


Design, Synthesis, and Antiproliferative Activity of Selective Histone Deacetylases 6 Inhibitors Containing a Tetrahydropyridopyrimidine Scaffold


Abstract
:
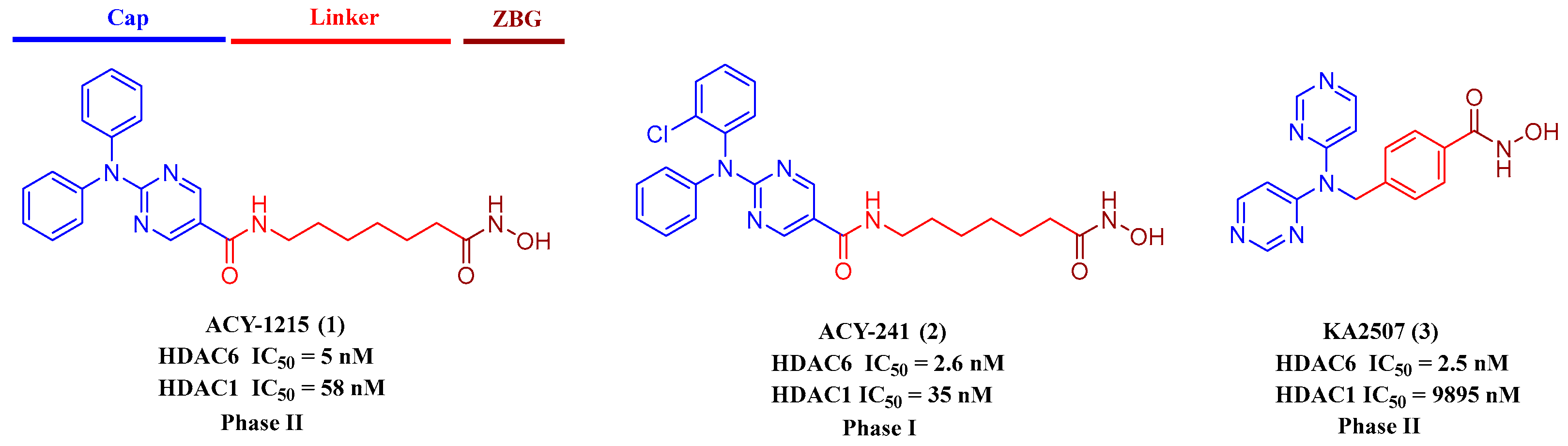
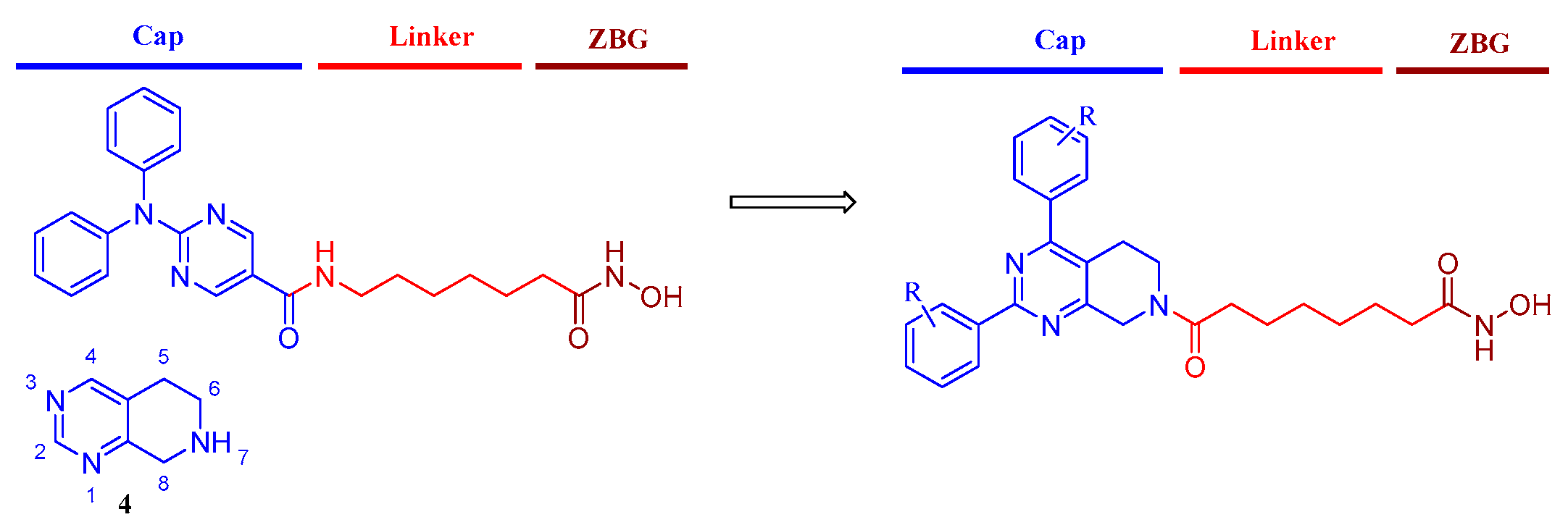
1. Introduction
2. Results and Discussion
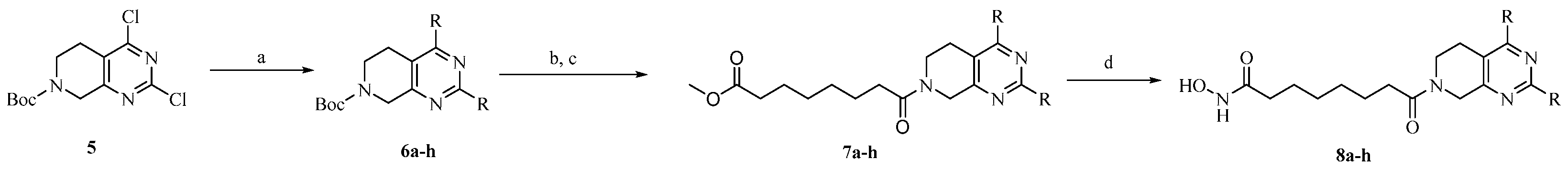
2.1. Chemistry
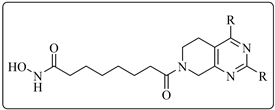
2.2. HDAC1, 6 Activities and SAR Study of the Target Compounds
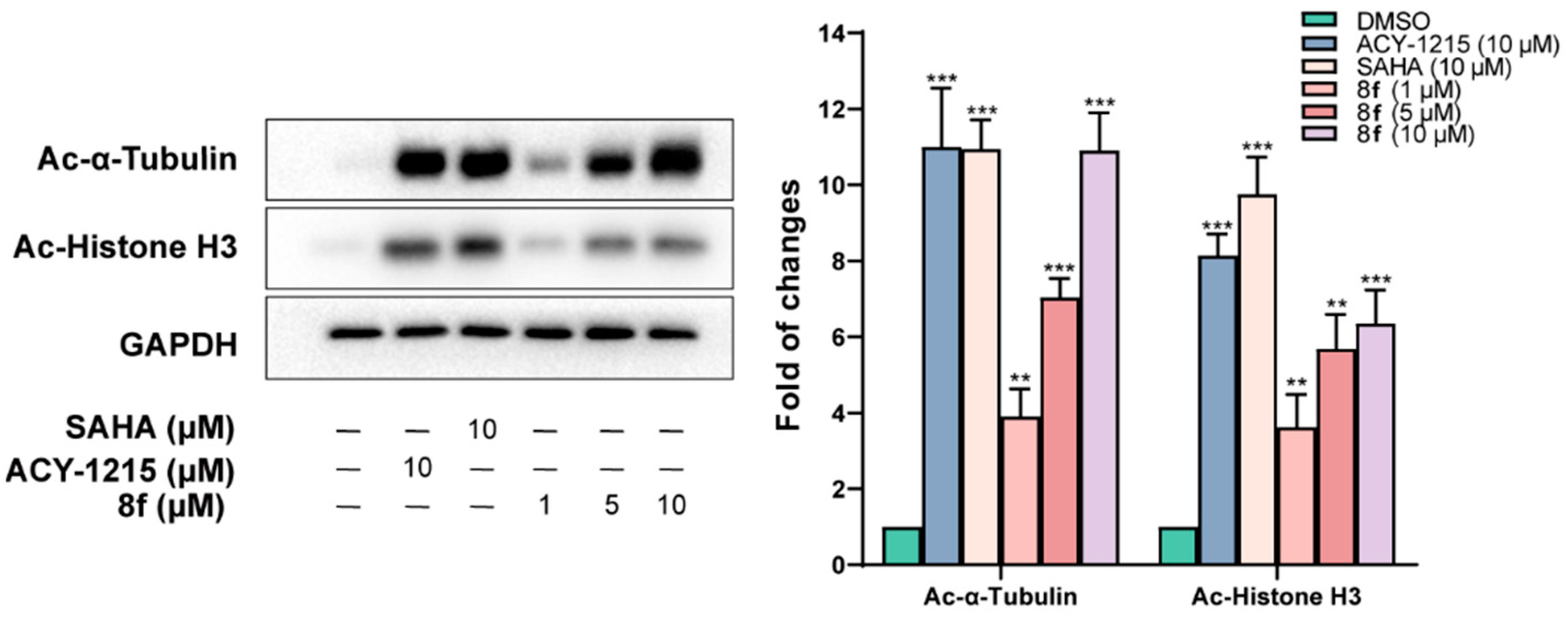
2.3. Western Blot Assay
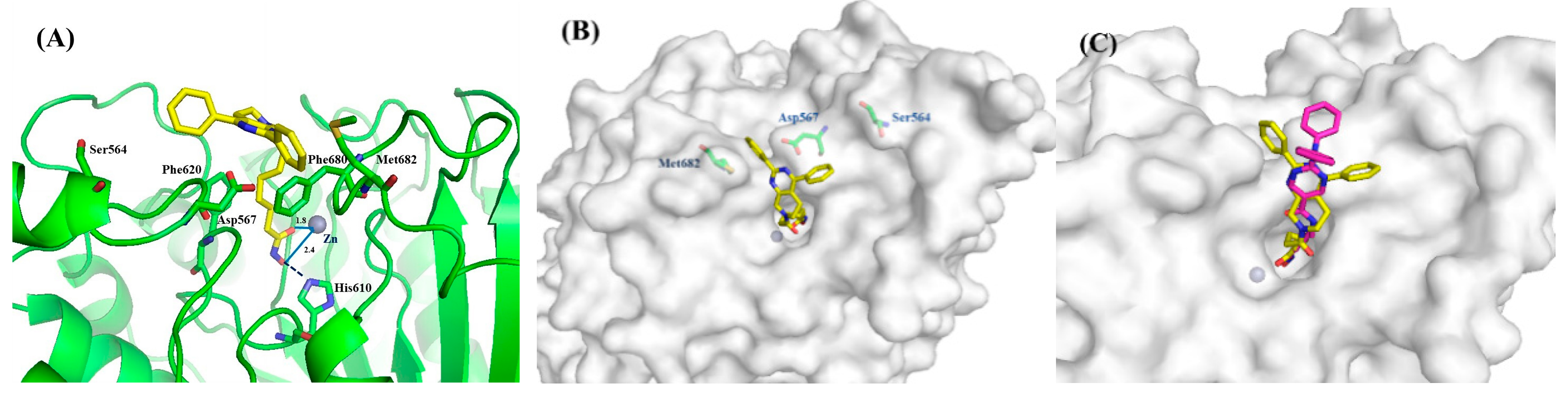
2.4. Molecular Simulation
2.5. Antiproliferative Activities of Representative Compounds
3. Experimental Section
3.1. Chemistry
3.1.1. Tert-Butyl 2, 4-Diphenyl-5, 8-dihydropyrido[3, 4-d]pyrimidine-7(6H)-carboxylate (6a)
3.1.2. Tert-Butyl 2, 4-di-p-tolyl-5, 8-dihydropyrido[3, 4-d]pyrimidine-7(6H)-carboxylate (6b)
3.1.3. Tert-Butyl 2, 4-Bis(4-methoxyphenyl)-5, 8-dihydropyrido[3, 4-d]pyrimidine-7(6H)-carboxylate (6c)
3.1.4. Tert-Butyl 2, 4-Di(furan-3-yl)-5, 8-dihydropyrido[3, 4-d]pyrimidine-7(6H)-carboxylate (6d)
3.1.5. Tert-Butyl 2, 4-Di(thiophen-3-yl)-5,8-dihydropyrido [3, 4-d]pyrimidine-7(6H)-carboxylate (6e)
3.1.6. Tert-Butyl 2, 4-Bis(3-methoxyphenyl)-5, 8-dihydropyrido [3, 4-d]pyrimidine-7(6H)-carboxylate (6f)
3.1.7. Tert-Butyl 2, 4-Di(thiophen-2-yl)-5, 8-dihydropyrido [3, 4-d]pyrimidine-7(6H)-carboxylate (6g)
3.1.8. Tert-Butyl 2, 4-Bis(4-(trifluoromethyl)phenyl)-5, 8-dihydropyrido [3, 4-d]pyrimidine-7(6H)-carboxylate (6h)
3.1.9. Methyl 8-(2, 4-Diphenyl-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-8-oxooctanoate (7a)
3.1.10. Methyl 8-(2, 4-di-p-tolyl-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-8-oxooctanoate (7b)
3.1.11. Methyl 8-(2, 4-Bis(4-methoxyphenyl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-8-oxooctanoate (7c)
3.1.12. Methyl 8-(2, 4-Di(furan-3-yl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-8-oxooctanoate (7d)
3.1.13. Methyl 8-(2, 4-Di(thiophen-3-yl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-8-oxooctanoate (7e)
3.1.14. Methyl 8-(2, 4-Bis(3-methoxyphenyl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-8-oxooctanoate (7f)
3.1.15. Methyl 8-(2, 4-Di(thiophen-2-yl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-8-oxooctanoate (7g)
3.1.16. Methyl 8-(2, 4-Bis(4-(trifluoromethyl)phenyl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-8-oxooctanoate (7h)
3.1.17. 8-(2, 4-Diphenyl-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-N-hydroxy-8-oxooctanamide (8a)
3.1.18. 8-(2, 4-di-p-tolyl-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-N-hydroxy-8-oxooctanamide (8b)
3.1.19. 8-(2, 4-Bis(4-methoxyphenyl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-N-hydroxy-8-oxooctanamide (8c)
3.1.20. 8-(2, 4-Di(furan-3-yl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-N-hydroxy-8-oxooctanamide (8d)
3.1.21. 8-(2, 4-Di(thiophen-3-yl)-5,8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-N-hydroxy-8-oxooctanamide (8e)
3.1.22. 8-(2, 4-Bis(3-methoxyphenyl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-N-hydroxy-8-oxooctanamide (8f)
3.1.23. 8-(2, 4-Di(thiophen-2-yl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-N-hydroxy-8-oxooctanamide (8g)
3.1.24. 8-(2, 4-Bis(4-(trifluoromethyl)phenyl)-5, 8-dihydropyrido [3, 4-d]pyrimidin-7(6H)-yl)-N-hydroxy-8-oxooctanamide (8h)
3.2. In Vitro HDAC Enzyme Assay
3.3. Cell Culture and Antiproliferative Assay
3.4. Western Blotting Assay
3.5. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Liang, T.; Xie, Z.; Dang, B.; Wang, J.; Zhang, T.; Luan, X.; Lu, T.; Cao, C.; Chen, X. Discovery of indole-piperazine derivatives as selective histone deacetylase 6 inhibitors with neurite outgrowth-promoting activities and neuroprotective activities. Bioorg. Med. Chem. Lett. 2023, 81, 129148. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Dasko, M.; de Pascual-Teresa, B.; Ortin, I.; Ramos, A. HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules 2022, 27, 715. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.Q.; Li, Z.B.; Newman, M.J.; Shan, S.; Wang, X.H.; Pan, D.S.; Zhang, J.; Dong, M.; Du, X.; Lu, X.P. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Sawas, A.; Radeski, D.; O’Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: A perspective review. Ther. Adv. Hematol. 2015, 6, 202–208. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, S.; Pisano, C.; Zunino, F. Development and therapeutic impact of HDAC6-selective inhibitors. Biochem. Pharmacol. 2012, 84, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Conte, M.; Altucci, L. Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid. Redox Signal. 2015, 23, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schluter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Kalin, J.H.; Bergman, J.A. Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J. Med. Chem. 2013, 56, 6297–6313. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P. HDAC6 in Diseases of Cognition and of Neurons. Cells 2020, 10, 12. [Google Scholar] [CrossRef]
- Rao, R.; Fiskus, W.; Ganguly, S.; Kambhampati, S.; Bhalla, K.N. HDAC inhibitors and chaperone function. Adv. Cancer Res. 2012, 116, 239–262. [Google Scholar]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef]
- Dos Santos Passos, C.; Simoes-Pires, C.A.; Carrupt, P.A.; Nurisso, A. Molecular dynamics of zinc-finger ubiquitin binding domains: A comparative study of histone deacetylase 6 and ubiquitin-specific protease 5. J. Biomol. Struct. Dyn. 2016, 34, 2581–2598. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.H.; Zhang, L.; Zhang, Y.J.; Zhang, J.; Xu, W.F. HDAC6: Physiological function and its selective inhibitors for cancer treatment. Drug Discov. Ther. 2013, 7, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Rajoria, P.; Chopra, M. HDAC6: A unique HDAC family member as a cancer target. Cell. Oncol. 2022, 45, 779–829. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.C.; Kang, H.Q.; Wang, B.; Zhu, Y.Z.; Mamun, M.A.A.; Zhao, L.F.; Nie, H.Q.; Liu, Y.; Zhao, L.J.; Zhang, X.N.; et al. Curriculum vitae of HDAC6 in solid tumors. Int. J. Biol. Macromol. 2023, 230, 123219. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liang, T.; Hou, X.; Fang, H. Recent Development of Novel HDAC6 Isoform-selective Inhibitors. Curr. Med. Chem. 2021, 28, 4133–4151. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, X.; Steimbach, R.R.; Wu, T.; Li, H.; Dan, W.; Shi, P.; Cao, C.; Li, D.; Miller, A.K.; et al. Novel 2, 5-diketopiperazine derivatives as potent selective histone deacetylase 6 inhibitors: Rational design, synthesis and antiproliferative activity. Eur. J. Med. Chem. 2020, 187, 111950. [Google Scholar] [CrossRef]
- Chen, X.; Gong, G.; Chen, X.; Song, R.; Duan, M.; Qiao, R.; Jiao, Y.; Qi, J.; Chen, Y.; Zhu, Y. Design, Synthesis and Biological Evaluation of Novel Benzoylimidazole Derivatives as Raf and Histone Deacetylases Dual Inhibitors. Chem. Pharm. Bull. 2019, 67, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Xie, F.; Qin, P.; Liu, Y.; Niu, H.; Sun, J.; Xue, H.; Zhao, Q.; Liu, J.; Wu, J. Recent development of selective inhibitors targeting the HDAC6 as anti-cancer drugs: Structure, function and design. Bioorg. Chem. 2023, 138, 106622. [Google Scholar] [CrossRef]
- Tavares, M.T.; Shen, S. Recent innovative advances in the discovery of selective HDAC6 inhibitors. Future Med. Chem. 2021, 13, 1017–1019. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, D.; Tan, Y. Role of HDAC6 and Its Selective Inhibitors in Gastrointestinal Cancer. Front. Cell Dev. Biol. 2021, 9, 719390. [Google Scholar] [CrossRef]
- Chen, X.; Wang, J.; Zhao, P.; Dang, B.; Liang, T.; Steimbach, R.R.; Miller, A.K.; Liu, J.; Wang, X.; Zhang, T.; et al. Tetrahydro-beta-carboline derivatives as potent histone deacetylase 6 inhibitors with broad-spectrum antiproliferative activity. Eur. J. Med. Chem. 2023, 260, 115776. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694–2707. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Beer, P.A.; Cartwright, C.A.; Haymaker, C.; Vo, H.H.; Kiany, S.; Cecil, A.R.L.; Dow, J.; Haque, K.; Silva, F.A.; et al. Preclinical Development and First-in-Human Study of KA2507, a Selective and Potent Inhibitor of Histone Deacetylase 6, for Patients with Refractory Solid Tumors. Clin. Cancer Res. 2021, 27, 3584–3594. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Blake, J.F.; Gaudino, J.J.; De Meese, J.; Mohr, P.; Chicarelli, M.; Tian, H.; Garrey, R.; Thomas, A.; Siedem, C.S.; Welch, M.B.; et al. Discovery of 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine inhibitors of Erk2. Bioorg. Med. Chem. Lett. 2014, 24, 2635–2639. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Yamane, Y.; Tsukamoto, S.; Murai, N.; Azuma, H.; Nagao, S.; Nishibata, K.; Fukushima, S.; Ichikawa, K.; Nakagawa, T.; et al. Discovery of 5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine derivatives as novel selective Axl inhibitors. Bioorg. Med. Chem. Lett. 2021, 48, 128247. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Targeting class I histone deacetylases in cancer therapy. Expert Opin. Ther. Targets 2013, 17, 29–41. [Google Scholar] [CrossRef]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Xing, L.; Gong, G.; Chen, X.; Chen, X. Discovery of Indole-Piperazine Hybrid Structures as Potent Selective Class I Histone Deacetylases Inhibitors. Chem. Pharm. Bull. 2023, 71, 206–212. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound | R | HDAC1 | HDAC6 | Selectivity Index |
| 8a |  | 565 | 16.2 | 35 |
| 8b |  | 640 | 24.4 | 41 |
| 8c |  | 472 | 9.6 | 49 |
| 8d | 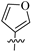 | 306 | 19.6 | 16 |
| 8e | 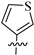 | 423 | 25.7 | 16 |
| 8f | 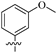 | 308 | 6.4 | 48 |
| 8g | 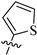 | 891 | 54.0 | 17 |
| 8h | 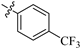 | 551 | 21.5 | 26 |
| 1 | / | 97.6 | 8.1 | 12 |
| SAHA | / | 4.3 | 7.4 | 0.6 |
| Compound | IC50 | ||||||
|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC5 | HDAC6 | HDAC8 | |
| 8f | 308 | 390 | 411 | >10,000 | >10,000 | 6.4 | 510 |
| SAHA | 4.2 | 11.5 | 3.5 | >10,000 | 8870 | 7.1 | 1033 |
| 1 | 62.5 | 47.5 | 50.0 | 7250 | 5100 | 5.1 | 150 |
| TMP269 | >10,000 | >10,000 | >10,000 | 0.133 | 0.090 | ND | ND |
| Compound | HL60 | HCT116 | RPMI-8226 |
|---|---|---|---|
| 8a | 12.5 | 23.4 | 16.3 |
| 8c | 3.78 | 4.72 | 4.63 |
| 8f | 3.20 | 3.25 | 2.80 |
| 1 | 4.10 | 6.50 | 1.70 |
| SAHA | 0.87 | 4.50 | 0.66 |
| Cancer Type | Cell Line | 8f | Cancer Type | Cell Line | 8f |
|---|---|---|---|---|---|
| Leukemia | CCRF-CEM | 85.0 | M14 | 90.5 | |
| HL60 | 100 | MDA-MB-435 | 95.8 | ||
| K-562 | 90.2 | SK-MEL-2 | 100 | ||
| MOLT-4 | 87.7 | SK-MEL-28 | 100 | ||
| RPMI-8226 | 88.7 | SK-MEL-5 | 100 | ||
| SR | 84.3 | UACC-257 | 98.7 | ||
| Non-Small Cell Lung Cancer | A549/ATCC | 87.7 | UACC-62 | 100 | |
| EKVX | 84.4 | Ovarian Cancer | IGROV1 | 100 | |
| HOP-62 | 99.1 | OVCAR-3 | 100 | ||
| HOP-92 | 100 | OVCAR-4 | 67.1 | ||
| NCI-H226 | 93.3 | OVCAR-5 | 100 | ||
| NCI-H23 | 100 | OVCAR-8 | 93.6 | ||
| NCI-H322M | 89.0 | NCI/ADR-RES | 100 | ||
| NCI-H460 | 95.5 | SK-OV-3 | 100 | ||
| NCI-H522 | 69.1 | Renal Cancer | 786-0 | 89.1 | |
| Colon Cancer | COLO 205 | 100 | A498 | 100 | |
| HCC-2998 | 100 | ACHN | 100 | ||
| HCT-116 | 96 | CAKI-1 | 100 | ||
| HCT-15 | 100 | SN12C | 100 | ||
| HT29 | 95.3 | TK-10 | 100 | ||
| KM12 | 88.4 | UO-31 | 100 | ||
| SW-620 | 100 | Prostate Cancer | PC-3 | 83.3 | |
| CNS Cancer | SF-268 | 92.2 | DU-145 | 89.8 | |
| SF-295 | 100 | Breast Cancer | MCF7 | 92.1 | |
| SF-539 | 99.0 | MDA-MB-231/ATCC | 100 | ||
| SNB-19 | 100 | HS 578T | 97.8 | ||
| SNB-75 | 71.0 | BT-549 | 81.9 | ||
| U251 | 100 | T-47D | 100 | ||
| Melanoma | LOX IMVI | 100 | MDA-MB-468 | 100 | |
| MALME-3M | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Liu, Y.; Zhang, L.; Wang, Y.; Li, Z.; Chen, X. Design, Synthesis, and Antiproliferative Activity of Selective Histone Deacetylases 6 Inhibitors Containing a Tetrahydropyridopyrimidine Scaffold. Molecules 2023, 28, 7323. https://doi.org/10.3390/molecules28217323
Wang B, Liu Y, Zhang L, Wang Y, Li Z, Chen X. Design, Synthesis, and Antiproliferative Activity of Selective Histone Deacetylases 6 Inhibitors Containing a Tetrahydropyridopyrimidine Scaffold. Molecules. 2023; 28(21):7323. https://doi.org/10.3390/molecules28217323
Chicago/Turabian StyleWang, Bin, Youcai Liu, Lejing Zhang, Yajuan Wang, Zhaoxi Li, and Xin Chen. 2023. "Design, Synthesis, and Antiproliferative Activity of Selective Histone Deacetylases 6 Inhibitors Containing a Tetrahydropyridopyrimidine Scaffold" Molecules 28, no. 21: 7323. https://doi.org/10.3390/molecules28217323
APA StyleWang, B., Liu, Y., Zhang, L., Wang, Y., Li, Z., & Chen, X. (2023). Design, Synthesis, and Antiproliferative Activity of Selective Histone Deacetylases 6 Inhibitors Containing a Tetrahydropyridopyrimidine Scaffold. Molecules, 28(21), 7323. https://doi.org/10.3390/molecules28217323