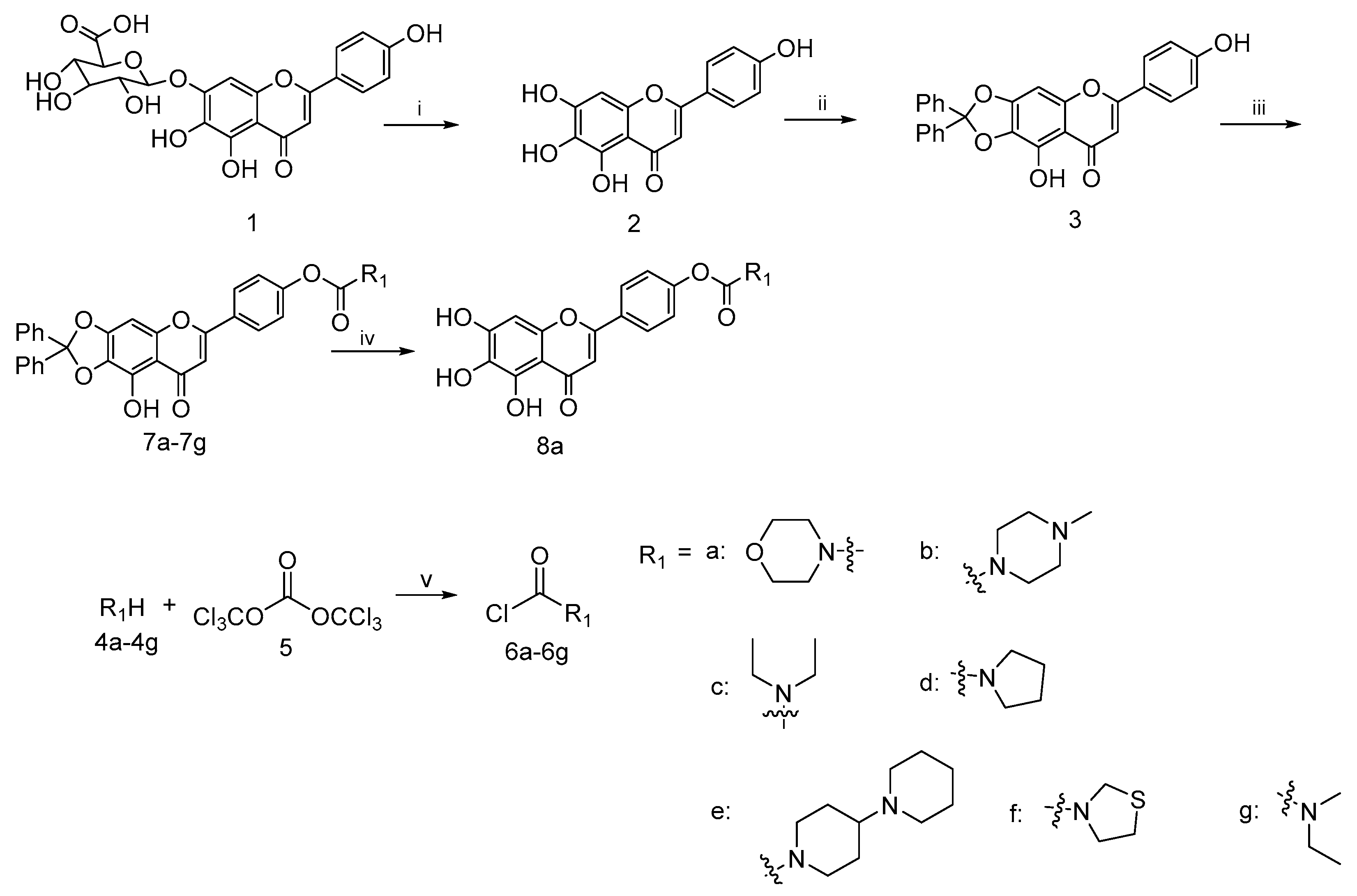
4.2.1. 5,6,7-Trihydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one (2)
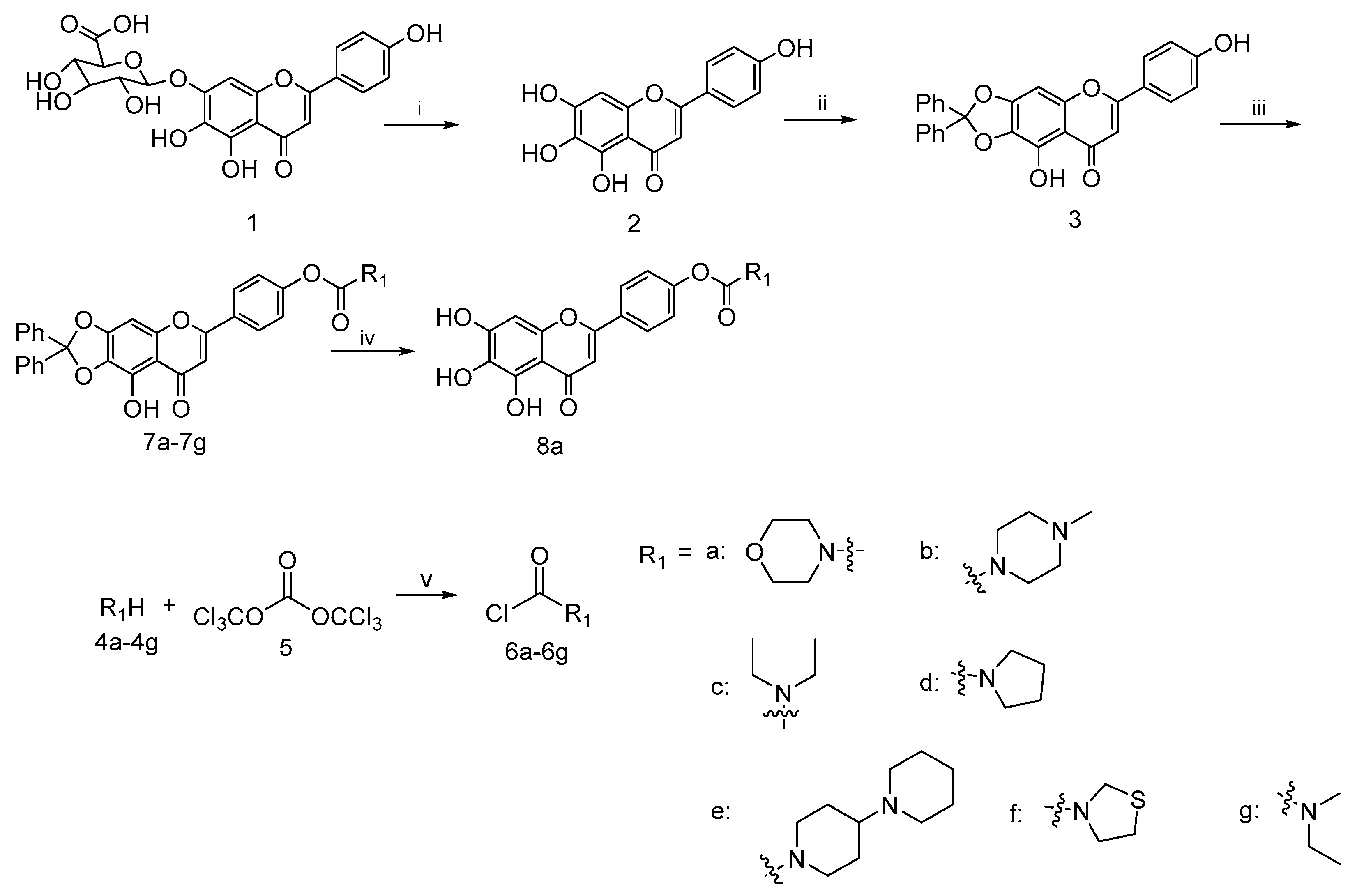
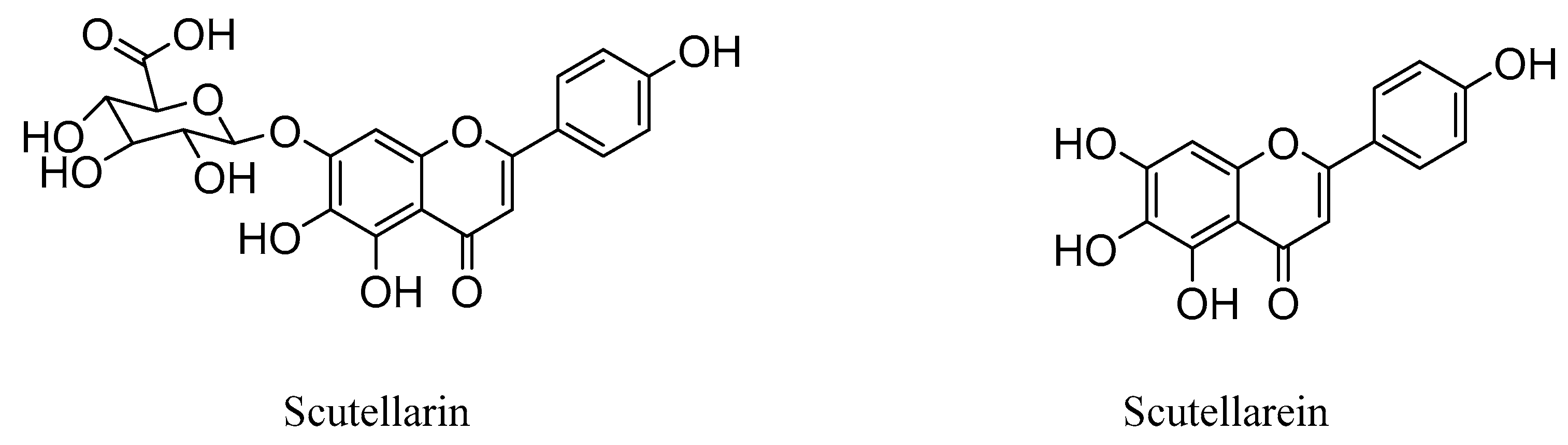
To a well-stirred mixture of scutellarin (1) (3.0 g, 6.4 mmol) in 95% ethanol (48 mL), solution of concentrated H2SO4 (18 mL) was added dropwise and the reaction was refluxed for 4 h under nitrogen. After that, excess water (50 mL) was added in portions to the reaction mixture. Meanwhile, the precipitates that appeared were filtered, washed with glacial acetic acid and dried in vacuo to afford scutellarein 2 (1.1 g, 59% yield) as a yellow solid; mp 324−335 °C; 1H NMR (400 MHz, CDCl3) δ 12.70 (s, 1H), 7.73–7.82 (m, 2H), 7.49–7.59 (m, 4H), 7.28–7.35 (m, 6H), 7.15–7.25 (m, 2H), 6.55 (s, 1H), 6.58 (s, 1H), 3.26–3.32 (m, 2H), 3.21 (q, J = 7.17 Hz, 2H) and 1.09–1.13 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 182.5, 164.0, 161.5, 153.8, 150.1, 147.5, 129.6, 128.8, 121.9, 116.4, 104.4, 102.7 and 94.3. HRMS(Q-TOF) m/z calculated for C15H10O6 [M + H]+: 287.0477 found: 287.0490.
4.2.2. 9-Hydroxy-6-(4-hydroxyphenyl)-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-8-one (3)
A mixture of scutellarein (2) (10.0 g, 34.94 mmol) and dichlorodiphenylmethane (10.0 g, 42.17 mmol) in diphenyl ether (200 mL) was stirred at 180 °C under nitrogen protection for 1.5 h. Then, the resulting dilution was allowed to slowly cool to room temperature. Petroleum ether (400 mL) was added dropwise to the solution to remove the diphenyl ether from the solid that appeared. Then, the filtered solid was purified by column chromatography on silica gel with 25% ethyl acetate in petroleum ether as eluent to afford 3 (10.7 g, 68%) as a yellow solid; mp 357−364 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.81–7.93 (m, 2H), 7.46–7.55 (m, 4H), 7.36–7.46 (m, 6H), 6.99 (s, 1H), 6.89 (d, J = 8.80 Hz, 2H) and 6.79 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 182.9, 164.7, 162.8, 162.2, 153.2, 152.9, 139.3, 130.2, 129.2, 129.2, 129.0, 126.3, 121.1, 118.7, 116.5, 107.4, 103.1, 36.3 and 31.2. HRMS(Q-TOF) m/z calculated for C28H18O6 [M + H]+: 451.1103 found: 451.1179.
4.2.3. General Procedure for the Synthesis of Compounds 7a–7g
To a well-stirred mixture of compound 4a–4g (10.3 mmol) in dichlorodiphenylmethane (10 mL) a solution of triphosgene (3.0 g, 10.1 mmol) in DCM (15 mL) was added dropwise at 0 °C and the reaction mixture was stirred for 6–8 h. Then it was allowed to warm slowly to room temperature. After the disappearance of starting material, as indicated by TLC, the reaction mixture was blown by nitrogen and filtered to afford the filtrate 6a–6g, respectively, which could be used directly in the next step.
To a well-stirred solution of compound 3 (3 g, 6.6 mmol) in anhydrous DMF (30 mL) at rt. the aforementioned fresh prepared carbamoyl chloride 6a–6g in the presence of K2CO3 (1.0 g, 7.2 mmol) and KI (1.0 g, 6.0 mmol) was added. The resulting solution was diluted with ethyl acetate, washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The crude residue was purified by flash column chromatography on silica gel using 50% ethyl acetate in petroleum ether as eluent to afford 7a–7g.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl morpholine-4-carboxylate (7a)
This compound (2.2 g, yield 54.0%) was prepared from important immediate compound 3 (3.0 g, 6.6 mmol), DMF (30 mL) and 4-morpholinylcarbonyl chloride 6a (1.2 g, 8 mmol) according to the general procedure described above. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 7.97–8.19 (m, J = 8.80 Hz, 2H), 7.47–7.55 (m, 5H), 7.39–7.45 (m, 6H), 7.28–7.34 (m, J = 8.80 Hz, 2H), 7.08 (s, 1H), 7.01 (s, 1H), 3.57–3.64 (m, 4H), 3.45–3.57 (m, 2H) and 3.38 (br. s., 2H). 13C NMR (101 MHz, DMSO-d6) δ 183.2, 163.5, 154.5, 153.2, 139.2, 130.3, 129.2, 128.3, 126.3, 123.0, 107.6, 22.6 and 19.4. HRMS(Q-TOF) m/z calculated for C33H25NO8 [M + H]+: 564.1580 found: 564.1654.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl 4-methylpiperazine-1-carboxylate (7b)
This compound (0.4 g, yield 62.6%) was prepared from important immediate compound 3 (0.5 g, 1.1 mmol), DMF (10 mL) and 4-methylpiperazine-1-carbonyl chloride 6b (0.18 g, 1.3 mmol) according to the general procedure described above. 1H NMR (400 MHz, DMSO-d6) δ 13.00 (br. s., 1H), 8.12 (d, J = 8.07 Hz, 2H), 7.53–7.62 (m, 4H), 7.48 (d, J = 5.62 Hz, 6H), 7.36 (d, J = 7.58 Hz, 2H), 7.14 (s, 1H), 7.07 (s, 1H), 3.62 (br. s., 2H), 3.45 (br. s., 2H), 2.41 (br. s., 4H) and 2.25 (s, 3H). 13C NMR (101 MHz, METHANOL-d4) δ 166.6, 152.1, 98.3, 94.2, 77.1, 76.5, 76.1, 73.0, 70.3, 50.3, 50.1 and 12.5. HRMS(Q-TOF) m/z calculated for C34H28N2O7 [M + H]+: 577.1897 found: 577.1964.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl diethylcarbamate (7c)
This compound (0.32 g, yield 52.5%) was prepared from important immediate compound 3 (0.5 g, 1.1 mmol), DMF (10 mL) and diethylcarbamic chloride 6c (0.18 g, 1.3 mmol) according to the general procedure described above. 1H NMR (400 MHz, CDCl3) δ 12.70 (s, 1H), 7.73–7.82 (m, 2H), 7.49–7.59 (m, 4H), 7.28–7.35 (m, 6H), 7.15–7.25 (m, 2H), 6.55 (s, 1H), 6.58 (s, 1H), 3.26–3.32 (m, 2H), 3.21 (q, J = 7.17 Hz, 2H) and 1.09–1.13 (m, 6H). 13C NMR (101 MHz, CHLOROFORM-d) δ 182.99, 163.42,154.44, 153.50, 153.27, 142.30, 139.25, 130.04, 129.48, 128.38, 127.86, 127.43, 126.33, 122.45, 119.22, 107.77, 105.23, 89.56, 42.44, 42.37, 14.22 and 13.11. HRMS(Q-TOF) m/z calculated for C33H27NO7 [M + H]+: 550.1788 found: 550.1859.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl pyrrolidine-1-carboxylate (7d)
This compound (0.19 g, yield 79.2%) was prepared from important immediate compound 3 (0.2 g, 0.4 mmol), DMF (10 mL) and pyrrolidine-1-carbonyl chloride 6d (0.13 g, 0.97 mmol) according to the general procedure described above. 1H NMR (400 MHz, CHLOROFORM-d) δ 12.69 (s, 1H), 7.70–7.88 (m, 2H), 7.46–7.59 (m, 4H), 7.27–7.37 (m, 6H), 7.20–7.23 (m, 2H), 6.55 (s, 1H), 6.58 (s, 1H), 3.28–3.44 (m, 4H) and 1.17–1.22 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 183.0, 163.4, 142.3, 139.3, 130.1, 129.5, 128.4, 127.9, 127.4, 126.3, 122.4 and 89.5. HRMS(Q-TOF) m/z calculated for C33H25NO7 [M + Na]+: 570.1631 found: 572.1684.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl [1,4′-bipiperidine]-1′-carboxylate (7e)
This compound (0.18 g, yield 64.3%) was prepared from important immediate compound 3 (0.2 g, 0.4 mmol), DMF (10 mL) and [1,4′-bipiperidine]-1′-carbonyl chloride 6e (0.23 g,0.99 mmol) according to the general procedure described above. 1H NMR (400 MHz, CDCl3) δ ppm 1.12–1.26 (m, 2 H), 1.33–1.42 (m, 2 H), 1.49–1.56 (m, 5 H), 1.82 (d, J = 12.72 Hz, 2 H), 2.35–2.42 (m, 1 H), 2.42–2.50 (m, 4 H), 2.77 (t, J = 12.23 Hz, 1 H), 2.91 (t, J = 12.23 Hz, 1 H), 4.14–4.40 (m, 2 H), 6.48–6.60 (m, 2 H), 7.14–7.23 (m, 2 H), 7.25–7.38 (m, 6 H), 7.45–7.61 (m, 4 H) and 7.69–7.84 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ 183.0, 163.3, 153.5, 153.3, 142.3, 139.3, 129.5, 128.4, 128.1, 127.5, 126.3, 122.4, 119.2, 107.8, 105.3, 89.5, 62.5, 50.3, 26.0 and 24.5. HRMS(Q-TOF) m/z calculated for C39H36N2O7 [M + H]+: 645.2523 found: 645.2678.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl thiazolidine-3-carboxylate (7f)
This compound (0.05 g, yield 71.4%) was prepared from important immediate compound 3 (0.06 g, 0.1 mmol), DMF (10 mL) and thiazolidine-3-carbonyl chloride 6f (0.04 g, 0.2 mmol) according to the general procedure described above. 1H NMR (400 MHz, CDCl3) δ ppm 3.05–3.19 (m, 2 H), 3.84–3.97 (m, 2 H), 4.61 (s, 1 H), 4.68 (s, 1 H), 6.62–6.65 (m, 1 H), 6.66 (s, 1 H), 7.31 (m, J = 8.80 Hz, 2 H), 7.38–7.43 (m, 6 H), 7.58–7.66 (m, 4 H), 7.81–7.93 (m, 2 H) and 12.75 (s, 1 H). 13C NMR (101 MHz, CDCl3) δ ppm 182.95, 163.17, 153.55, 153.28, 142.31, 139.24, 130.09, 129.48, 128.38, 127.55, 126.34, 122.35, 119.26, 107.79, 105.42 and 89.53. HRMS(Q-TOF) m/z calculated for C32H23NO7S [M + H]+:566.5925 found: 566.1265.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl ethyl(methyl)carbamate (7g)
This compound (0.16 g, yield 71.4%) was prepared from important immediate compound 3 (0.2 g, 0.4 mmol), DMF (10 mL) and ethyl(methyl)carbamic chloride 6g (0.12 g, 0.9 mmol) according to the general procedure described above. 1H NMR (400 MHz, CDCl3) δ 12.67 (s, 1H), 7.77–7.84 (m, 2H), 7.52–7.59 (m, 4H), 7.31–7.36 (m, 6H), 7.21–7.27 (m, J = 8.80 Hz, 2H), 6.52–6.60 (m, 2H), 4.60 (s, 1H), 4.52 (s, 1H), 3.76–3.89 (m, 2H) and 2.99–3.09 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 183.0, 164.0, 162.6, 153.3, 153.2, 142.4, 139.3, 129.4, 128.4, 128.0, 126.3, 114.5, 107.7, 104.0 and 89.4. HRMS(Q-TOF) m/z calculated for C32H25NO7 [M + H]+: 536.1631 found: 536.1702.
4.2.4. Synthesis of 4-(5,6,7-Trihydroxy-4-oxo-4H-chromen-2-yl) phenyl morpholine-4-carboxylate (8a)
Compound 7a (0.3 g, 0.5 mmol) was stirred with 10% Pd/C catalyst (0.03 g, 0.2 mmol) in a mixture solution of EtOH (10 mL) and THF (10 mL) for 12 h under hydrogen atmosphere, followed by filtration over celite for removal of catalyst. Then the filtrate was evaporated in vacuo and the resulting residue was purified by column chromatography on silica gel using 20% ethyl acetate in petroleum ether as eluent to afford 8a (0.15 g, 75.0% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 3.59–3.77 (m, 8 H), 6.58 (s, 1 H), 6.86 (s, 1 H), 7.24–7.41 (m, 2 H) and 7.97–8.16 (m, 2 H). HRMS(Q-TOF) m/z calculated for C20H17NO8 [M + H]+: 400.1184 found: 400.1215.
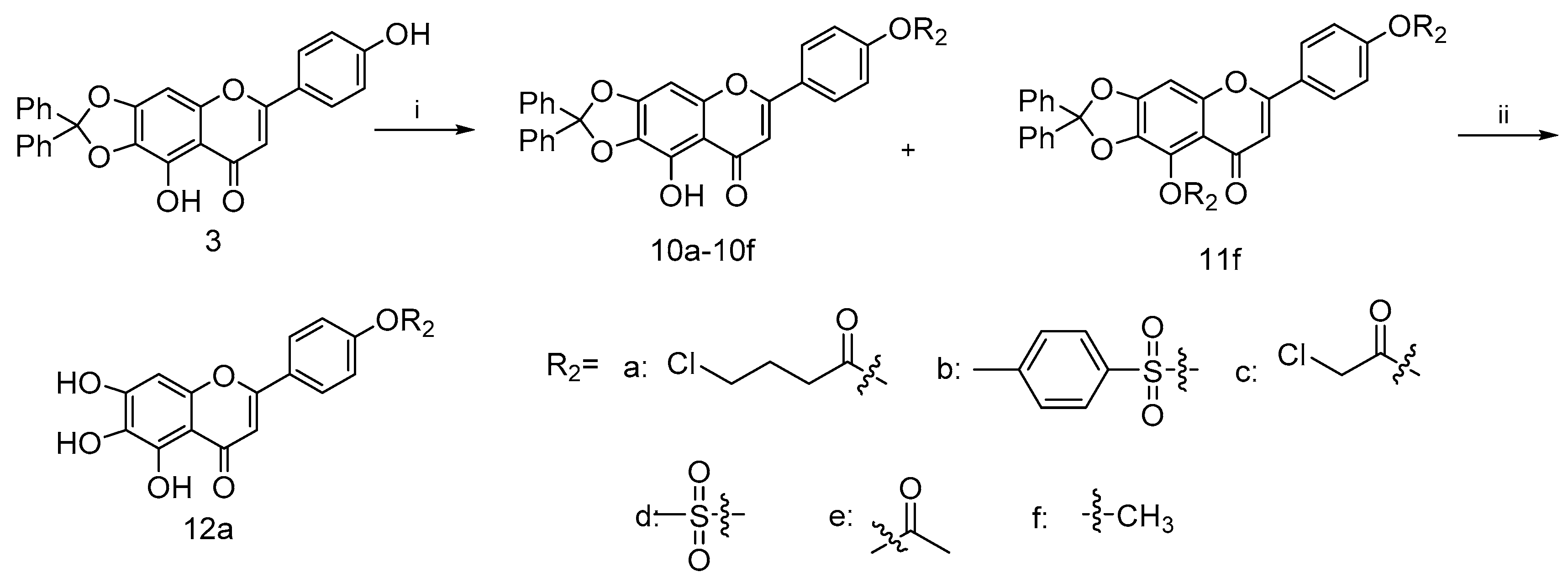
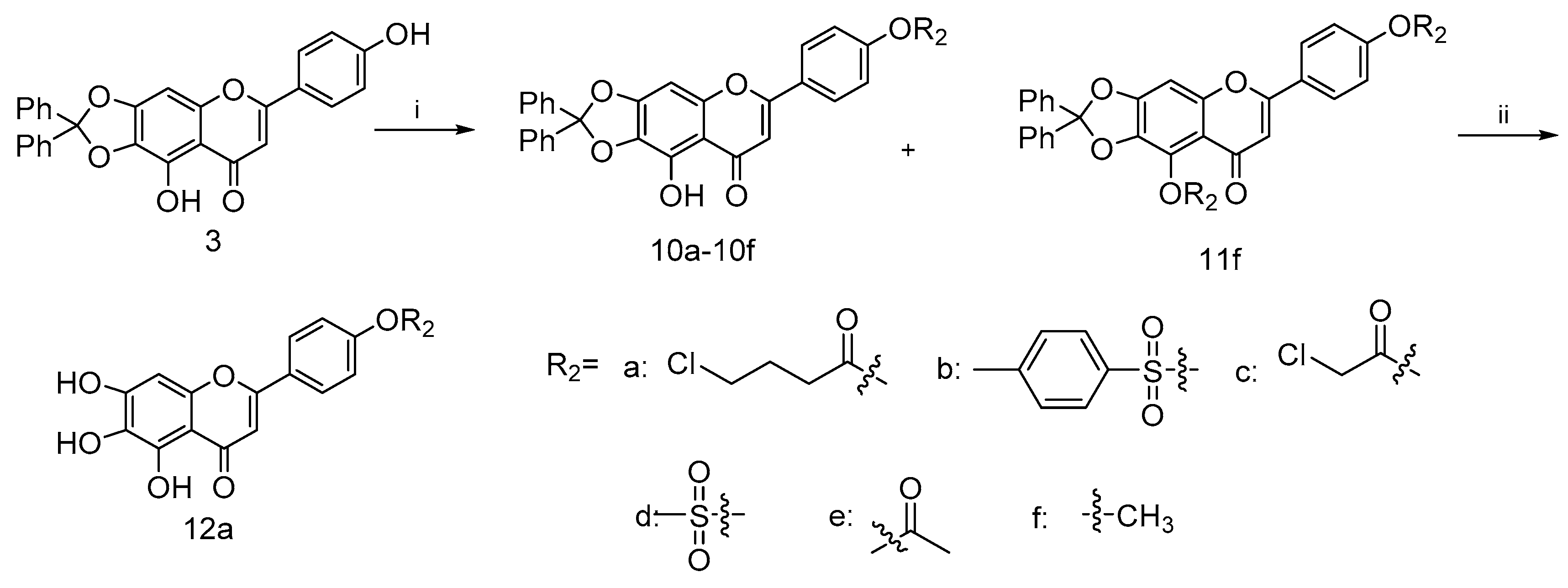
4.2.5. General Procedure for the Synthesis of Compounds 10a–10e
To a mixture solution of compound 3 and Et3N in anhydrous DCM (20 mL) at 0 °C, commercially available carbonyl chlorides 9a–9e (0.29 mmol) were added dropwise, stirring well over 12 h. Then, the reaction was diluted with water (20 mL). The collected organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Purification of the crude residue was performed by flash column chromatography on silica gel using 25% ethyl acetate in petroleum ether as eluent to afford the desired compound 10a–10f.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl 4-chlorobutanoate (10a)
This compound (0.34 g, yield 34.7%) was prepared from important immediate compound 3 (0.8 g, 1.7 mmol), Et3N (0.35 g, 3.4 mmol) and 4-chlorobutyrylchloride 9a (0.3 g, 2.1 mmol) according to the general procedure described above. 1H NMR (400 MHz, DMSO-d6) δ 12.98 (s, 1H), 8.08–8.23 (m, 2H), 7.54–7.60 (m, 4H), 7.46–7.52 (m, 6H), 7.36–7.42 (m, 2H), 7.15 (s, 1H), 7.09 (s, 1H), 3.76 (t, J = 6.48 Hz, 2H), 2.79 (t, J = 7.34 Hz, 2H) and 2.11 (t, J = 7.09 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 183.2, 171.3, 163.4, 153.8, 153.4, 141.8, 139.2, 130.3, 129.4, 129.2, 128.6, 128.5, 126.3, 123.2, 118.9, 107.6, 90.8, 44.9, 31.4, 27.8 and 19.4. HRMS(Q-TOF) m/z calculated for C32H23ClO7 [M + H]+: 555.1132 found: 555.1964.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl 4-methylbenzenesulfonate (10b)
This compound (0.3 g, yield 75.0%) was prepared from important immediate compound 3 (0.3 g, 0.6 mmol), Et3N (0.88 g, 8.6 mmol) and tosyl chloride 9b (0.19 g, 0.99 mmol) according to the general procedure described above. 1H NMR (400 MHz, DMSO-d6) δ 12.90 (s, 1H), 8.06–8.16 (m, 2H), 7.66–7.85 (m, 2H), 7.53–7.62 (m, 4H), 7.41–7.53 (m, 9H), 7.19–7.29 (m, 2H), 7.06 (s, 1H), 7.10 (s, 1H) and 2.42 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 183.1, 162.7, 153.3, 153.3, 151.9, 146.6, 141.8, 139.2, 131.5, 130.8, 130.3, 130.1, 129.4, 129.2, 128.9, 128.8, 126.3, 123.3, 107.7, 90.7 and 21.7. HRMS(Q-TOF) m/z calculated for C35H24O8S [M + H]+: 605.1192 found: 605.1261.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl 2-chloroacetate (10c)
This compound (0.37 g, yield 46.3%) was prepared from important immediate compound 3 (0.5 g, 1.1 mmol), Et3N (0.28 g, 2.7 mmol) and chloroacetyl chloride 9c (0.15 g, 1.3 mmol) according to the general procedure described above. With 46.3% yield, 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H), 8.07–8.32 (m, J = 8.80 Hz, 2H), 7.53–7.62 (m, 4H), 7.46–7.53 (m, 7H), 7.40–7.46 (m, J = 8.80 Hz, 2H), 7.14 (s, 1H) and 7.09 (s, 1H), 4.75 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 183.2, 166.6, 163.3, 153.4, 153.4, 153.2, 141.8, 139.2, 130.3, 129.4, 129.2, 128.6, 126.3, 122.9, 118.9, 107.7, 105.6, 90.7 and 41.9. HRMS(Q-TOF) m/z calculated for C30H19ClO7 [M + H]+: 527.0819 found: 527.0889.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl methanesulfonate (10d)
This compound (41 mg, yield 70.6%) was prepared from important immediate compound 3 (50 mg, 0.1 mmol), Et3N (20 mg, 0.1 mmol) and methanesulfonyl chloride 9d (10 mg, 0.08 mmol) according to the general procedure described above. 1H NMR (400 MHz, DMSO-d6) δ 12.87 (s, 1H), 8.15 (d, J = 8.80 Hz, 2H), 7.47–7.56 (m, 6H), 7.35–7.47 (m, 6H), 7.08 (d, J = 7.34 Hz, 2H) and 3.42 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ ppm 183.18, 162.94, 153.39, 152.04, 141.80, 139.16, 130.25, 130.05, 129.19, 129.16, 129.06, 126.31, 118.98, 107.70, 106.01, 90.77, 38.20 and 19.34. HRMS(Q-TOF) m/z calculated for C29H20O8S [M + H]+: 529.0879 found: 529.0951.
4-(9-Hydroxy-8-oxo-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-6-yl)phenyl acetate (10e)
This compound (0.02 g, yield 40.6%) was prepared from important immediate compound 3 (0.15 g, 0.1 mmol), Et3N (0.08 g, 0.7 mmol) and acetyl chloride 9e (0.07 g, 0.9 mmol) according to the general procedure described above. 1H NMR (400 MHz, DMSO-d6) δ 12.93–13.22 (m, 1H), 8.19 (d, J = 7.82 Hz, 2H), 7.59–7.68 (m, 4H), 7.49–7.59 (m, 6H), 7.41 (d, J = 7.83 Hz, 2H), 7.15–7.20 (m, 1H), 7.11 (s, 1H) and 2.36 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 183.2, 169.4, 163.4, 153.8, 153.3, 141.8, 139.2, 130.2, 129.4, 129.2, 128.5, 128.4, 126.3, 123.2, 107.6, 105.5, 90.7 and 21.4. HRMS(Q-TOF) m/z calculated for C30H20O7 [M + H]+: 493.1209 found: 493.1279.
4.2.6. 9-Hydroxy-6-(4-methoxyphenyl)-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-8-one (10f) and 9-methoxy-6-(4-methoxyphenyl)-2,2-diphenyl-8H-[1,3]dioxolo[4,5-g]chromen-8-one (11f)
A mixture of compound 3 (0.2 g, 0.4 mmol), methyl iodide (0.14 g, 0.9 mmol) and excess potassium carbonate (0.25 g, 1.8 mmol) in dry DMF (20 mL) was stirred in ice bath under nitrogen atmosphere for 30 min in the presence of catalytic amount of KI. The solution was allowed to slowly warm to room temperature with stirring overnight. Then, the reaction was diluted with brine (20 mL). The collected organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Subsequent purification of the crude mixture was performed using the flash column chromatography on silica gel with gradient elution (EA/PE, 50% to 60%) to afford the target compound 10f (0.03 g, 50.1% yield) and a side product compound 11f (0.1 g, 49.8% yield). For 10f: 1H NMR (400 MHz, CDCl3) δ 12.79 (s, 1H), 7.65–7.85 (m, 2H), 7.49–7.63 (m, 4H), 7.25–7.43 (m, 6H), 6.86–6.98 (m, 2H), 6.55 (s, 1H), 6.49 (s, 1H) and 3.80 (s, 3H). 13C NMR (101 MHz, CDCl3) d 183.0, 164.0, 162.6, 153.3, 153.2, 142.4, 139.3, 129.4, 128.4, 128.0, 126.3, 114.5, 107.7, 104.0, 89.4 and 55.5. HRMS(Q-TOF) m/z calculated for C29H20O6 [M + H]+: 465.1260 found: 465.1529. For 11f: 1H NMR (400 MHz, CDCl3) δ 7.67–7.76 (m, 2H), 7.47–7.56 (m, 4H), 7.24–7.39 (m, 6H), 6.87–6.97 (m, 2H), 6.70 (s, 1H), 6.47 (s, 1H), 4.13 (s, 3H) and 3.79 (s, 3H). 13C NMR (101 MHz, CDCl3) d 162.1, 160.8, 154.7, 139.2, 129.6, 128.4, 127.6, 126.3, 123.8, 118.6, 114.4, 107.1, 93.3, 61.2 and 55.5. HRMS(Q-TOF) m/z calculated for C30H22O6 [M + H]+: 479.1416 found: 479.1498.
4.2.7. 4-(5,6,7-Trihydroxy-4-oxo-4H-chromen-2-yl)phenyl 4-chlorobutanoate (12a)
Compound 10a (0.3 g) was stirred with 10% Pd/C catalyst (0.02 g, 0.1 mmol) in EtOH (10 mL) and THF (10 mL) for 12 h under hydrogen at atmospheric pressure, then the Pd/C catalyst was removed by filtration over celite. After the filtrate was evaporated, the crude material was purified by column chromatography on silica gel using 50% ethyl acetate in petroleum ether as eluent to afford 12a. With 90% yield, 1H NMR (400 MHz, DMSO-d6) δ 8.03–8.29 (m, 2H), 7.29–7.44 (m, 2H), 6.84–7.05 (m, 1H), 6.54–6.74 (m, 1H), 3.76 (t, J = 6.48 Hz, 2H), 2.79 (t, J = 7.21 Hz, 2H) and 2.12 (t, J = 7.09 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.3, 171.3, 162.4, 153.5, 150.3, 147.5, 129.8, 129.2, 128.3, 128.3, 123.1, 104.7, 44.9, 31.5 and 27.8. HRMS(Q-TOF) m/z calculated for C19H15ClO7 [M − H]+: 389.0506 found: 389.6161.
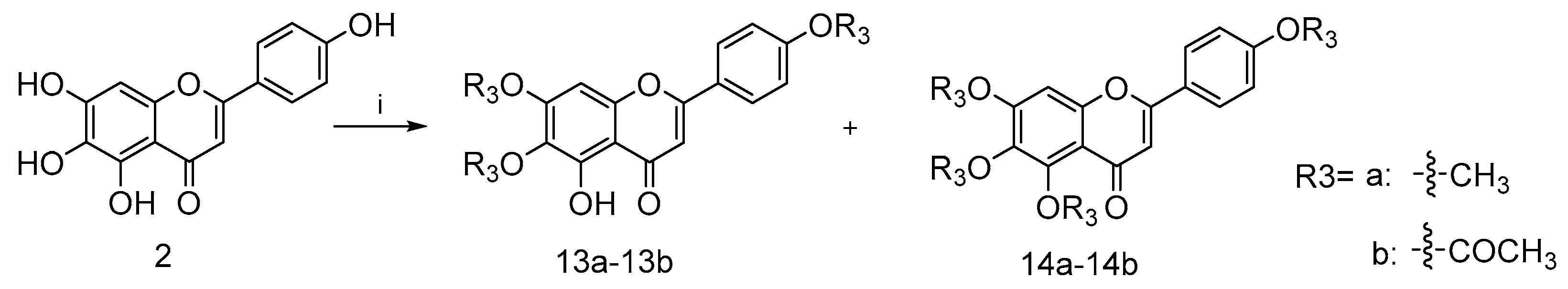
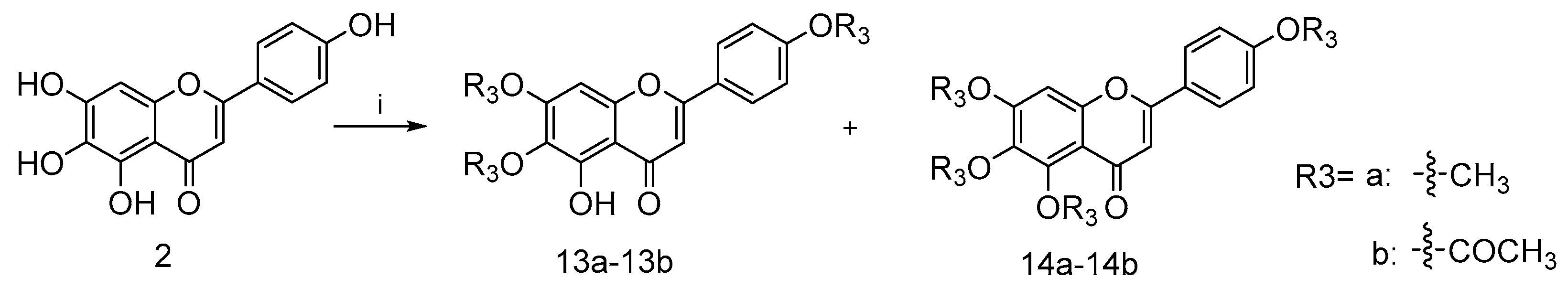
4.2.8. 5-Hydroxy-6,7-dimethoxy-2-(4-methoxyphenyl)-4H-chromen-4-one (13a) and 5,6,7-trimethoxy-2-(4-methoxyphenyl)-4H-chromen-4-one (14a)
To a mixture solution of compound 2 (0.2 g, 0.7 mmol) and NaH (0.17 g, 7.1 mmol) in anhydrous DMF (15 mL) at 170 ℃ under nitrogen atmosphere commercially available methyl iodide (0.69 g, 4.8 mmol) was added dropwise, stirring well over 12 h. After the disappearance of starting material, as indicated by TLC, the reaction mixture was diluted with EA and water. The collected organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Purification of the crude residue was performed by flash column chromatography on silica gel using 50% ethyl acetate in petroleum ether as eluent to afford the desired compound 13a (0.19 g, 82.9% yield) and a side product 14a (0.18 g, 80.1% yield). For 13a: 1H NMR (400 MHz, CDCl3) δ ppm 3.90 (s, 3 H) 3.93 (s, 3 H), 3.97 (s, 3 H), 6.59 (s, 1 H), 6.55 (s, 1 H), 7.02 (m, J = 9.05 Hz, 2 H), 7.85 (m, J = 9.05 Hz, 2 H) and 12.78 (s, 1 H). 13C NMR (101 MHz, CDCl3) δ ppm 182.68, 164.02, 162.62, 158.73, 153.23, 153.09, 128.00, 123.58, 114.53, 106.16, 104.15, 90.57, 60.86, 56.32 and 55.55. HRMS(Q-TOF) m/z calculated for 328.32 [M + Na]+: 351.3160 found: 351.0843. For 14a: 1H NMR (400 MHz, CDCl3) δ ppm 3.87 (s, 3 H), 3.91 (s, 3 H), 3.97 (s, 3 H), 3.98 (s, 3 H), 6.57 (s, 1 H), 6.79 (s, 1 H), 6.95–7.03 (m, 2 H) and 7.75–7.87 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ ppm 177.18, 162.13, 161.14, 157.61, 154.49, 152.57, 127.63, 123.86, 114.39, 112.87, 107.05, 96.26, 62.19, 61.53, 56.28 and 55.48. HRMS(Q-TOF) m/z calculated for C19H18O6 [M + Na]+: 365.3426 found: 365.0996.
4.2.9. 2-(4-Acetoxyphenyl)-5-hydroxy-4-oxo-4H-chromene-6,7-diyl diacetate (13b) and 2-(4-acetoxyphenyl)-4-oxo-4H-chromene-5,6,7-triyl triacetate (14b)
Compound 2 (0.2 g, 0.6 mmol) was dissolved in pyridine (15 mL) and mixed with acetic anhydride (0.32 g, 3.1 mmol) and a small amount of DMAP as catalyst under nitrogen atmosphere. The reaction mixture was stirred at room temperature overnight. After the disappearance of starting material, as indicated by TLC, the reaction mixture was diluted with DCM and brine. The collected organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Purification of the crude residue was performed by flash column chromatography on silica gel using 50% ethyl acetate in petroleum ether as eluent to afford the desired compound 13b (1.78 g, 56.2% yield) and a side product 14b (0.26 g, 57.1% yield). For 13b: 1H NMR (400 MHz, CDCl3) δ ppm 2.28 (s, 6 H), 2.30 (s, 3 H), 6.63 (s, 1 H), 6.89 (s, 1 H), 7.13–7.31 (m, 3 H), 7.84 (d, J = 8.80 Hz, 2 H) and 12.83 (s, 1 H). 13C NMR (101 MHz, CDCl3) δ ppm 182.68, 164.02, 162.62, 158.73, 153.23, 153.09, 128.00, 123.58, 114.53, 106.16, 104.15, 90.57, 60.86, 56.32 and 55.55. HRMS(Q-TOF) m/z calculated for C21H16O9 [M + Na]+: 435.3463 found: 435.0687. For 14b: 1H NMR (400 MHz, CDCl3) δ ppm 2.25–2.30 (m, 10 H), 2.37 (s, 3 H), 6.55 (s, 1 H), 7.19 (t, J = 4.40 Hz, 3 H), 7.42 (s, 1 H) and 7.81 (d, J = 9.05 Hz, 2 H). 13CNMR (101 MHz, CDCl3) δ ppm 176.08, 168.89, 168.30, 167.25, 166.99, 161.93, 154.18, 153.42, 146.95, 142.19, 132.80, 128.54, 127.63, 122.44, 115.56, 110.27, 108.25, 21.15, 20.82 and 20.09. HRMS(Q-TOF) m/z calculated for C23H18O10 [M + H]+: 455.0900 found: 455.0976.
4.5. Patch Clamp Assay for Nav1.5, Cav1.2 and hERK
4.5.1. Cell Culture
Human embryonic kidney cells (HEK293) that showed no macroscopic sodium currents were used for patch clamp assay and purchased from the Shanghai Institute of Biological Sciences, Chinese Academy of Sciences (Shanghai, China). HEK293 cells were cultured in medium consisting of 90% DMEM, 10% fetal bovine serum, 1% penicillin and streptomycin at 37 °C and 5% CO2 atmosphere.
4.5.2. Cell Transfection
The Nav1.5 (SCN5A) plasmid was purchased from Addgene and the GFP plasmid was purchased from Shenzhen Yanming Biotechnology. According to the manufacturer’s instructions, HEK293 cells were transfected with GFP plasmids using pCDNA3.1hSCN5A for 6–8 h. Subsequently, patch clamp assays were carried out in whole-cell mode after about 24 h transfection.
The Cav1.2 (CACNA2D) plasmid was purchased from Youbio and the GFP plasmid was purchased from Beijing Bozhiyuan Biotechnology Co., Ltd. (Beijing, China). According to the manufacturer’s instructions, HEK293 cells were transfected with GFP plasmids using pCDNA3.1hCACNA2D for 6–8 h. Subsequently, patch clamp assays were carried out in whole-cell mode after about 24 h transfection.
The HEK293 cell lines were transfected with pcmv-kcnh2-egfp-neo plasmid purchased from Shenzhen Dingke Biotechnology Co., Ltd. (Shenzhen, China) using jetPRIME (polyplus, 101000046) when cell confluence reached 60–80%. Electrophysiological experiments were performed 24–48 h after cell transfection.
4.5.3. Patch Clamp Measurements for Nav1.5
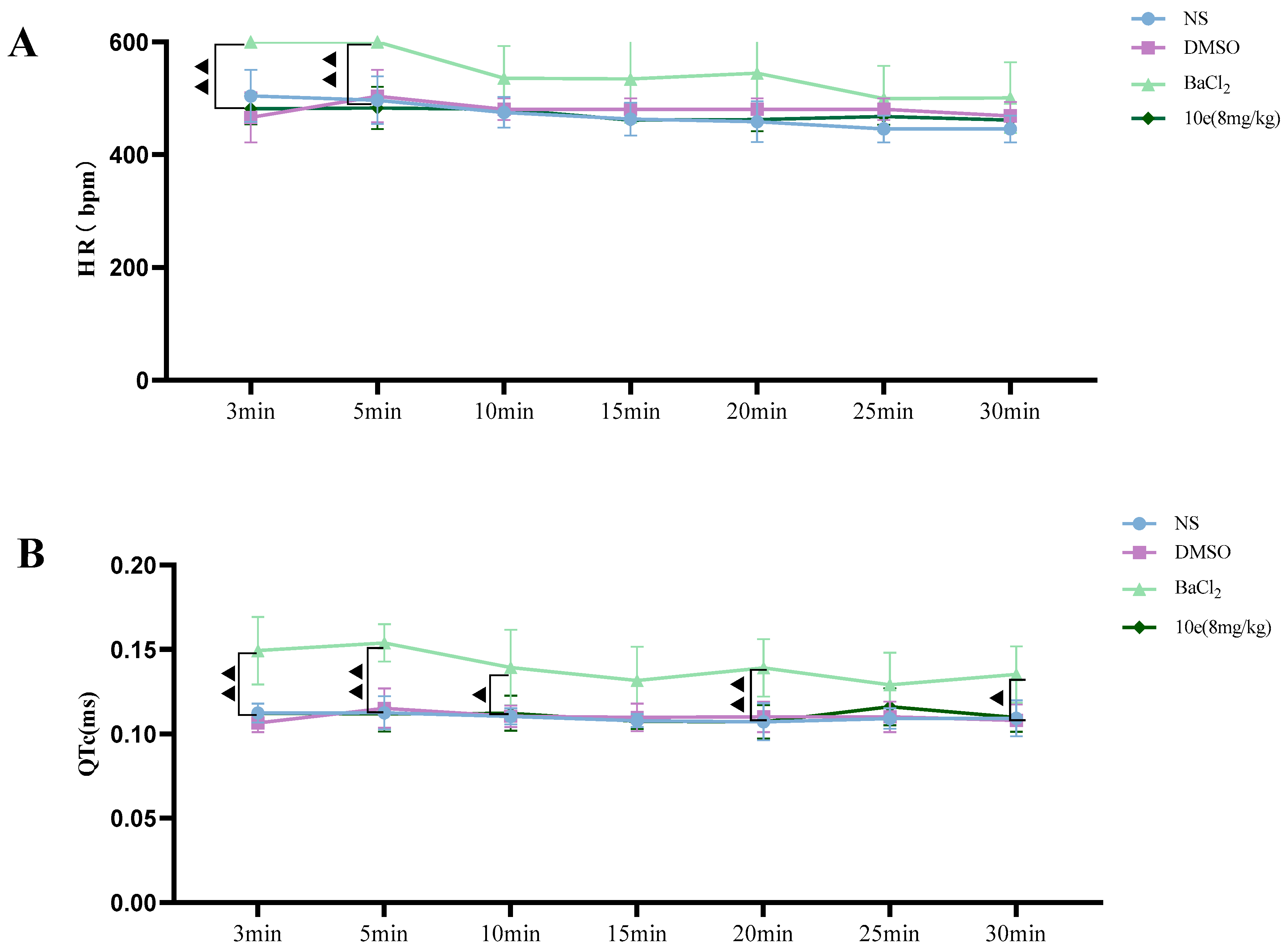
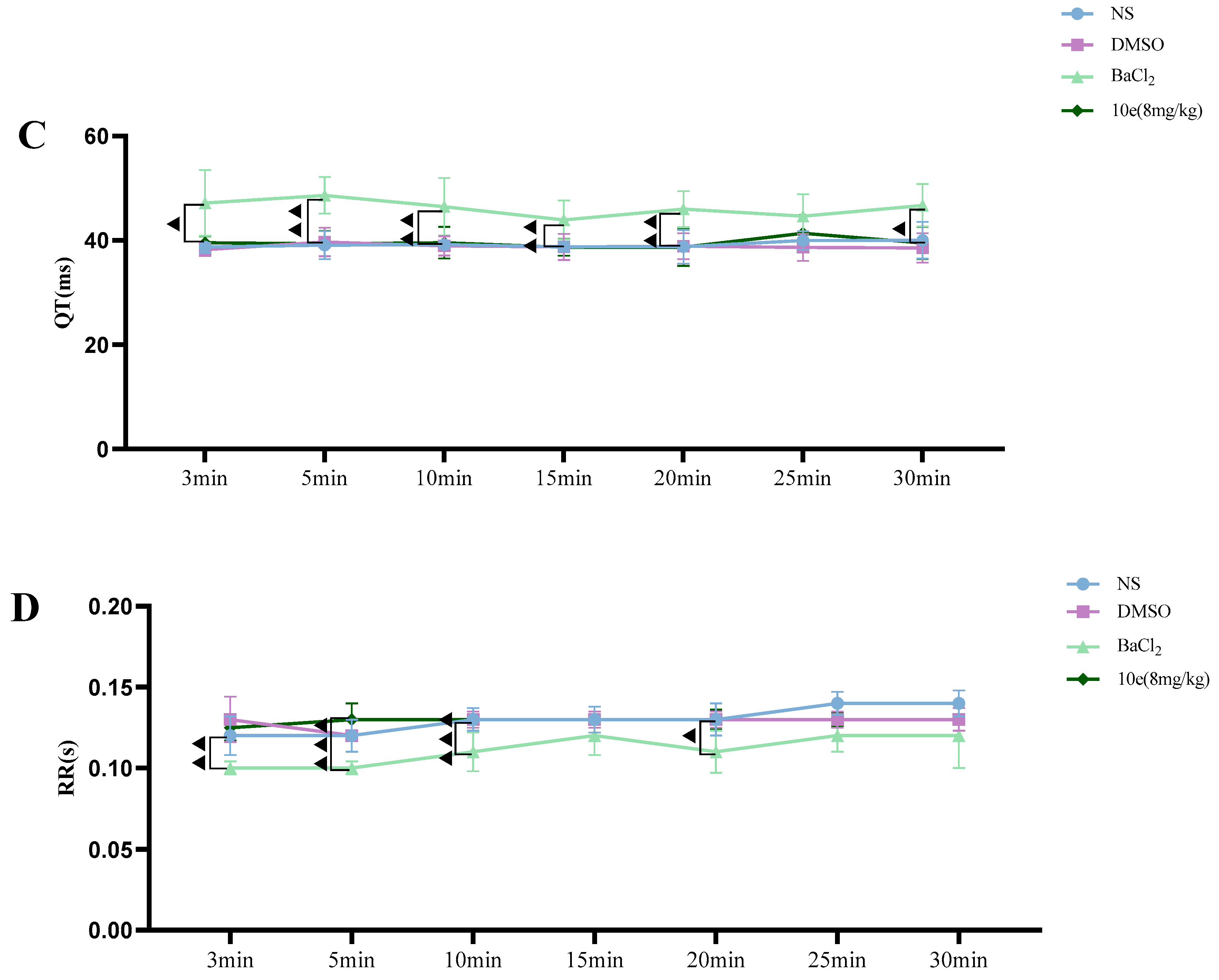
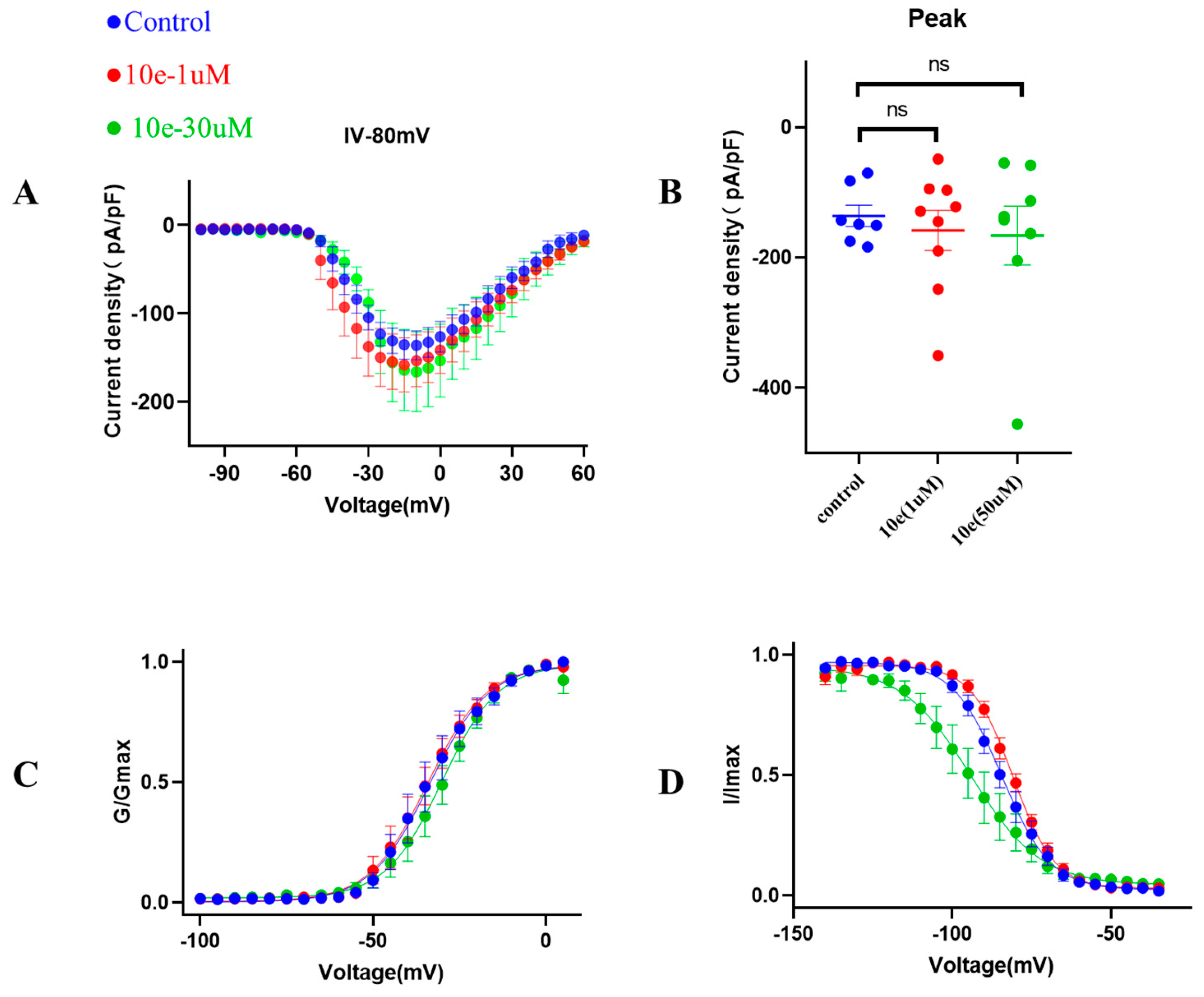
Patch clamp experiments were conducted by using an EPC-10.2 patch-clamp amplifier (HEKA Elektronik, Reutlingen, Germany) and patchmaster v2x91 acquisition software in transfected HEK293 cells with heterologous expression of human Nav1.5 channels. Before membrane Nav1.5 currents recorded, cells were replated on glass coverslips and mounted on a recording chamber. The extracellular recording solution for recording sodium current consisted of (in mM) 140 NaCl, 30 tetraethylammonium chloride, 10 D-glucose, 3 KCl, 1 CaCl2, 0.5 CdCl2, 1 MgCl2 and 10 HEPES. The pH of the solution was adjusted to 7.3 by using NaOH, meanwhile the osmolality was maintained at 310–315 mOsm/L. The composition of the intracellular recording solution was (in mM) 140 CsF, 10 NaCl, 15 HEPES and 1.1 Cs-EGTA, in which pH was adjusted to 7.3 with CsOH. HEK293 cells were used for all records. The current voltage (I-V) measurement was recorded by 50 ms square pulses ranging from −100 to +60 mV, with 5 mV increment from a holding potential of −120 mV. The corresponding current density was obtained. Voltage dependency for sodium current activation was recorded by 50 ms square pulses ranging from −100 to +40 mV, with 5 mV increment from a holding potential of −120 mV. Data were fitted with the modified Boltzman function , where V1/2 was the voltage in which half of the current is activated. Em was the membrane potential and K was the slope factor. Voltage-dependent inactivation was measured in a 50 ms duration test pulse to −30 mV amplitude immediately after 5000 ms conditioning pulse ranging from −140 mV to −35 mV, with 5 mV increment from a holding potential of −120 mV. Peak currents from the test pulse were normalized by maximum current (I/IMAX) and also fitted with the modified Boltzman function to calculate the potential Vh related to the half of inactivation and slope factor kh.
4.5.4. Patch Clamp Measurements for Cav1.2
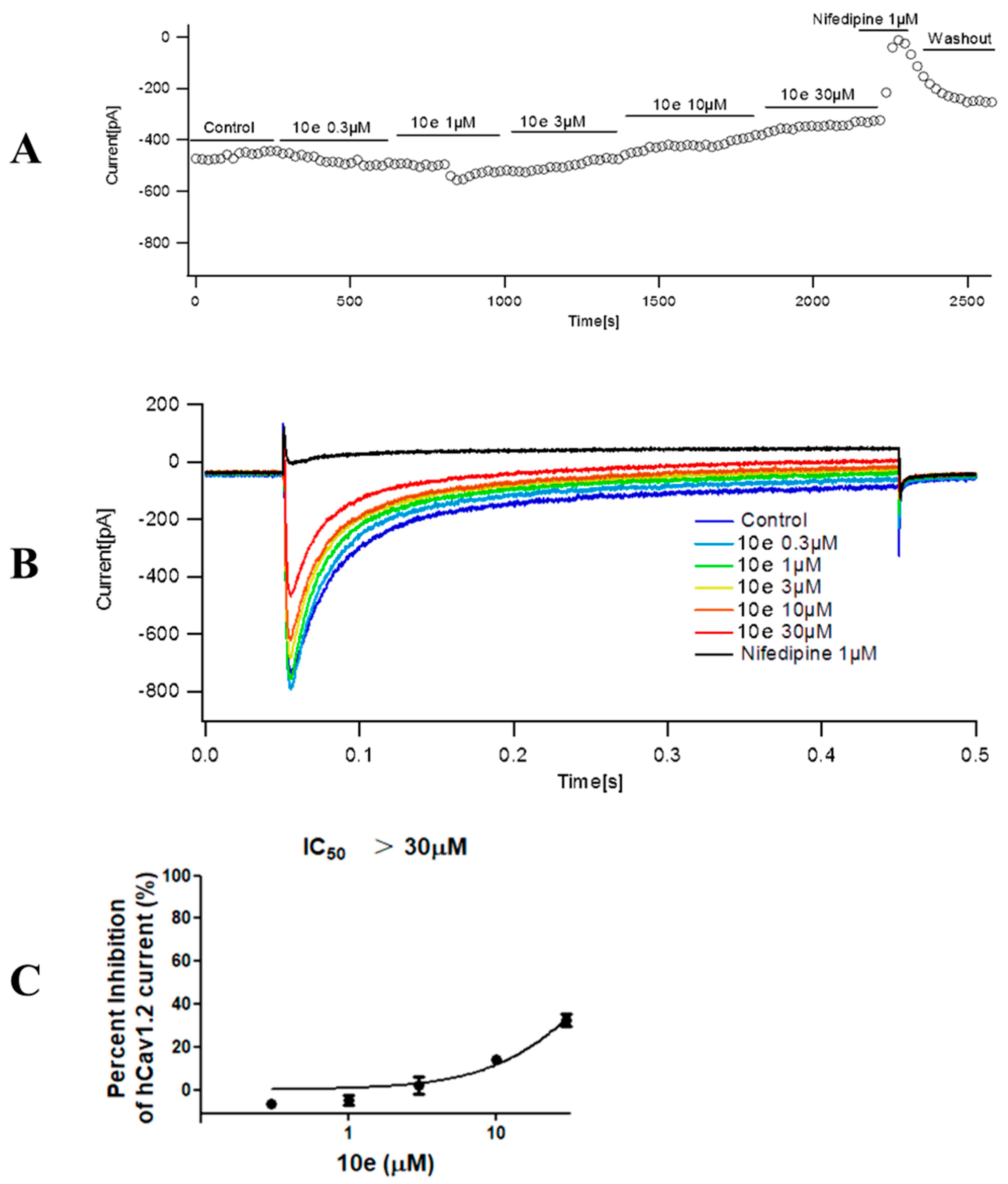
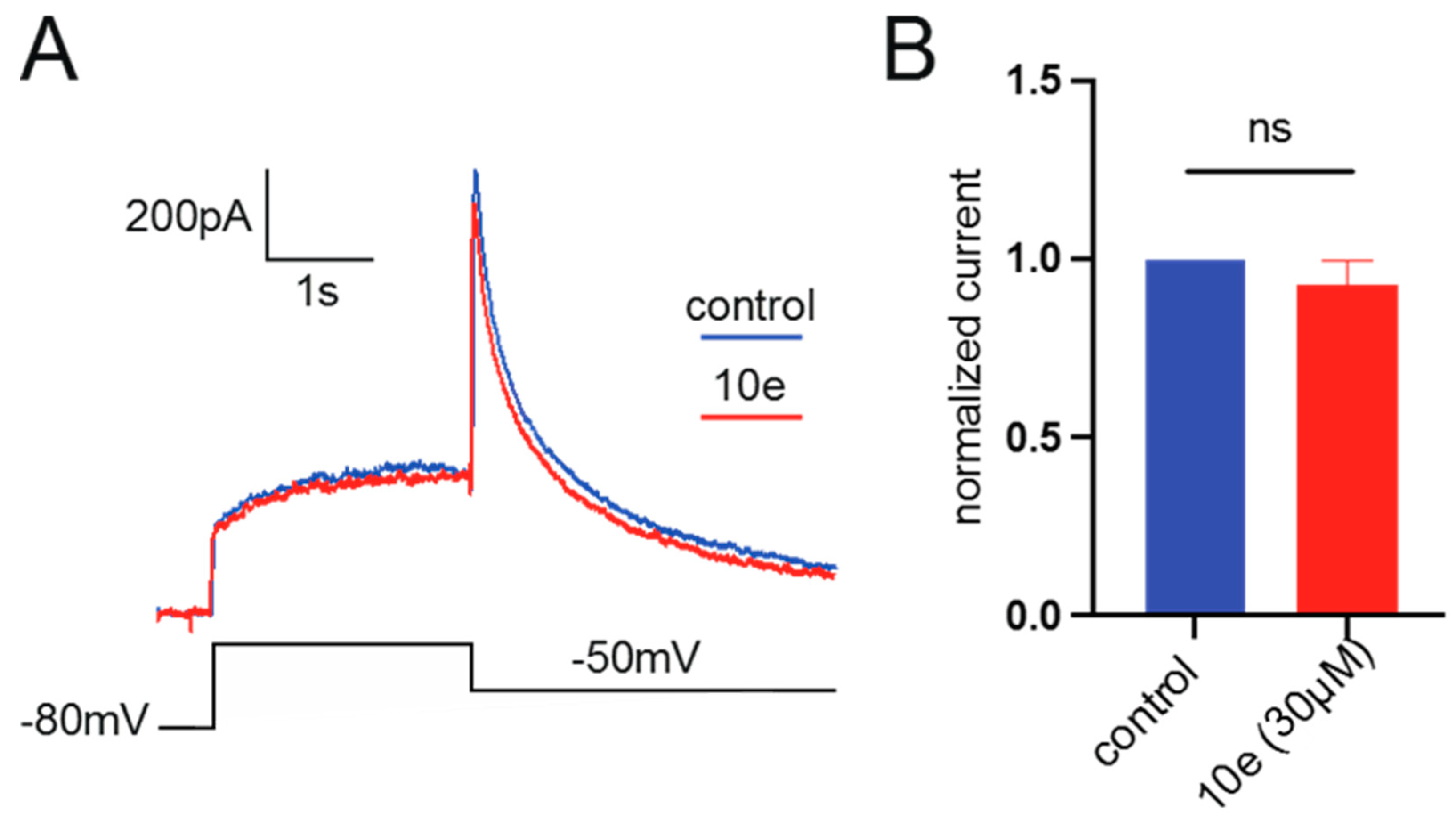
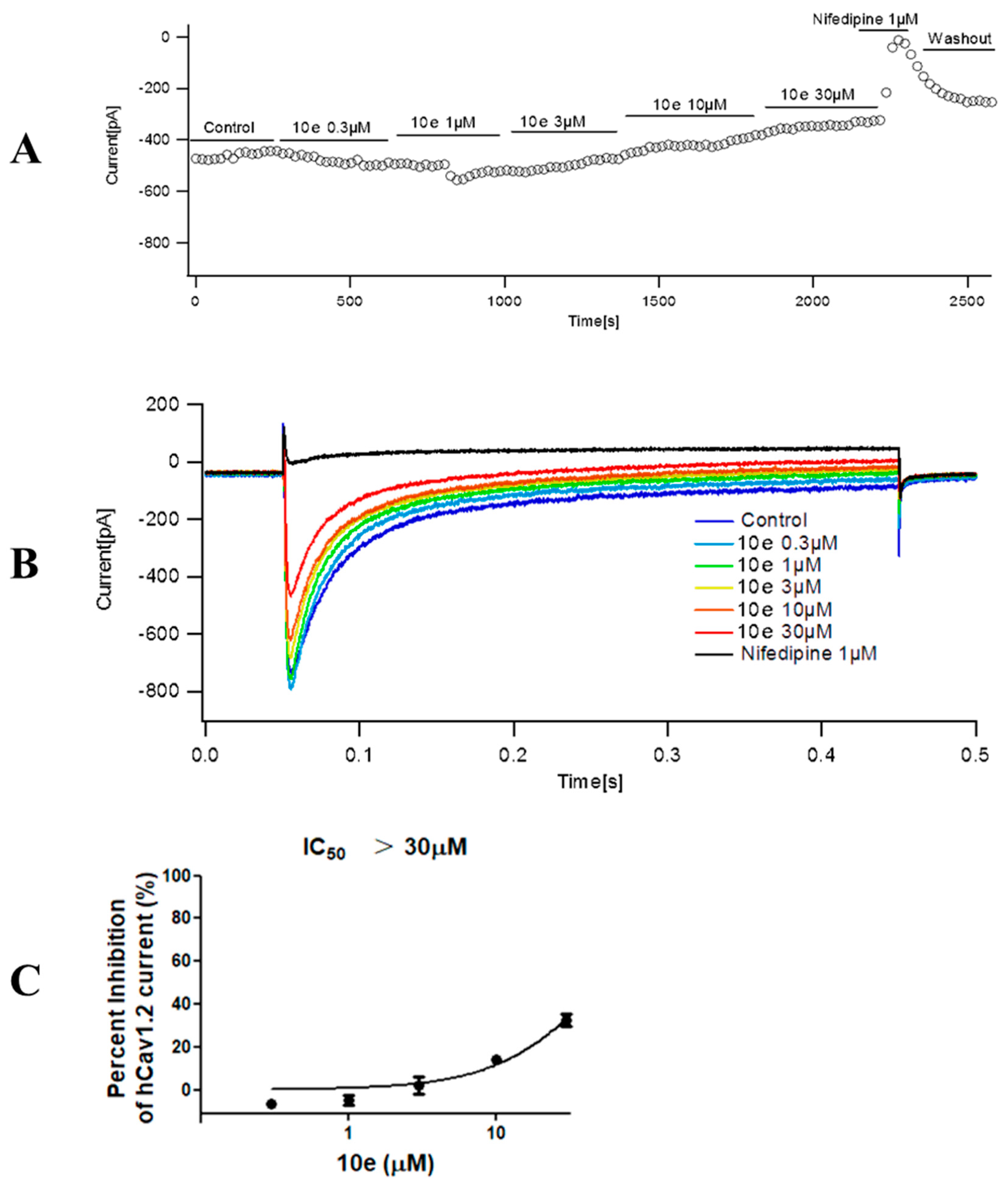
The extracellular recording solution for recording calcium current consisted of (in mM) 140 TEA-Cl, 1 MgCl2•6H2O, 4 KCl, 10 CaCl2•2H2O, 10 D-Glucose and 5 mM HEPES. The pH of the solution was adjusted to 7.4 using TEA-OH. The composition of the intracellular recording solution was (in mM) 110 CsCl, 10 EGTA, 5 HEPES and 5 Mg-ATP, in which pH was adjusted to 7.2 with CsOH. Cav1.2 cells were used for all records. The stimulation procedure is shown below. The clamping voltage was set to −80 mV, then depolarized to 10 mV over a time range of 400 ms, then repolarized to −80 mV. The current was recorded every 20 s. The original data Cav1.2 current peak was extracted from the PatchMaster v2x91 software, and the current inhibition rate was calculated as follows: peak calcium current inhibition rate = 1 − (Peak current compound/Peak current vehicle). The mean and standard error were calculated for each concentration and the concentration–effect relationship was obtained. The analysis statistics were completed using Graphpad Prism 5.0 software.
4.5.5. Patch Clamp Measurements for hERG
The extracellular recording solution for recording calcium current consisted of (in mM) 140 NaCl, 3.5 KCl, 1 MgCl2•6H2O, 2 CaCl2•2H2O, 10 D-Glucose, 10 HEPES and 1.25 NaH2PO4•2H2O. The pH of the solution was adjusted to 7.4 using NaOH. The composition of the intracellular recording solution was (in mM) 10 NaCl, 50 CsCl, 60 CsF, 10 HEPES and 20 EGTA, in which pH was adjusted to 7.2 with CsOH. The current was recorded by depolarizing the voltage to +40 mV for 2.5 s from the holding potential at −80 mV, followed by a hyperpolarization step to −50 mV lasting for 4 s, which elicited the peak tail current of hERG channel. The analysis statistics were completed by using Graphpad Prism 5.0 software.


 and
and 
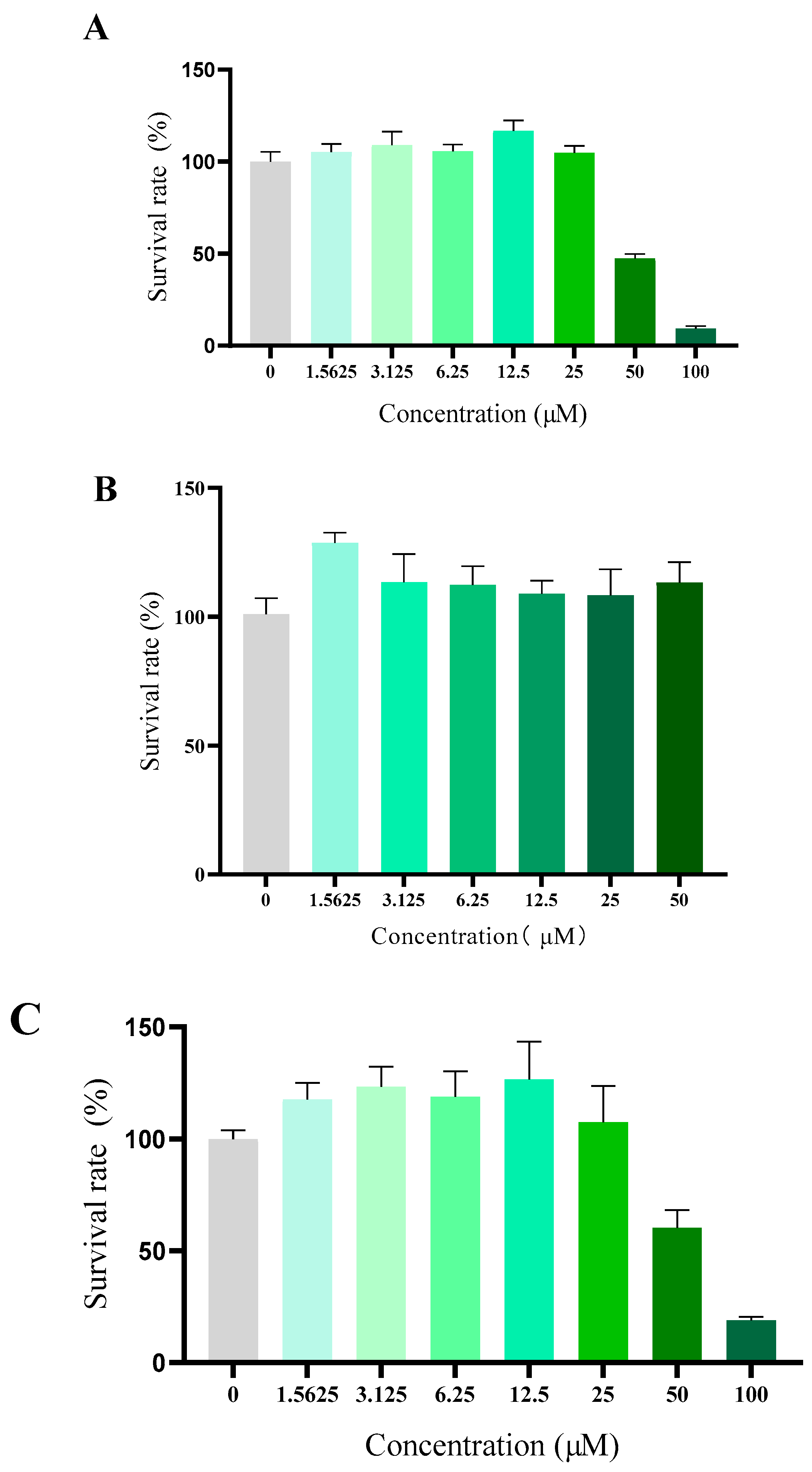
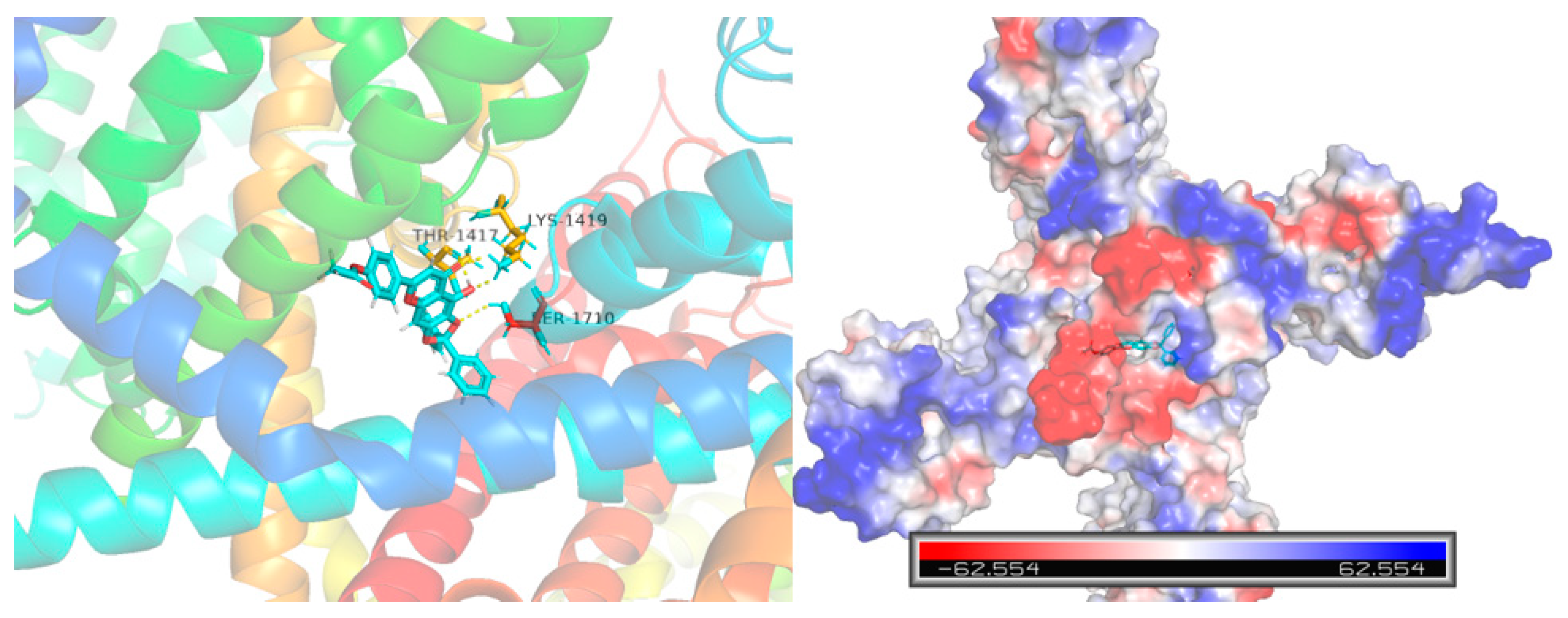
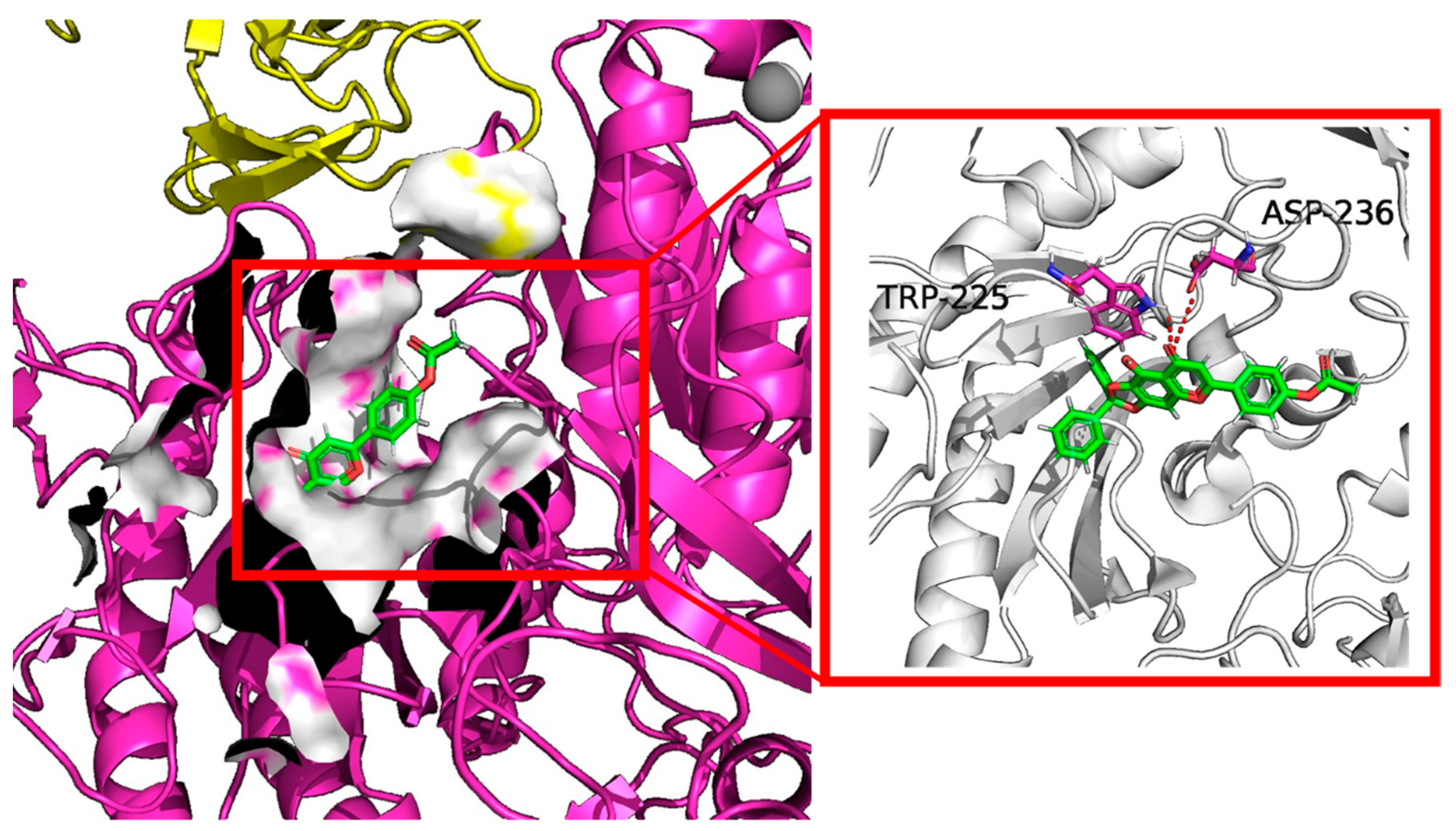
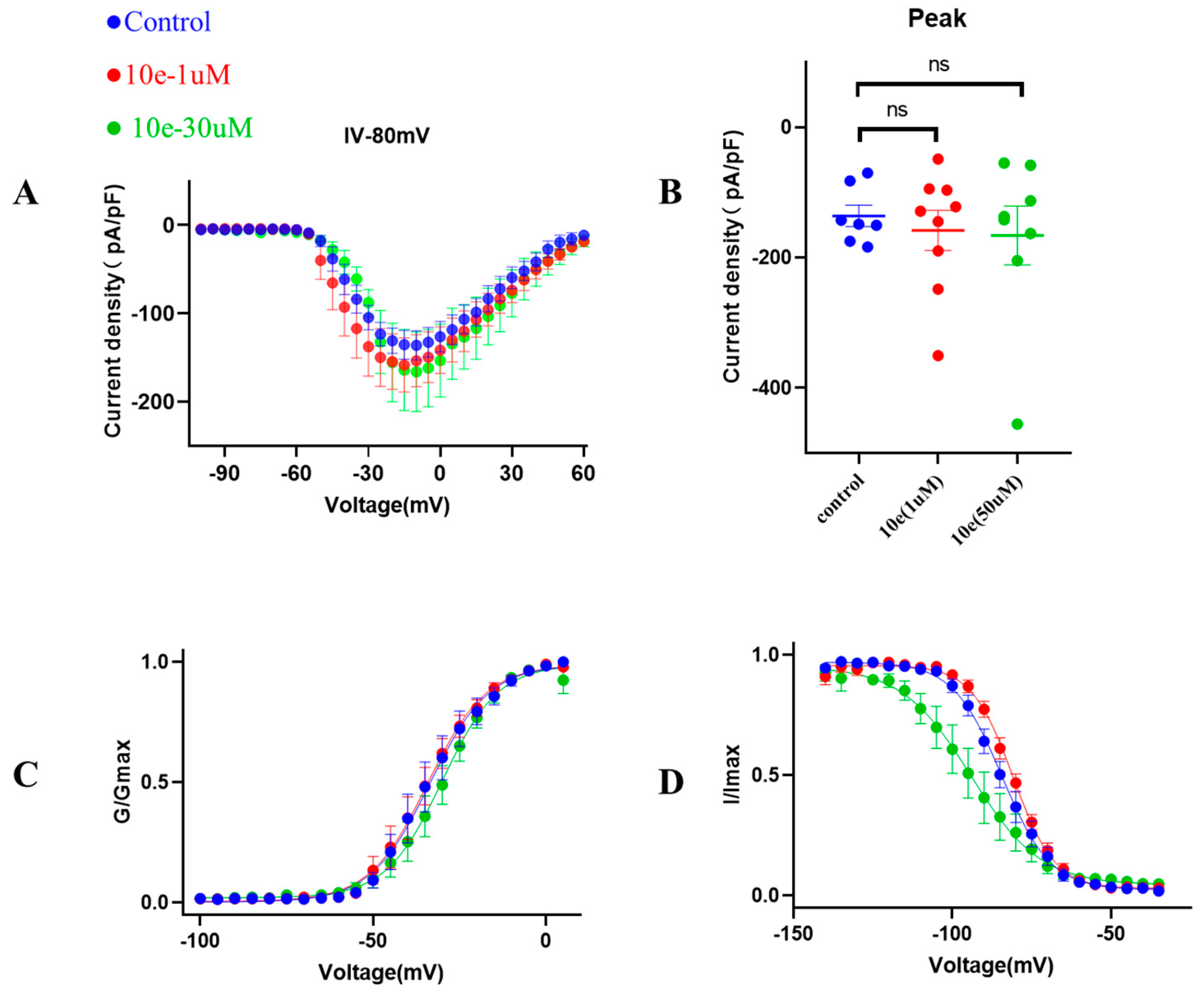



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}