
Improving the Activity of Tryptophan Synthetase via a Nucleic Acid Scaffold

 ,
,
Abstract
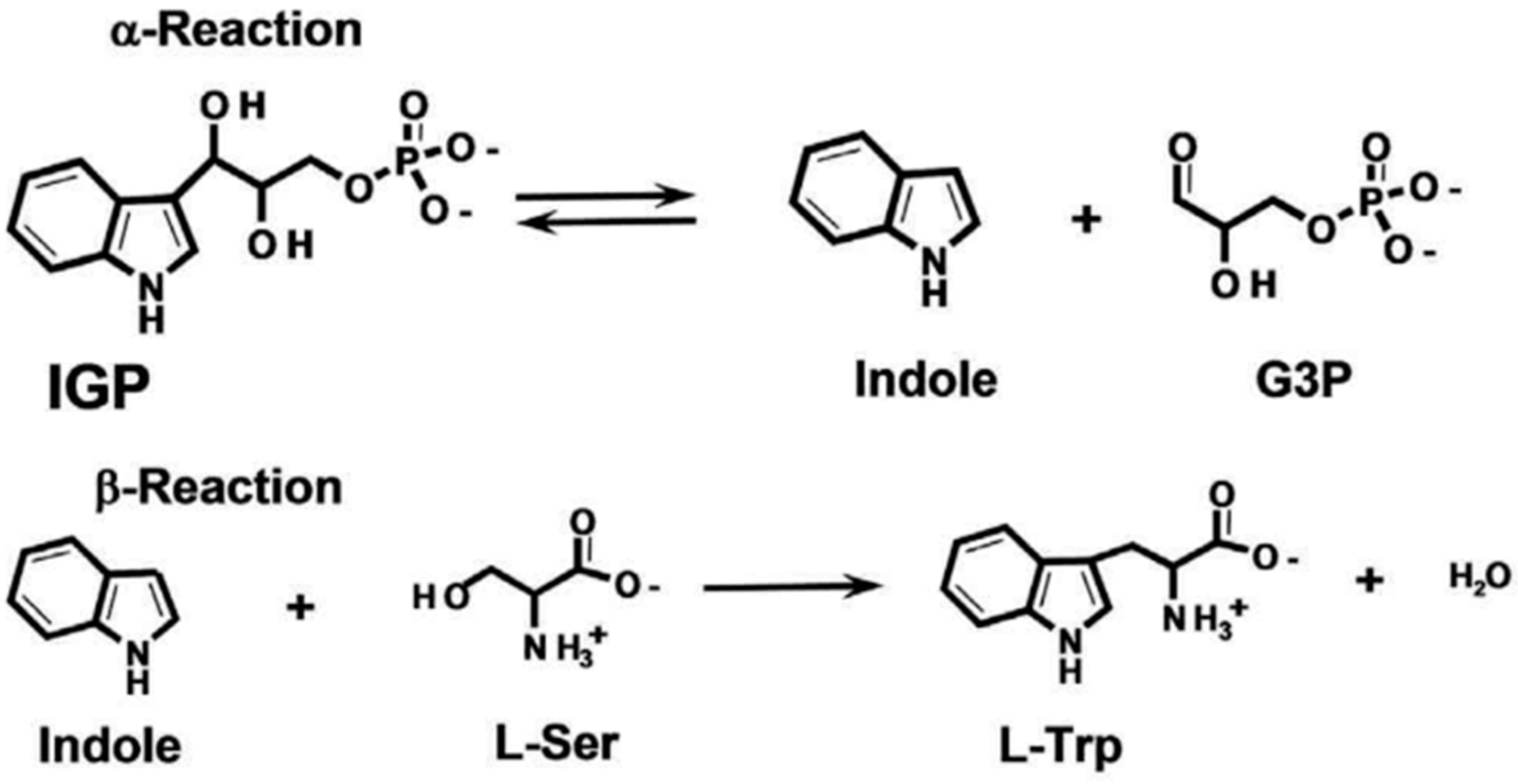
:1. Introduction
2. Results and Discussion
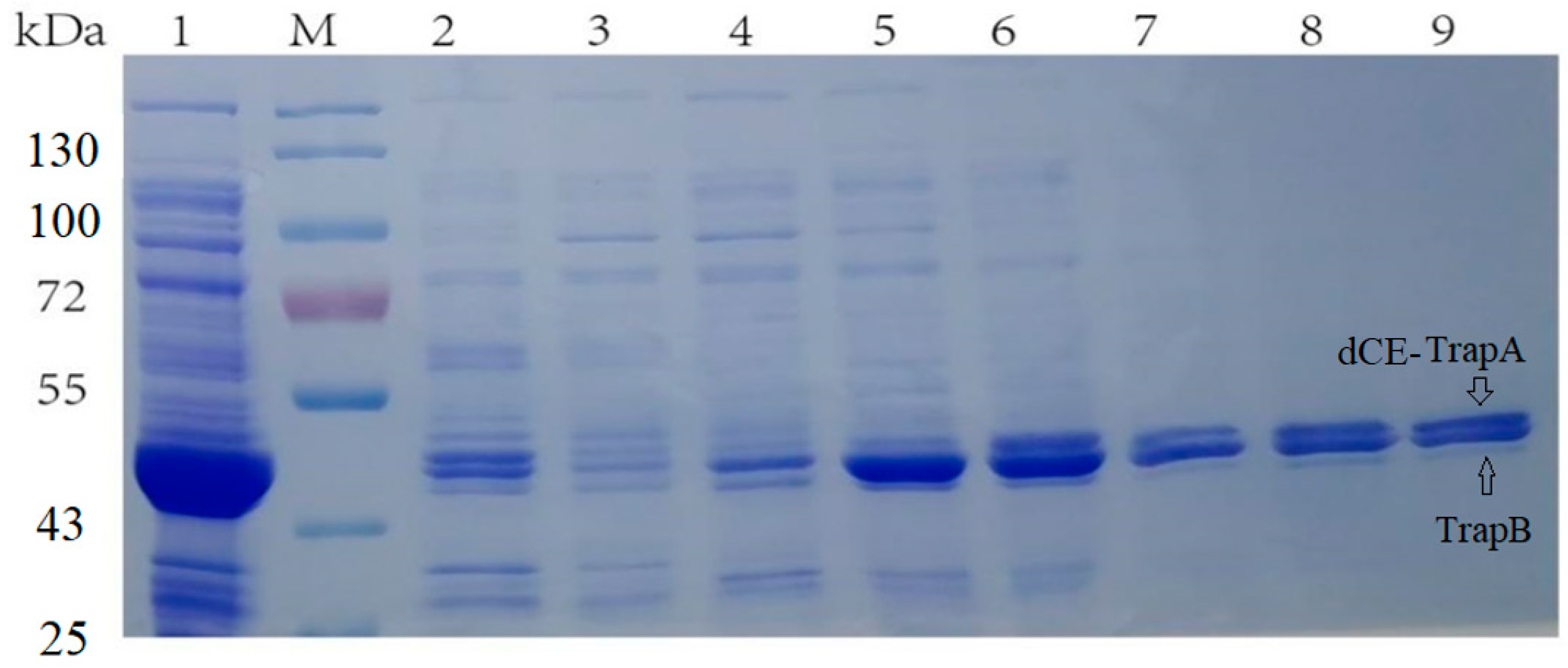
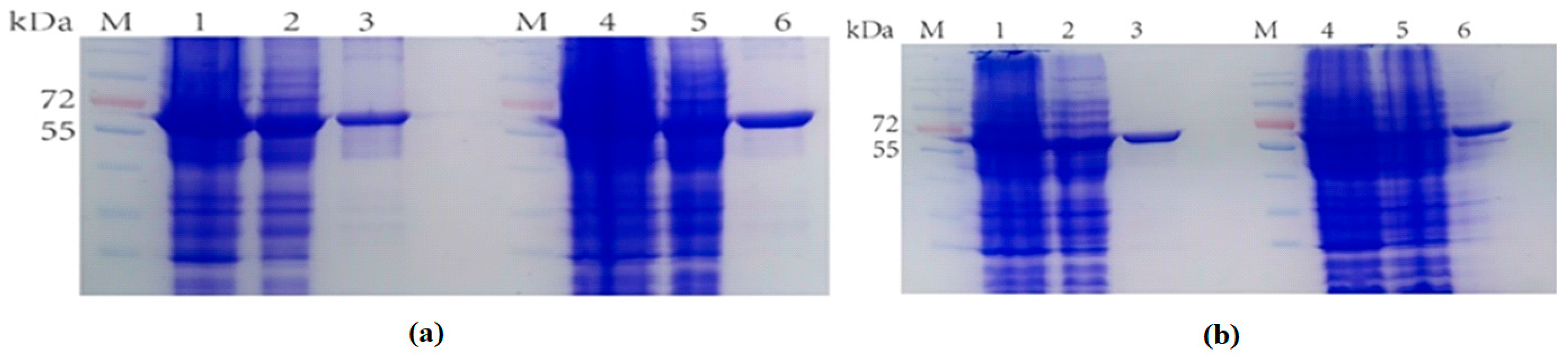
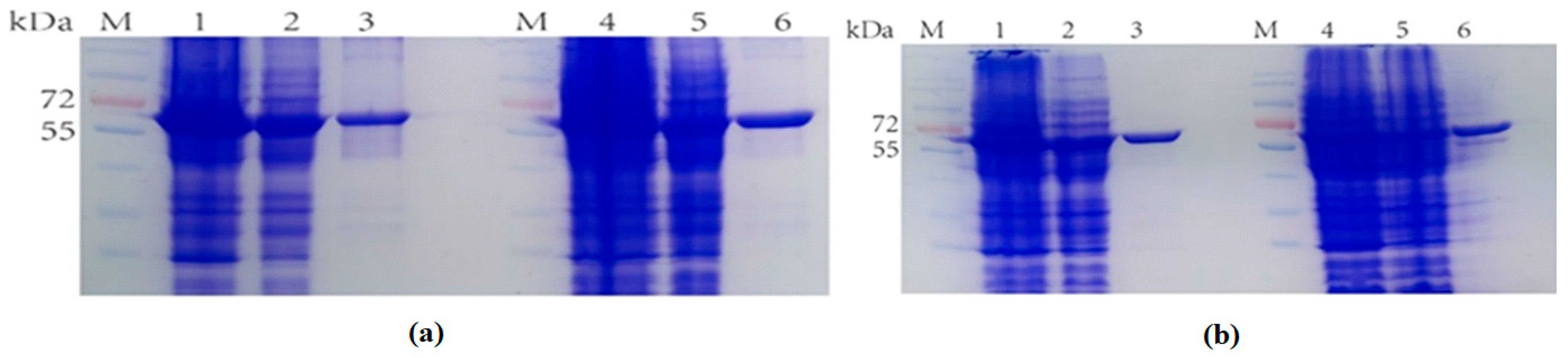
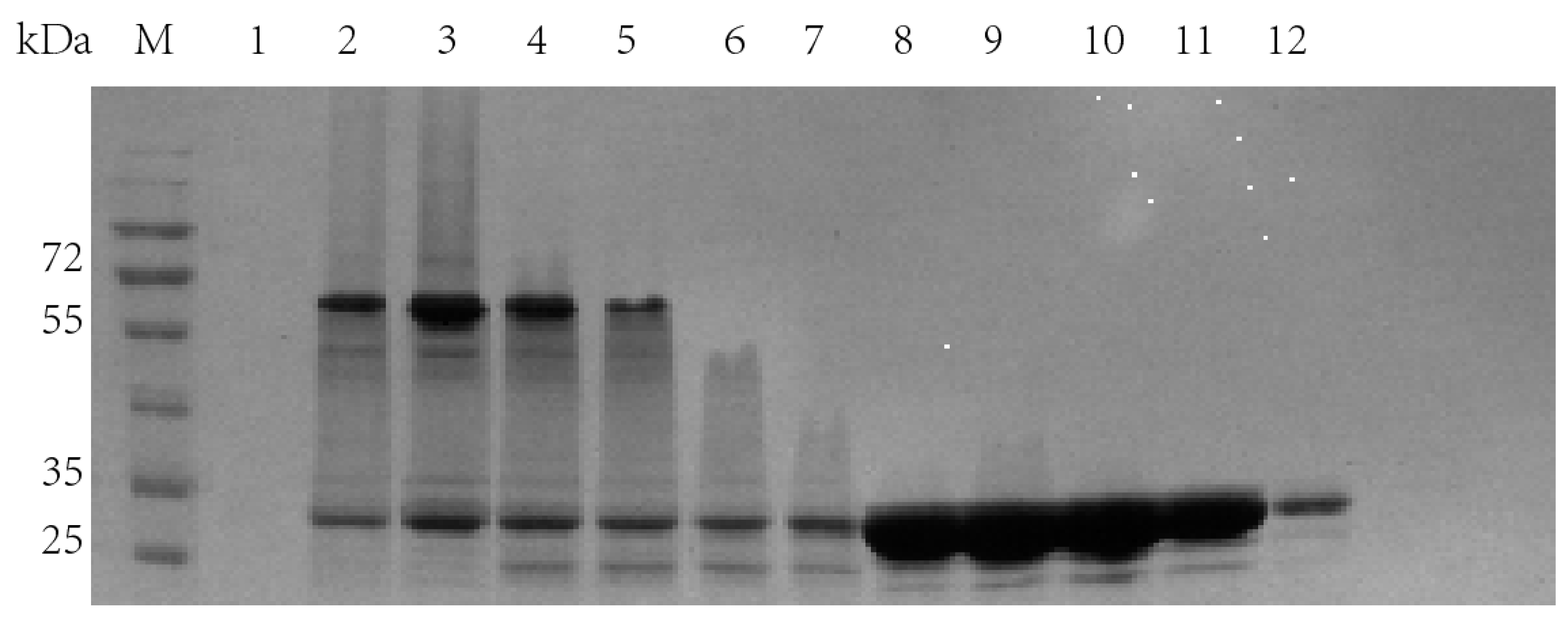
2.1. The Expression of Fusion Proteins Ectrp-dCEs
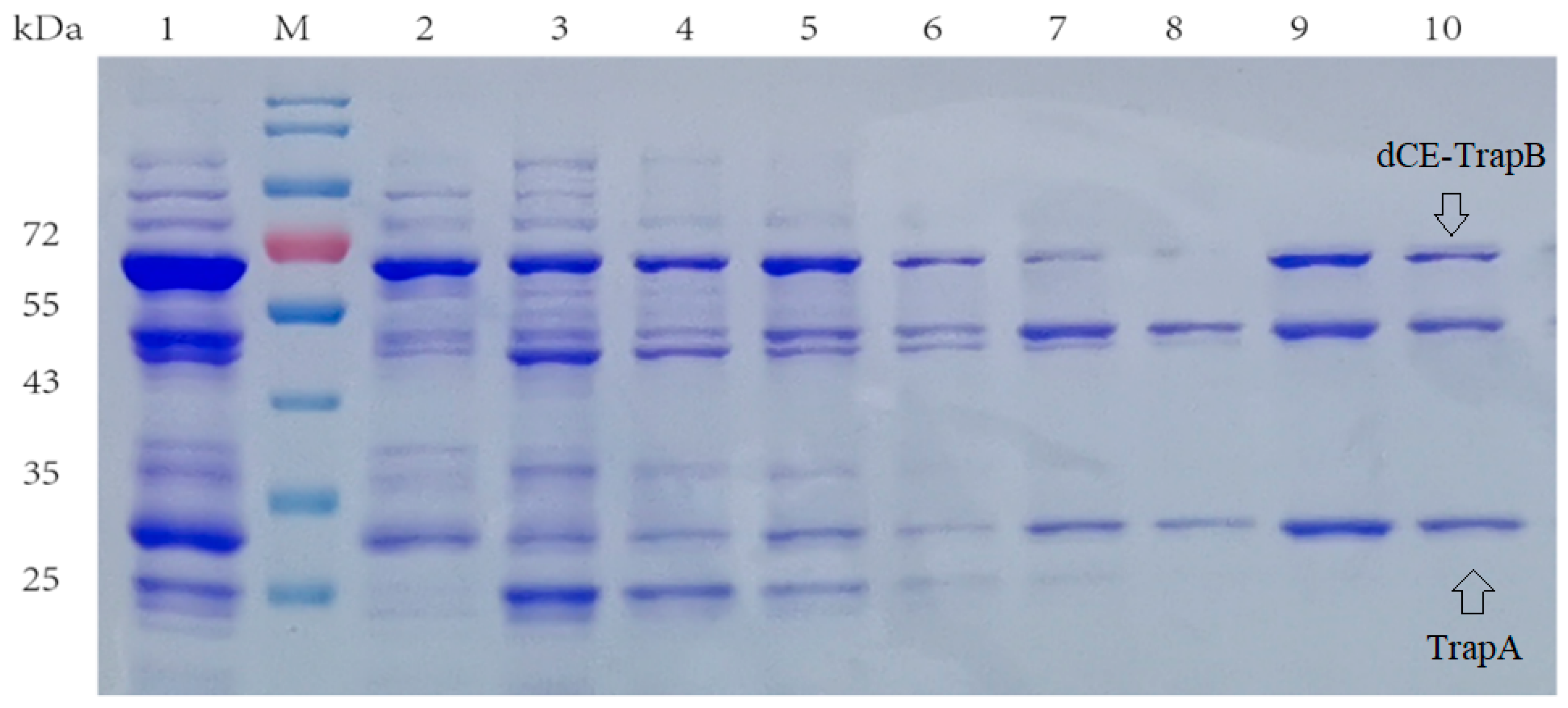
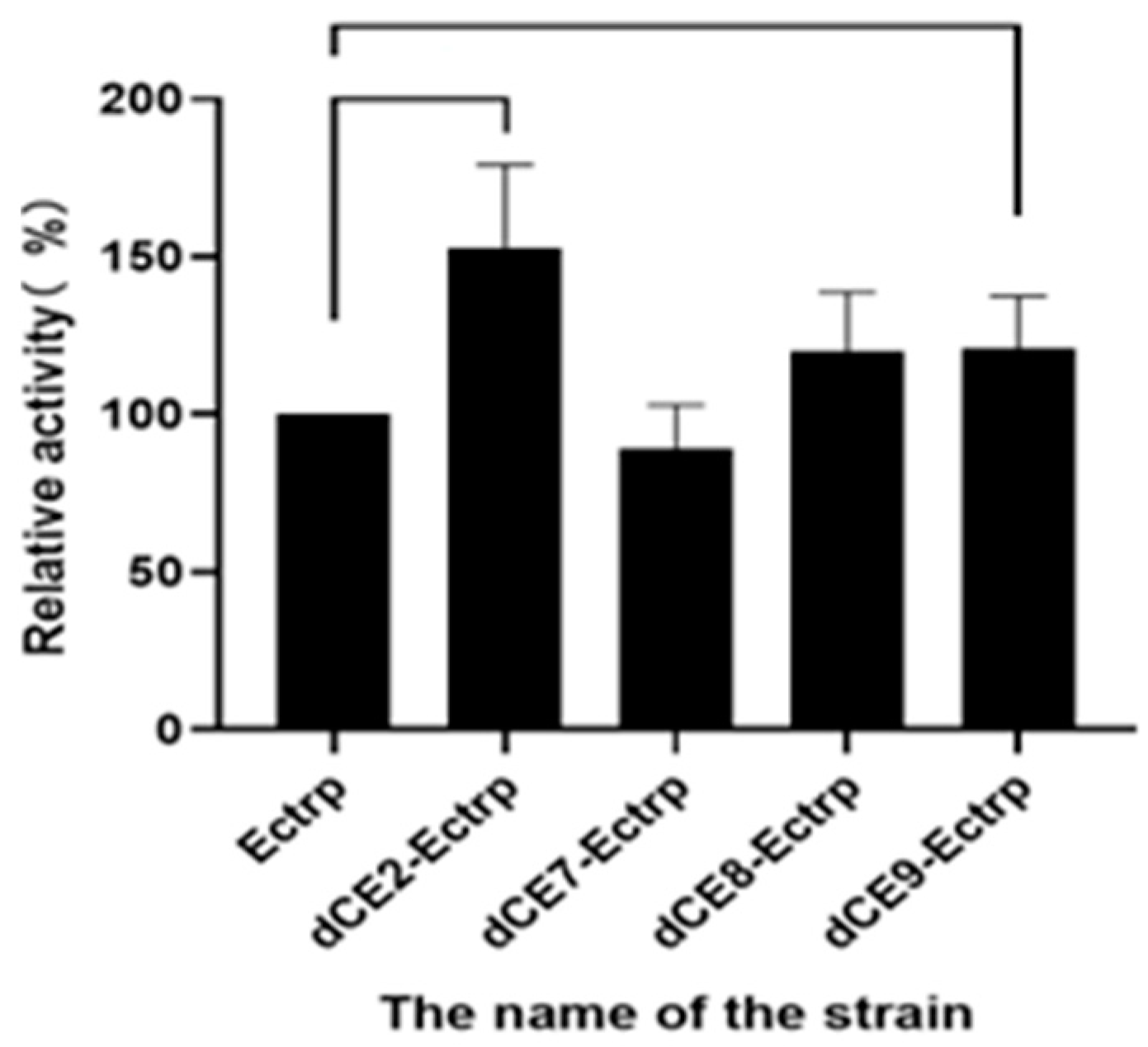
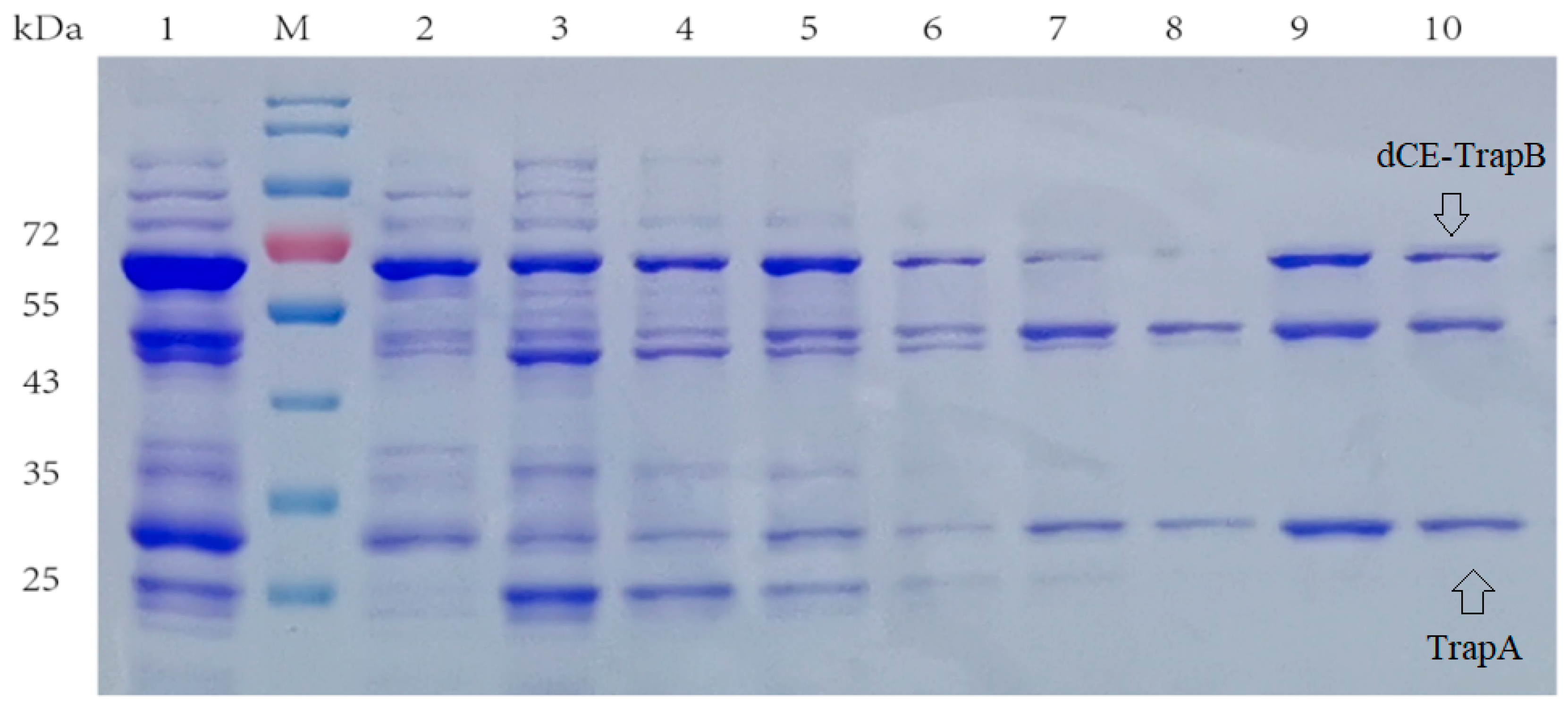
2.2. The Expression of Fusion Proteins dCEs-Ectrp
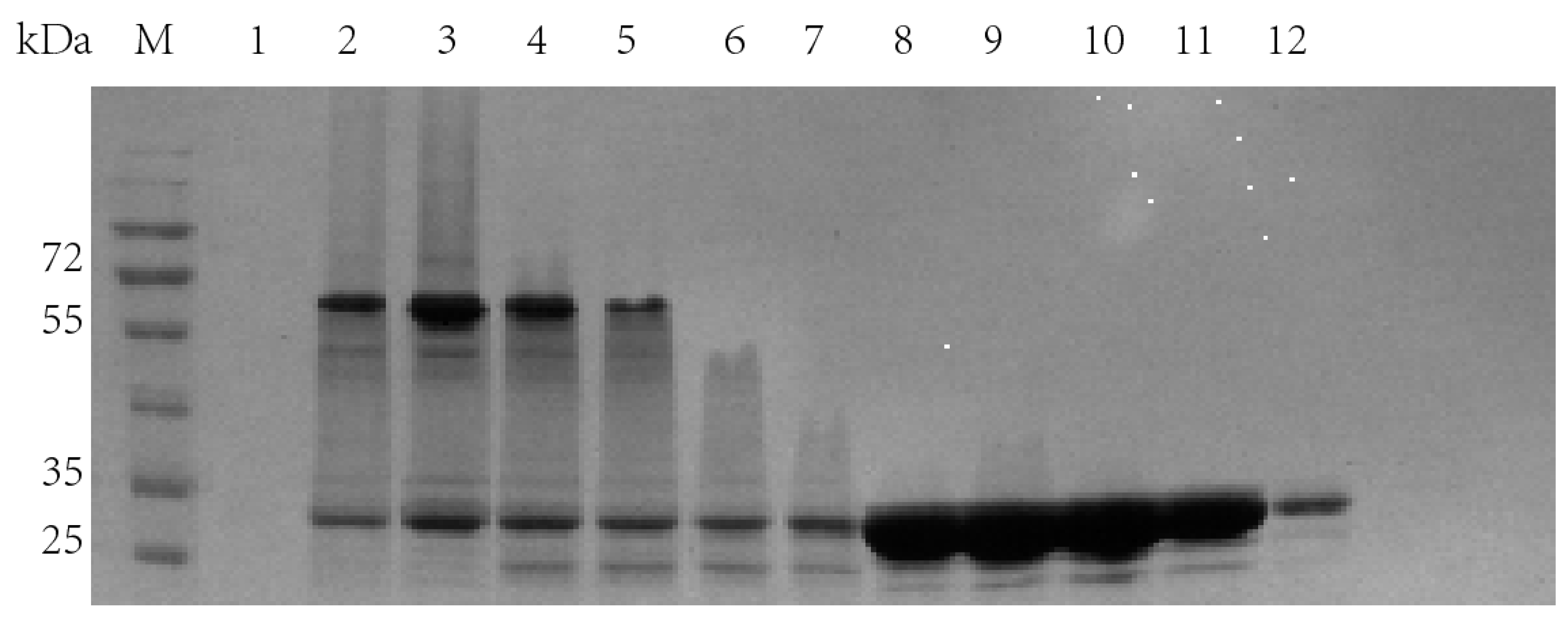
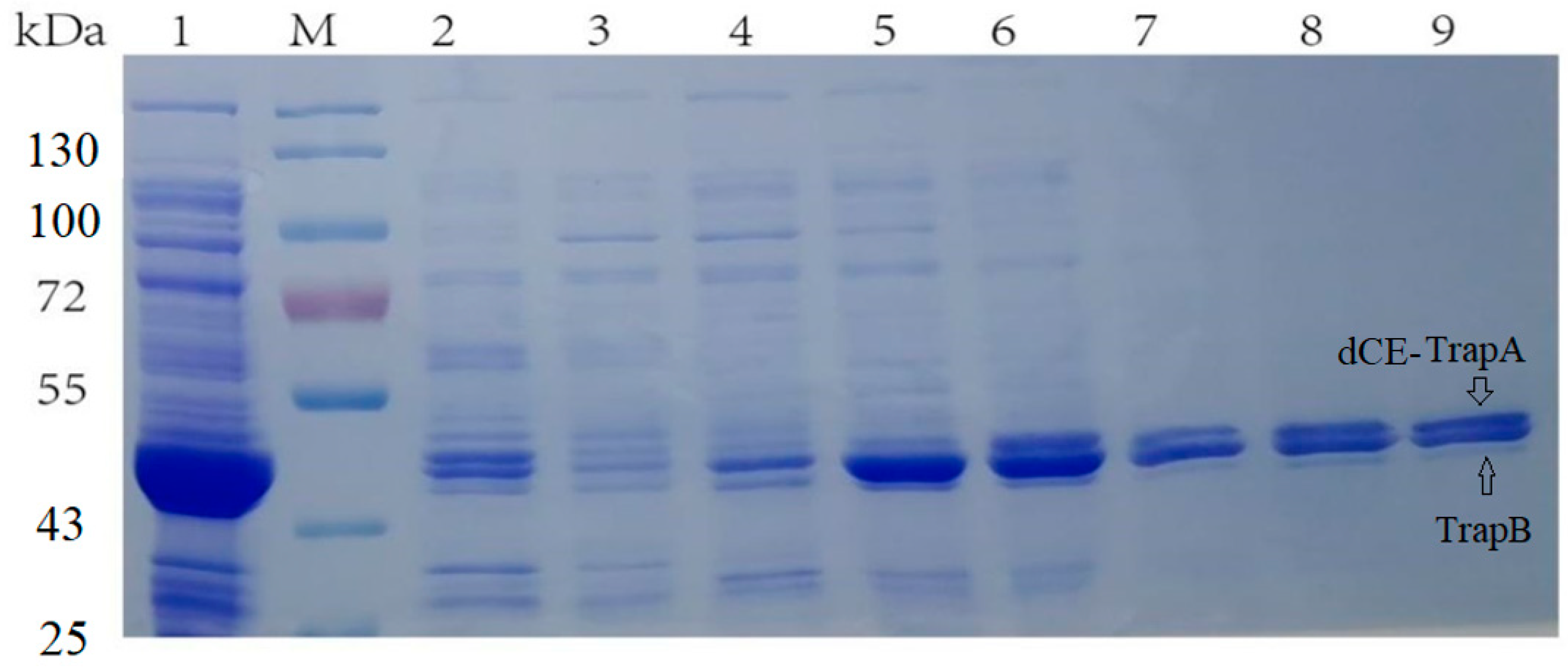
2.3. Separate Expression of TrapA and dCE-Labeled TrapB
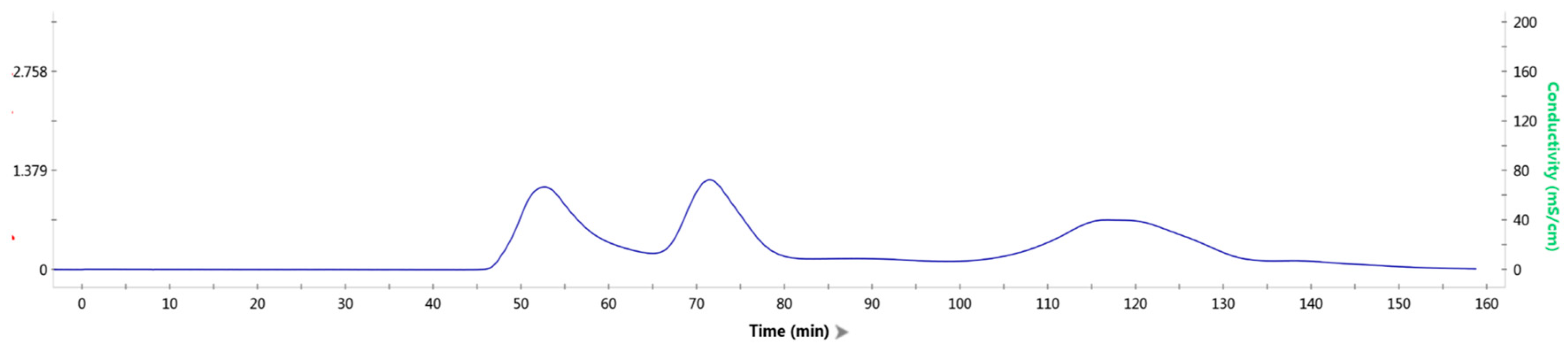
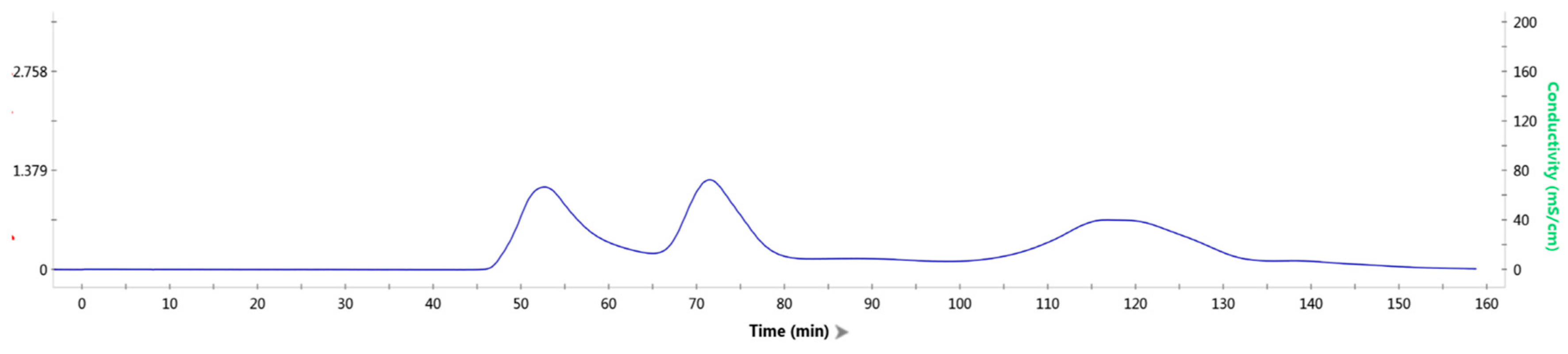
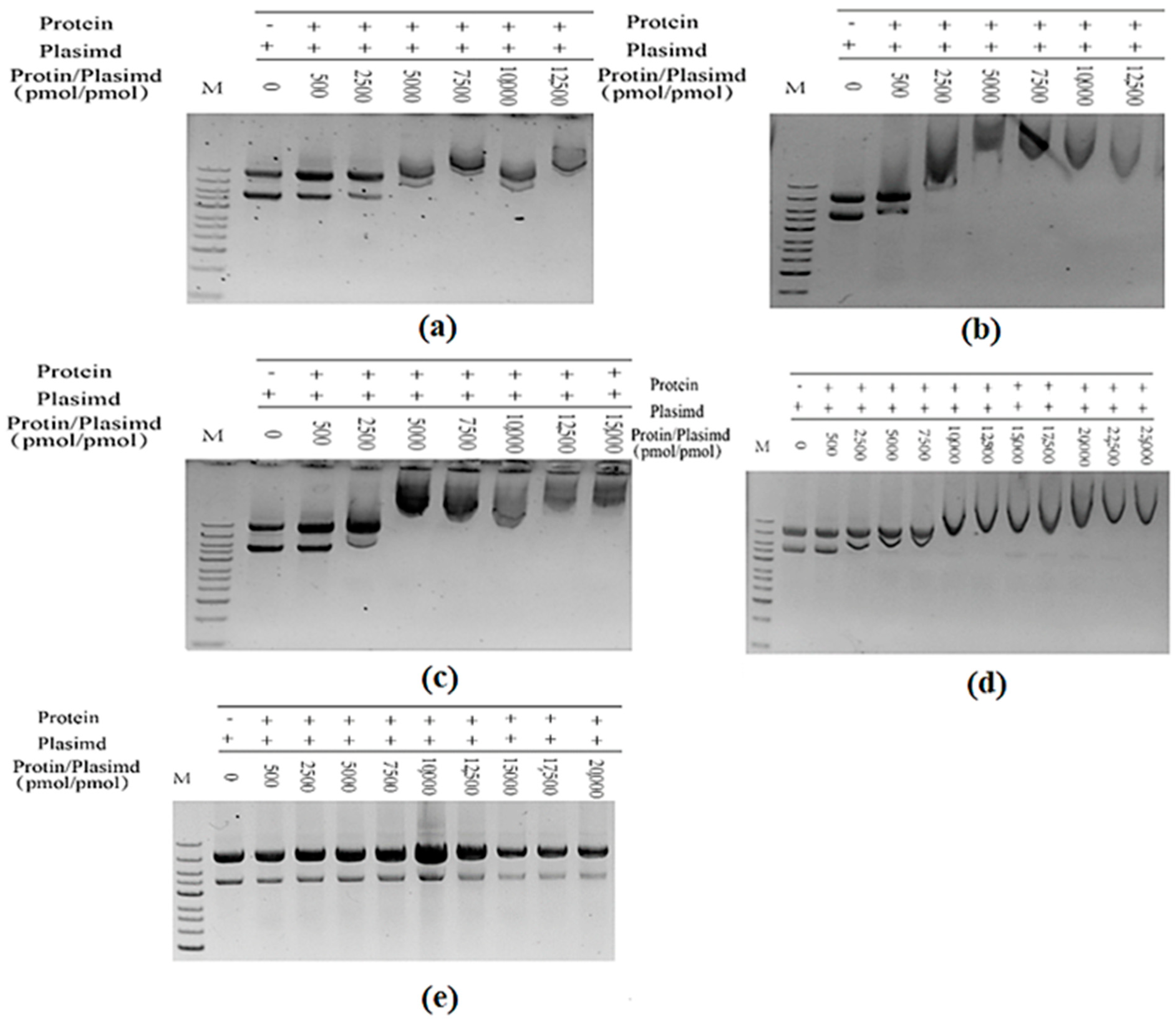
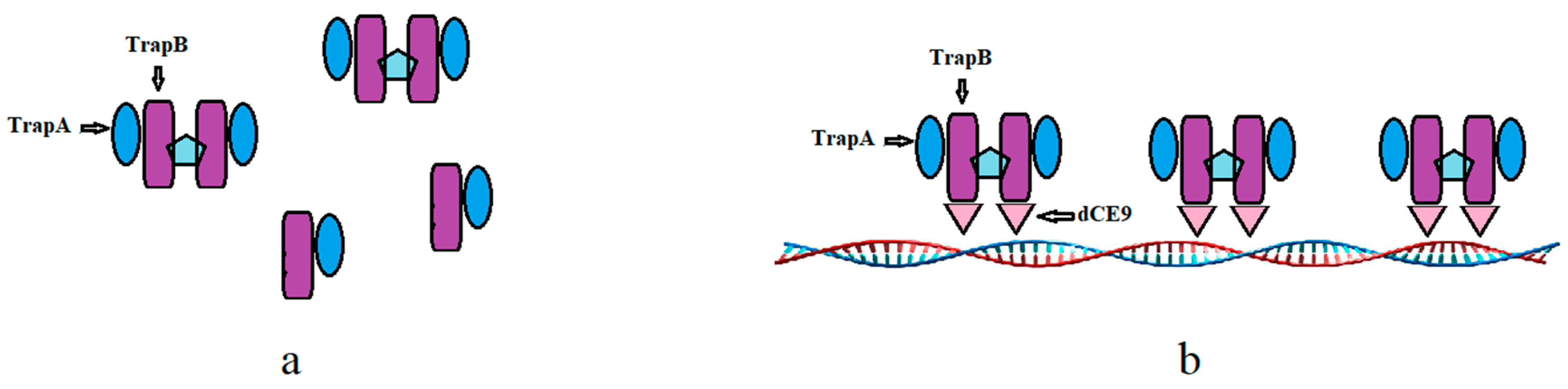
2.4. In Vitro Assembly of Tryptophan Synthetase
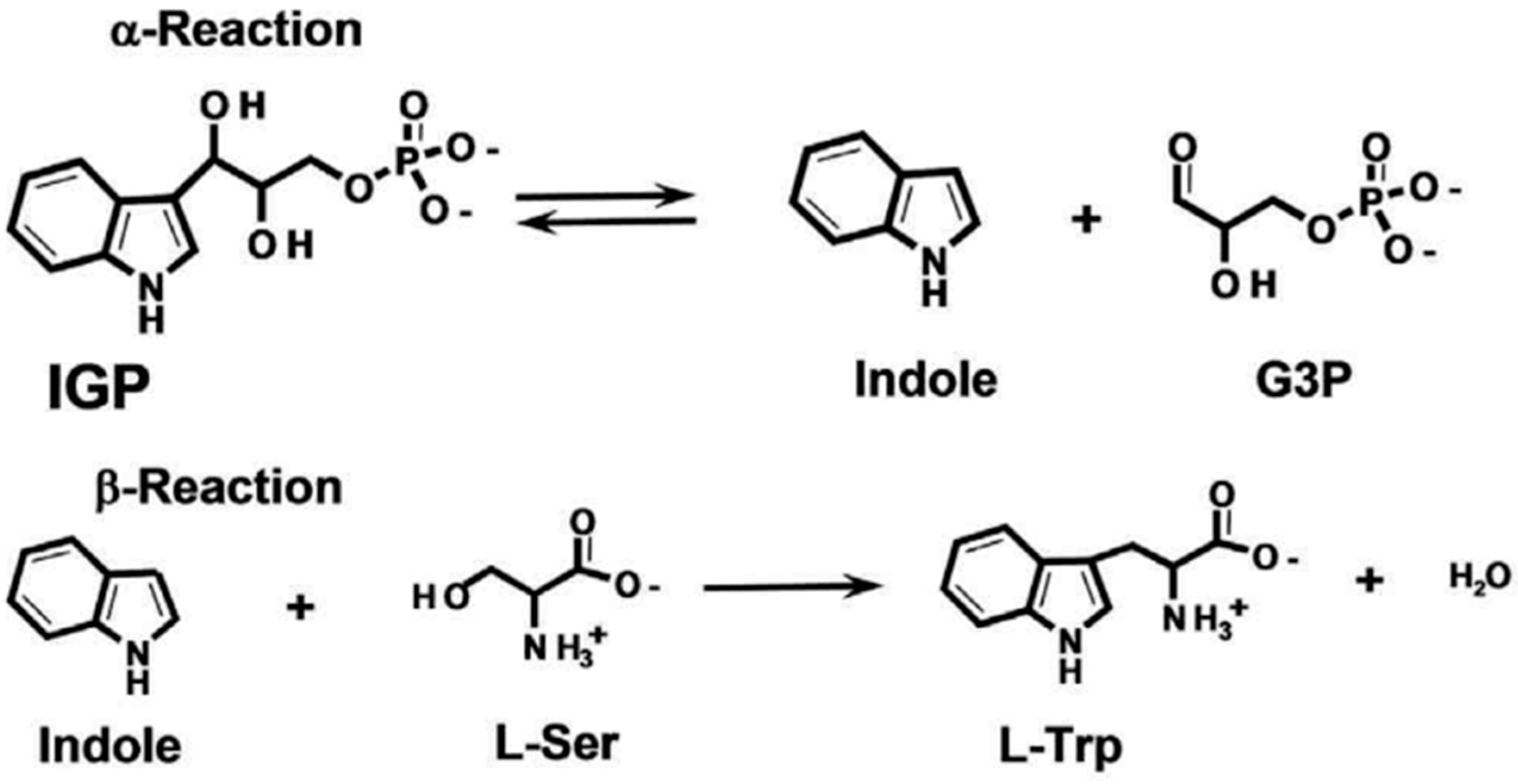
2.5. Reaction and Detection of Assembled-TSase
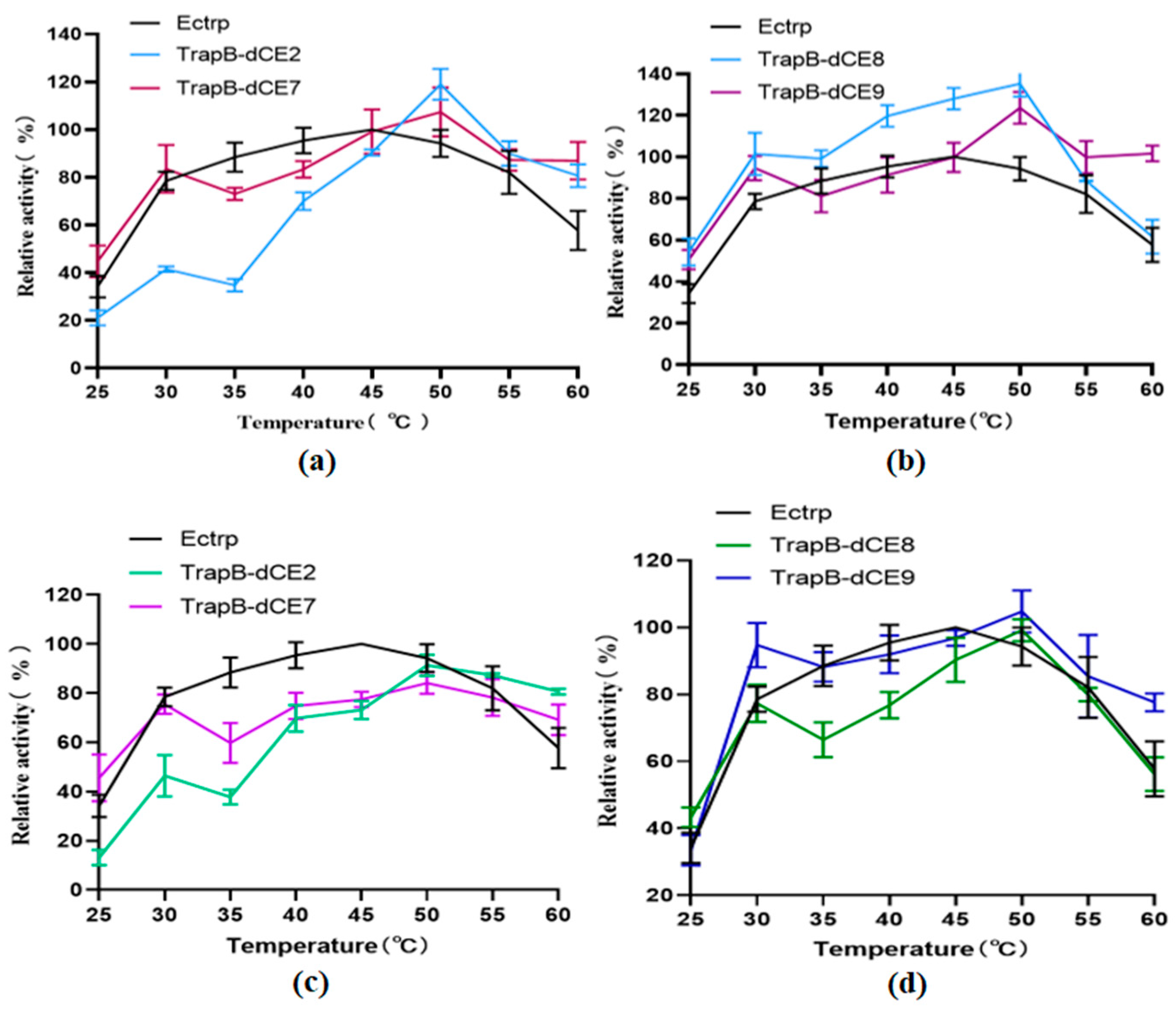
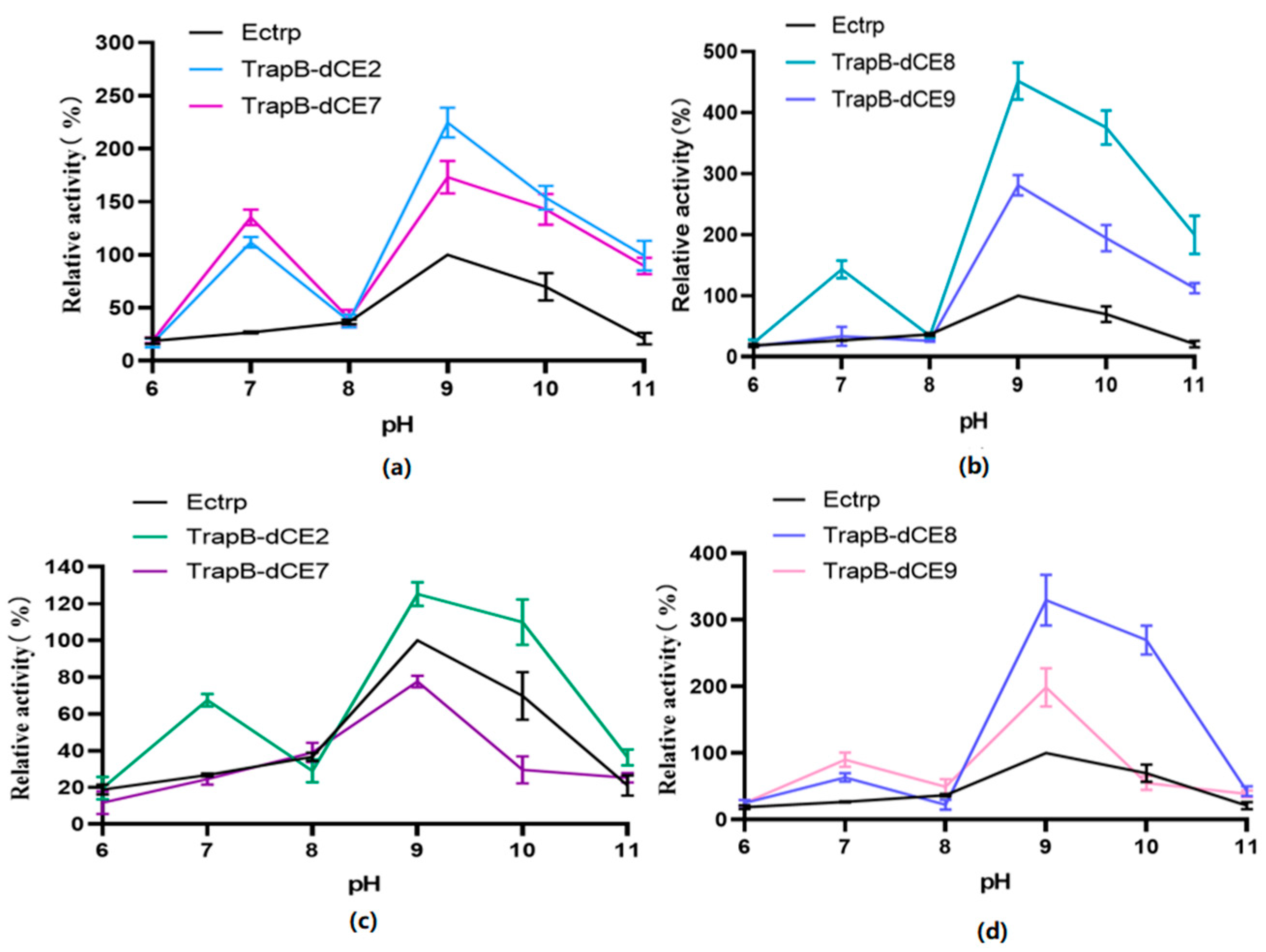
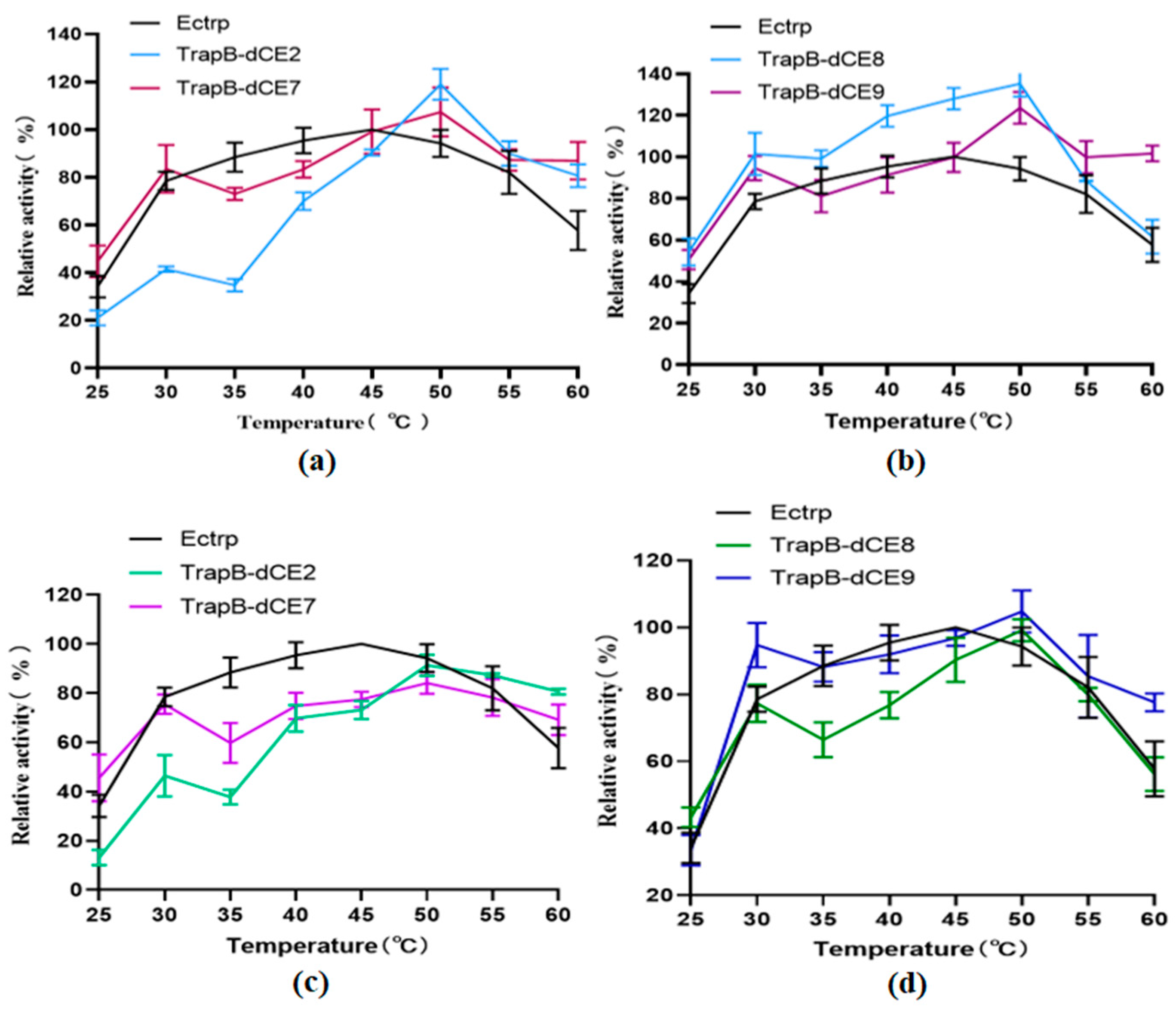
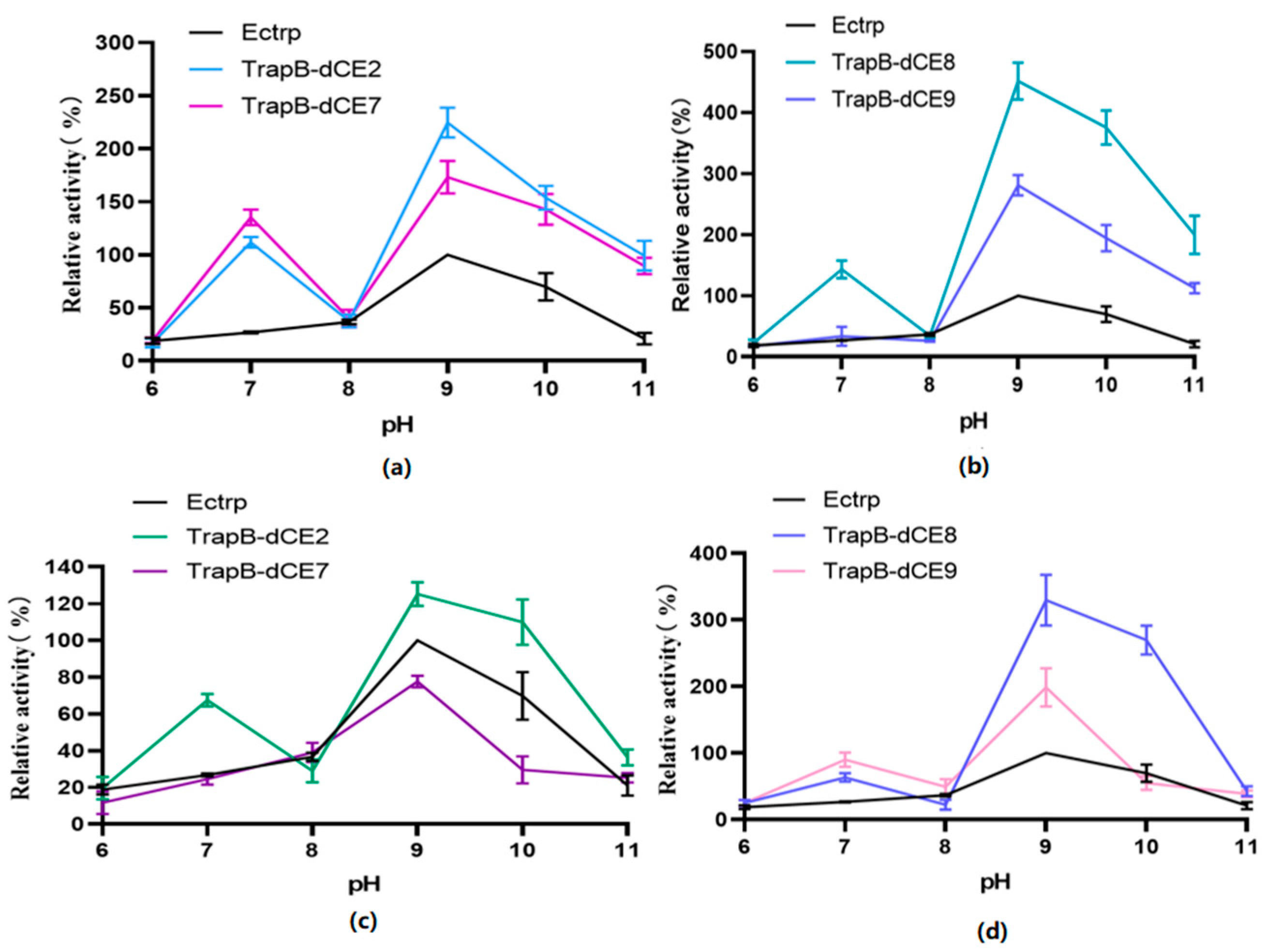
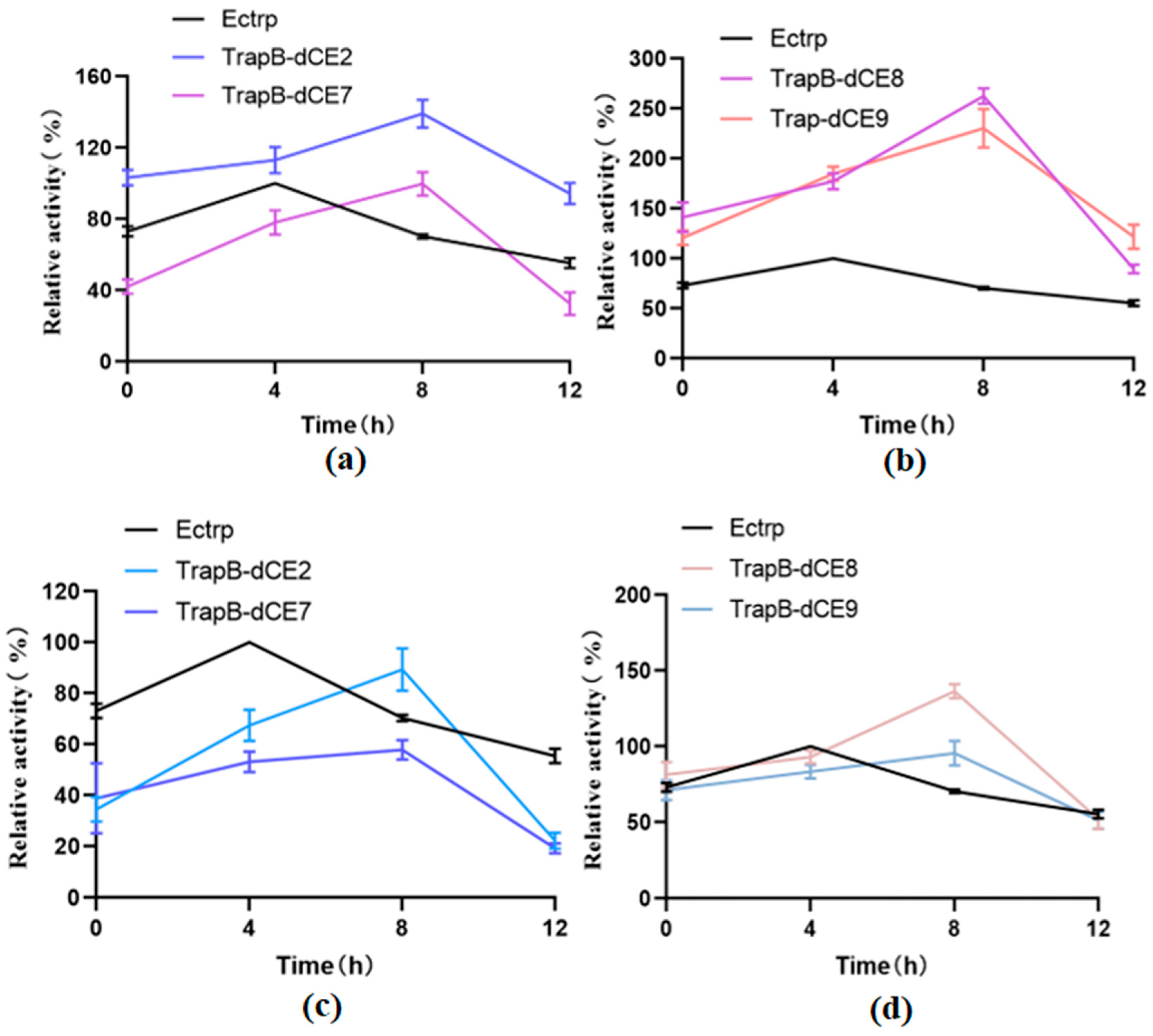
2.6. Characterization of Assembled Tryptophan Synthetase
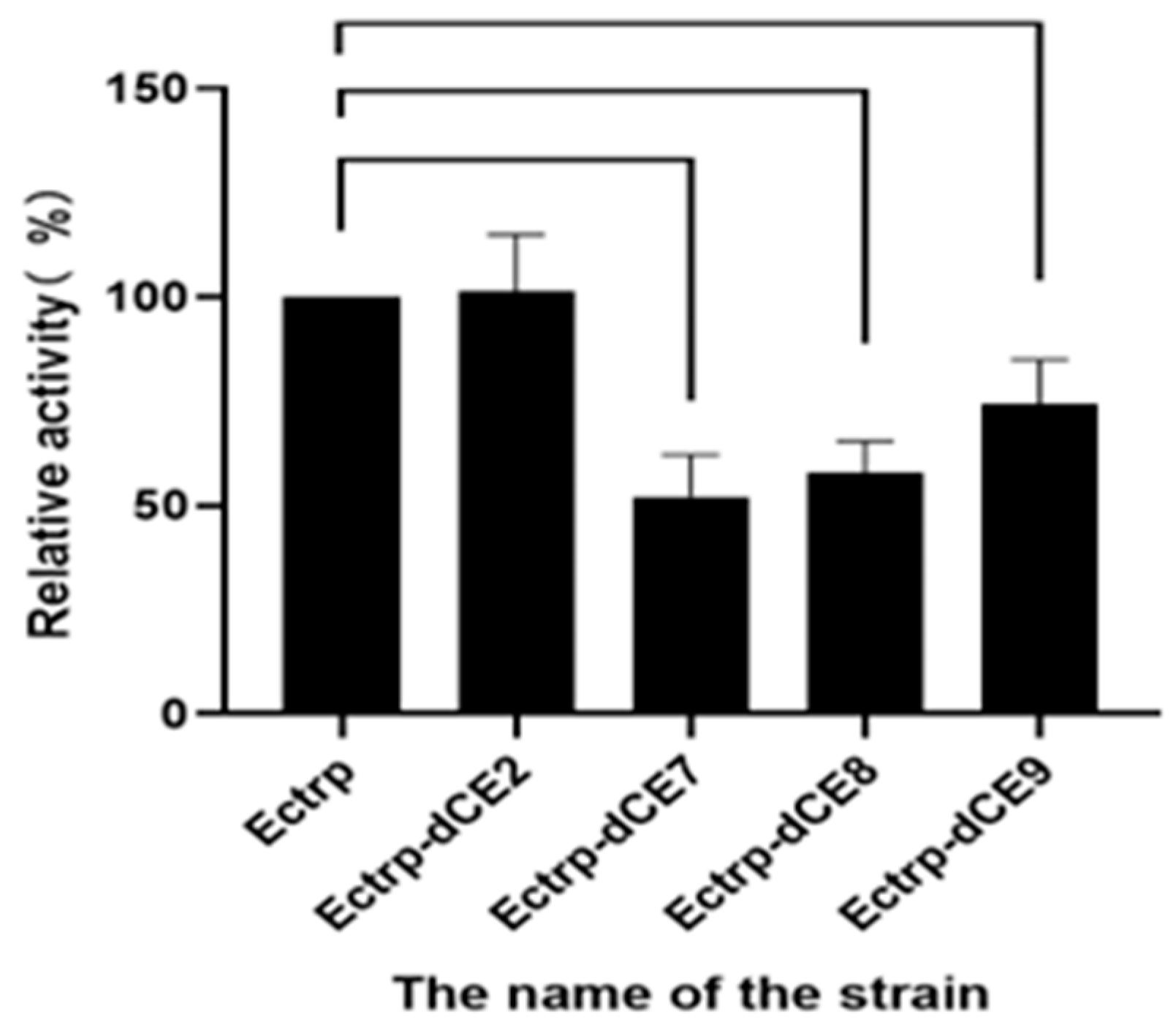
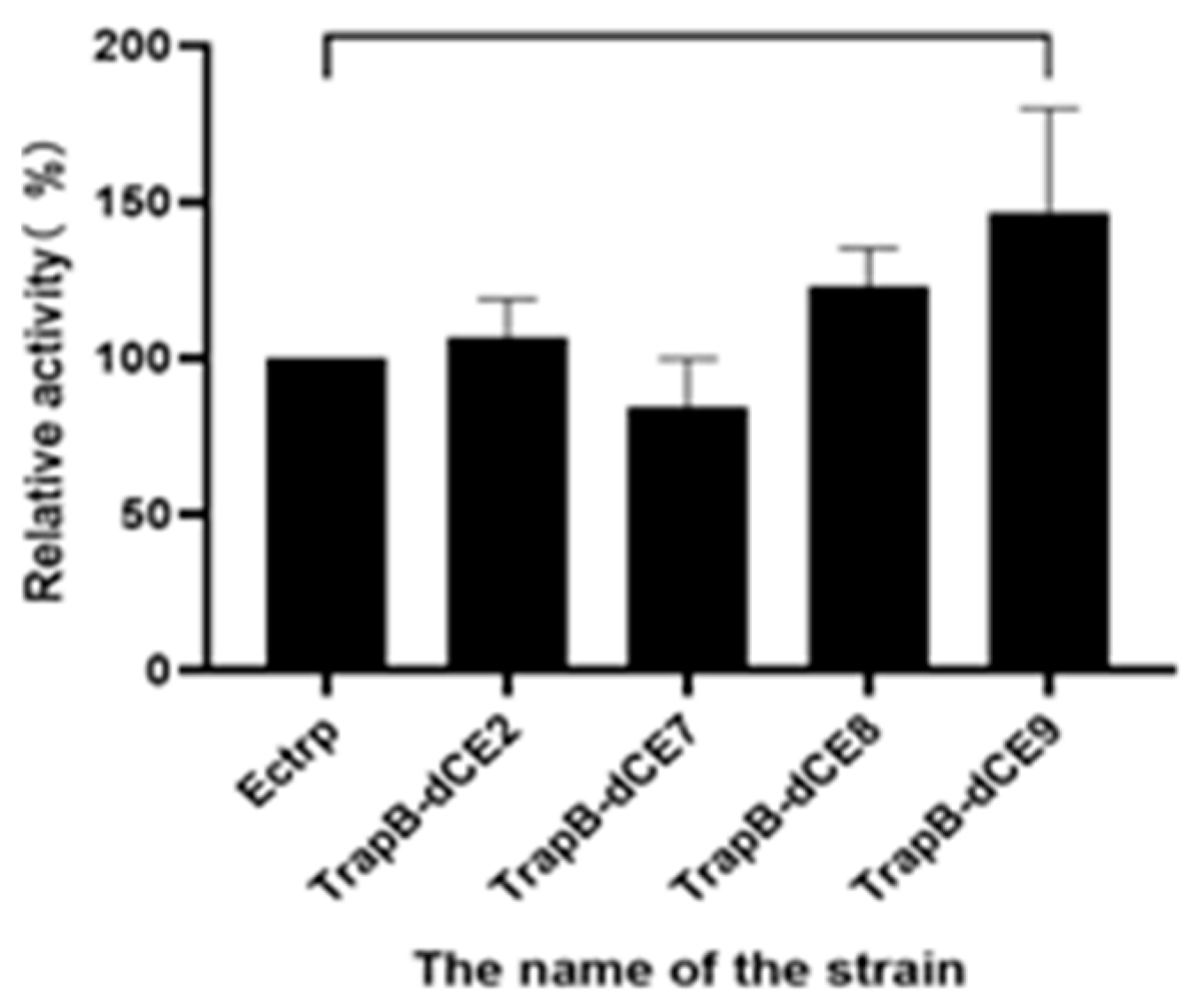
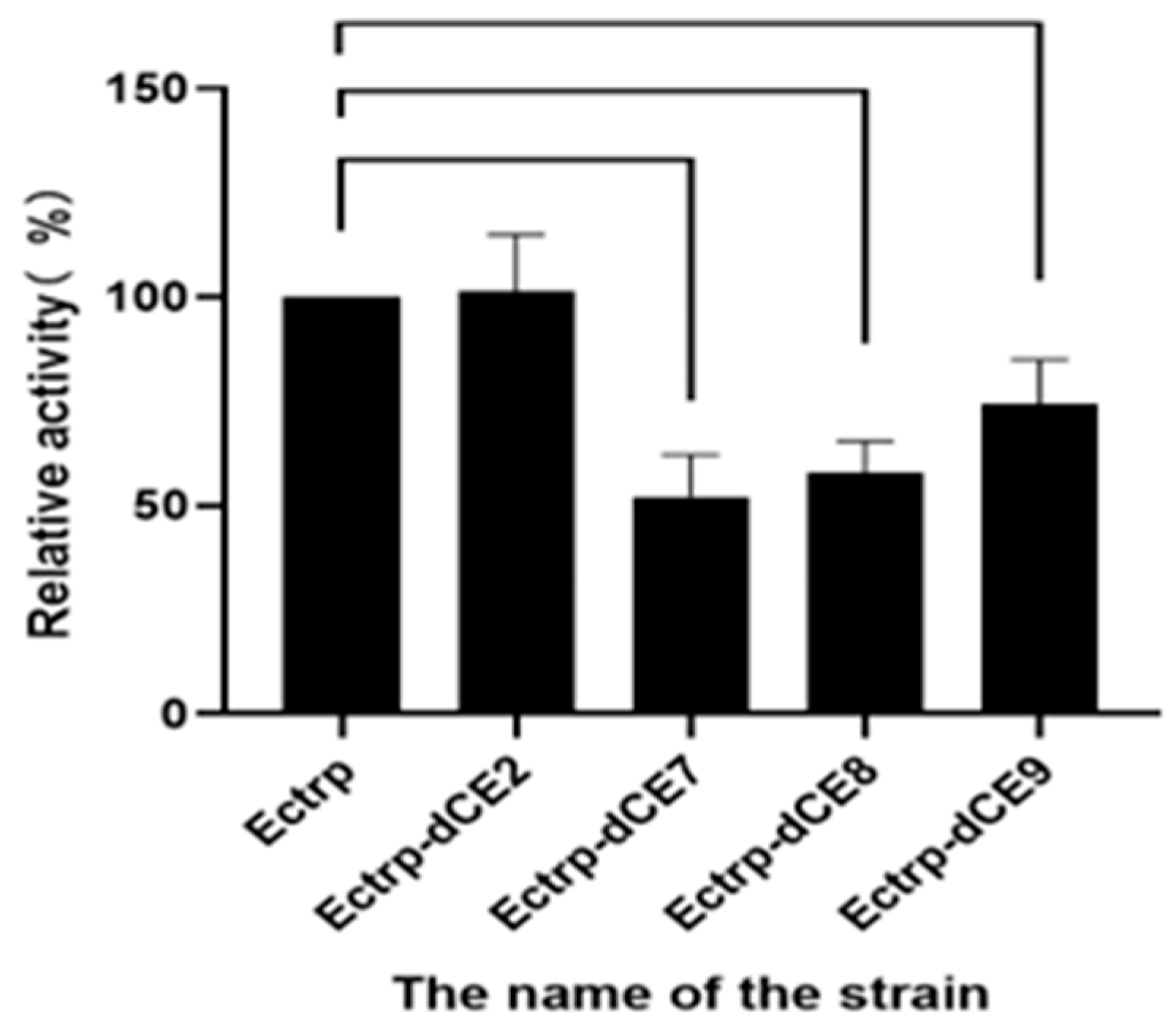
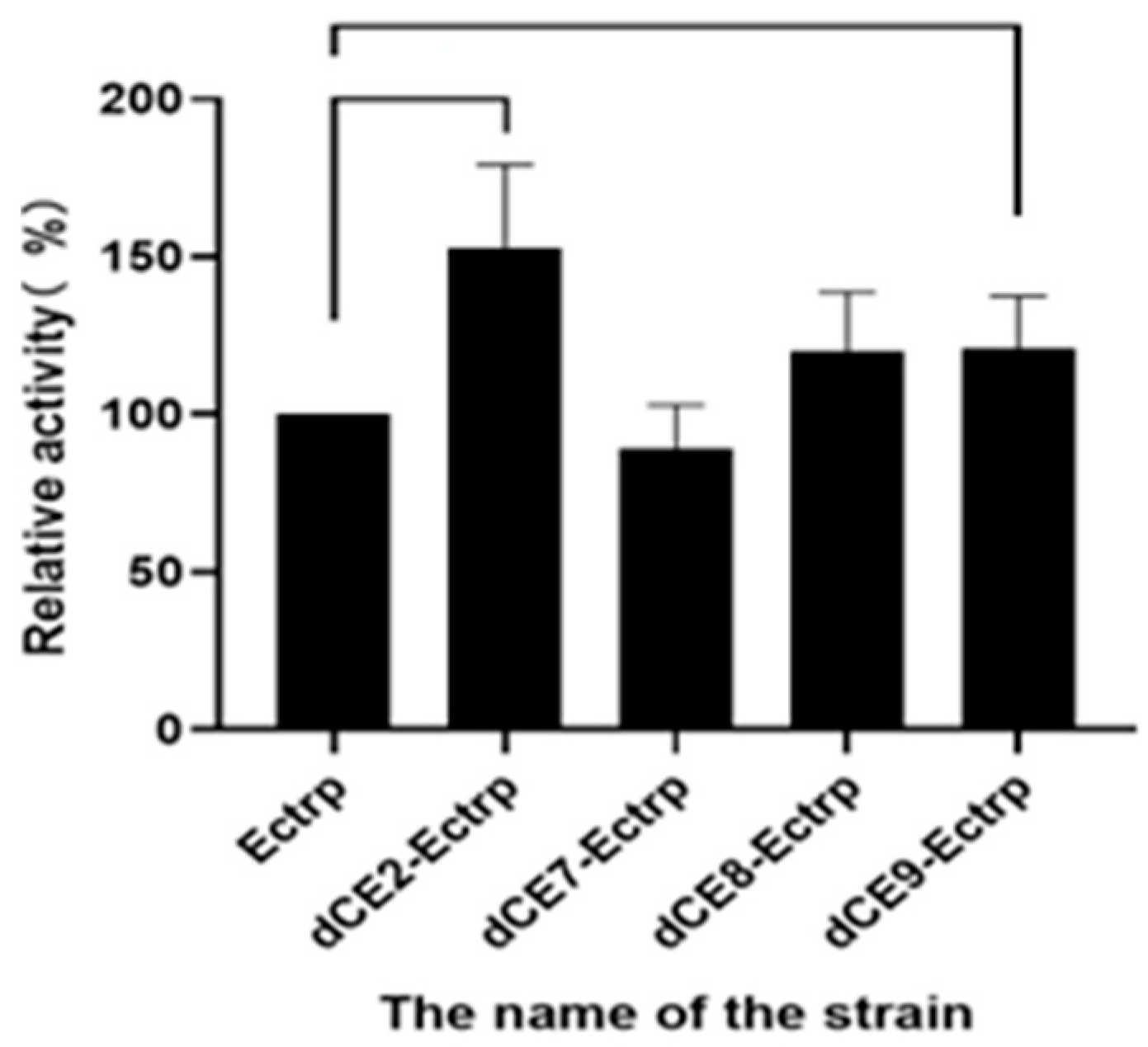
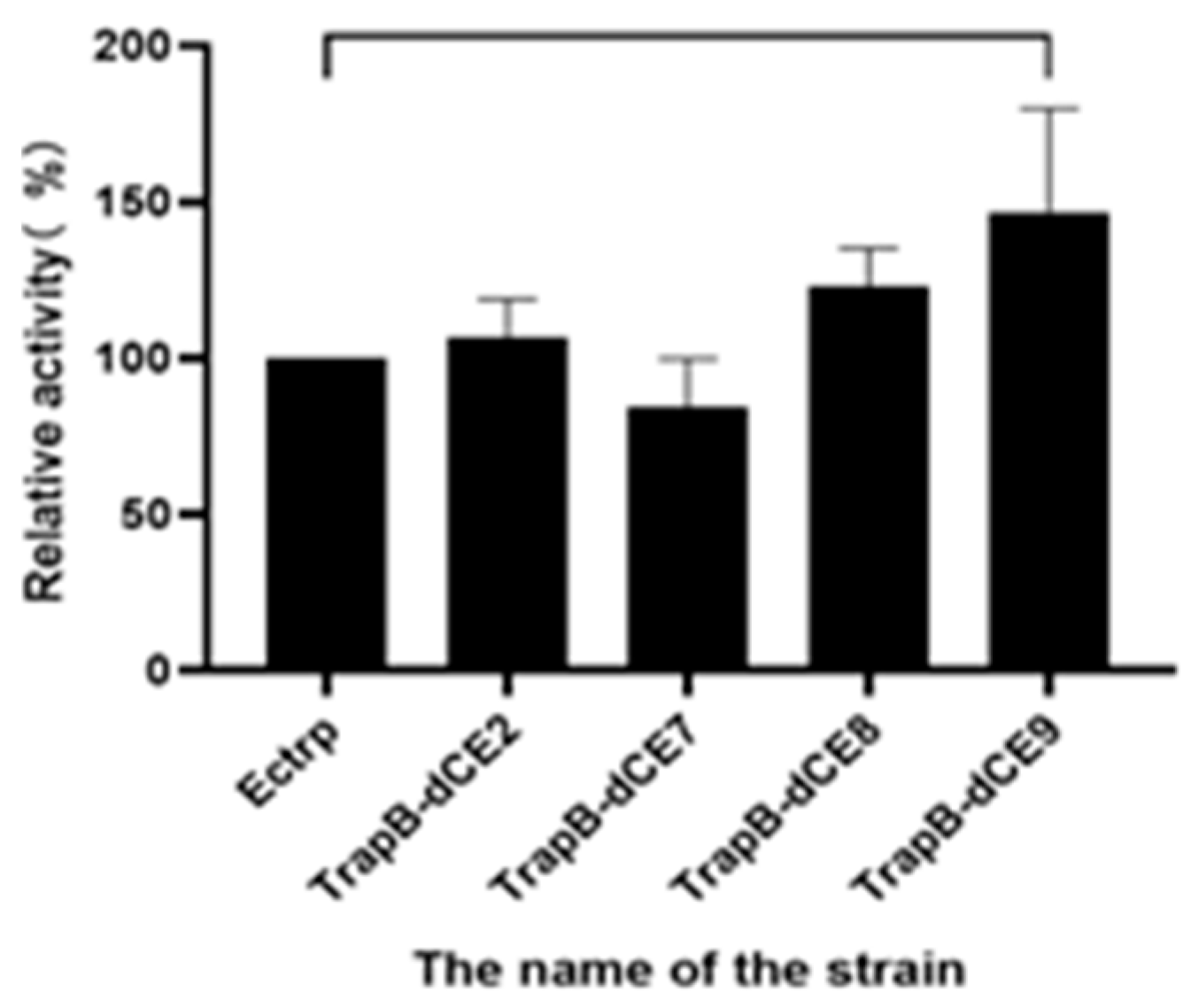
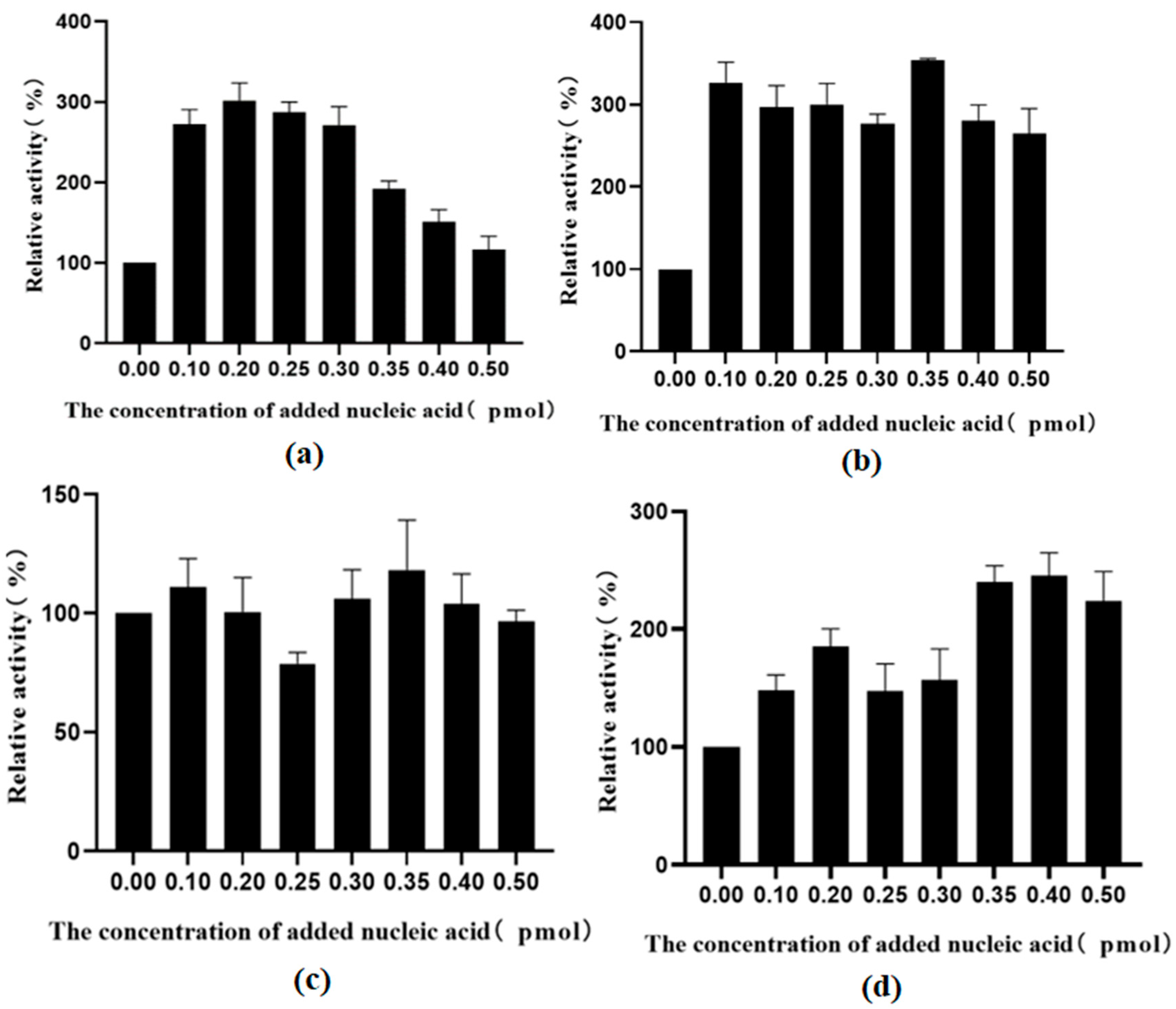
2.7. Using Exogenous Nucleic Acids to Improve the Enzyme Activity of Fusion Protein
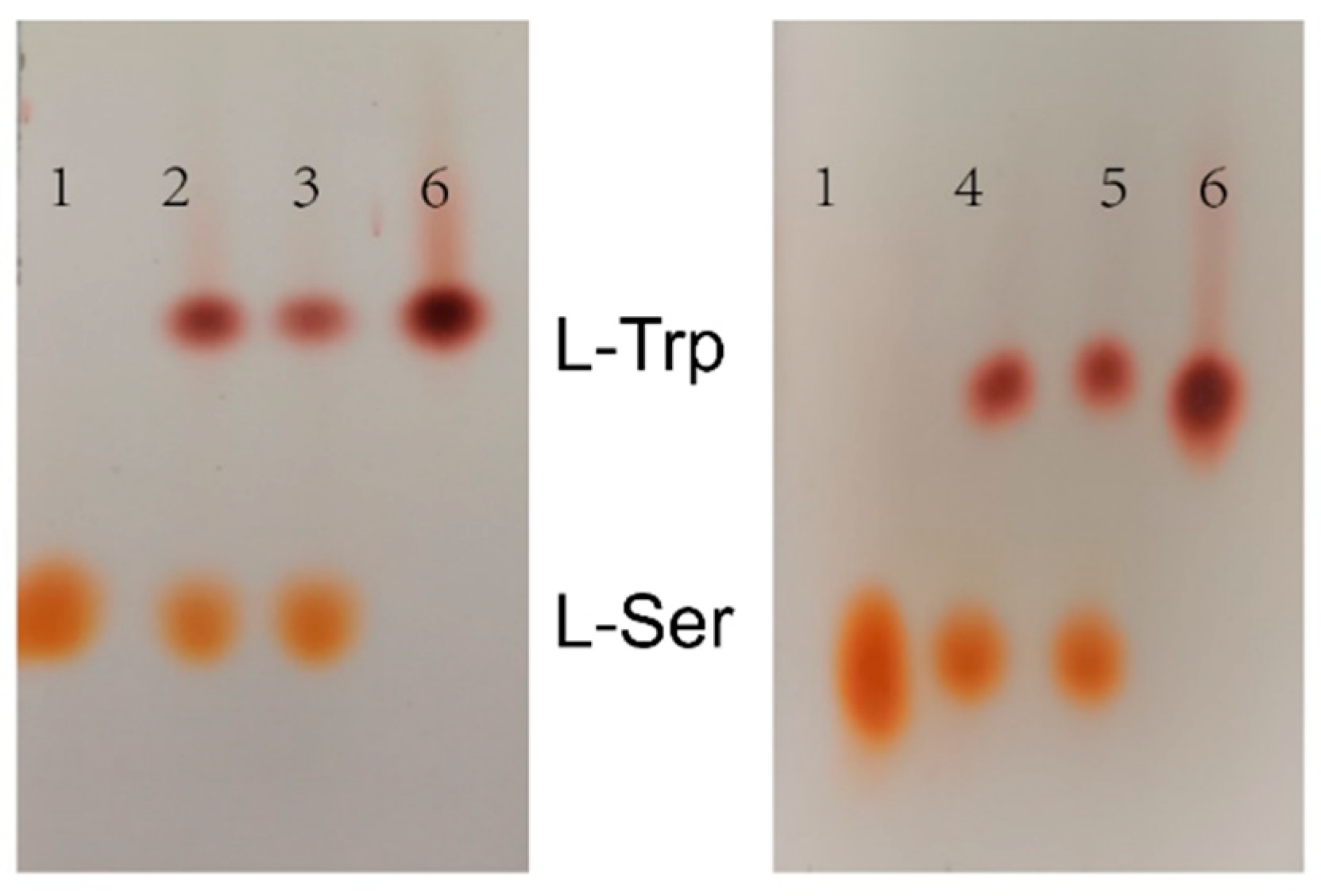
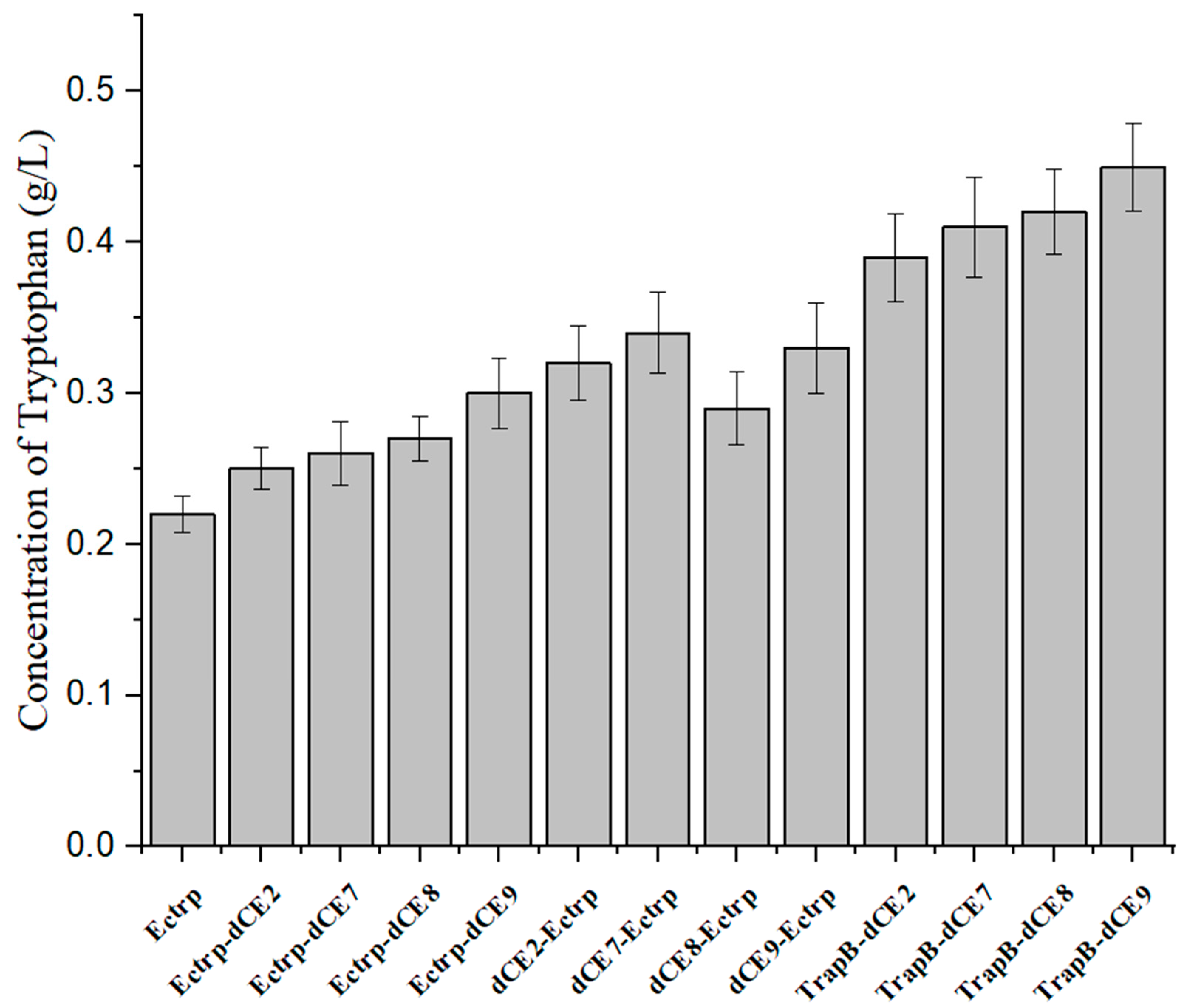
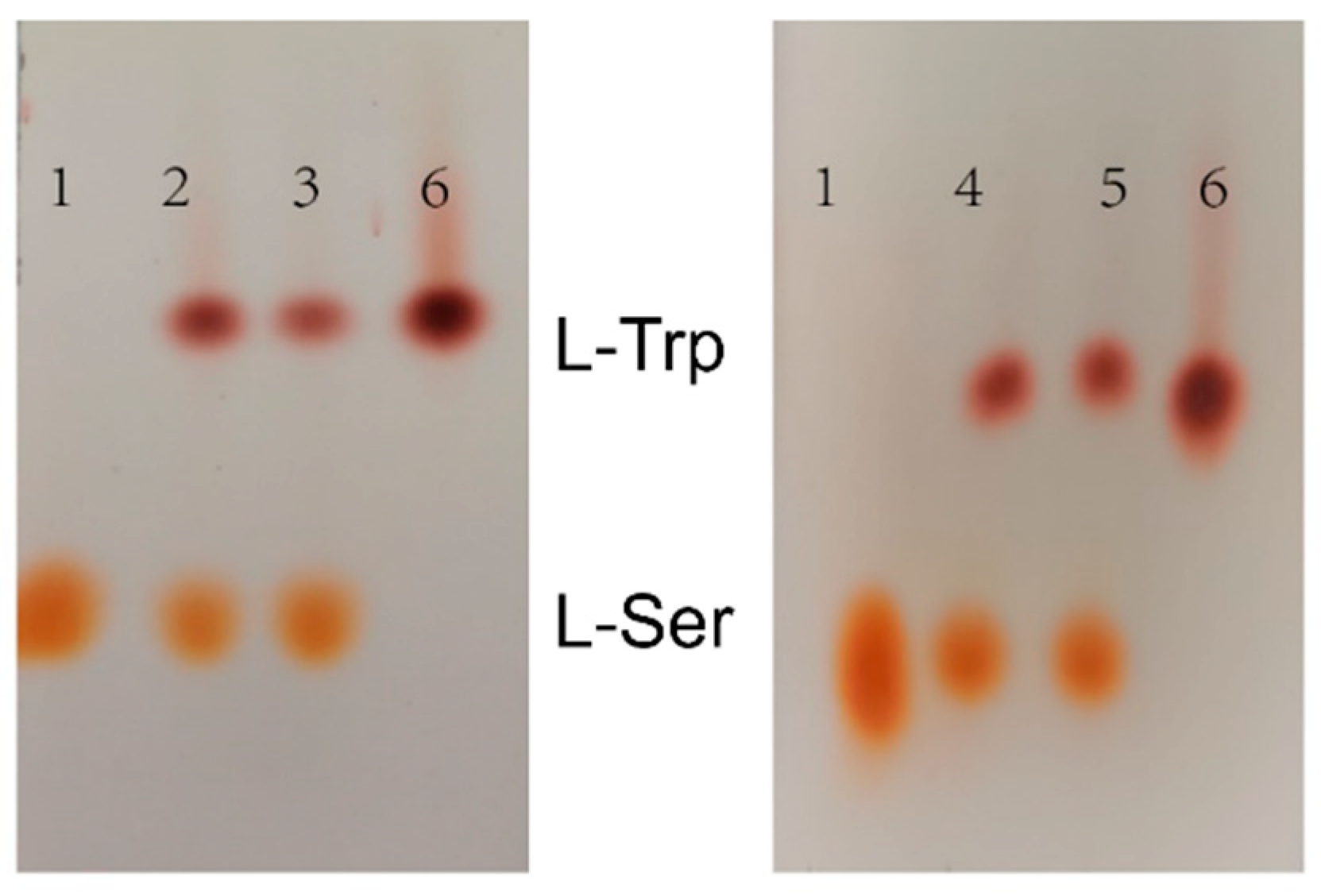
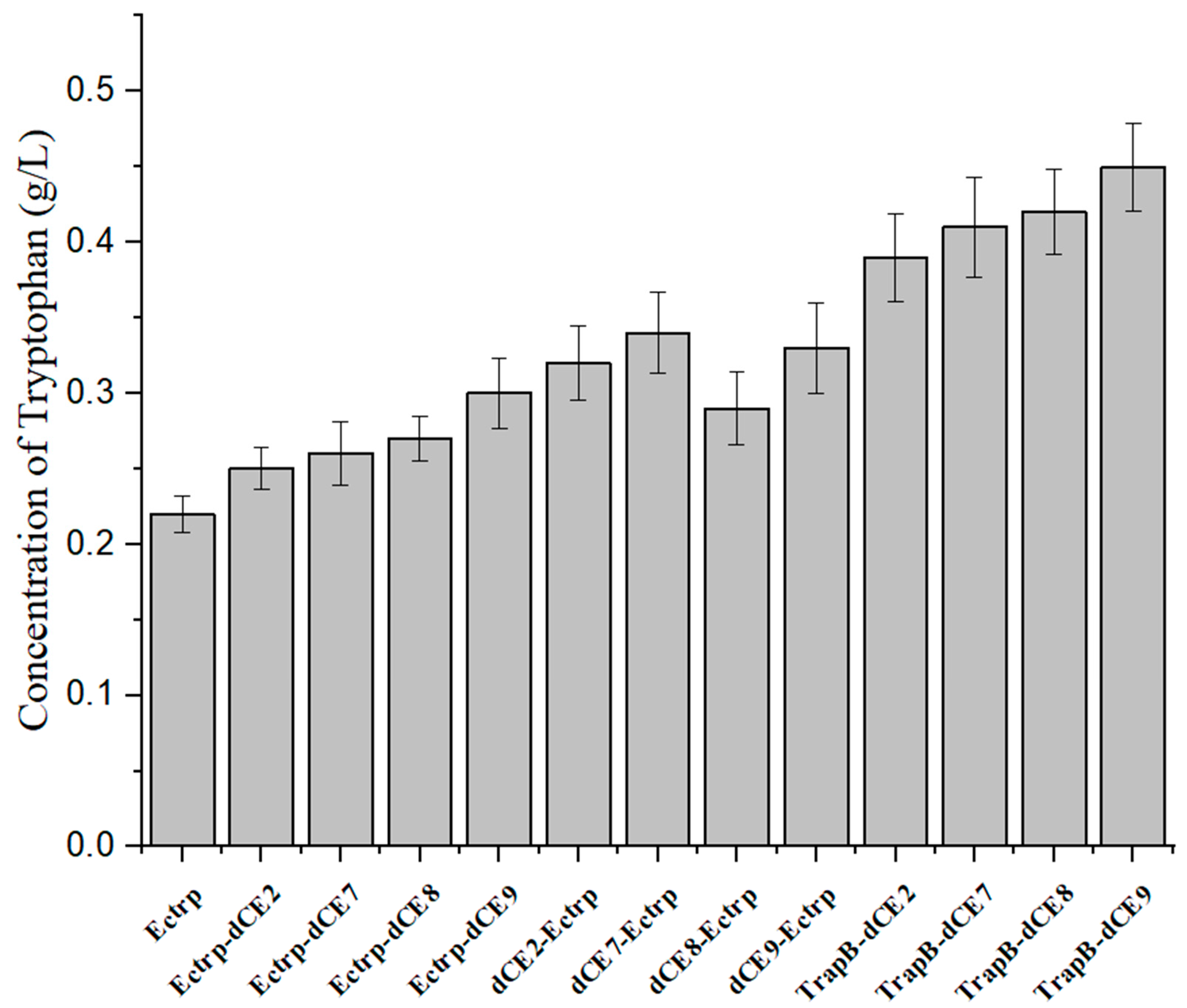
2.8. Synthesis of Tryptophan by Assembled Enzymes
3. Materials and Methods
3.1. Strains and Plasmids
3.2. Recombinant Plasmid Construction
3.3. Protein Expression and Purification
3.4. Enzymatic Characterization of Tryptophan Synthetase
3.5. Determination of L-Tryptophan Levels
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hyde, C.C.; Ahmed, S.A.; Padlan, E.A.; Miles, E.W.; Davies, D.R. Three-dimensional structure of the tryptophan synthase α2 β2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem. 1988, 263, 17857–17871. [Google Scholar] [CrossRef] [PubMed]
- Weyand, M.; Schlichting, I. Crystal Structure of Wild-Type Tryptophan Synthase Complexed with the Natural Substrate Indole-3-glycerol Phosphate. Biochemistry 1999, 38, 16469–16480. [Google Scholar] [CrossRef] [PubMed]
- Miles, E.W. Tryptophan synthase: A multienzyme complex with an intramolecular tunnel. Chem. Rec. 2001, 1, 140–151. [Google Scholar] [PubMed]
- Buller, A.R.; Van Roye, P.; Cahn, J.K.; Scheele, R.A.; Herger, M.; Arnold, F.H. Directed evolution mimics allosteric activation by step-wise tuning of the conformational ensemble. J. Am. Chem. Soc. 2018, 140, 7256–7266. [Google Scholar] [CrossRef]
- Banik, U.; Ahmed, S.A.; McPhie, P.; Miles, E.W. Subunit assembly in the tryptophan synthase α2/β2 complex, stabilization by pyridoxal phosphate aldimine intermediates. J. Biol. Chem. 1995, 270, 7944–7949. [Google Scholar] [CrossRef]
- Huang, Y.M.; You, W.; Caulkins, B.G.; Dunn, M.F.; Mueller, L.J.; Chang, C.A. Protonation states and catalysis: Molecular dynamics studies of intermediates in tryptophan synthase. Protein Sci. 2016, 25, 166–183. [Google Scholar] [CrossRef]
- Henning, U.; Yanofsky, C. An electrophoretic study of mutationally altered a proteins of the tryptophan synthetase of Escherichia coli. J. Mol. Biol. 1963, 6, 16-IN1. [Google Scholar] [CrossRef]
- Dunn, M.F. Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch. Biochem. Biophys. 2012, 519, 154–166. [Google Scholar] [CrossRef]
- Schupfner, M.; Straub, K.; Busch, F.; Merkl, R.; Sterner, R. Analysis of allosteric communication in a multienzyme complex by ancestral sequence reconstruction. Proc. Natl. Acad. Sci. USA 2019, 117, 346–354. [Google Scholar] [CrossRef]
- Banik, U.; Zhu, D.-M.; Chock, P.B.; Miles, E.W. The Tryptophan Synthase α2β2 Complex: Kinetic Studies with a Mutant Enzyme (β. K87T) To Provide Evidence for Allosteric Activation by an Aminoacrylate Intermediate. Biochemistry 1995, 34, 12704–12711. [Google Scholar] [CrossRef]
- Lee, S.J.; Ogasahara, K.; Ma, J.; Nishio, K.; Ishida, M.; Yamagata, Y.; Tsukihara, T.; Yutani, K. Conformational changes in the tryptophan synthase from a hyperthermophile upon α2β2 complex formation: Crystal structure of the complex. Biochemistry 2005, 44, 11417–11427. [Google Scholar] [PubMed]
- Dunathan, H.C. Advances in Enzymology and Related Areas of Molecular Biology; Meister, A., Ed.; Wiley: Hoboken, NJ, USA, 1971; pp. 79–134. [Google Scholar]
- Blumenstein, L.; Domratcheva, T.; Niks, D.; Ngo, H.; Seidel, R.; Dunn, M.F.; Schlichting, I. Kinetics and crystal structure of the tryptophan synthase bienzyme complex from Pyrococcus furiosus. Biochemistry 2007, 46, 14100–14116. [Google Scholar] [PubMed]
- Smith, D.R.; Willemse, T.; Gkotsi, D.S.; Schepens, W.; Maes, B.U.; Ballet, S.; Goss, R.J. The first one-pot synthesis of l-7-iodo tryptophan from 7-iodo indole and serine, and an improved synthesis of other l-7-halo tryptophans. Org. Lett. 2014, 16, 2622–2625. [Google Scholar] [CrossRef]
- Buller, A.R.; Brinkmann-Chen, S.; Romney, D.K.; Herger, M.; Murciano-Calles, J.; Arnold, F.H. Directed evolution of the tryptophan synthase β-subunit for stand-alone function recapitulates allosteric activation. Proc. Natl. Acad. Sci. USA 2015, 112, 14599–14604. [Google Scholar] [PubMed]
- Zhao, G.; Liu, J.; Dong, K.; Zhang, F.; Zhang, H.; Liu, Q.; Jiao, Q. Enzymatic synthesis of l-tryptophan from hair acid hydrolysis industries wastewater with tryptophan synthase. Bioresour. Technol. 2011, 102, 3554–3557. [Google Scholar] [CrossRef]
- Francis, D.; Winn, M.; Latham, J.; Greaney, M.F.; Micklefield, J. An Engineered Tryptophan Synthase Opens New Enzymatic Pathways to β-Methyltryptophan and Derivatives. ChemBioChem 2017, 18, 382–386. [Google Scholar] [CrossRef]
- Martínez-Rodríguez, S.; Torres, J.M.; Sánchez, P.; Ortega, E. Overview on Multienzymatic Cascades for the Production of Non-canonical α-Amino Acids. Front. Bioeng. Biotechnol. 2020, 8, 887. [Google Scholar] [CrossRef]
- Budisa, N. Engineering the Genetic Code: Expanding the Amino Acid Repertoire for the Design of Novel Proteins; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Calcuttawala, F.; Pal, A.; Nath, P.; Kar, R.; Hazra, D.; Pal, R. Structural and functional insights into colicin: A new paradigm in drug discovery. Arch. Microbiol. 2021, 204, 37. [Google Scholar] [CrossRef]
- Cursino, L.; Marda, J.; Chartone-Souza, E.; Nascimento, A.M. Recent updated aspects of colicins of Enterobacteriaceae. Braz. J. Microbiol. 2002, 33, 185–195. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Bishop, C.M.; Bishop, T.C.; Wimley, W.C.; Wiener, M.C. Enzymatic E-colicins Bind to Their Target Receptor BtuB by Presentation of a Small Binding Epitope on a Coiled-coil Scaffold. J. Biol. Chem. 2003, 278, 40953–40958. [Google Scholar] [CrossRef]
- Jakes, K.S. Translocation trumps receptor binding in colicin entry into Escherichia coli. Biochem. Soc. Trans. 2012, 40, 1443–1448. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmids | Features | Source |
|---|---|---|
| pET28a-Ectrp | Promoter T7, PBR322 Ori, Kanr, N-His | Our lab |
| pET23a-dCE2- | Promoter T7, PBR322 Ori, Ampr | Our lab |
| pET23a-dCE7 | Promoter T7, PBR322 Ori, Ampr | Our lab |
| pET23a-dCE8 | Promoter T7, PBR322 Ori, Ampr | Our lab |
| pET23a-dCE9 | Promoter T7, PBR322 Ori, Ampr | Our lab |
| pET23a-Ectrp | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | Our lab |
| pET23a-Ectrp-dCE2 | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | This paper |
| pET23a-Ectrp-dCE7 | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | This paper |
| pET23a-Ectrp-dCE8 | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | This paper |
| pET23a-Ectrp-dCE9 | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | This paper |
| pET23a-dCE2-Ectrp | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | This paper |
| pET23a-dCE7-Ectrp | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | This paper |
| pET23a-dCE8-Ectrp | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | This paper |
| pET23a-dCE9-Ectrp | Promoter T7, PBR322 Ori, Ampr, N-His, C-His | This paper |
| pET23a-TrapA | Promoter T7, PBR322 Ori, Ampr, C-His | This paper |
| pET23a-TrapB-dCE2 | Promoter T7, PBR322 Ori, Ampr, N-His | This paper |
| pET23a-TrapB-dCE7 | Promoter T7, PBR322 Ori, Ampr, N-His | This paper |
| pET23a-TrapB-dCE8 | Promoter T7, PBR322 Ori, Ampr, N-His | This paper |
| pET23a-TrapB-dCE9 | Promoter T7, PBR322 Ori, Ampr, N-His | This paper |
| pET23a-dCE2-TrapB | Promoter T7, PBR322 Ori, Ampr, C-His | This paper |
| pET23a-dCE7-TrapB | Promoter T7, PBR322 Ori, Ampr, C-His | This paper |
| pET23a-dCE8-TrapB | Promoter T7, PBR322 Ori, Ampr, C-His | This paper |
| pET23a-dCE9-TrapB | Promoter T7, PBR322 Ori, Ampr, C-His | This paper |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, X.; Niu, S.; Cheng, W.; Liu, X.; Min, Y.; Qiu, Y.; Ma, L.; Rao, B.; Zhu, L. Improving the Activity of Tryptophan Synthetase via a Nucleic Acid Scaffold. Molecules 2023, 28, 7272. https://doi.org/10.3390/molecules28217272
Wang Y, Wang X, Niu S, Cheng W, Liu X, Min Y, Qiu Y, Ma L, Rao B, Zhu L. Improving the Activity of Tryptophan Synthetase via a Nucleic Acid Scaffold. Molecules. 2023; 28(21):7272. https://doi.org/10.3390/molecules28217272
Chicago/Turabian StyleWang, Yaping, Xiangyi Wang, Shuhui Niu, Wei Cheng, Xiaoyan Liu, Yong Min, Yimin Qiu, Lixin Ma, Ben Rao, and Lei Zhu. 2023. "Improving the Activity of Tryptophan Synthetase via a Nucleic Acid Scaffold" Molecules 28, no. 21: 7272. https://doi.org/10.3390/molecules28217272
APA StyleWang, Y., Wang, X., Niu, S., Cheng, W., Liu, X., Min, Y., Qiu, Y., Ma, L., Rao, B., & Zhu, L. (2023). Improving the Activity of Tryptophan Synthetase via a Nucleic Acid Scaffold. Molecules, 28(21), 7272. https://doi.org/10.3390/molecules28217272