

Repurposing Drugs for Inhibition against ALDH2 via a 2D/3D Ligand-Based Similarity Search and Molecular Simulation

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Virtual Screening of World-Approved Drugs via the 2D/3D Similarity Search
2.2. Assessment via Molecular Docking
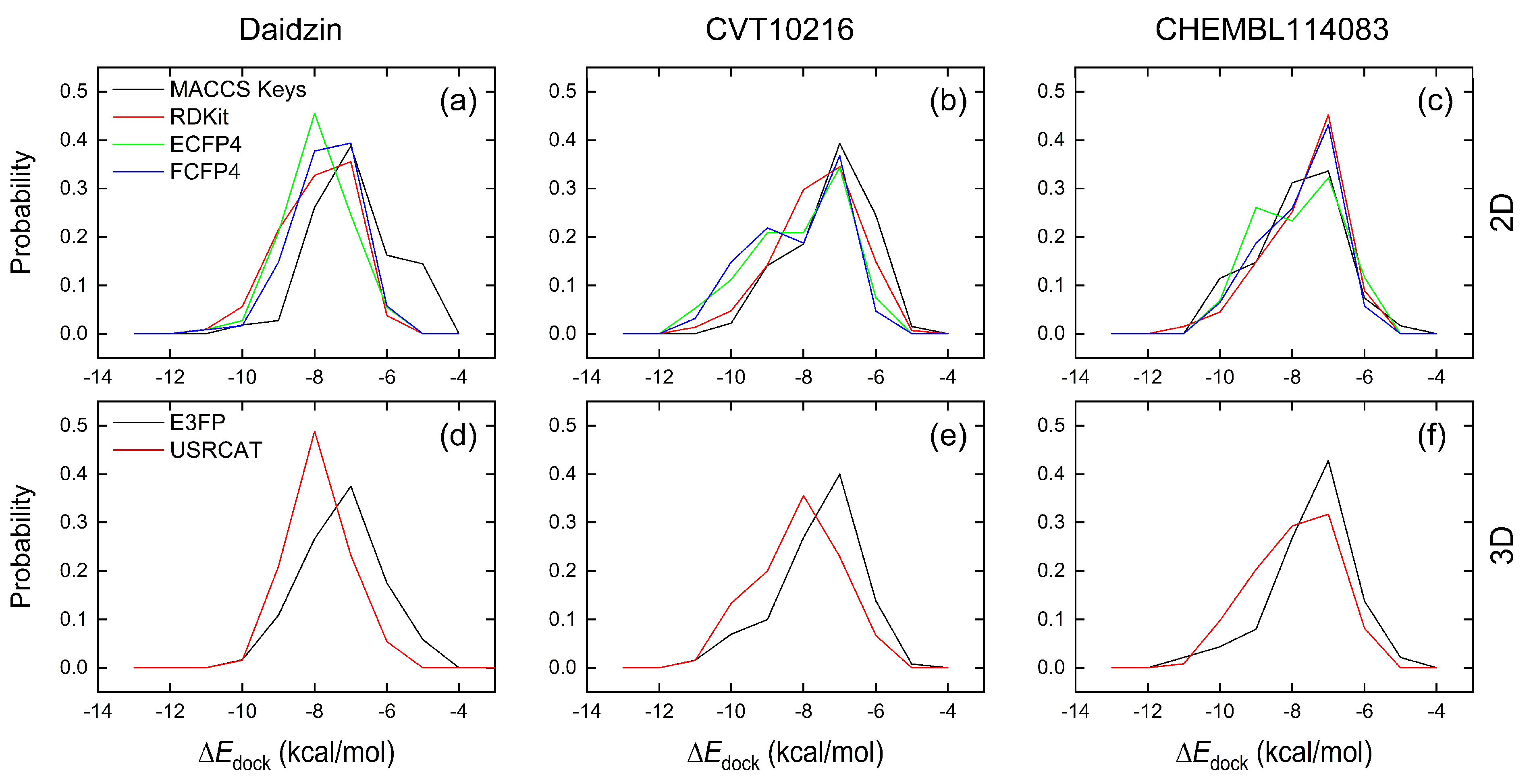
2.2.1. Comparison of Similarity-Search Methods
2.2.2. Molecular Docking Prediction
2.3. Assessment via a Toxicity Evaluation
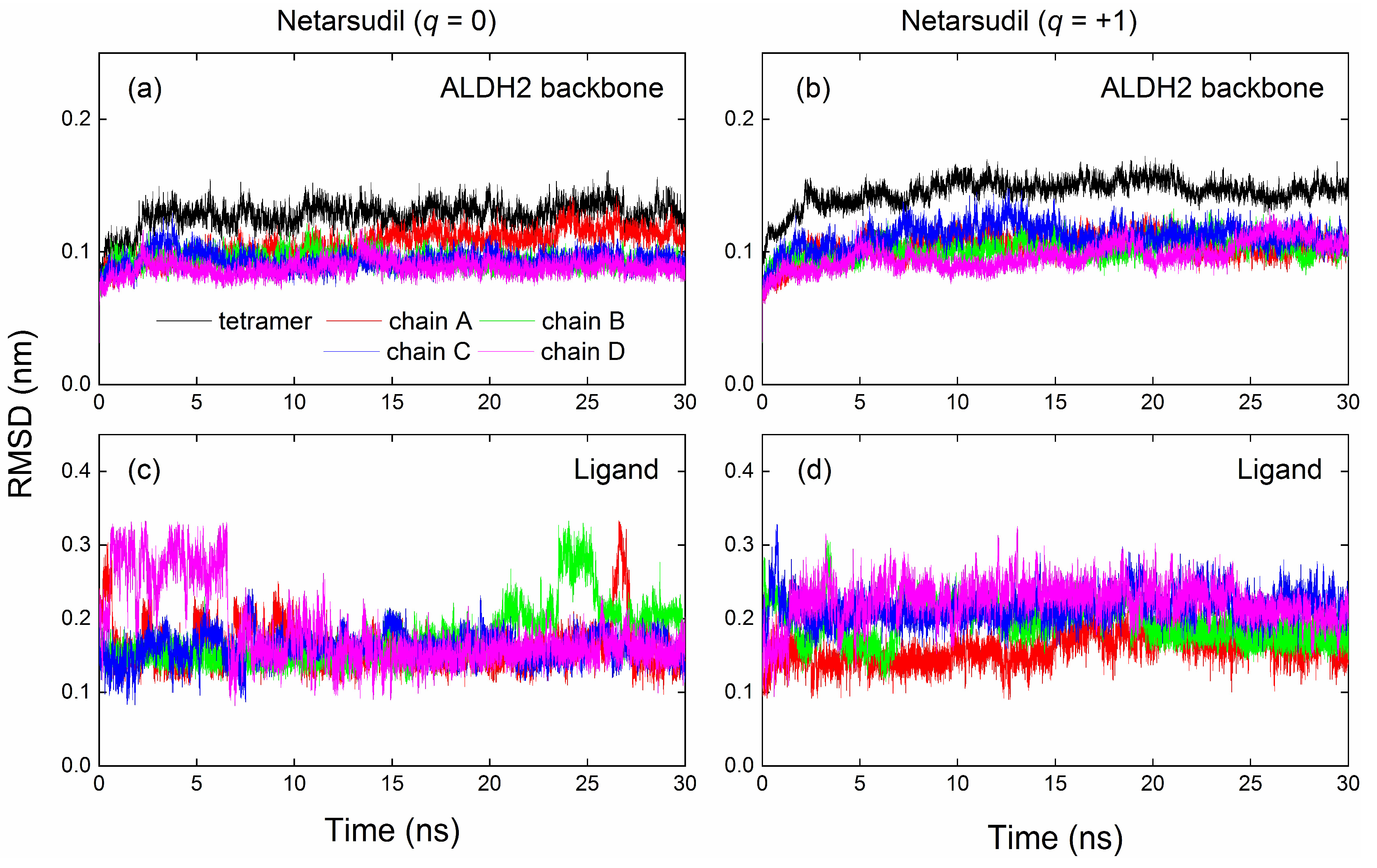
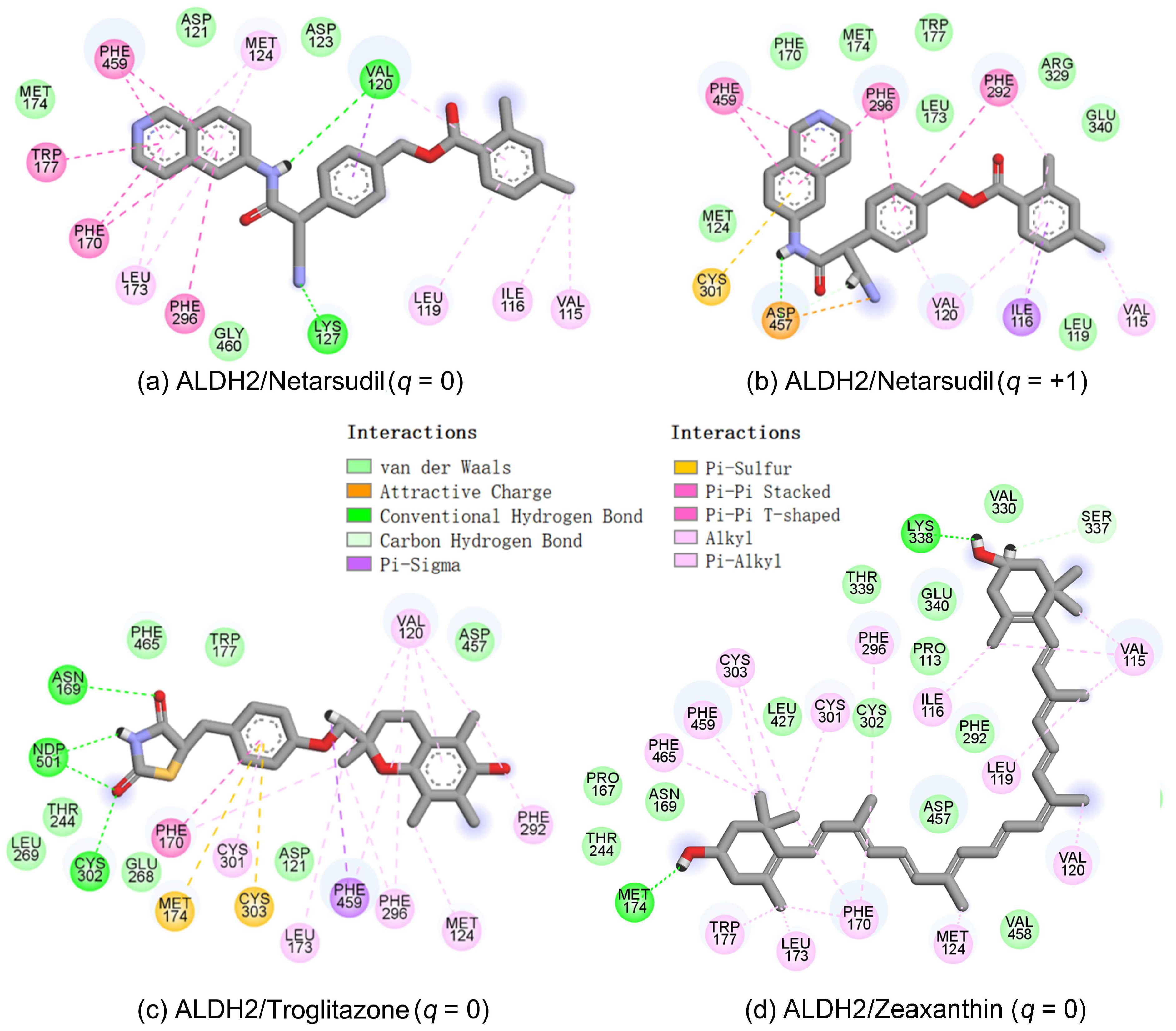
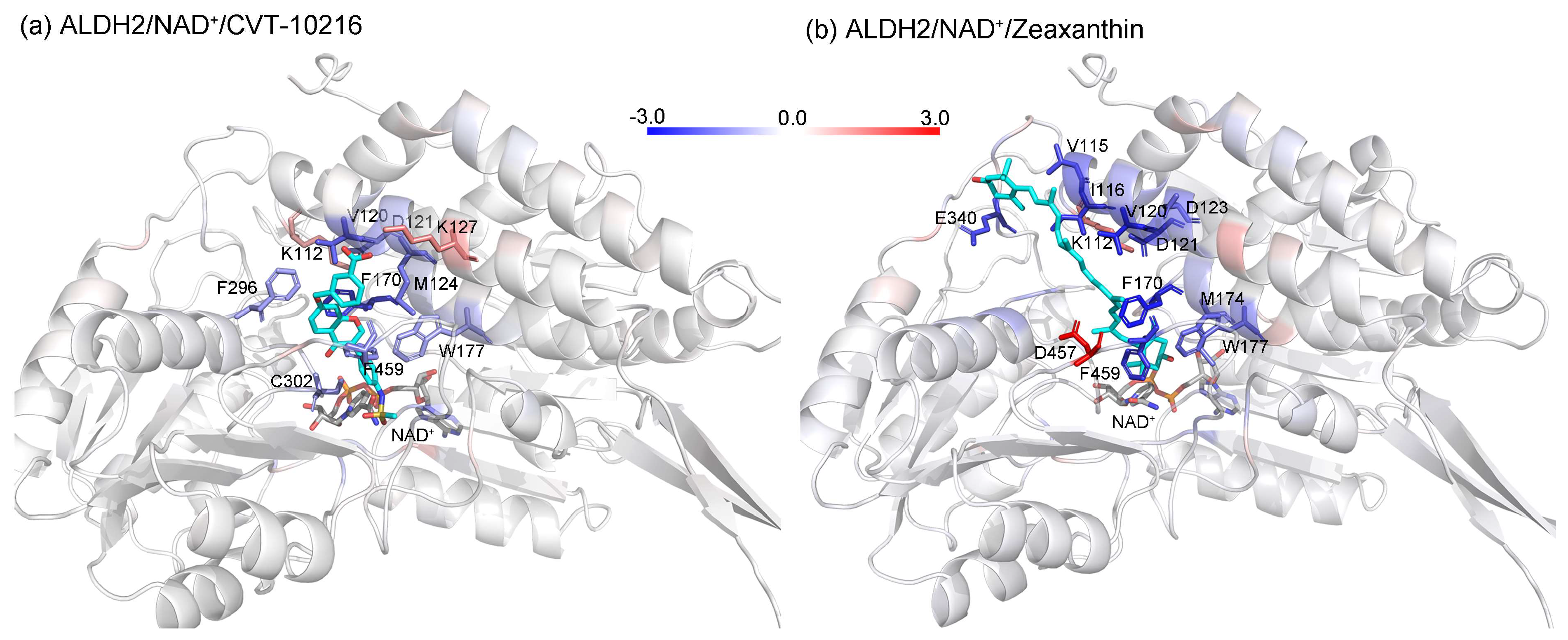
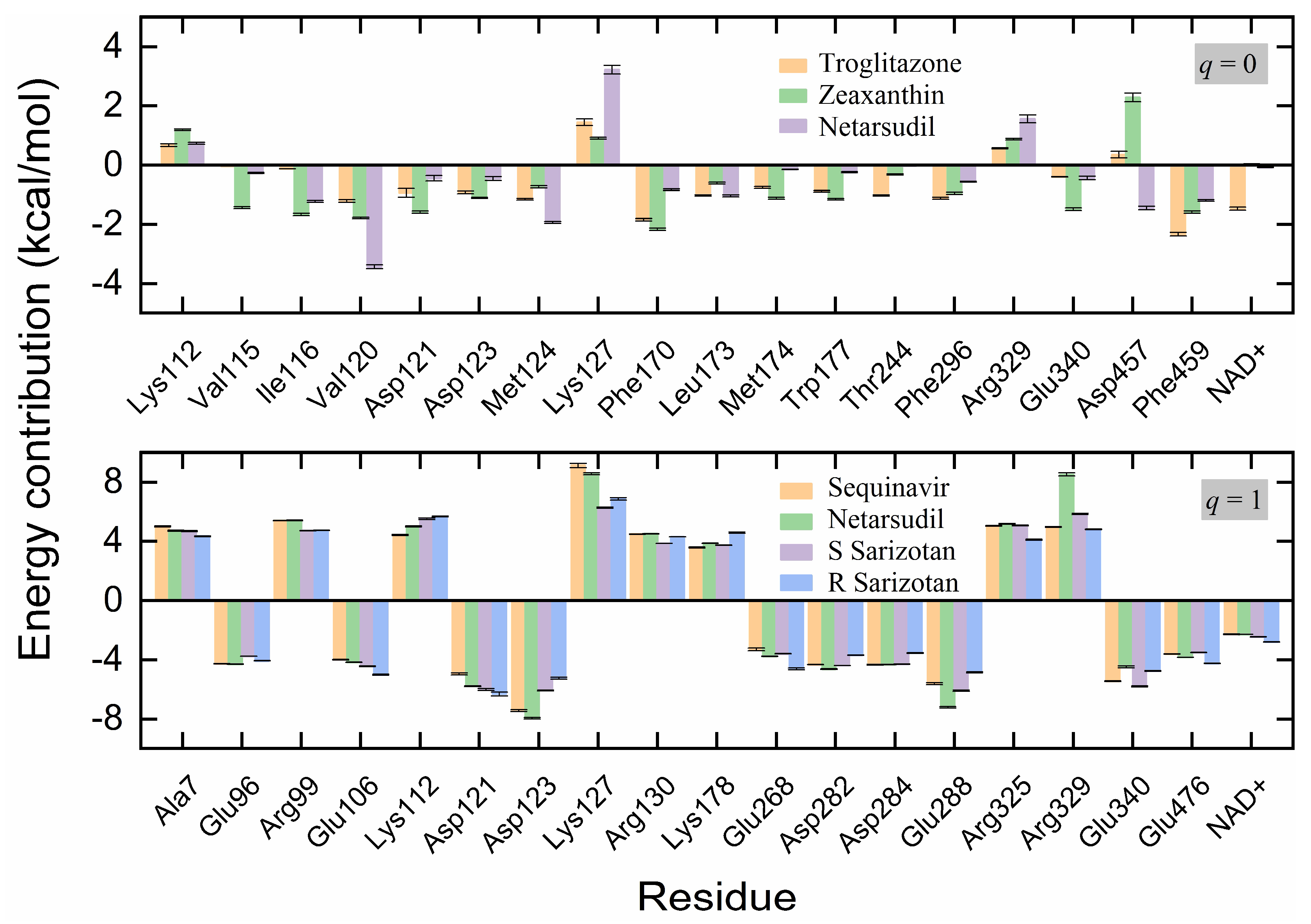
2.4. MD Simulation and Binding Energy Calculation
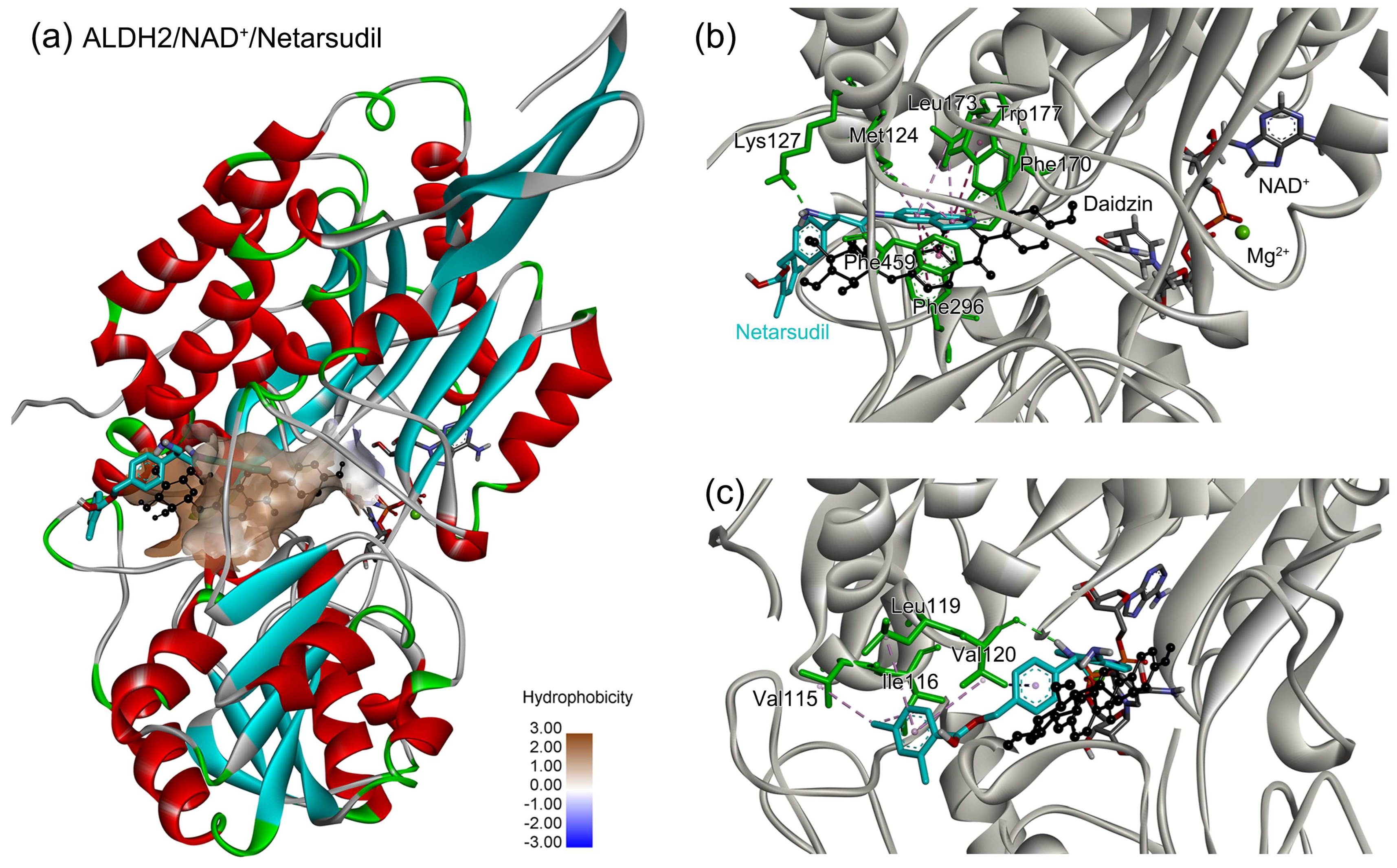
2.5. Identification of the Key Residues for Ligand Binding
3. Computational Methods
3.1. Ligand-Based Similarity Search
3.1.1. Reference Molecule and Drug Database for the Similarity Search
3.1.2. The 2D/3D Similarity Search
3.2. Docking Protocol
3.2.1. Ligand and Receptor Preparations
3.2.2. Docking Calculation
3.3. Toxicity Prediction
3.4. Molecular Simulation Protocol
3.5. MM–PBSA Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hasin, D.S.; O’brien, C.P.; Auriacombe, M.; Borges, G.; Bucholz, K.; Budney, A.; Compton, W.M.; Crowley, T.; Ling, W.; Petry, N.M.; et al. DSM-5 criteria for substance use disorders: Recommendations and rationale. Am. J. Psychiatry 2013, 170, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Ren, J. Alcohol and acetaldehyde in public health: From marvel to menace. Int. J. Environ. Res. Public Health 2010, 7, 1285–1301. [Google Scholar] [CrossRef] [PubMed]
- Melo, C.O.d.A.; Vieira, T.C.; Gigonzac, M.A.D.; Fortes, J.S.; Duarte, S.S.M.; da Cruz, A.D.; Silva, D.d.M.e. Evaluation of polymorphisms in repair and detoxification genes in alcohol drinkers and non-drinkers using capillary electrophoresis. Electrophoresis 2020, 41, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Wold, L.E. Mechanisms of alcoholic heart disease. Ther. Adv. Cardiovasc. Dis. 2008, 2, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Siggins, R.W.; McTernan, P.M.; Simon, L.; Souza-Smith, F.M.; Molina, P.E. Mitochondrial Dysfunction: At the Nexus between Alcohol-Associated Immunometabolic Dysregulation and Tissue Injury. Int. J. Mol. Sci. 2023, 24, 8650. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef] [PubMed]
- Boye, A.; Zou, Y.H.; Yang, Y. Metabolic derivatives of alcohol and the molecular culprits of fibro-hepatocarcinogenesis: Allies or enemies? World J. Gastroenterol. 2016, 22, 50–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Chen, Y.-C.; Chen, S.-J.; Lee, C.-H.; Cheng, C.-M. Alcohol Addiction, Gut Microbiota, and Alcoholism Treatment: A Review. Int. J. Mol. Sci. 2020, 21, 6413. [Google Scholar] [CrossRef]
- Xiao, L.; Xiang, J.; Liu, X.; Yang, L.; Wei, Y.; Fang, S.; Li, J.; Ye, Y. Lipidomic changes of cerebral cortex in aldehyde dehydrogenase-2 knock-in heterozygote mice after chronic alcohol exposure. Front. Mol. Neurosci. 2022, 15, 1053411. [Google Scholar] [CrossRef]
- Attilia, F.; Perciballi, R.; Rotondo, C.; Capriglione, I.; Iannuzzi, S.; Attilia, M.L.; Coriale, G.; Vitali, M.; Cereatti, F.; Fiore, M.; et al. Alcohol withdrawal syndrome: Diagnostic and therapeutic methods. Riv. Psichiatr. 2018, 53, 118–122. [Google Scholar] [CrossRef]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 aldehyde oxidizing enzymes: The aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Ferreira, J.C.B.; Gross, E.R.; Mochly-Rosen, D.; Gray, J.P.; Burgos, D.Z.; Yuan, T.; Seeram, N.; Rebar, R.; Follmer, R.; et al. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.-C.; Lo, W.-C.; Chang, Y.-W.; Lee, S.-S.; Chang, C.-C. Design, synthesis and the structure-activity relationship of agonists targeting on the ALDH2 catalytic tunnel. Bioorganic Chem. 2020, 104, 104166. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.C.; Liou, S.R.; Tsai, S.F.; Yin, S.J. Dominance of the mutant ALDH2(2) allele in the expression of human stomach aldehyde dehydrogenase-2 activity. Proc. Natl. Sci. Counc. Repub. China B 1993, 17, 98–102. [Google Scholar] [PubMed]
- Wang, Q.; Chang, B.; Li, X.; Zou, Z. Role of ALDH2 in Hepatic Disorders: Gene Polymorphism and Disease Pathogenesis. J. Clin. Transl. Hepatol. 2021, 9, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, D.H.; Knapp, D.J.; Breese, G.R.; Diamond, I. A selective ALDH-2 inhibitor reduces anxiety in rats. Pharmacol. Biochem. Behav. 2009, 94, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Bocarsly, M.E.; Hoebel, B.G.; Paredes, D.; von Loga, I.; Murray, S.M.; Wang, M.; Arolfo, M.P.; Yao, L.; Diamond, I.; Avena, N.M. GS 455534 selectively suppresses binge eating of palatable food and attenuates dopamine release in the accumbens of sugar-bingeing rats. Behav. Pharmacol. 2014, 25, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Arolfo, M.P.; Overstreet, D.H.; Yao, L.; Fan, P.; Lawrence, A.J.; Tao, G.; Keung, W.-M.; Vallee, B.L.; Olive, M.F.; Gass, J.T.; et al. Suppression of heavy drinking and alcohol seeking by a selective ALDH-2 inhibitor. Alcohol. Clin. Exp. Res. 2009, 33, 1935–1944. [Google Scholar] [CrossRef]
- Yao, L.; Fan, P.; Arolfo, M.; Jiang, Z.; Olive, M.F.; Zablocki, J.; Sun, H.-L.; Chu, N.; Lee, J.; Kim, H.-Y.; et al. Inhibition of aldehyde dehydrogenase-2 suppresses cocaine seeking by generating THP, a cocaine use–dependent inhibitor of dopamine synthesis. Nat. Med. 2010, 16, 1024–1028. [Google Scholar] [CrossRef]
- Kim, S.; Jang, E.Y.; Song, S.-H.; Kim, J.S.; Ryu, I.S.; Jeong, C.-H.; Lee, S. Brain Microdialysis Coupled to LC-MS/MS Revealed That CVT-10216, a Selective Inhibitor of Aldehyde Dehydrogenase 2, Alters the Neurochemical and Behavioral Effects of Methamphetamine. ACS Chem. Neurosci. 2021, 12, 1552–1562. [Google Scholar] [CrossRef]
- Douaihy, A.B.; Kelly, T.M.; Sullivan, C. Medications for substance use disorders. Soc. Work. Public Health 2013, 28, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Fischler, P.V.; Soyka, M.; Seifritz, E.; Mutschler, J. Off-label and investigational drugs in the treatment of alcohol use disorder: A critical review. Front. Pharmacol. 2022, 13, 927703. [Google Scholar] [CrossRef] [PubMed]
- Mason, B.J.; Heyser, C.J. Alcohol Use Disorder: The Role of Medication in Recovery. Alcohol Res. Curr. Rev. 2021, 41, 7. [Google Scholar] [CrossRef] [PubMed]
- Guillot, A.; Ren, T.; Jourdan, T.; Pawlosky, R.J.; Han, E.; Kim, S.-J.; Zhang, L.; Koob, G.F.; Gao, B. Targeting liver aldehyde dehydrogenase-2 prevents heavy but not moderate alcohol drinking. Proc. Natl. Acad. Sci. USA 2019, 116, 25974–25981. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.A.; Baker, H.M.; Blythe, T.J.; Kitson, K.E.; Kitson, T.M.; Baker, E.N. Sheep liver cytosolic aldehyde dehydrogenase: The structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. Structure 1998, 6, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.-H.; Zhang, H.-F.; Yang, Z.-B.; Li, T.-B.; Liu, B.; Lou, Z.; Ma, Q.-L.; Luo, X.-J.; Peng, J. Alda-1 reduces cerebral ischemia/reperfusion injury in rat through clearance of reactive aldehydes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Perez-Miller, S.; Younus, H.; Vanam, R.; Chen, C.-H.; Mochly-Rosen, D.; Hurley, T.D. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat. Struct. Mol. Biol. 2010, 17, 159–164. [Google Scholar] [CrossRef]
- Mali, V.R.; Deshpande, M.; Pan, G.; Thandavarayan, R.A.; Palaniyandi, S.S. Impaired ALDH2 activity decreases the mitochondrial respiration in H9C2 cardiomyocytes. Cell Signal. 2016, 28, 1–6. [Google Scholar] [CrossRef]
- Gao, G.-Y.; Li, D.-J.; Keung, W.M. Synthesis of potential antidipsotropic isoflavones: Inhibitors of the mitochondrial monoamine oxidase−aldehyde dehydrogenase pathway. J. Med. Chem. 2001, 44, 3320–3328. [Google Scholar] [CrossRef]
- Lowe, E.D.; Gao, G.-Y.; Johnson, L.N.; Keung, W.M. Structure of daidzin, a naturally occurring anti-alcohol-addiction agent, in complex with human mitochondrial aldehyde dehydrogenase. J. Med. Chem. 2008, 51, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Advances in the Development of Shape Similarity Methods and Their Application in Drug Discovery. Front. Chem. 2018, 6, 315. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Kuang, G.; Xiao, W.; Li, W.; Liu, G.; Tang, Y. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J. Chem. Inf. Model. 2012, 52, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.; Hirte, S.; Kirchmair, J. Maximizing the Performance of Similarity-Based Virtual Screening Methods by Generating Synergy from the Integration of 2D and 3D Approaches. Int. J. Mol. Sci. 2022, 23, 7747. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, K.; Flachner, B.; Hajdú, I.; Szaszkó, M.; Dobi, K.; Lőrincz, Z.; Cseh, S.; Dormán, G. Rapid Identification of Potential Drug Candidates from Multi-Million Compounds’ Repositories. Combination of 2D Similarity Search with 3D Ligand/Structure Based Methods and In Vitro Screening. Molecules 2021, 26, 5593. [Google Scholar] [CrossRef] [PubMed]
- Pavadai, E.; Kaur, G.; Wittlin, S.; Chibale, K. Identification of steroid-like natural products as antiplasmodial agents by 2D and 3D similarity-based virtual screening. MedChemComm 2017, 8, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Tanbin, S.; Fuad, F.A.A.; Hamid, A.A.A. Virtual Screening for Potential Inhibitors of Human Hexokinase II for the Development of Anti-Dengue Therapeutics. BioTech 2020, 10, 1. [Google Scholar] [CrossRef]
- Patil, S.P.; Ballester, P.J.; Kerezsi, C.R. Prospective virtual screening for novel p53–MDM2 inhibitors using ultrafast shape recognition. J. Comput. Mol. Des. 2014, 28, 89–97. [Google Scholar] [CrossRef]
- Distinto, S.; Esposito, F.; Kirchmair, J.; Cardia, M.C.; Gaspari, M.; Maccioni, E.; Alcaro, S.; Markt, P.; Wolber, G.; Zinzula, L.; et al. Identification of HIV-1 reverse transcriptase dual inhibitors by a combined shape-, 2D-fingerprint- and pharmacophore-based virtual screening approach. Eur. J. Med. Chem. 2012, 50, 216–229. [Google Scholar] [CrossRef]
- Wang, B.; Buchman, C.D.; Li, L.; Hurley, T.D.; Meroueh, S.O. Enrichment of chemical libraries docked to protein conformational ensembles and application to aldehyde dehydrogenase 2. J. Chem. Inf. Model. 2014, 54, 2105–2116. [Google Scholar] [CrossRef]
- Yang, J.; Cai, Y.; Zhao, K.; Xie, H.; Chen, X. Concepts and applications of chemical fingerprint for hit and lead screening. Drug Discov. Today 2022, 27, 103356. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Bajorath, J. ccbmlib−A Python package for modeling Tanimoto similarity value distributions [version 2; peer review: 2 approved]. F1000Research 2020, 9, Chem Inf Sci-100. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org/ (accessed on 30 June 2023).
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Axen, S.D.; Huang, X.-P.; Cáceres, E.L.; Gendelev, L.; Roth, B.L.; Keiser, M.J. A Simple Representation of Three-Dimensional Molecular Structure. J. Med. Chem. 2017, 60, 7393–7409. [Google Scholar] [CrossRef]
- Schreyer, A.M.; Blundell, T. USRCAT: Real-time ultrafast shape recognition with pharmacophoric constraints. J. Chemin. 2012, 4, 27. [Google Scholar] [CrossRef]
- Ballester, P.J.; Richards, W.G. Ultrafast shape recognition to search compound databases for similar molecular shapes. J. Comput. Chem. 2007, 28, 1711–1723. [Google Scholar] [CrossRef]
- Ballester, P.J.; Finn, P.W.; Richards, W.G. Ultrafast shape recognition: Evaluating a new ligand-based virtual screening technology. J. Mol. Graph. Model. 2009, 27, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Schreyer, A.; Blundell, T. CREDO: A Protein-Ligand Interaction Database for Drug Discovery. Chem. Biol. Drug Des. 2009, 73, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Dobi, K.; Hajdú, I.; Flachner, B.; Fabó, G.; Szaszkó, M.; Bognár, M.; Magyar, C.; Simon, I.; Szisz, D.; Lőrincz, Z.; et al. Combination of 2D/3D ligand-based similarity search in rapid virtual screening from multimillion compound repositories. Selection and biological evaluation of potential PDE4 and PDE5 inhibitors. Molecules 2014, 19, 7008–7039. [Google Scholar] [CrossRef] [PubMed]
- Bechelane-Maia, E.H.; Assis, L.C.; Alves de Oliveira, T.; Marques da Silva, A.; Gutterres Taranto, A. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.-H.; Zhu, X.; Bures, M.G.; Kihara, D. Three-dimensional compound comparison methods and their application in drug discovery. Molecules 2015, 20, 12841–12862. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Nguyen, D.D.; Sresht, V.; Mathiowetz, A.M.; Tu, M.; Wei, G.W. Are 2D fingerprints still valuable for drug discovery? Phys. Chem. Chem. Phys. 2020, 22, 8373–8390. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, Y.; Jiang, W.; Zhang, H. Virtual Screening of FDA-Approved Drugs for Enhanced Binding with Mitochondrial Aldehyde Dehydrogenase. Molecules 2022, 27, 8773. [Google Scholar] [CrossRef]
- Zhang, H.; Tan, T.; Hetényi, C.; van der Spoel, D. Quantification of Solvent Contribution to the Stability of Noncovalent Complexes. J. Chem. Theory Comput. 2013, 9, 4542–4551. [Google Scholar] [CrossRef]
- Zhang, H.; Tan, T.; Hetényi, C.; Lv, Y.; van der Spoel, D. Cooperative Binding of Cyclodextrin Dimers to Isoflavone Analogues Elucidated by Free Energy Calculations. J. Phys. Chem. C 2014, 118, 7163–7173. [Google Scholar] [CrossRef] [PubMed]
- Spoel, D.; Zhang, J.; Zhang, H. Quantitative predictions from molecular simulations using explicit or implicit interactions. WIREs Comput. Mol. Sci. 2022, 12, e1560. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, Y.; Zhang, H. Computational Investigation of Structural Basis for Enhanced Binding of Isoflavone Analogues with Mitochondrial Aldehyde Dehydrogenase. ACS Omega 2022, 7, 8115–8127. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Genet. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Walker, R.C.; de Souza, M.M.; Mercer, I.P.; Gould, I.R.; Klug, D.R. Large and Fast Relaxations inside a Protein: Calculation and Measurement of Reorganization Energies in Alcohol Dehydrogenase. J. Phys. Chem. B 2002, 106, 11658–11665. [Google Scholar] [CrossRef]
- Pavelites, J.J.; Gao, J.; Bash, P.A.; Mackerell, A.D. A molecular mechanics force field for NAD+ NADH, and the pyrophosphate groups of nucleotides. J. Comput. Chem. 1997, 18, 221–239. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1998, 79, 926–935. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2017, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
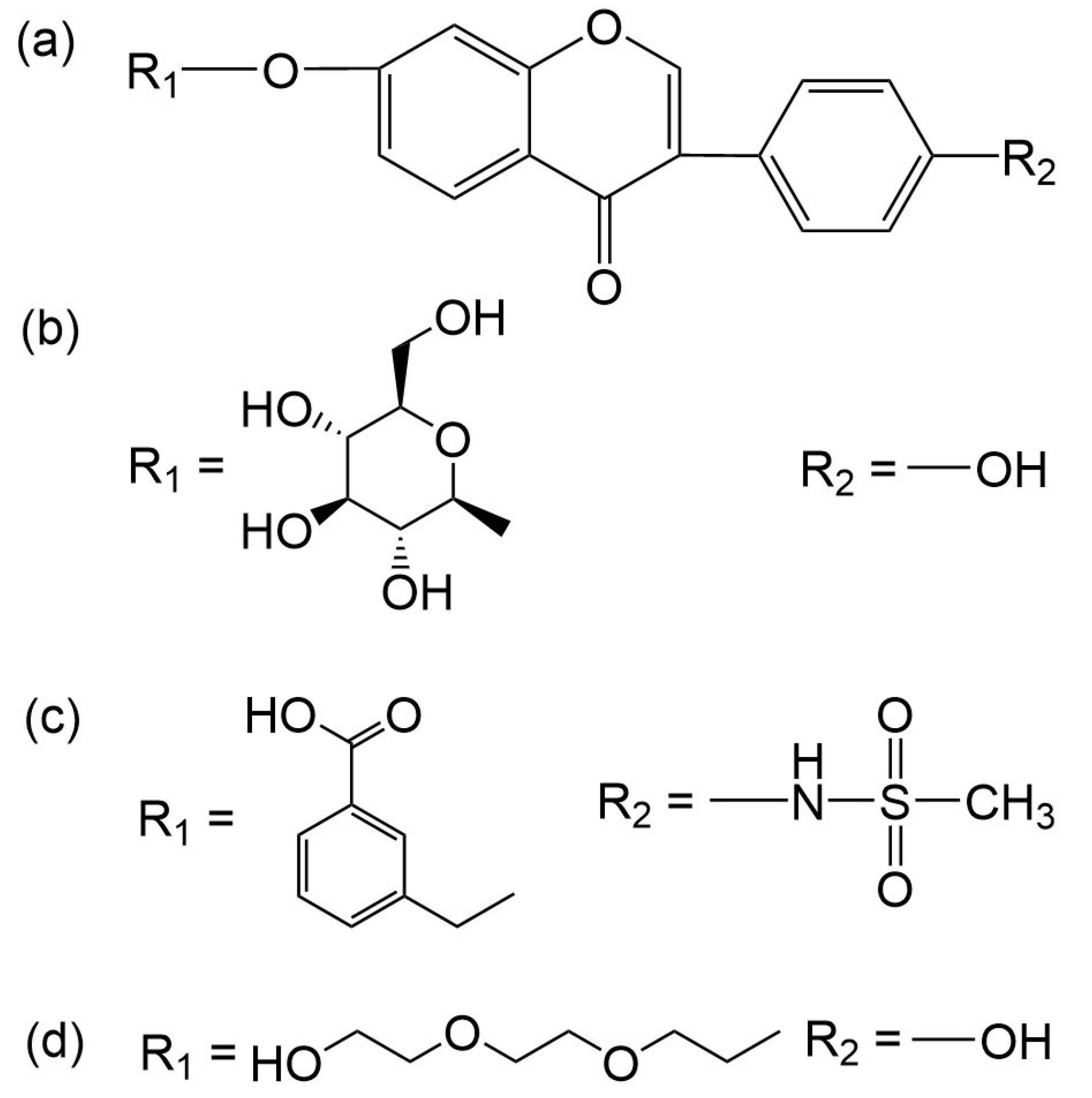

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | 2D | 3D | Total | ||||
|---|---|---|---|---|---|---|---|
| MACCS Keys | RDKit | ECFP4 | FCFP4 | E3FP | USRCAT | ||
| Daidzin | 0 (0) | 2 (0) | 1 (0) | 2 (0) | 0 (0) | 1 (1) | 4 (1) |
| CVT-10216 | 1 (0) | 4 (0) | 11 (2) | 11 (2) | 4 (1) | 11 (4) | 24 (9) |
| CHEMBL114083 | 4 (1) | 3 (0) | 3 (0) | 4 (0) | 3 (0) | 7 (1) | 17 (2) |
| Total | 5 (1) | 6 (0) | 14 (2) | 14 (2) | 5 (1) | 13 (6) | 33 (12) |
| ZINC ID | Molecular Structure | Name | q | ∆Edock | MACCS Keys | RDKit | ECFP4 | FCFP4 | E3FP | USRCAT | Hit |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ZINC011679756 | 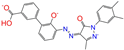 | Eltrombopag | −3 | −11.2 | 0.456 | 0.371 | 0.285 | 0.370 | 0.114 | 0.097 | N/Y/N |
| ZINC049783754 |  | Indacaterol-8-O-Glucuronide | 0 | −11.2 | 0.451 | 0.379 | 0.190 | 0.240 | 0.132 | 0.078 | Y/N/Y |
| ZINC011679756 |  | Eltrombopag | −2 | −11.0 | 0.456 | 0.371 | 0.285 | 0.370 | 0.114 | 0.097 | N/Y/N |
| ZINC001542146 |  | Pranlukast-IA | −1 | −10.9 | 0.494 | 0.424 | 0.318 | 0.411 | 0.153 | 0.104 | N/Y/Y |
| ZINC001542146 |  | Pranlukast-IB | −1 | −10.9 | 0.494 | 0.424 | 0.318 | 0.411 | 0.153 | 0.104 | N/Y/Y |
| ZINC095618662 | 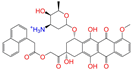 | ZINC095618662 | 1 | −10.8 | 0.527 | 0.404 | 0.208 | 0.266 | 0.109 | 0.134 | N/Y/N |
| ZINC019632618 | 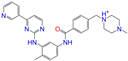 | Imatinib-I | 1 | −10.8 | 0.292 | 0.318 | 0.256 | 0.322 | 0.104 | 0.083 | N/Y/N |
| ZINC019632618 | 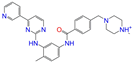 | Imatinib-II | 1 | −10.7 | 0.292 | 0.318 | 0.256 | 0.322 | 0.104 | 0.083 | N/Y/N |
| ZINC003824921 | 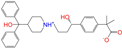 | Fexofenadine-I | 0 | −10.7 | 0.312 | 0.235 | 0.186 | 0.253 | 0.082 | 0.137 | N/Y/Y |
| ZINC021981222 | 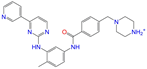 | N-Desmethyl Imatinib | 1 | −10.7 | 0.287 | 0.317 | 0.259 | 0.327 | 0.098 | 0.092 | N/Y/N |
| ZINC150339055 | 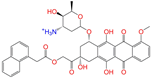 | ZINC150339055 | 1 | −10.6 | 0.527 | 0.404 | 0.208 | 0.266 | 0.115 | 0.135 | N/Y/N |
| ZINC008220175 | 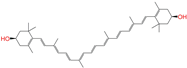 | Zeaxanthin | 0 | −10.6 | 0.197 | 0.132 | 0.035 | 0.020 | 0.064 | 0.114 | N/N/Y |
| ZINC077313075 | 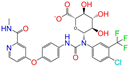 | Sorafenib Beta-D-Glucuronide | −1 | −10.5 | 0.469 | 0.385 | 0.238 | 0.323 | 0.091 | 0.124 | N/Y/N |
| ZINC113149554 | 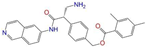 | Netarsudil | 0 | −10.5 | 0.425 | 0.298 | 0.284 | 0.397 | 0.127 | 0.125 | N/Y/Y |
| ZINC001493878 | 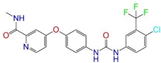 | Sorafenib | 0 | −10.5 | 0.427 | 0.298 | 0.264 | 0.348 | 0.092 | 0.089 | N/Y/N |
| ZINC000968278 | 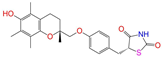 | Troglitazone | 0 | −10.4 | 0.481 | 0.311 | 0.193 | 0.234 | 0.089 | 0.172 | N/Y/Y |
| ZINC000968278 | 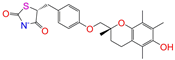 | Troglitazone | −1 | −10.3 | 0.481 | 0.311 | 0.193 | 0.234 | 0.089 | 0.172 | N/Y/Y |
| ZINC013449462 |  | 5-O-Desmethyldonepezil-I | 0 | −10.3 | 0.403 | 0.279 | 0.188 | 0.213 | 0.097 | 0.108 | N/N/Y |
| ZINC003872566 |  | Fexofenadine-II | 0 | −10.3 | 0.312 | 0.235 | 0.186 | 0.253 | 0.079 | 0.142 | N/Y/Y |
| ZINC000057674 |  | Flavone | 0 | −10.3 | 0.322 | 0.427 | 0.233 | 0.375 | 0.096 | 0.062 | Y/Y/Y |
| ZINC000021067 |  | R Sarizotan | 1 | −10.3 | 0.256 | 0.305 | 0.223 | 0.360 | 0.058 | 0.069 | N/Y/N |
| ZINC006037085 | 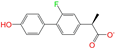 | (R)-4′-Hydroxyflurbipron | −1 | −10.3 | 0.312 | 0.247 | 0.250 | 0.295 | 0.078 | 0.077 | N/N/Y |
| ZINC068202099 | 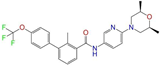 | Erismodegib | 0 | −10.3 | 0.393 | 0.349 | 0.291 | 0.394 | 0.106 | 0.131 | N/Y/Y |
| ZINC003817152 | 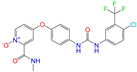 | Sorafenib N-Oxide | 0 | −10.3 | 0.469 | 0.309 | 0.260 | 0.324 | 0.104 | 0.087 | N/Y/N |
| ZINC000896717 | 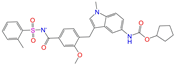 | Accolate | −1 | −10.2 | 0.639 | 0.368 | 0.290 | 0.357 | 0.122 | 0.103 | N/Y/N |
| ZINC001550477 |  | Lapatinib | 1 | −10.2 | 0.551 | 0.410 | 0.359 | 0.387 | 0.160 | 0.083 | N/Y/N |
| ZINC013515303 |  | 17-Alpha-Estradiol-3-Glucuronide | −1 | −10.2 | 0.368 | 0.341 | 0.110 | 0.159 | 0.103 | 0.095 | Y/N/Y |
| ZINC015919406 |  | Pranlukast-IIA | −1 | −10.2 | 0.494 | 0.434 | 0.354 | 0.432 | 0.174 | 0.099 | N/Y/Y |
| ZINC013449412 |  | 6-O-Desmethyldonepeil | 0 | −10.1 | 0.403 | 0.277 | 0.188 | 0.213 | 0.118 | 0.093 | N/N/Y |
| ZINC013449412 | 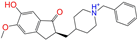 | 6-O-Desmethyldonepeil | 1 | −10.1 | 0.403 | 0.277 | 0.188 | 0.213 | 0.118 | 0.093 | N/N/Y |
| ZINC006030312 | 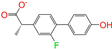 | (S)-4′-Hydroxyflurbipron | −1 | −10.1 | 0.312 | 0.247 | 0.250 | 0.295 | 0.075 | 0.077 | N/N/Y |
| ZINC113149554 | 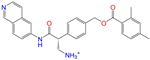 | Netarsudil | 1 | −10.1 | 0.425 | 0.298 | 0.284 | 0.397 | 0.127 | 0.125 | N/Y/Y |
| ZINC015919406 | 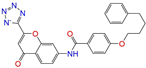 | Pranlukast-IIB | −1 | −10.1 | 0.494 | 0.434 | 0.354 | 0.432 | 0.174 | 0.099 | N/Y/Y |
| ZINC013449462 | 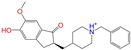 | 5-O-Desmethyldonepezil-I | 1 | −10.1 | 0.403 | 0.279 | 0.188 | 0.213 | 0.097 | 0.108 | N/N/Y |
| ZINC013449465 | 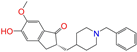 | 5-O-Desmethyldonepezil-II | 0 | −10.0 | 0.403 | 0.279 | 0.188 | 0.213 | 0.111 | 0.101 | N/N/Y |
| ZINC000006990 | 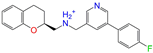 | S Sarizotan | 1 | −10.0 | 0.256 | 0.305 | 0.223 | 0.360 | 0.074 | 0.089 | N/Y/N |
| ZINC000105216 | 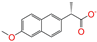 | Naproxen | −1 | −10.0 | 0.328 | 0.204 | 0.197 | 0.308 | 0.161 | 0.062 | N/Y/N |
| ZINC256630457 |  | ZINC256630457 | 1 | −10.0 | 0.527 | 0.404 | 0.208 | 0.266 | 0.118 | 0.101 | Y/N/N |
| ZINC256630463 |  | ZINC256630463 | 1 | −10.0 | 0.527 | 0.404 | 0.208 | 0.266 | 0.119 | 0.131 | N/Y/N |
| ZINC028639340 |  | Posaconazole | 0 | −10.0 | 0.426 | 0.393 | 0.194 | 0.263 | 0.075 | 0.125 | N/Y/Y |
| ZINC026985532 |  | Sequinavir | 0 | −10.0 | 0.323 | 0.349 | 0.173 | 0.255 | 0.095 | 0.121 | N/Y/N |
| ZINC026985532 | 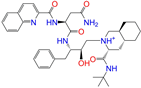 | Sequinavir | 1 | −10.0 | 0.323 | 0.349 | 0.173 | 0.255 | 0.095 | 0.121 | N/Y/N |
| ZINC ID | Name | q | Toxicity | FDA | ||||
|---|---|---|---|---|---|---|---|---|
| Dill | Carcino | Immuno | Mutagen | Cyto | ||||
| ZINC011679756 | Eltrombopag | −3 | Y (0.67) | N (0.57) | N (0.72) | N (0.56) | N (0.84) | yes |
| ZINC049783754 | Indacaterol-8-O-Glucuronide | 0 | N (0.71) | N (0.61) | Y (0.87) | N (0.59) | N (0.54) | no |
| ZINC011679756 | Eltrombopag | −2 | Y (0.67) | N (0.57) | N (0.72) | N (0.56) | N (0.84) | yes |
| ZINC001542146 | Pranlukast-IA | −1 | Y (0.57) | N (0.72) | N (0.87) | Y (0.53) | N (0.77) | no |
| ZINC001542146 | Pranlukast-IB | −1 | Y (0.57) | N (0.72) | N (0.87) | Y (0.53) | N (0.77) | no |
| ZINC095618662 | ZINC095618662 | 1 | N (0.85) | N (0.85) | Y (0.99) | Y (0.94) | Y (0.79) | no |
| ZINC019632618 | Imatinib-I | 1 | Y (0.71) | N (0.67) | Y (0.66) | N (0.73) | N (0.52) | yes |
| ZINC019632618 | Imatinib-II | 1 | Y (0.71) | N (0.67) | Y (0.66) | N (0.73) | N (0.52) | yes |
| ZINC003824921 | Fexofenadine-I | 0 | N (0.99) | Y (0.50) | N (0.86) | N (0.85) | N (0.81) | yes |
| ZINC021981222 | N-Desmethyl Imatinib | 1 | N (0.61) | N (0.62) | Y (0.66) | N (0.69) | N (0.60) | no |
| ZINC150339055 | ZINC150339055 | 1 | N (0.85) | N (0.85) | Y (0.99) | Y (0.94) | Y (0.79) | no |
| ZINC008220175 | Zeaxanthin | 0 | N (0.79) | N (0.67) | N (0.92) | N (0.81) | N (0.89) | no |
| ZINC077313075 | Sorafenib Beta-D-Glucuronide | −1 | Y (0.65) | N (0.60) | Y (0.92) | N (0.74) | N (0.63) | no |
| ZINC113149554 | Netarsudil | 0 | N (0.72) | N (0.52) | N (0.95) | N (0.58) | N (0.59) | no |
| ZINC001493878 | Sorafenib | 0 | Y (0.82) | N (0.50) | Y (0.92) | N (0.79) | Y (0.77) | yes |
| ZINC000968278 | Troglitazone | 0 | N (0.62) | N (0.62) | N (0.90) | N (0.58) | N (0.61) | no |
| ZINC000968278 | Troglitazone | −1 | N (0.62) | N (0.62) | N (0.90) | N (0.58) | N (0.61) | no |
| ZINC013449462 | 5-O-Desmethyldonepezil-I | 0 | N (0.97) | N (0.55) | Y (0.98) | N (0.55) | Y (0.58) | no |
| ZINC003872566 | Fexofenadin-II | 0 | N (0.99) | Y (0.50) | N (0.86) | N (0.85) | N (0.81) | yes |
| ZINC000057674 | Flavone | 0 | N (0.70) | Y (0.69) | N (0.99) | N (0.54) | Y (0.75) | no |
| ZINC000021067 | R Sarizotan | 1 | N (0.71) | N (0.62) | N (0.87) | N (0.62) | N (0.62) | no |
| ZINC006037085 | (R)-4′-Hydroxyflurbipron | −1 | Y (0.68) | N (0.66) | N (0.99) | N (0.85) | N (0.54) | no |
| ZINC068202099 | Erismodegib | 0 | N (0.52) | N (0.60) | Y (0.85) | N (0.67) | N (0.69) | yes |
| ZINC003817152 | Sorafenib N-Oxide | 0 | Y (0.67) | N (0.58) | Y (0.76) | Y (0.54) | Y (0.54) | no |
| ZINC000896717 | Accolate | −1 | Y (0.76) | N (0.57) | N (0.65) | N (0.67) | N (0.56) | yes |
| ZINC001550477 | Lapatinib | 1 | Y (0.80) | N (0.55) | Y (0.96) | N (0.51) | Y (0.76) | yes |
| ZINC013515303 | 17-Alpha-Estradiol-3-Glucuronide | −1 | N (0.84) | N (0.70) | Y (0.99) | N (0.78) | N (0.58) | no |
| ZINC015919406 | Pranlukast-IIA | −1 | Y (0.57) | N (0.72) | N (0.87) | Y (0.53) | N (0.77) | no |
| ZINC013449412 | 6-O-Desmethyldonepeil | 0 | N (0.98) | N (0.54) | Y (0.98) | N (0.54) | Y (0.65) | no |
| ZINC013449412 | 6-O-Desmethyldonepeil | 1 | N (0.98) | N (0.54) | Y (0.98) | N (0.54) | Y (0.65) | no |
| ZINC006030312 | (S)-4′-Hydroxyflurbipron | −1 | Y (0.68) | N (0.66) | N (0.99) | N (0.85) | N (0.54) | no |
| ZINC113149554 | Netarsudil | 1 | N (0.72) | N (0.52) | N (0.95) | N (0.58) | N (0.59) | no |
| ZINC015919406 | Pranlukast-IIB | −1 | Y (0.57) | N (0.72) | N (0.87) | Y (0.53) | N (0.77) | no |
| ZINC013449462 | 5-O-Desmethyldonepezil-I | 1 | N (0.97) | N (0.55) | Y (0.98) | N (0.55) | Y (0.58) | no |
| ZINC013449465 | 5-O-Desmethyldonepezil-II | 0 | N (0.97) | N (0.55) | Y (0.98) | N (0.55) | Y (0.58) | no |
| ZINC000006990 | S Sarizotan | 1 | N (0.71) | N (0.62) | N (0.87) | N (0.62) | N (0.62) | no |
| ZINC000105216 | Naproxen | −1 | Y (0.51) | N (0.53) | N (0.85) | N (0.74) | N (0.80) | yes |
| ZINC256630457 | ZINC256630457 | 1 | N (0.85) | N (0.85) | Y (0.99) | Y (0.94) | Y (0.79) | no |
| ZINC256630463 | ZINC256630463 | 1 | N (0.85) | N (0.85) | Y (0.99) | Y (0.94) | Y (0.79) | no |
| ZINC028639340 | Posaconazole | 0 | Y (0.86) | N (0.62) | Y (0.99) | N (0.56) | N (0.75) | yes |
| ZINC026985532 | Sequinavir | 0 | N (0.60) | N (0.63) | N (0.97) | N (0.79) | N (0.80) | yes |
| ZINC026985532 | Sequinavir | 1 | N (0.60) | N (0.63) | N (0.97) | N (0.79) | N (0.80) | yes |
| Name | q | Chain A | Chain B | Chain C | Chain D | Tetramer |
| RMSD (nm) | ||||||
| Sequinavir | 1 | 0.15 | 0.13 | 0.14 | 0.13 | 0.15 |
| R Sarizotan | 1 | 0.17 | 0.12 | 0.13 | 0.13 | 0.15 |
| S Sarizotan | 1 | 0.18 | 0.12 | 0.13 | 0.16 | 0.17 |
| Netarsudil | 1 | 0.10 | 0.11 | 0.11 | 0.11 | 0.15 |
| Zeaxanthin | 0 | 0.14 | 0.11 | 0.13 | 0.13 | 0.14 |
| Troglitazone | 0 | 0.11 | 0.11 | 0.11 | 0.09 | 0.15 |
| Sequinavir | 0 | 0.19 | 0.11 | 0.13 | 0.12 | 0.15 |
| Netarsudil | 0 | 0.11 | 0.09 | 0.09 | 0.09 | 0.13 |
| Fexofenadine-II | 0 | 0.17 | 0.12 | 0.13 | 0.12 | 0.14 |
| Troglitazone | −1 | 0.14 | 0.13 | 0.13 | 0.13 | 0.15 |
| Pranlukast-IA | −1 | 0.14 | 0.10 | 0.13 | 0.17 | 0.15 |
| Pranlukast-IIB | −1 | 0.20 | 0.11 | 0.15 | 0.15 | 0.16 |
| Pranlukast-IIA | −1 | 0.19 | 0.12 | 0.13 | 0.12 | 0.16 |
| Pranlukast-IB | −1 | 0.14 | 0.13 | 0.13 | 0.13 | 0.14 |
| Naproxen | −1 | 0.14 | 0.12 | 0.14 | 0.18 | 0.16 |
| Daidzin | 0 | 0.09 | 0.11 | 0.10 | 0.09 | 0.12 |
| CVT-10216 | 0 | 0.18 | 0.11 | 0.16 | 0.13 | 0.16 |
| CHEMBL114083 | 0 | 0.17 | 0.12 | 0.14 | 0.15 | 0.16 |
| ligand-free | 0.10 | 0.10 | 0.12 | 0.11 | 0.13 | |
| Name | q | Chain A | Chain B | Chain C | Chain D | <∆Ebind> |
| ∆Ebind (kcal/mol) | ||||||
| Sequinavir | 1 | −56.6 ± 2.4 | −61.1 ± 1.2 | −65.4 ± 0.5 | −43.1 ± 1.7 | −65.4 ± 0.5 |
| R Sarizotan | 1 | −54.5 ± 1.5 | −56.9 ± 0.6 | −60.8 ± 0.3 | −55.8 ± 1.6 | −60.8 ± 0.3 |
| S Sarizotan | 1 | −59.3 ± 1.8 | −55.6 ± 0.6 | −57.4 ± 0.7 | −60.3 ± 1.2 | −60.1 ± 1.3 |
| Netarsudil | 1 | −52.7 ± 2.3 | −59.7 ± 1.1 | −57.3 ± 0.9 | −57.1 ± 2.7 | −59.6 ± 1.6 |
| Zeaxanthin | 0 | −45.9 ± 1.3 | −42.4 ± 0.8 | −34.1 ± 1.7 | −38.2 ± 0.8 | −45.9 ± 1.3 |
| Troglitazone | 0 | −40.3 ± 1.8 | −25.5 ± 1.0 | −28.2 ± 1.3 | −26.3 ± 1.5 | −40.3 ± 1.8 |
| Sequinavir | 0 | −22.0 ± 2.0 | −27.5 ± 1.4 | −18.4 ± 1.6 | −14.1 ± 2.4 | −27.5 ± 1.4 |
| Netarsudil | 0 | −21.3 ± 2.4 | −21.4 ± 1.5 | −24.6 ± 0.9 | −19.8 ± 0.4 | −24.5 ± 0.6 |
| Fexofenadine-II | 0 | −19.8 ± 4.1 | −9.2 ± 1.1 | −21.7 ± 1.5 | −5.7 ± 1.3 | −21.7 ± 1.7 |
| Troglitazone | −1 | −0.2 ± 2.0 | 12.9 ± 1.1 | 12.0 ± 1.3 | 16.8 ± 1.8 | −0.2 ± 2.0 |
| Pranlukast-IA | −1 | 14.4 ± 0.8 | 3.1 ± 1.6 | 0.7 ± 0.2 | 16.6 ± 0.7 | 0.7 ± 0.2 |
| Pranlukast-IIB | −1 | 9.7 ± 1.5 | 18.9 ± 2.2 | 11.2 ± 6.0 | 5.5 ± 3.6 | 5.5 ± 1.3 |
| Pranlukast-IIA | −1 | 12.4 ± 2.2 | 12.6 ± 0.9 | 18.4 ± 2.9 | 17.6 ± 2.8 | 12.5 ± 0.8 |
| Pranlukast-IB | −1 | 20.2 ± 1.2 | 16.3 ± 1.4 | 19.4 ± 2.7 | 15.6 ± 1.5 | 15.7 ± 1.7 |
| Naproxen | −1 | 22.1 ± 0.3 | 22.9 ± 0.7 | 28.9 ± 3.5 | 16.3 ± 1.0 | 16.3 ± 1.0 |
| Daidzin | 0 | −19.0 ± 2.4 | −22.0 ± 1.1 | −26.3 ± 1.1 | −20.8 ± 1.2 | −26.3 ± 1.5 |
| CVT-10216 | 0 | −27.3 ± 1.7 | −25.1 ± 1.2 | −35.5 ± 1.1 | −29.3 ± 1.1 | −35.5 ± 1.1 |
| CHEMBL114083 | 0 | −23.5 ± 0.5 | −22.1 ± 1.1 | −23.6 ± 1.1 | −12.9 ± 1.5 | −23.5 ± 0.5 |
| Name | FDA | q | pH | ΔEvdW | ΔEelec | ΔEMM | ΔGpolar | ΔGnonpolar | ΔGsol | ΔEbind |
|---|---|---|---|---|---|---|---|---|---|---|
| Sequinavir | yes | 1 | ref | −50.9 ± 1.1 | −70.3 ± 1.2 | −121.2 ± 0.8 | 61.2 ± 0.5 | −5.3 ± 0.1 | 55.9 ± 0.5 | −65.4 ± 0.5 |
| R Sarizotan | no | 1 | ref | −40.5 ± 0.4 | −89.1 ± 1.6 | −129.6 ± 1.7 | 73.4 ± 1.7 | −4.6 ± 0.0 | 68.8 ± 1.7 | −60.8 ± 0.3 |
| S Sarizotan | no | 1 | ref | −38.1 ± 0.9 | −84.6 ± 1.7 | −122.7 ± 1.6 | 66.8 ± 2.2 | −4.4 ± 0.0 | 62.4 ± 2.1 | −60.3 ± 1.2 |
| Netarsudil | no | 1 | ref | −47.6 ± 0.4 | −88.5 ± 1.3 | −136.1 ± 1.5 | 81.7 ± 1.5 | −5.3 ± 0.1 | 76.4 ± 1.5 | −59.7 ± 1.1 |
| Zeaxanthin | no | 0 | ref | −65.7 ± 2.1 | −3.9 ± 0.7 | −69.7 ± 2.2 | 31.0 ± 1.4 | −7.2 ± 0.1 | 23.8 ± 1.4 | −45.9 ± 1.3 |
| Troglitazone | no | 0 | lo | −57.2 ± 0.9 | −7.1 ± 0.5 | −64.3 ± 0.9 | 29.3 ± 1.2 | −5.3 ± 0.1 | 24.0 ± 1.2 | −40.3 ± 1.8 |
| Sequinavir | yes | 0 | hi | −48.6 ± 2.7 | −10.8 ± 2.2 | −59.4 ± 4.8 | 37.0 ± 4.4 | −5.2 ± 0.3 | 31.9 ± 4.1 | −27.5 ± 1.4 |
| Netarsudil | no | 0 | hi | −42.6 ± 1.1 | −12.3 ± 1.4 | −54.9 ± 1.7 | 35.1 ± 1.6 | −4.8 ± 0.0 | 30.3 ± 1.5 | −24.6 ± 0.9 |
| Fexofenadine-II | yes | 0 | ref | −45.2 ± 0.8 | −59.4 ± 1.2 | −104.6 ± 1.3 | 88.0 ± 2.3 | −5.2 ± 0.1 | 82.9 ± 2.3 | −21.7 ± 1.5 |
| Troglitazone | no | −1 | ref | −57.5 ± 1.0 | 8.8 ± 0.6 | −48.7 ± 1.6 | 53.6 ± 0.7 | −5.2 ± 0.0 | 48.4 ± 0.7 | −0.2 ± 2.0 |
| Pranlukast-IA | no | −1 | mid | −66.4 ± 0.7 | 19.0 ± 1.2 | −47.4 ± 1.7 | 54.1 ± 1.6 | −6.1 ± 0.1 | 48.1 ± 1.6 | 0.7 ± 0.2 |
| Pranlukast-IIB | no | −1 | mid | −50.5 ± 3.1 | 19.6 ± 4.2 | −30.9 ± 7.0 | 41.2 ± 6.0 | −4.8 ± 0.2 | 36.4 ± 5.8 | 5.5 ± 3.6 |
| Pranlukast-IIA | no | −1 | ref | −48.4 ± 1.6 | 44.6 ± 1.6 | −3.9 ± 0.2 | 21.4 ± 1.0 | −4.9 ± 0.1 | 16.5 ± 0.9 | 12.6 ± 0.9 |
| Pranlukast-IB | no | −1 | ref | −47.7 ± 1.4 | 53.7 ± 4.0 | 6.0 ± 4.7 | 14.6 ± 3.7 | −5.0 ± 0.1 | 9.6 ± 3.7 | 15.6 ± 1.5 |
| Naproxen | yes | −1 | ref | −32.8 ± 0.5 | 9.5 ± 1.4 | −23.2 ± 1.6 | 42.8 ± 1.2 | −3.3 ± 0.0 | 39.5 ± 1.2 | 16.3 ± 1.0 |
| Daidzin | no | 0 | −53.3 ± 0.9 | −15.6 ± 0.6 | −68.9 ± 0.8 | 47.3 ± 0.9 | −4.7 ± 0.0 | 42.5 ± 0.9 | −26.3 ± 1.1 | |
| CVT-10216 | no | 0 | −62.3 ± 0.8 | −23.2 ± 0.6 | −85.5 ± 1.0 | 55.6 ± 0.8 | −5.6 ± 0.0 | 50.0 ± 0.8 | −35.5 ± 1.1 | |
| CHEMBL114083 | no | 0 | −44.8 ± 1.3 | −7.8 ± 1.1 | −52.6 ± 1.2 | 33.7 ± 1.8 | −4.6 ± 0.1 | 29.1 ± 1.7 | −23.5 ± 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Chen, J.; Zhang, P.; Zheng, N.; Ma, L.; Zhang, Y.; Zhang, H. Repurposing Drugs for Inhibition against ALDH2 via a 2D/3D Ligand-Based Similarity Search and Molecular Simulation. Molecules 2023, 28, 7325. https://doi.org/10.3390/molecules28217325
Jiang W, Chen J, Zhang P, Zheng N, Ma L, Zhang Y, Zhang H. Repurposing Drugs for Inhibition against ALDH2 via a 2D/3D Ligand-Based Similarity Search and Molecular Simulation. Molecules. 2023; 28(21):7325. https://doi.org/10.3390/molecules28217325
Chicago/Turabian StyleJiang, Wanyun, Junzhao Chen, Puyu Zhang, Nannan Zheng, Le Ma, Yongguang Zhang, and Haiyang Zhang. 2023. "Repurposing Drugs for Inhibition against ALDH2 via a 2D/3D Ligand-Based Similarity Search and Molecular Simulation" Molecules 28, no. 21: 7325. https://doi.org/10.3390/molecules28217325
APA StyleJiang, W., Chen, J., Zhang, P., Zheng, N., Ma, L., Zhang, Y., & Zhang, H. (2023). Repurposing Drugs for Inhibition against ALDH2 via a 2D/3D Ligand-Based Similarity Search and Molecular Simulation. Molecules, 28(21), 7325. https://doi.org/10.3390/molecules28217325