
Revealing Allosteric Mechanism of Amino Acid Binding Proteins from Open to Closed State


Abstract
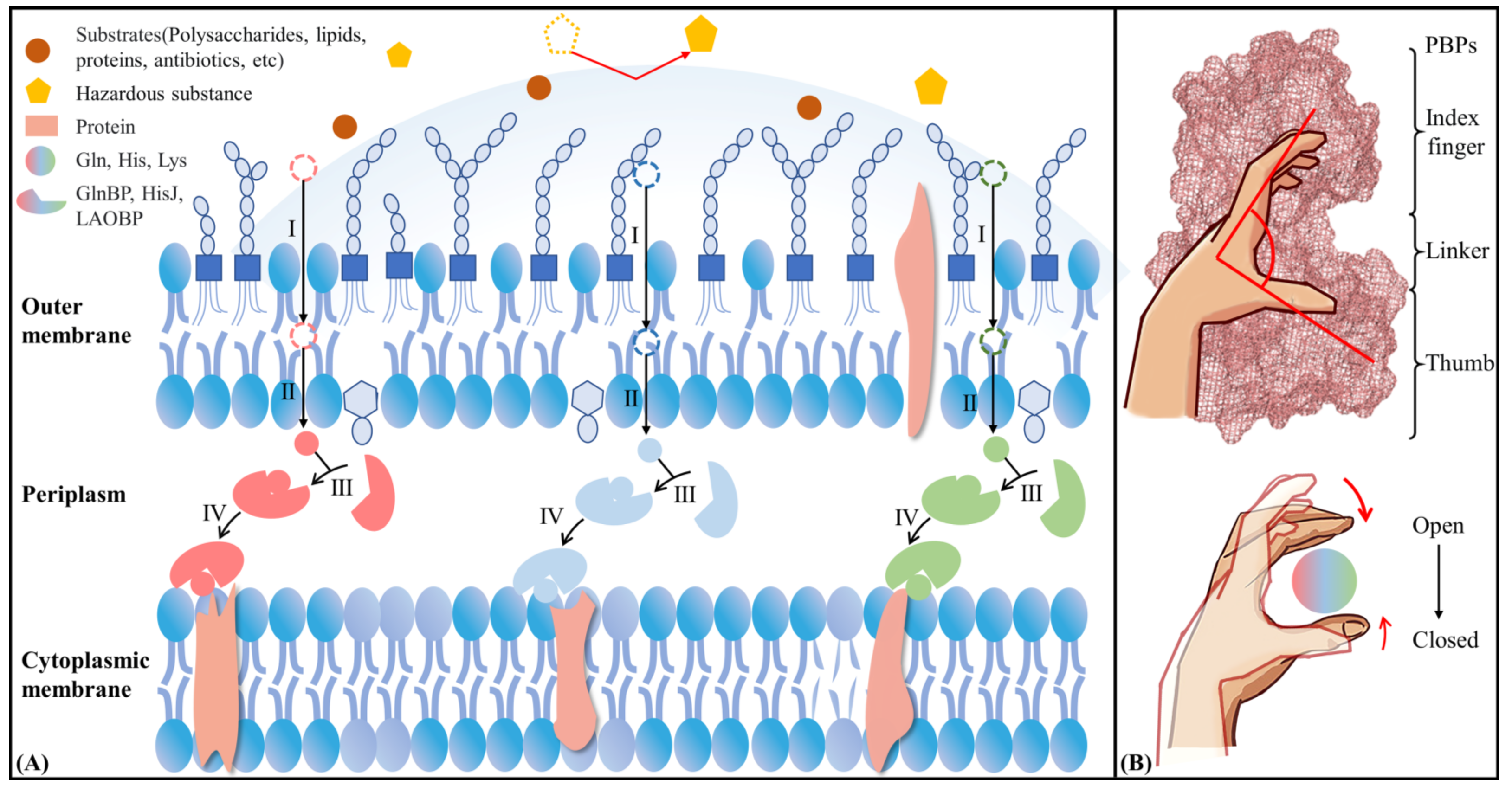
:1. Introduction
2. Results and Discussion
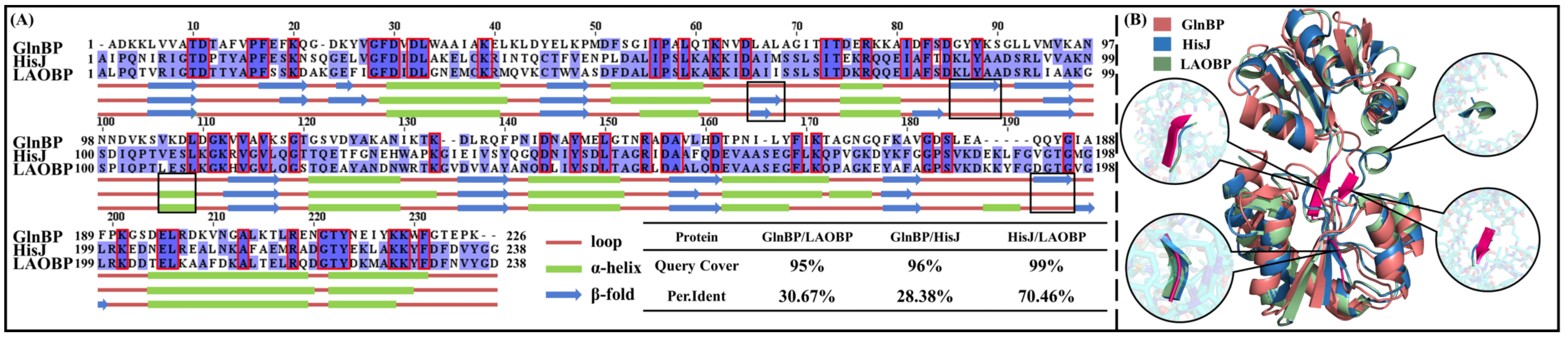
2.1. GlnBP, HisJ and LAOBP Showing High Structural Similarity
2.2. Allosteric Effects Revealed by Elastic Network Models
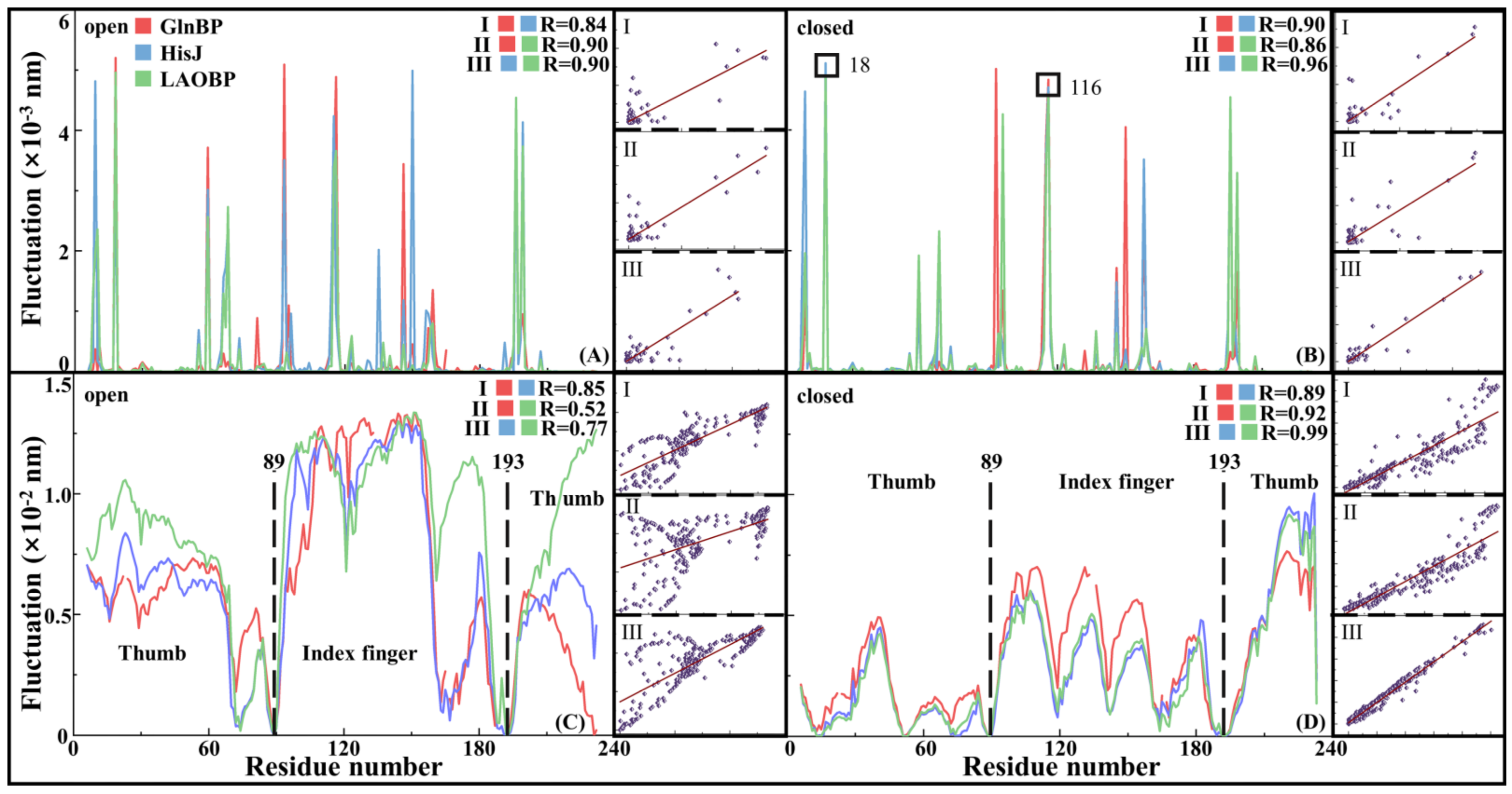
- Fast Motion Patterns Revealing Topological Characteristics of AABPs
- Flexible Distribution of AABPs Slow Motion
- Motion Correlation of AABPs
- The motion patterns of AABPs contributing to capture of amino acid substrates
2.3. Interaction of Protein Fragments Inferred from Neural Relational Inference Molecular Dynamics
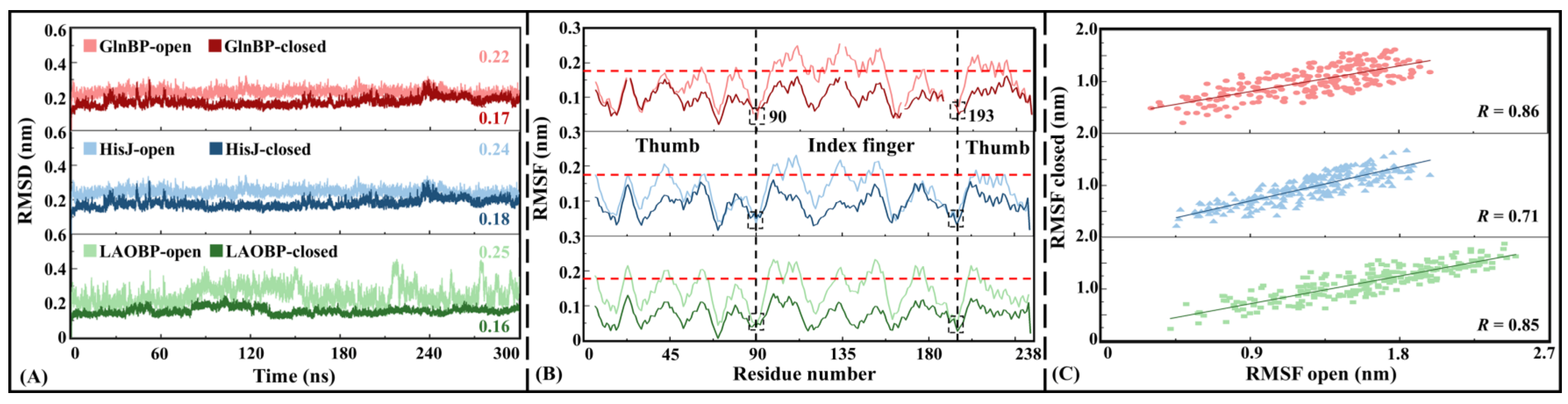
- Convergent cMD simulations are a prerequisite for NRI-MD training
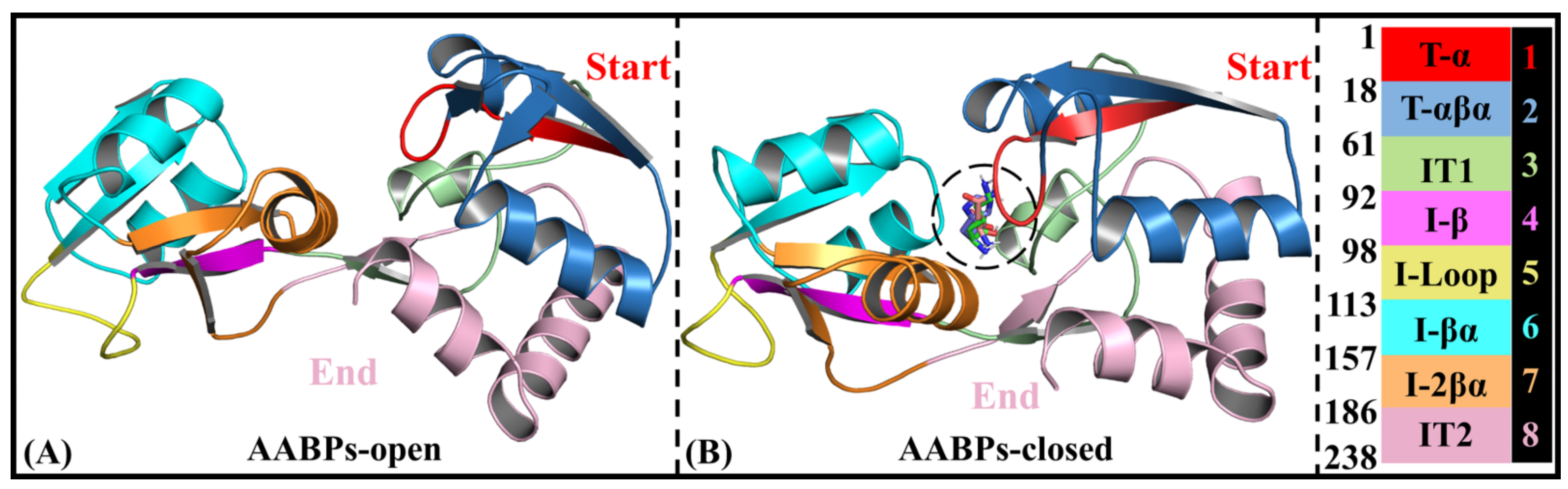
- Rational domain partitions for subsequent NRI-MD analysis
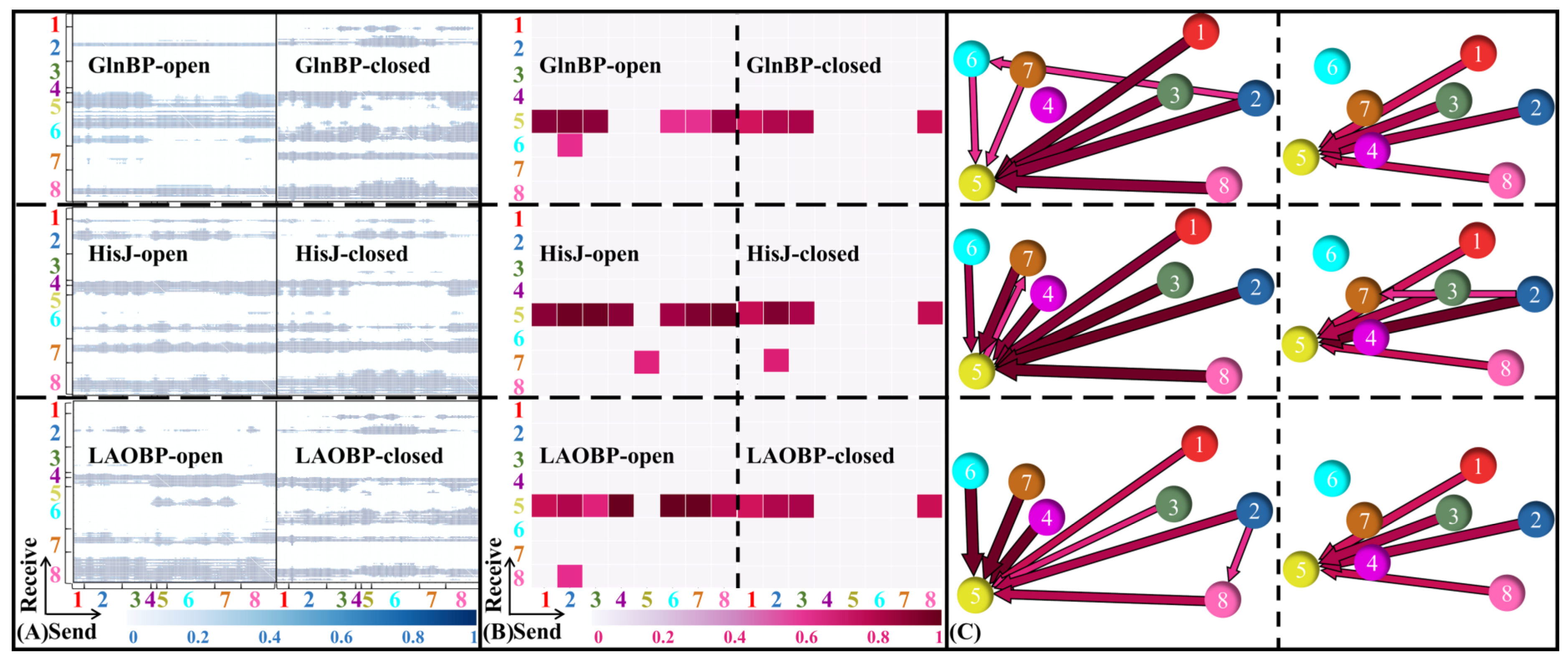
- Domain communications of three AABPs’ systems
2.4. Communication Pathways in the Allosteric Process of AABPs
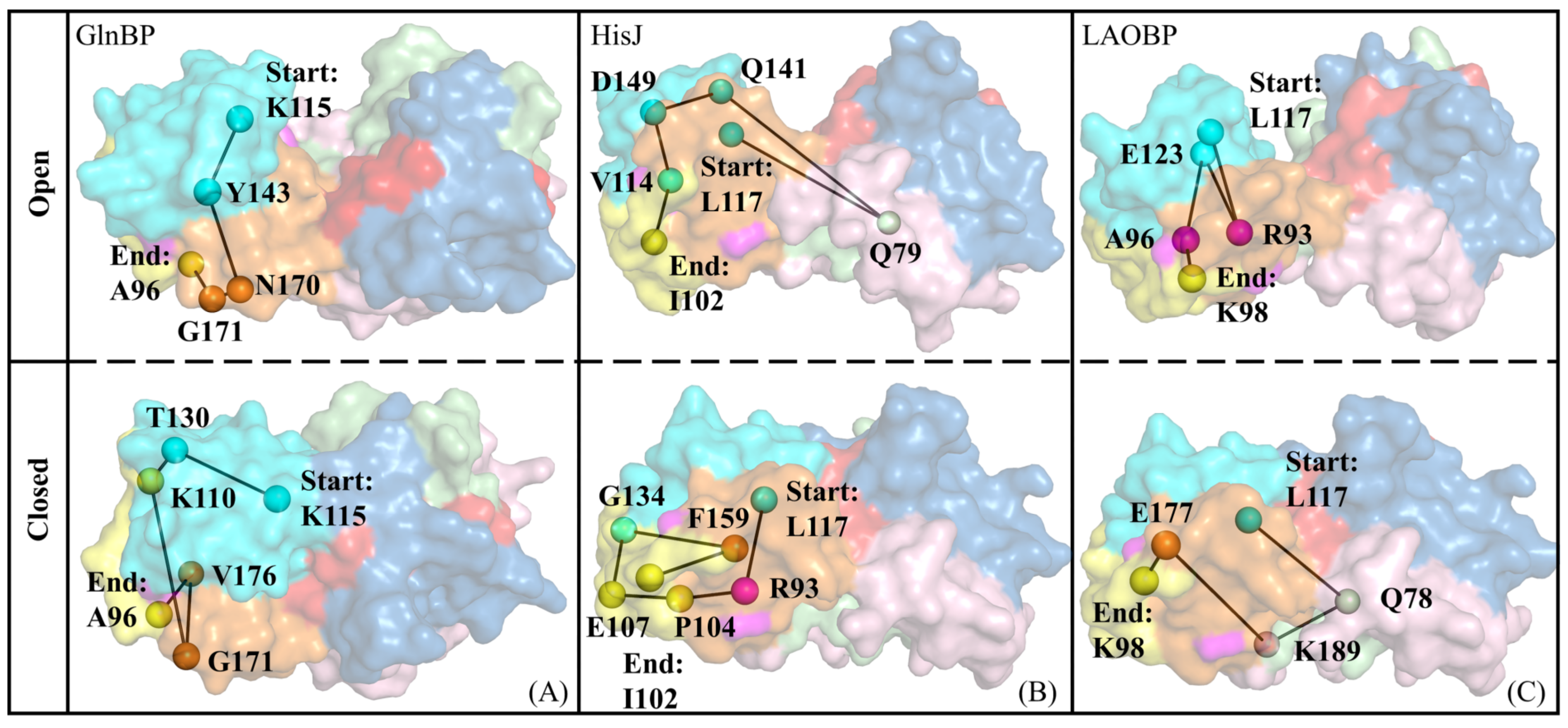
- Key residues for receptor–ligand recognition
- Signal transmission pathways for conformational closure of AABPs
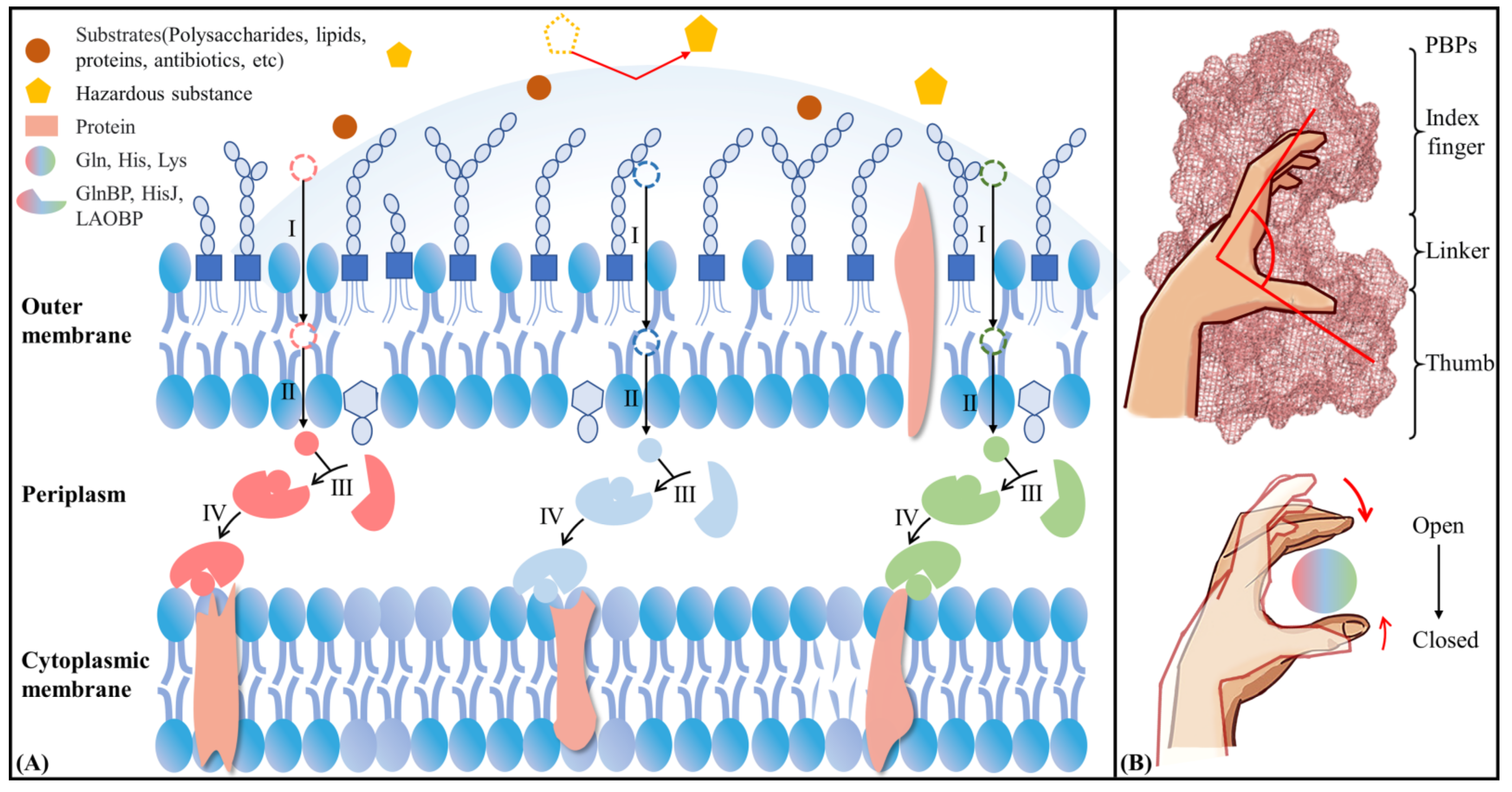
2.5. Possible Allosteric Mechanism of AABPs’ Binding Substrate
3. Materials and Methods
3.1. Preparation of Simulation Systems
3.2. Multiple Sequence Alignment
3.3. Gaussian Network Model (GNM)
3.4. Anisotropic Network Model (ANM)
3.5. Molecular Dynamics Simulation
3.6. Neural Relational Inference Molecular Dynamics (NRI-MD)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bladen, H.A.; Mergenhagen, S.E. Ultrastructure of Veillonella and morphological correlation of an outer membrane with particles associated with endotoxic activity. J. Bacteriol. 1964, 88, 1482–1492. [Google Scholar] [CrossRef]
- Ames, G.F.-L. Bacterial periplasmic transport systems: Structure, mechanism, and evolution. Annu. Rev. Biochem. 1986, 55, 397–425. [Google Scholar] [CrossRef]
- Brown, K. Formation of aromatic amino acid pools in Escherichia coli K-12. J. Bacteriol. 1970, 104, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.; Lesk, A.M.; Chothia, C. Structural mechanisms for domain movements in proteins. Biochemistry 1994, 33, 6739–6749. [Google Scholar] [CrossRef] [PubMed]
- Neu, H.C.; Heppel, L.A. The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J. Biolog. Chem. 1965, 240, 3685–3692. [Google Scholar] [CrossRef]
- Friesen, J.D. Escherichia coli and Salmonella typhimurium: Cellular and molecular biology. Science 1988, 240, 1678–1681. [Google Scholar] [CrossRef] [PubMed]
- Piperno, J.; Oxender, D. Amino acid transport systems in Escherichia coli K12. J. Biolog. Chem. 1968, 243, 5914–5920. [Google Scholar] [CrossRef]
- Rosen, B.P. Basic Amino Acid Transport in Escherichia coli: II. purification and properties of an arginine-specific binding protein. J. Biolog. Chem. 1973, 248, 1211–1218. [Google Scholar] [CrossRef]
- Pang, A.; Arinaminpathy, Y.; Sansom, M.S.; Biggin, P.C. Interdomain dynamics and ligand binding: Molecular dynamics simulations of glutamine binding protein. FEBS Lett. 2003, 550, 168–174. [Google Scholar] [CrossRef]
- Cortes-Hernandez, P.; Domínguez-Ramírez, L. Role of cis-trans proline isomerization in the function of pathogenic enterobacterial Periplasmic Binding Proteins. PLoS ONE 2017, 12, e0188935. [Google Scholar] [CrossRef]
- Kang, C.; Shin, W.; Yamagata, Y.; Gokcen, S.; Ames, G.; Kim, S. Crystal structure of the lysine-, arginine-, ornithine-binding protein (LAO) from Salmonella typhimurium at 2.7-A resolution. J. Biolog. Chem. 1991, 266, 23893–23899. [Google Scholar] [CrossRef]
- Blein, S.; Hawrot, E.; Barlow, P. The metabotropic GABA receptor: Molecular insights and their functional consequences. Cell Mol. Life Sci. 2000, 57, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997, 79, 3090. [Google Scholar] [CrossRef]
- Quiocho, F. Atomic structures of periplasmic binding proteins and the high-affinity active transport systems in bacteria. Philos. Trans. R. Soc. London. B Biol. Sci. 1990, 326, 341–352. [Google Scholar] [CrossRef]
- Hsiao, C.-D.; Suna, Y.J.; Rosec, J.; Wang, B.-C. The crystal structure of glutamine-binding protein from Escherichia coli. J. Mol. Biolog. 1996, 262, 225–242. [Google Scholar] [CrossRef]
- Lv, D.; Gong, W.; Zhang, Y.; Liu, Y.; Li, C. A coarse-grained method to predict the open-to-closed behavior of glutamine binding protein. Chem. Phys. 2017, 493, 166–174. [Google Scholar] [CrossRef]
- Weiner, J.H.; Heppel, L.A. A binding protein for glutamine and its relation to active transport in Escherichia coli. J. Biol. Chem. 1971, 246, 6933–6941. [Google Scholar] [CrossRef]
- Masters, P.S.; Hong, J. Genetics of the glutamine transport system in Escherichia coli. J. Bacteriol. 1981, 147, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-J.; Rose, J.; Wang, B.-C.; Hsiao, C.-D. The structure of glutamine-binding protein complexed with glutamine at 1.94 Å resolution: Comparisons with other amino acid binding proteins. J. Mol. Biol. 1998, 278, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.G.; Hu, J.P.; Li, C.H.; Zu Chen, W.; Wang, C.X. A molecular dynamics simulation study of glutamine-binding protein. J. Mol. Struct. Theochem 2005, 725, 9–16. [Google Scholar] [CrossRef]
- Su, J.G.; Jiao, X.; Sun, T.G.; Li, C.H.; Zu Chen, W.; Wang, C.X. Analysis of domain movements in glutamine-binding protein with simple models. Biophys. J. 2007, 92, 1326–1335. [Google Scholar] [CrossRef]
- Chu, B.C.; DeWolf, T.; Vogel, H.J. Role of the two structural domains from the periplasmic Escherichia coli histidine-binding protein HisJ. J. Biol. Chem. 2013, 288, 31409–31422. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Chan, D.I.; DeWolf, T.; Periole, X.; Vogel, H.J. Molecular dynamics simulations reveal that apo-HisJ can sample a closed conformation. Proteins Struct. Funct. Bioinform. 2014, 82, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Banda-Vázquez, J.; Shanmugaratnam, S.; Rodríguez-Sotres, R.; Torres-Larios, A.; Höcker, B.; Sosa-Peinado, A. Redesign of LAOBP to bind novel l-amino acid ligands. Protein Sci. 2018, 27, 957–968. [Google Scholar] [CrossRef]
- Kundu, S.; Jernigan, R.L. Molecular mechanism of domain swapping in proteins: An analysis of slower motions. Biophys. J. 2004, 86, 3846–3854. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Jernigan, R.L.; Bahar, I. Proteins with similar architecture exhibit similar large-scale dynamic behavior. Biophys. J. 2000, 78, 2093–2106. [Google Scholar] [CrossRef] [PubMed]
- Haliloglu, T.; Keskin, O.; Ma, B.; Nussinov, R. How similar are protein folding and protein binding nuclei? Examination of vibrational motions of energy hot spots and conserved residues. Biophys. J. 2005, 88, 1552–1559. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, J.; Han, W.; Xu, D. Neural relational inference to learn long-range allosteric interactions in proteins from molecular dynamics simulations. Nat. Commun. 2022, 13, 1661. [Google Scholar] [CrossRef] [PubMed]
- Staudenmaier, H.; Van Hove, B.; Yaraghi, Z.; Braun, V. Nucleotide sequences of the fecBCDE genes and locations of the proteins suggest a periplasmic-binding-protein-dependent transport mechanism for iron (III) dicitrate in Escherichia coli. J. Bacteriol. 1989, 171, 2626–2633. [Google Scholar] [CrossRef]
- Dwyer, M.A.; Hellinga, H.W. Periplasmic binding proteins: A versatile superfamily for protein engineering. Curr. Opin. Struc. Biol. 2004, 14, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.-F.; Doolittle, R.F. Progressive sequence alignment as a prerequisitetto correct phylogenetic trees. J. Mol. Evol. 1987, 25, 351–360. [Google Scholar] [CrossRef]
- Wang, L.-B.; Wang, Y.-C.; He, R.; Zhuang, A.; Wang, X.; Zeng, J.; Hou, J.G. A new nanobiocatalytic system based on allosteric effect with dramatically enhanced enzymatic performance. J. Am. Chem. Soc. 2013, 135, 1272–1275. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Durell, S.; Jernigan, R.; Demirel, M.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Paul, S.; Banerjee, S.; Vogel, H.J. Ligand binding specificity of the Escherichia coli periplasmic histidine binding protein, HisJ. Protein Sci. 2017, 26, 268–279. [Google Scholar] [CrossRef]
- Oh, B.; Pandit, J.; Kang, C.; Nikaido, K.; Gokcen, S.; Ames, G.; Kim, S. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J. Biol. Chem. 1993, 268, 11348–11355. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, H.H.; Kitao, A. Collective dynamics of periplasmic glutamine binding protein upon domain closure. Biophysical 2009, 97, 2541–2549. [Google Scholar] [CrossRef]
- Mnih, V.; Heess, N.; Graves, A. Recurrent models of visual attention. Adv. Neural Inf. Process Syst. 2014, 2, 2204–2212. [Google Scholar]
- Bermejo, G.A.; Strub, M.-P.; Ho, C.; Tjandra, N. Ligand-free open− closed transitions of periplasmic binding proteins: The case of glutamine-binding protein. Biochemistry 2010, 49, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Duan, H.; Cheng, Y.; Wang, Y.; Hu, J.; Shi, H. Omicron-included mutation-induced changes in epitopes of SARS-CoV-2 spike protein and effectiveness assessments of current antibodies. Mol. Biomed. 2022, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.X.; Okimoto, N.; Morimoto, G.; Taiji, M. Free-energy landscapes of protein domain movements upon ligand binding. J. Phys. Chem. B. 2011, 115, 7629–7636. [Google Scholar] [CrossRef]
- Edgar, R.C.; Batzoglou, S. Multiple sequence alignment. Curr. Opin. Struc. Biol. 2006, 16, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36 (Suppl. S2), W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Zhou, Y.; Shi, X.; Luo, Q.; Gao, J.; Liang, L.; Liu, W.; Peng, L.; Deng, D.; Hu, J. Allosteric and transport modulation of human concentrative nucleoside transporter 3 at the atomic scale. Phys. Chem. Chem. Phys. 2021, 23, 25401–25413. [Google Scholar] [CrossRef]
- Eyal, E.; Yang, L.-W.; Bahar, I. Anisotropic network model: Systematic evaluation and a new web interface. Bioinformatics 2006, 22, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar] [CrossRef]
- Kipf, T.; Fetaya, E.; Wang, K.C.; Welling, M.; Zemel, R. Neural relational inference for interacting systems. In Proceedings of the International Conference on Machine Learning, Stockholm, Sweden, 10–15 July 2018. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | Residues | EVDW a | EELE b | EGB c | EGBSUR d | ETOT |
|---|---|---|---|---|---|---|
| GlnBP | 117 (K115) | −0.44 ± 0.31 | −6.97 ± 4.61 | 1.33 ± 3.8 | −0.02 ± 0.02 | −6.10 ± 0.52 |
| 161 (D157) | −0.69 ± 0.8 | −3.89 ± 3.44 | 2.57 ± 0.19 | −0.04 ± 0.02 | −2.04 ± 1.35 | |
| 69 (A67) | −0.23 ± 0.33 | −2.92 ± 0.37 | 1.53 ± 0.34 | −0.02 ± 0.01 | −1.64 ± 0.39 | |
| 11 (D10) | −0.45 ± 0.01 | −2.70 ± 0.22 | 1.72 ± 0.22 | 0.01 ± 0.00 | −1.43 ± 0.01 | |
| 70 (G68) | −0.54 ± 0.01 | −1.99 ± 1.32 | 1.24 ± 0.37 | −0.03 ± 0.01 | −1.33 ± 0.45 | |
| 121 (G119) | −0.22 ± 0.13 | −1.61 ± 0.26 | 0.75 ± 0.25 | −0.06 ± 0.01 | −1.14 ± 0.12 | |
| HisJ | 11 (D11) | 0.11 ± 0.09 | −9.80 ± 0.45 | 5.11 ± 0.56 | −0.05 ± 0 | −4.63 ± 0.02 |
| 117 (L117) | −1.44 ± 0.06 | −1.24 ± 1.70 | 0.90 ± 0.02 | −0.07 ± 0.03 | −1.85 ± 0.38 | |
| 69 (S69) | −0.37 ± 0.41 | −1.13 ± 0.91 | −0.14 ± 0.29 | −0.05 ± 0.02 | −1.68 ± 0.23 | |
| 14 (Y14) | −1.26 ± 0.09 | −0.56 ± 0.90 | 0.43 ± 0.36 | −0.08 ± 0.02 | −1.45 ± 0.66 | |
| 52 (L52) | −0.95 ± 0.19 | −0.09 ± 0.02 | 0.16 ± 0.01 | −0.12 ± 0.01 | −1.01 ± 0.17 | |
| LAOBP | 117 (L117) | −0.91 ± 0 | 0.40 ± 0.33 | −1.64 ± 0.14 | −0.08 ± 0.01 | −2.26 ± 0.16 |
| 11 (D11) | −0.59 ± 0.18 | −1.73 ± 0.04 | 0.21 ± 0.06 | 0.05 ± 0.02 | −2.11 ± 0.23 | |
| 52 (F52) | −1.5 ± 0.07 | −0.57 ± 0.04 | 0.17 ± 0.08 | −0.07 ± 0.01 | −1.98 ± 0.01 | |
| 72 (S72) | −1.17 ± 0.03 | −0.86 ± 0.50 | 0.11 ± 0.04 | −0.03 ± 0 | −1.94 ± 0.09 | |
| 77 (R77) | −0.06 ± 0.04 | −1.08 ± 0.50 | −0.41 ± 0.02 | −0.01 ± 0.01 | −1.56 ± 0.16 | |
| 70 (S70) | −1.12 ± 0.08 | −0.38 ± 0.01 | 0.40 ± 0.01 | 0.06 ± 0.02 | −1.04 ± 0.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Q.; Liu, L.; Duan, H.; Jiang, Y.; Luo, W.; Sun, G.; Ge, Y.; Liang, L.; Liu, W.; Shi, H.; et al. Revealing Allosteric Mechanism of Amino Acid Binding Proteins from Open to Closed State. Molecules 2023, 28, 7139. https://doi.org/10.3390/molecules28207139
Shi Q, Liu L, Duan H, Jiang Y, Luo W, Sun G, Ge Y, Liang L, Liu W, Shi H, et al. Revealing Allosteric Mechanism of Amino Acid Binding Proteins from Open to Closed State. Molecules. 2023; 28(20):7139. https://doi.org/10.3390/molecules28207139
Chicago/Turabian StyleShi, Quanshan, Ling Liu, Huaichuan Duan, Yu Jiang, Wenqin Luo, Guangzhou Sun, Yutong Ge, Li Liang, Wei Liu, Hubing Shi, and et al. 2023. "Revealing Allosteric Mechanism of Amino Acid Binding Proteins from Open to Closed State" Molecules 28, no. 20: 7139. https://doi.org/10.3390/molecules28207139
APA StyleShi, Q., Liu, L., Duan, H., Jiang, Y., Luo, W., Sun, G., Ge, Y., Liang, L., Liu, W., Shi, H., & Hu, J. (2023). Revealing Allosteric Mechanism of Amino Acid Binding Proteins from Open to Closed State. Molecules, 28(20), 7139. https://doi.org/10.3390/molecules28207139