
Development and Validation of a Stable Isotope Dilution Headspace–SPME–GC/MS Method for the Determination of Vanillin in Fragrant Vegetable Oils
 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Optimization of the HS–SPME–GC/MS Method
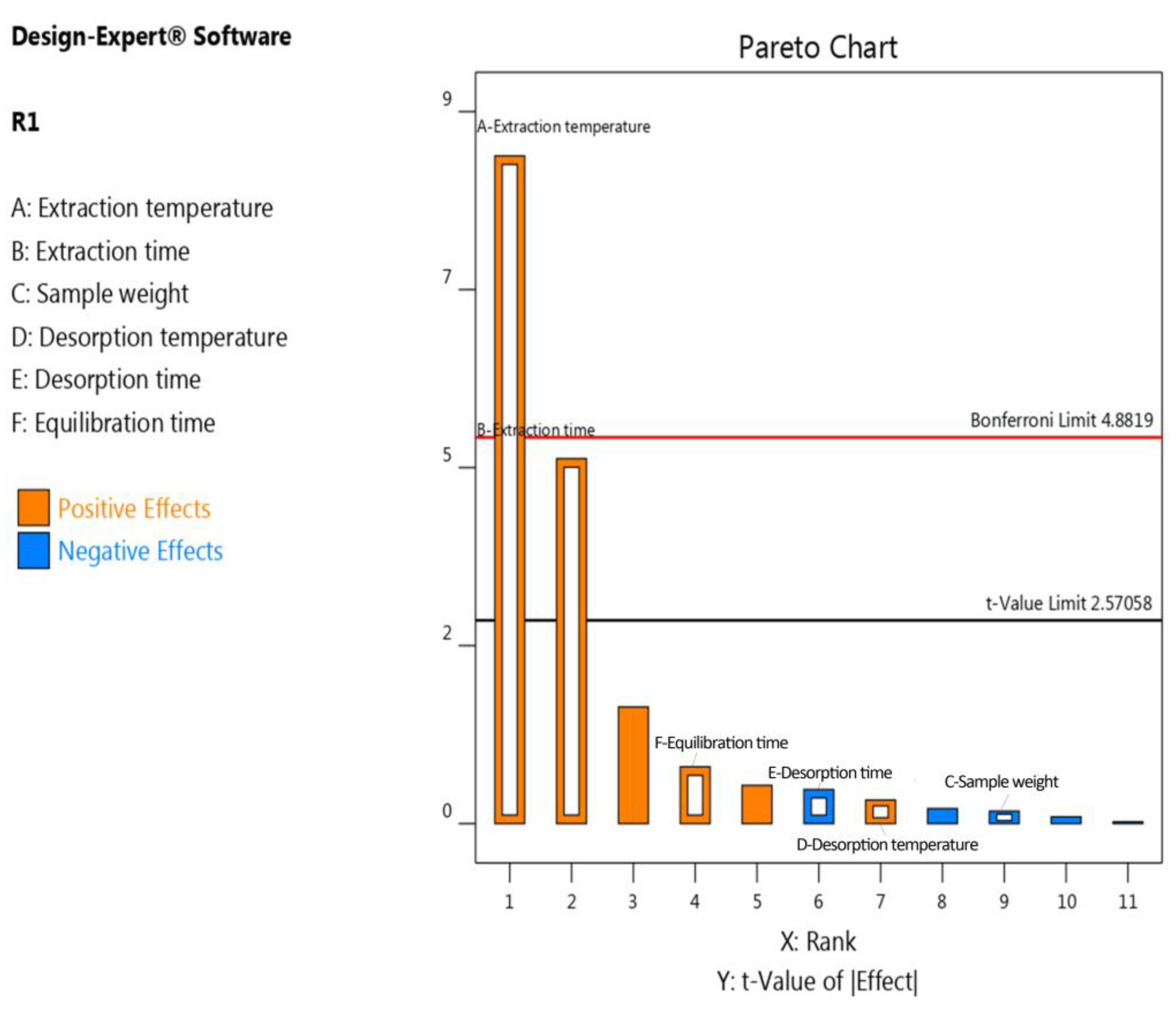
2.1.1. Screening of Significant Factors (Plackett–Burman Design)
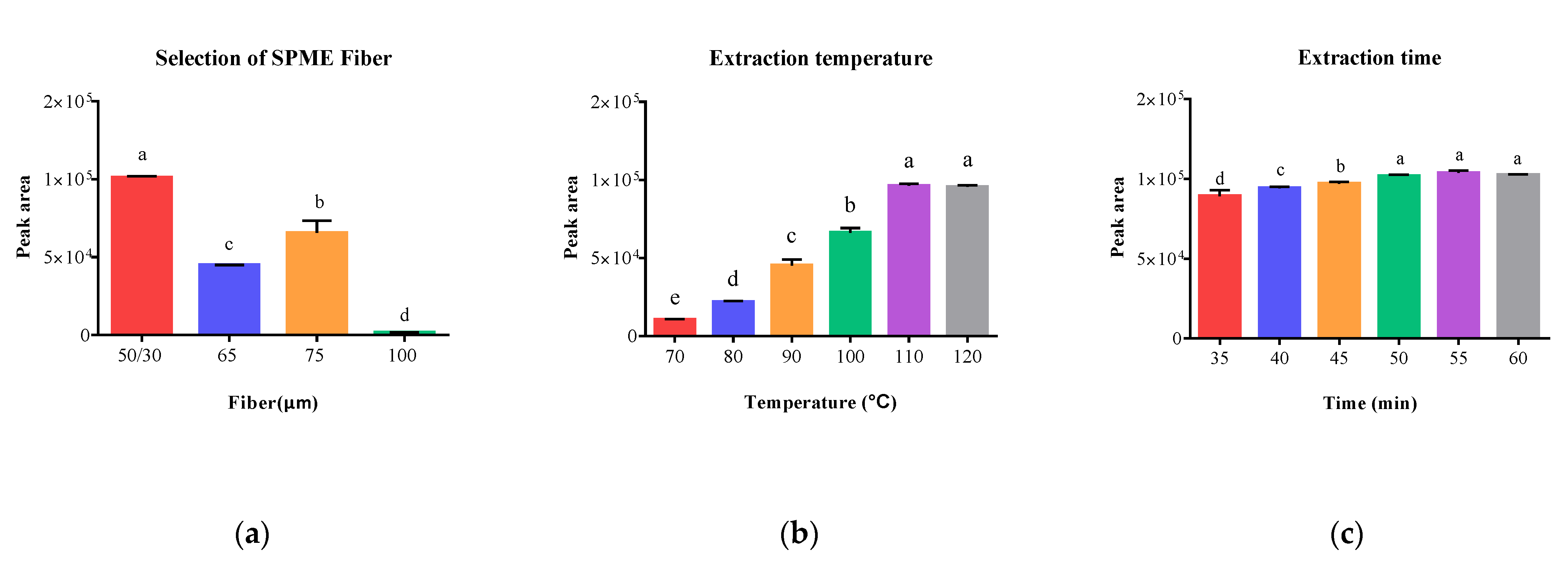
2.1.2. Single-Factor Experiment
2.1.3. Optimization of Significant Factors
2.2. Method Validation
2.2.1. Calibration, LOD, and LOQ
2.2.2. Precision and Recovery
2.2.3. Comparison of Vanillin Content in SSO Determined by Two Methods
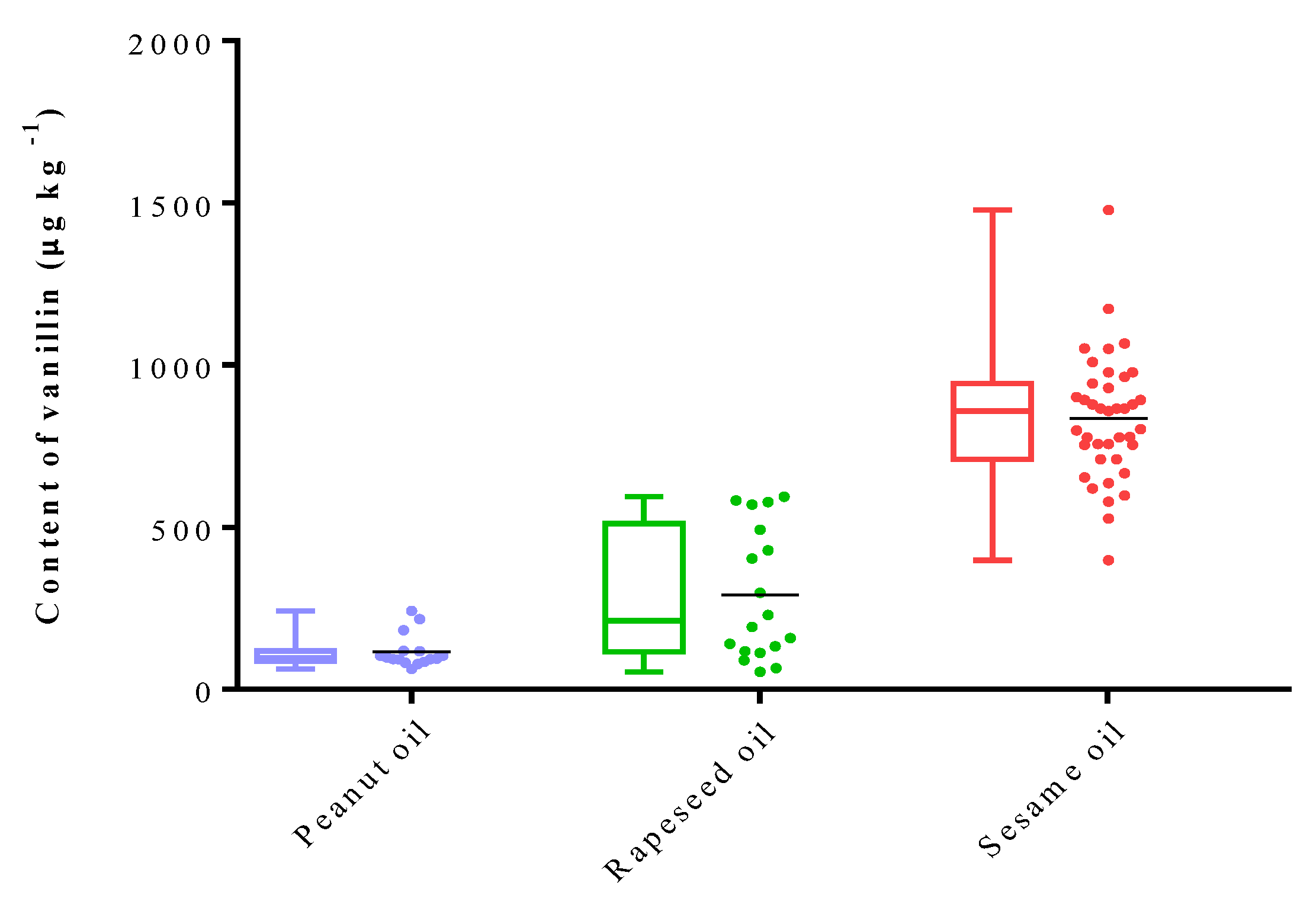
2.3. Quantification of Vanillin in Fragrant Vegetable Oils
3. Materials and Methods
3.1. Materials and Reagents
3.2. Preparation of Oil Samples
3.3. Optimization of the HS–SPME–GC/MS Method
3.3.1. SPME Optimization (Experimental Design Approach)
- Screening of significant factors (Plackett–Burman design)
- Single-factor experiment
- Optimization of significant factors
3.3.2. HS–SPME–GC/MS Analysis
3.4. Method Validation
3.4.1. Calibration, Limit of Detection (LOD) and Limit of Quantification (LOQ)
3.4.2. Precision and Recovery
3.4.3. UPLC–MS/MS Analysis
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sinha, A.; Sharma, U.; Sharma, N. A comprehensive review on vanilla flavor: Extraction, isolation and quantification of vanillin and others constituents. Int. J. Food Sci. Nutr. 2008, 59, 29–326. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Ito, M. Appetite-enhancing effects of curry oil. Biol. Pharm. Bull. 2016, 39, 1559–1563. [Google Scholar] [CrossRef][Green Version]
- Pérez-Esteve, É.; Lerma-García, M.J.; Fuentes, A.; Palomares, C.; Baratet, J.M. Control of undeclared flavoring of cocoa powders by the determination of vanillin and ethyl vanillin by HPLC. Food Control 2016, 67, 171–176. [Google Scholar] [CrossRef]
- Chi, Q.; Li, X.; He, J.; Wang, K.; Liu, C. Simulaneous determination of maltol, ethyl maltol, vanillin, methyl vanillin and ethyl vanillin in soya bean milk by high performance liquid chromatography. Food Saf. Qual. 2016, 7, 2690–2695. [Google Scholar]
- Wang, Z.; Zeng, G.; Wei, X.; Ding, B.; Huang, C.; Xu, B. Determination of Vanillin and Ethyl-Vanillin in Milk Powder by Headspace Solid-Phase Microextraction Coupled with Gas Chromatography-Mass Spectrometry. Food Anal. Methods 2016, 9, 3360. [Google Scholar] [CrossRef]
- Khoyratty, S.; Kodja, H.; Verpoorte, R. Vanilla flavor production methods: A review. Ind. Crops Prod. 2018, 125, 433–442. [Google Scholar] [CrossRef]
- Chen, J.; Duan, G.; Liu, L.; Li, C.; Lv, Z.; Wu, L.; Liu, C.; Hong, Y. Rapid Determination of Four Vanillin Compounds in Milk and Dairy Products by High Performance Liquid Chromatography. J. Dairy Sci. 2020, 43, 19–24. [Google Scholar]
- Santos, J.; Graf, U.; Reguly, M.; Andrade, H. The synergistic effects of vanillin on recombination predominate over its antimu-tagenic action in relation to MMC-induced lesions in somatic cells of Drosophila melanogaster. Mutat. Res. 1999, 444, 355–365. [Google Scholar] [CrossRef]
- Fitzgerald, D.; Stratford, M.; Gasson, M.; Ueckert, J.; Bos, A.; Narbad, A. Mode of antimicrobial action of vanillin against Escherichia coli, Lactobacillus plantarum and Listeria innocua. J. Appl. Microbiol. 2004, 97, 104–113. [Google Scholar] [CrossRef]
- Ni, Y.; Zhang, G.; Kokot, S. Simultaneous spectrophotometric determination of maltol, ethyl maltol, vanillin and ethyl vanillin in foods by multivariate calibration and artificial neural networks. Food Chem. 2005, 89, 465–473. [Google Scholar] [CrossRef]
- Peng, J.; Wei, M.; Hu, Y.; Yang, Y.; Guo, Y.; Zhang, F. Simultaneous Determination of Maltol, Ethyl Maltol, Vanillin, and Ethyl Vanillin in Foods by Isotope Dilution Headspace Solid-Phase Microextraction Coupled with Gas Chromatography-Mass Spectrometry. Food Anal. Methods 2019, 12, 1725–1735. [Google Scholar] [CrossRef]
- Yang, X.; Feng, J.; Xiao, X.; Wang, J.; Zhou, T.; Xiang, S. Simultaneous determination of maltol, ethyl maltol, vanillin, methyl vanillin and ethyl vanillin in edible vegetable oil by liquid chromatography-mass spectrometry. China Oils Fats 2023, 48, 103. [Google Scholar]
- Ning, X.; He, H.; Jin, S.; Cao, J.; Ding, H. Simultaneous determination of vanillin, methyl vanillin, ethyl vanillin, maltol and ethyl maltol in foods by ultra performance liquid chromatography-tandem mass spectrometry. J. Food Saf. Food Qual. 2017, 8, 2555–2562. [Google Scholar]
- Liu, C.; Lin, H.; Xiao, Q.; Wan, Q.; Dai, Y. The corroborative study of endogenous vanillin in sesame oil. China Oils Fats 2023, 48, 110–115. [Google Scholar]
- GB 2760-2014; Chinese National Food Safety Standard—Standard for the Use of Food Additives. National Health Commission of the Peoples Republic of China: Beijing, China, 2014.
- Franco, M.N.; Galeano-Díaz, T.; López, O.; Fernández-Bolaños, J.; Sánchez, J.; Miguel, C.; Gil, M.V.; Martín-Vertedor, D. Phenolic compounds and antioxidant capacity of virgin olive oil. Food Chem. 2014, 163, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Herchi, W.; Sawalha, S.; Arráez-Román, D.; Boukhchina, S.; Segura-Carretero, A.; Kallel, H.; Fernández-Gutierrez, A. Determination of phenolic and other polar compounds in flaxseed oil using liquid chromatography coupled with time-of-flight mass spectrometry. Food Chem. 2011, 126, 332–338. [Google Scholar] [CrossRef]
- Yang, M.; Yi, J.; Tu, F.; Wei, C.; Lu, Y.; Wang, S.; Yang, Z.; Zhao, L.; Jiang, X. One-step purification of solid phase micro-extraction based on MIL-101(Cr) for the determination of vanillin, methyl vanillin and ethyl vanillin in edible vegetable oil samples. Food Control 2023, 154, 461–468. [Google Scholar] [CrossRef]
- Zheng, L.; Zheng, P.; Sun, Z.; Bai, Y.; Wang, J.; Guo, X. Production of vanillin from waste residue of rice bran oil by Aspergillus niger and Pycnoporus cinnabarinus. Bioresour. Technol. 2007, 98, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Waliszewski, K.; Pardio, V.; Ovando, S. A simple and rapid HPLC technique for vanillin determination in alcohol extract. Food Chem. 2006, 101, 783–794. [Google Scholar] [CrossRef]
- Alkan, D.; Figen, T.; Banu, O. Phenolic Characterization and Geographical Classification of Commercial Extra Virgin Olive Oils Produced in Turkey. J. Am. Oil. Chem. Soc. 2011, 89, 261–268. [Google Scholar] [CrossRef]
- Qu, B.; Dai, X.; Zhang, J.; Liu, C.; Mao, X.; Jiang, J.; Pang, L. Determination of vanillin, methyl vanillin and ethyl vanillin in vegetable oil by high performance liquid chromatography tandem mass spectrometry. J. Food Saf. Food Qual. 2018, 9, 935–940. [Google Scholar]
- Xiao, F.; Zhang, Y.; Chen, P.; Hou, L.; Yi, B.; Yue, Z. Simultaneous determination of vanillin, methyl vanillin and ethyl vanillin in infant formulas by high performance liquid chromatography-tandem mass spectrometry method. J. Food Saf. Food Qual. 2014, 5, 3431–3435. [Google Scholar]
- Chen, L.; Zheng, F.; Fan, T.; Zhang, J.; Yang, Q. Research progress on detection methods of vanillin in food. China Condiment 2022, 47, 206. [Google Scholar]
- Wang, M.; Yang, Y.; Lu, Y.; Zhang, L.; Guan, Z.; Qiu, X.; Chen, Z. Content level of vanillin, methyl vanillin and ethyl vanillin in commercial sesame oil. Chin. J. Oil Crop Sci. 2023, 45, 413–418. [Google Scholar]
- Pawliszyn, J.; Vuckovic, D.; Mirnaghi, F.; Risticevic, S. Handbook of Solid Phase Microextraction, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 5, pp. 201–249. [Google Scholar]
- Reilly, J.; Wang, Q.; Pawliszyn, J. Automation of solid-phase microextraction. J. Sep. Sci. 2005, 28, 2010–2022. [Google Scholar] [CrossRef]
- Ouyang, G.; Jiang, R. Solid Phase Microextraction Recent Developments and Applications, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 1–27. [Google Scholar]
- Reyes-Garcés, N.; Gionfriddo, E.; Gómez-Ríos, G.E.; Alam, M.N.; Boyacl, E.; Bojko, B.; Pawliszyn, J. Advances in Solid Phase Microextraction and Perspective on Future Directions. J. Anal. Chem. 2018, 90, 302–360. [Google Scholar] [CrossRef]
- Yu, X.; Yu, L.; Ma, F.; Li, P. Quantification of phenolic compounds in vegetable oils by mixed-mode solid-phase extraction isotope chemical labeling coupled with UHPLC-MS/MS. Food Chem. 2021, 334, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Wenzl, T.; Haedrich, J.; Schaechtele, A.; Robouch, P.; Stroka, J. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food; EUR 28099; Publications Office of the European Union: Luxembourg, 2016; ISBN 978-92-79-61768-3. [Google Scholar] [CrossRef]
- De-Jager, L.S.; Perfetti, G.A.; Diachenko, G.W. Comparison of headspace-SPME-GC–MS and LC–MS for the detection and quantification of coumarin, vanillin, and ethyl vanillin in vanilla extract products. Food Chem. 2008, 107, 1701–1709. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Samples | Concentration (μg kg−1) | Intra-Day Precision (%RSD) | Inter-Day Precision (%RSD) | Recovery (%) |
|---|---|---|---|---|
| SSO-25 | 950.1 | 6.40 | 2.07 | 95.13 |
| 1190.0 | 2.09 | 6.59 | 97.48 | |
| 1860.4 | 4.36 | 6.09 | 101.90 | |
| SSO-31 | 1350.3 | 2.34 | 7.46 | 89.14 |
| 1850.1 | 3.81 | 2.30 | 90.23 | |
| 2760.2 | 1.55 | 1.62 | 92.79 |
| Samples | Vanillin (μg kg−1) | ||
|---|---|---|---|
| LC–MS/MS | HS–SPME–GC/MS | Difference (%) | |
| Sesame oil 1 | 947.0a ± 15.5 | 963.7a ± 8.0 | 1.80 |
| Sesame oil 9 | 641.3a ± 11.1 | 636.3a ± 14.6 | 0.85 |
| Sesame oil 12 | 865.6a ± 20.0 | 858.3a ± 16.5 | 0.88 |
| Sesame oil 27 | 1082.0a ± 55.2 | 1113.3a ± 55.1 | 2.82 |
| Sesame oil 28 | 886.0a ± 16.0 | 879.0a ± 16.0 | 0.79 |
| Rapeseed oil 4 | 212.3a ± 12.7 | 229.0a ± 2.7 | 7.74 |
| Rapeseed oil 8 | 550.6a ± 15.1 | 577.0a ± 7.0 | 4.68 |
| Rapeseed oil 10 | 394.3a ± 13.0 | 403.2a ± 11.6 | 2.26 |
| Rapeseed oil 12 | 105.2a ± 2.6 | 113.4a ± 5.8 | 7.50 |
| Rapeseed oil 17 | 131.3a ± 2.1 | 140.0a ± 7.0 | 6.48 |
| Peanut oil 3 | 109.4a ± 8.4 | 118.3a ± 2.9 | 8.41 |
| Peanut oil 6 | 95.9a ± 2.3 | 99.3a ± 4.5 | 3.79 |
| Peanut oil 10 | 110.5a ± 2.9 | 118.7a ± 2.3 | 7.58 |
| Peanut oil 15 | 262.0a ± 5.0 | 242.3a ± 2.1 | 7.75 |
| Peanut oil 19 | 89.4a ± 3.7 | 93.0a ± 6.1 | 4.05 |
| Factor | Experimental Domain | ||
|---|---|---|---|
| −1 | 0 | 1 | |
| Extraction temperature (°C) | 80 | 100 | 120 |
| Extraction time (min) | 10 | 30 | 50 |
| Sample weight (g) | 3 | 5 | 7 |
| Desorption temperature (°C) | 240 | 250 | 260 |
| Desorption time (min) | 3 | 5 | 7 |
| Equilibration time (min) | 5 | 15 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, F.; Wang, H.; Zhang, Y.; Zhang, M.; Zhou, S.; Shi, H.; Jiang, Y. Development and Validation of a Stable Isotope Dilution Headspace–SPME–GC/MS Method for the Determination of Vanillin in Fragrant Vegetable Oils. Molecules 2023, 28, 7288. https://doi.org/10.3390/molecules28217288
Mei F, Wang H, Zhang Y, Zhang M, Zhou S, Shi H, Jiang Y. Development and Validation of a Stable Isotope Dilution Headspace–SPME–GC/MS Method for the Determination of Vanillin in Fragrant Vegetable Oils. Molecules. 2023; 28(21):7288. https://doi.org/10.3390/molecules28217288
Chicago/Turabian StyleMei, Fangyi, Hongling Wang, Yuquan Zhang, Mei Zhang, Shuai Zhou, Haiming Shi, and Yuanrong Jiang. 2023. "Development and Validation of a Stable Isotope Dilution Headspace–SPME–GC/MS Method for the Determination of Vanillin in Fragrant Vegetable Oils" Molecules 28, no. 21: 7288. https://doi.org/10.3390/molecules28217288
APA StyleMei, F., Wang, H., Zhang, Y., Zhang, M., Zhou, S., Shi, H., & Jiang, Y. (2023). Development and Validation of a Stable Isotope Dilution Headspace–SPME–GC/MS Method for the Determination of Vanillin in Fragrant Vegetable Oils. Molecules, 28(21), 7288. https://doi.org/10.3390/molecules28217288