

The Construction of Surface-Frustrated Lewis Pair Sites to Improve the Nitrogen Reduction Catalytic Activity of In2O3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
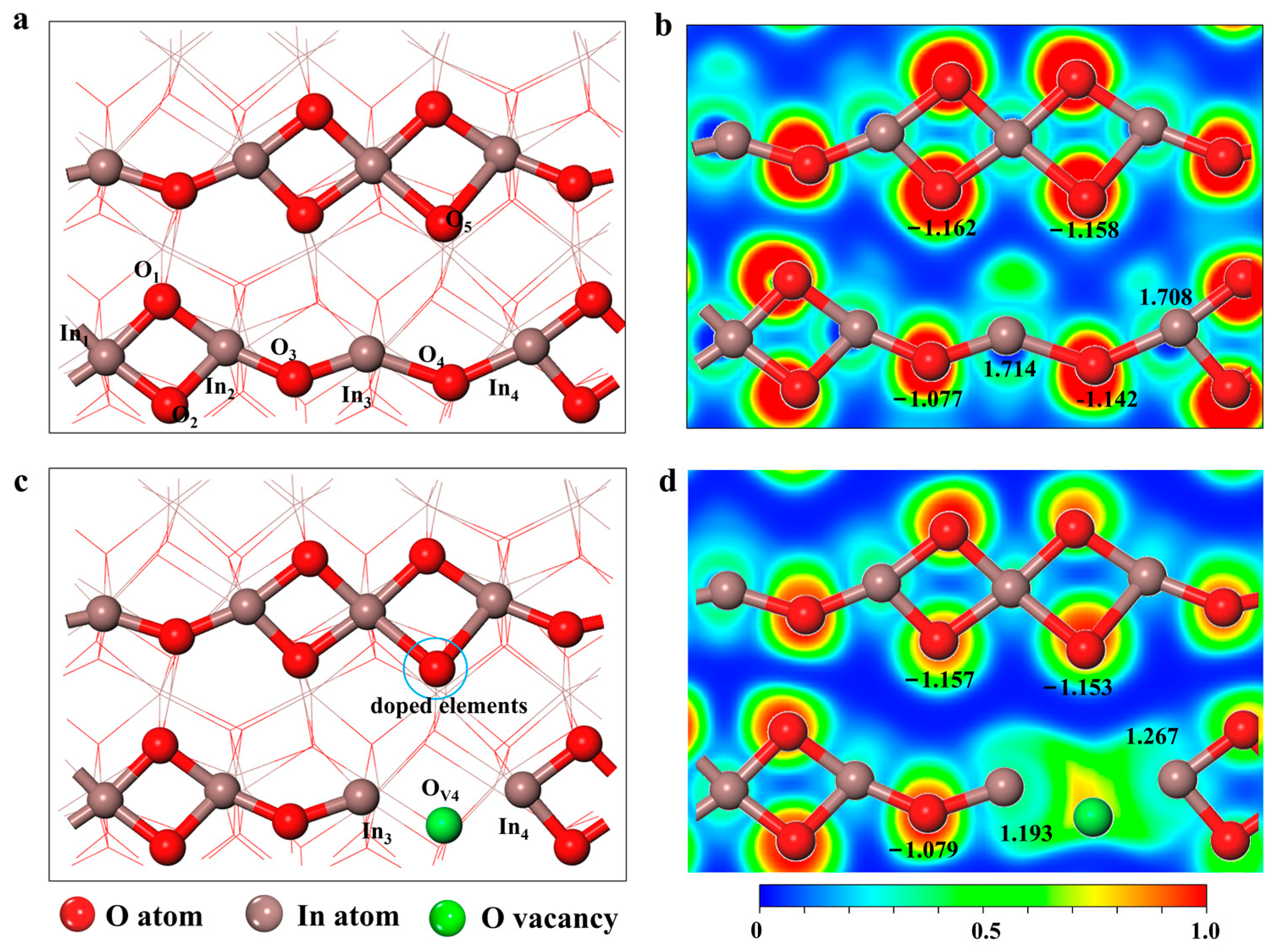
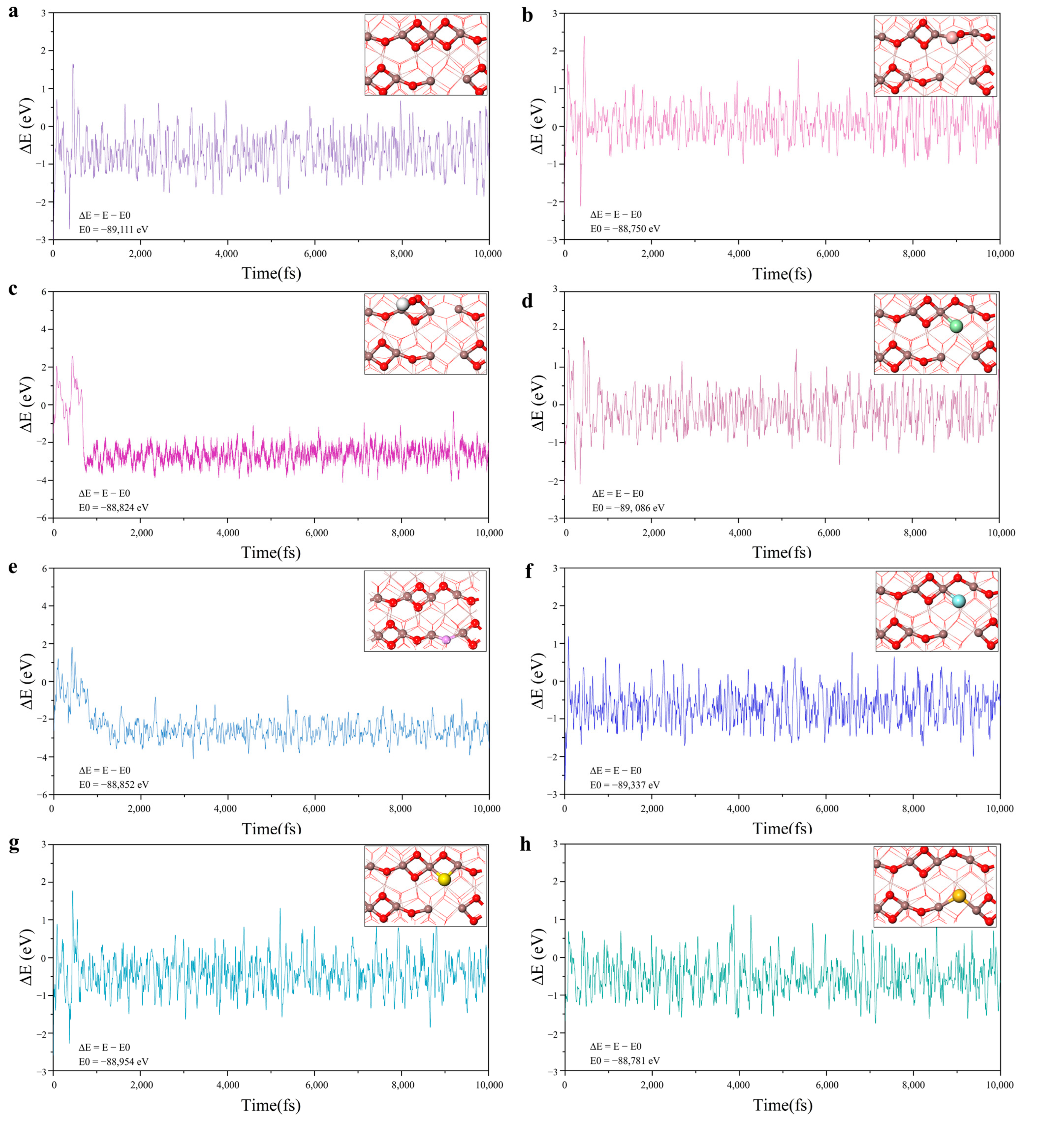
2.1. Geometric Structure of Doping V-In2O3
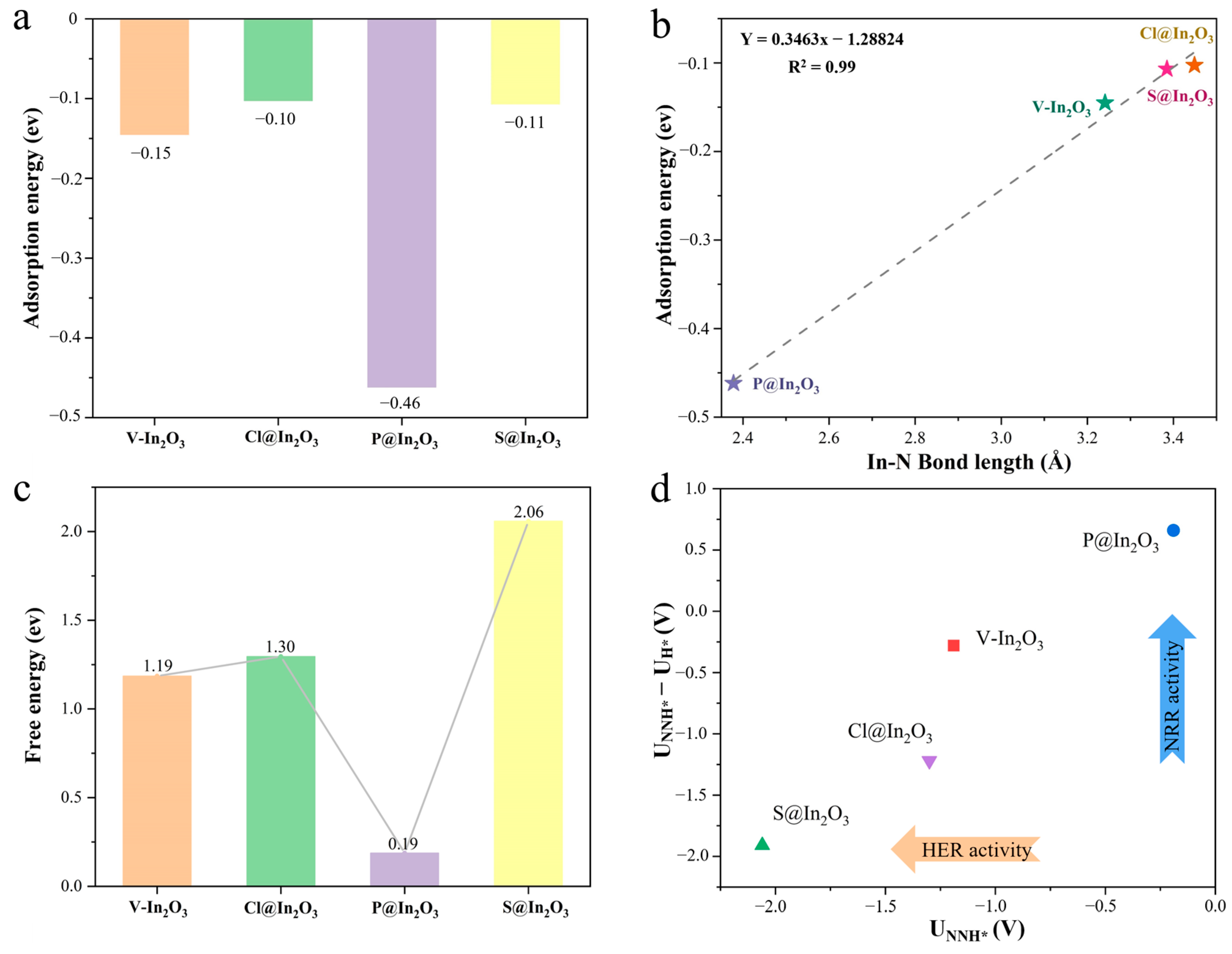
2.2. Adsorption and Protonation of Nitrogen
2.3. Catalytic Mechanism in Solution
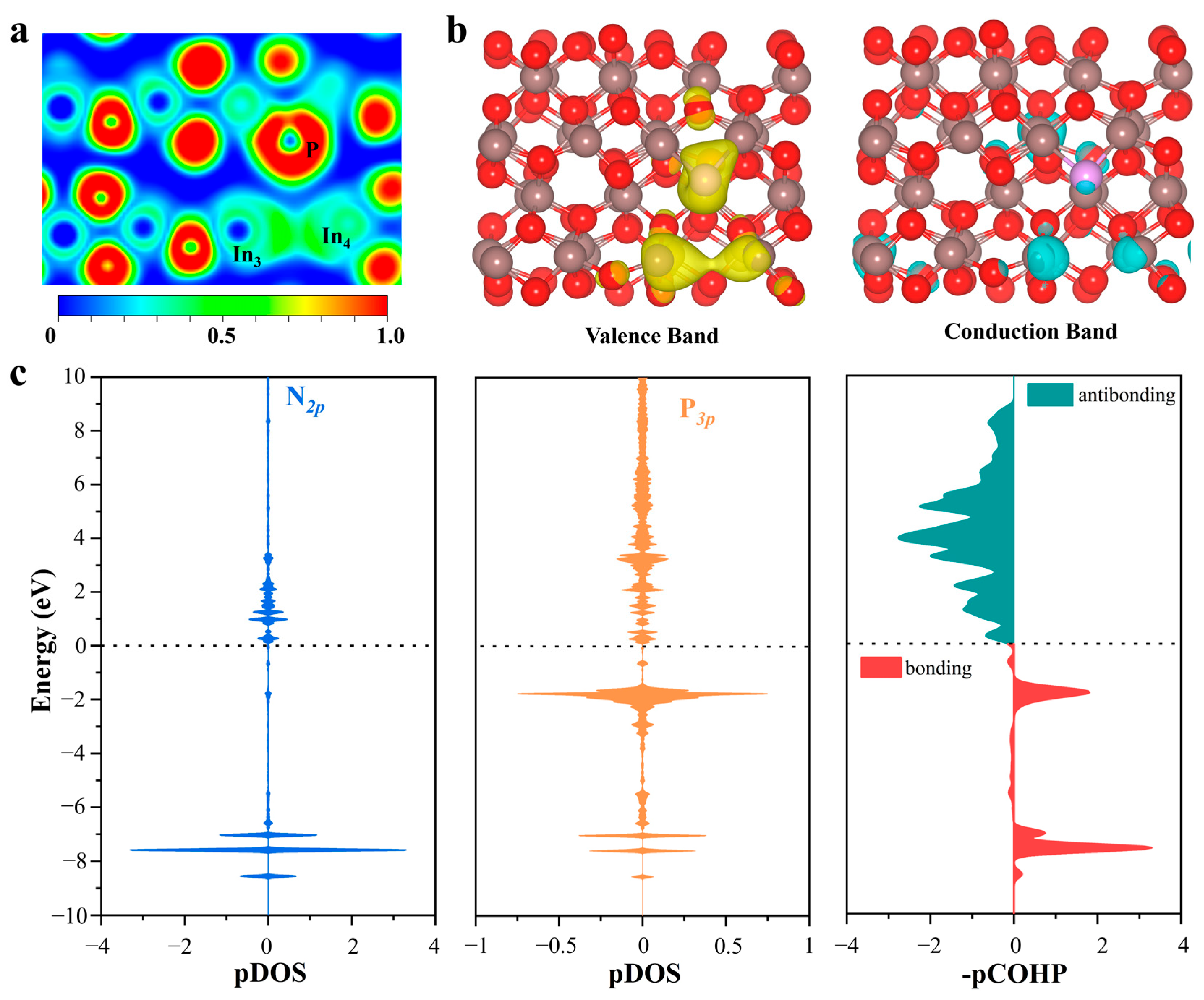
2.4. Origin of Catalytic Activity
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cui, X.; Tang, C.; Zhang, Q. A Review of Electrocatalytic Reduction of Dinitrogen to Ammonia under Ambient Conditions. Adv. Energy Mater. 2018, 8, 1800369. [Google Scholar] [CrossRef]
- Shi, L.; Yin, Y.; Wang, S.; Sun, H. Rational Catalyst Design for N2 Reduction under Ambient Conditions: Strategies toward Enhanced Conversion Efficiency. ACS Catal. 2020, 10, 6870–6899. [Google Scholar] [CrossRef]
- Zhu, X.; Mou, S.; Peng, Q.; Liu, Q.; Luo, Y.; Chen, G.; Gao, S.; Sun, X. Aqueous electrocatalytic N2 reduction for ambient NH3 synthesis: Recent advances in catalyst development and performance improvement. J. Mater. Chem. A 2020, 8, 1545–1556. [Google Scholar] [CrossRef]
- Wan, Y.; Xu, J.; Lv, R. Heterogeneous electrocatalysts design for nitrogen reduction reaction under ambient conditions. Mater. Today 2019, 27, 69–90. [Google Scholar] [CrossRef]
- Huang, Z.; Rafiq, M.; Woldu, A.R.; Tong, Q.-X.; Astruc, D.; Hu, L. Recent progress in electrocatalytic nitrogen reduction to ammonia (NRR). Coord. Chem. Rev. 2023, 478, 214981. [Google Scholar] [CrossRef]
- Zhao, X.; Hu, G.; Chen, G.-F.; Zhang, H.; Zhang, S.; Wang, H. Comprehensive Understanding of the Thriving Ambient Electrochemical Nitrogen Reduction Reaction. Adv. Mater. 2021, 33, 2007650. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yan, Q. Electrochemical reduction of nitrogen to ammonia: Progress, challenges and future outlook. Curr. Opin. Electrochem. 2021, 29, 100808. [Google Scholar] [CrossRef]
- Légaré, M.-A.; Bélanger-Chabot, G.; Dewhurst, R.D.; Welz, E.; Krummenacher, I.; Engels, B.; Braunschweig, H. Nitrogen fixation and reduction at boron. Science 2018, 359, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Q.; Guo, X.; Kong, X.; Ke, J.; Chi, M.; Li, Q.; Geng, Z.; Zeng, J. A Highly Efficient Metal-Free Electrocatalyst of F-Doped Porous Carbon toward N2 Electroreduction. Adv. Mater. 2020, 32, 1907690. [Google Scholar] [CrossRef]
- Lv, C.; Zhong, L.; Yao, Y.; Liu, D.; Kong, Y.; Jin, X.; Fang, Z.; Xu, W.; Yan, C.; Dinh, K.N.; et al. Boosting Electrocatalytic Ammonia Production through Mimicking “π Back-Donation”. Chem 2020, 6, 2690–2702. [Google Scholar] [CrossRef]
- Yuan, M.; Chen, J.; Xu, Y.; Liu, R.; Zhao, T.; Zhang, J.; Ren, Z.; Liu, Z.; Streb, C.; He, H.; et al. Highly selective electroreduction of N2 and CO2 to urea over artificial frustrated Lewis pairs. Energy Environ. Sci. 2021, 14, 6605–6615. [Google Scholar] [CrossRef]
- Lin, W.; Chen, H.; Lin, G.; Yao, S.; Zhang, Z.; Qi, J.; Jing, M.; Song, W.; Li, J.; Liu, X.; et al. Creating Frustrated Lewis Pairs in Defective Boron Carbon Nitride for Electrocatalytic Nitrogen Reduction to Ammonia. Angew. Chem. Int. Ed. 2022, 61, e202207807. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, Y.; Liu, D.; Qi, R.; Xia, C.; Li, M.; You, B.; Xia, B.Y. Carbon-Confined Indium Oxides for Efficient Carbon Dioxide Reduction in a Solid-State Electrolyte Flow Cell. Angew. Chem. Int. Ed. 2022, 61, e202200552. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ghoussoub, M.; Wang, H.; Shao, Y.; Sun, W.; Tountas, A.A.; Wood, T.E.; Li, H.; Loh, J.Y.Y.; Dong, Y.; et al. Photocatalytic Hydrogenation of Carbon Dioxide with High Selectivity to Methanol at Atmospheric Pressure. Joule 2018, 2, 1369–1381. [Google Scholar] [CrossRef]
- Du, X.; Qin, Y.; Gao, B.; Wang, K.; Li, D.; Li, Y.; Ding, S.; Song, Z.; Su, Y.; Xiao, C. Plasma-assisted and oxygen vacancy-engineered In2O3 films for enhanced electrochemical reduction of CO2. Appl. Surf. Sci. 2021, 563, 150405. [Google Scholar] [CrossRef]
- Dostagir, N.H.M.D.; Thompson, C.; Kobayashi, H.; Karim, A.M.; Fukuoka, A.; Shrotri, A. Rh promoted In2O3 as a highly active catalyst for CO2 hydrogenation to methanol. Catal. Sci. Technol. 2020, 10, 8196–8202. [Google Scholar] [CrossRef]
- Ye, J.; Liu, C.; Mei, D.; Ge, Q. Active Oxygen Vacancy Site for Methanol Synthesis from CO2 Hydrogenation on In2O3(110): A DFT Study. ACS Catal. 2013, 3, 1296–1306. [Google Scholar] [CrossRef]
- Agoston, P.; Albe, K. Thermodynamic stability, stoichiometry, and electronic structure of bcc- In2O3 surfaces. Phys. Rev. B 2011, 84, 045311. [Google Scholar] [CrossRef]
- Walsh, A.; Catlow, C.R.A. Structure, stability and work functions of the low index surfaces of pure indium oxide and Sn-doped indium oxide (ITO) from density functional theory. J. Mater. Chem. 2010, 20, 10438–10444. [Google Scholar] [CrossRef]
- Hagleitner, D.R.; Menhart, M.; Jacobson, P.; Blomberg, S.; Schulte, K.; Lundgren, E.; Kubicek, M.; Fleig, J.; Kubel, F.; Puls, C.; et al. Bulk and surface characterization of In2O3 (001) single crystals. Phys. Rev. B 2012, 85, 115441. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, W.; Chen, Y. Insight of DFT and ab initio atomistic thermodynamics on the surface stability and morphology of In2O3. Appl. Surf. Sci. 2018, 434, 1344–1352. [Google Scholar] [CrossRef]
- Yan, T.; Li, N.; Wang, L.; Ran, W.; Duchesne, P.N.; Wan, L.; Nguyen, N.T.; Wang, L.; Xia, M.; Ozin, G.A. Bismuth atom tailoring of indium oxide surface frustrated Lewis pairs boosts heterogeneous CO2 photocatalytic hydrogenation. Nat. Commun. 2020, 11, 6095. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhang, H.; Xu, Y.; Liu, R.; Wang, R.; Zhao, T.; Zhang, J.; Liu, Z.; He, H.; Yang, C.; et al. Artificial frustrated Lewis pairs facilitating the electrochemical N2 and CO2 conversion to urea. Chem Catal. 2022, 2, 309–320. [Google Scholar] [CrossRef]
- Wang, L.; Yan, T.; Song, R.; Sun, W.; Dong, Y.; Guo, J.; Zhang, Z.; Wang, X.; Ozin, G.A. Room-Temperature Activation of H2 by a Surface Frustrated Lewis Pair. Angew. Chem. Int. Ed. 2019, 58, 9501–9505. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Li, S. First principles investigation of dissociative adsorption of H2 during CO2 hydrogenation over cubic and hexagonal In2O3 catalysts. Phys. Chem. Chem. Phys. 2020, 22, 3390–3399. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shi, W.; Fu, Y.-Q.; Yu, H.; Wang, Z.; Li, Z. The novel π–d conjugated TM2B3N3S6 (TM = Mo, Ti and W) monolayers as highly active single-atom catalysts for electrocatalytic synthesis of ammonia. J. Colloid Interface Sci. 2023, 650, 1–12. [Google Scholar] [CrossRef]
- Hongzhiwei Technology, Device Studio, Version 2021A, China. 2021. Available online: https://iresearch.net.cn/cloud-software (accessed on 15 October 2023).
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar]
- Blochl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. LOBSTER: A Tool to Extract Chemical Bonding from Plane-Wave Based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Dronskowski, R.; Bloechl, P.E. Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Wang, W.; Chen, Y.; Zhang, M. Facet effect of In2O3 for methanol synthesis by CO2 hydrogenation: A mechanistic and kinetic study. Surf. Interfaces 2021, 25, 101244. [Google Scholar] [CrossRef]
- Ye, J.; Liu, C.; Ge, Q. DFT Study of CO2 Adsorption and Hydrogenation on the In2O3 Surface. J. Phys. Chem. C 2012, 116, 7817–7825. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zheng, M.; Sima, Y.; Lv, C.; Zhou, X. The Construction of Surface-Frustrated Lewis Pair Sites to Improve the Nitrogen Reduction Catalytic Activity of In2O3. Molecules 2023, 28, 7130. https://doi.org/10.3390/molecules28207130
Wang M, Zheng M, Sima Y, Lv C, Zhou X. The Construction of Surface-Frustrated Lewis Pair Sites to Improve the Nitrogen Reduction Catalytic Activity of In2O3. Molecules. 2023; 28(20):7130. https://doi.org/10.3390/molecules28207130
Chicago/Turabian StyleWang, Mingqian, Ming Zheng, Yuchen Sima, Chade Lv, and Xin Zhou. 2023. "The Construction of Surface-Frustrated Lewis Pair Sites to Improve the Nitrogen Reduction Catalytic Activity of In2O3" Molecules 28, no. 20: 7130. https://doi.org/10.3390/molecules28207130
APA StyleWang, M., Zheng, M., Sima, Y., Lv, C., & Zhou, X. (2023). The Construction of Surface-Frustrated Lewis Pair Sites to Improve the Nitrogen Reduction Catalytic Activity of In2O3. Molecules, 28(20), 7130. https://doi.org/10.3390/molecules28207130