
Critical Influence of Water on the Polymorphism of 1,3-Dimethylurea and Other Heterogeneous Equilibria

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
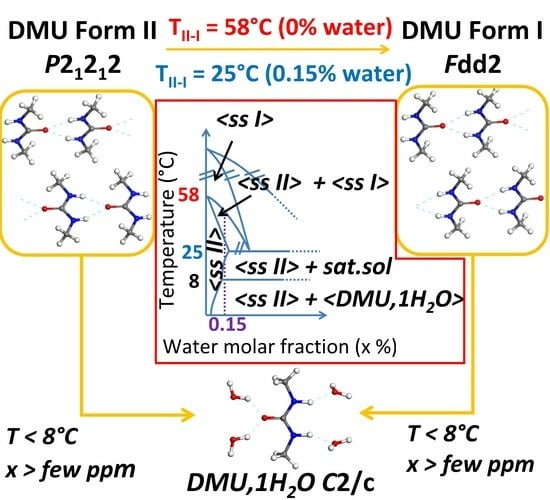
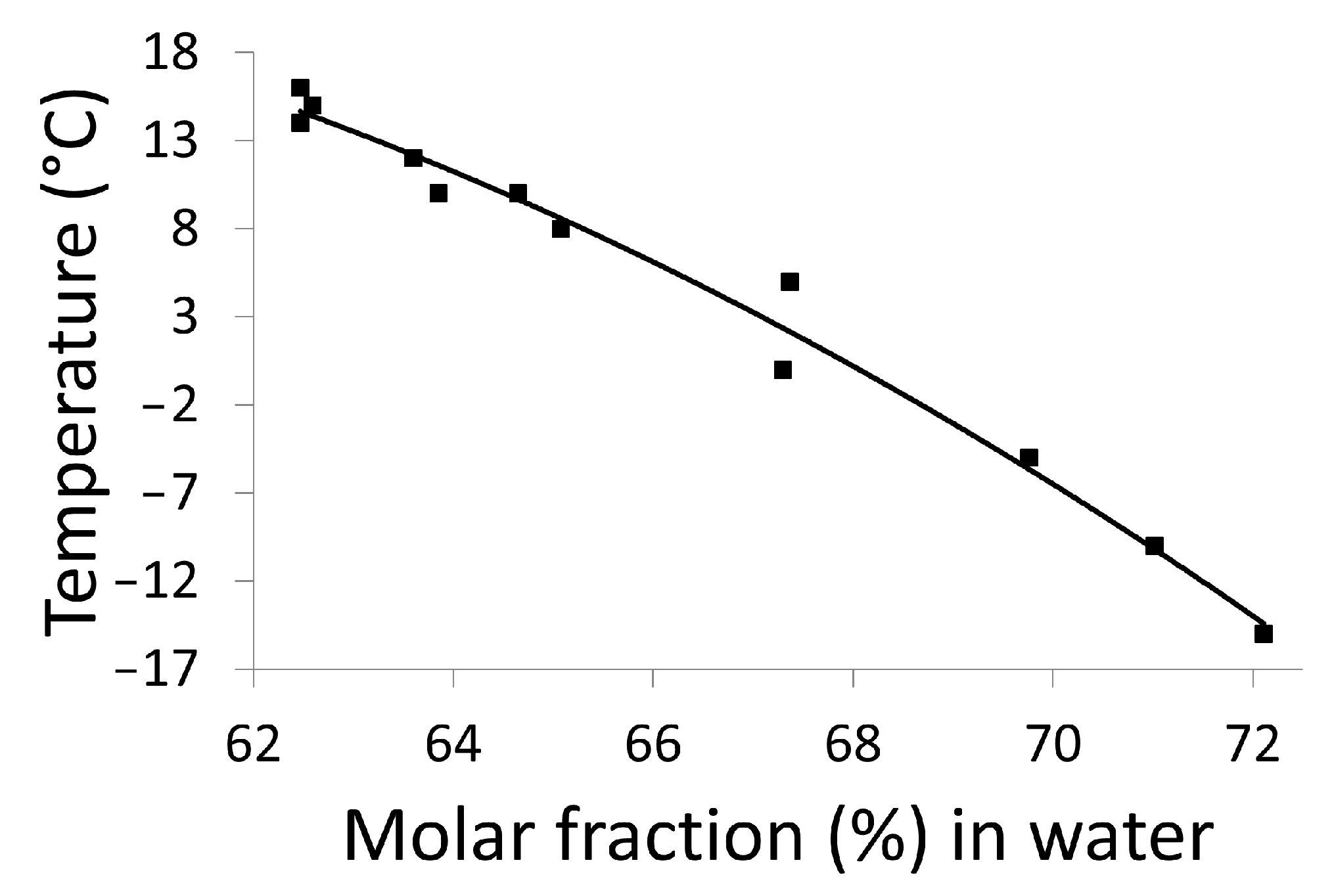
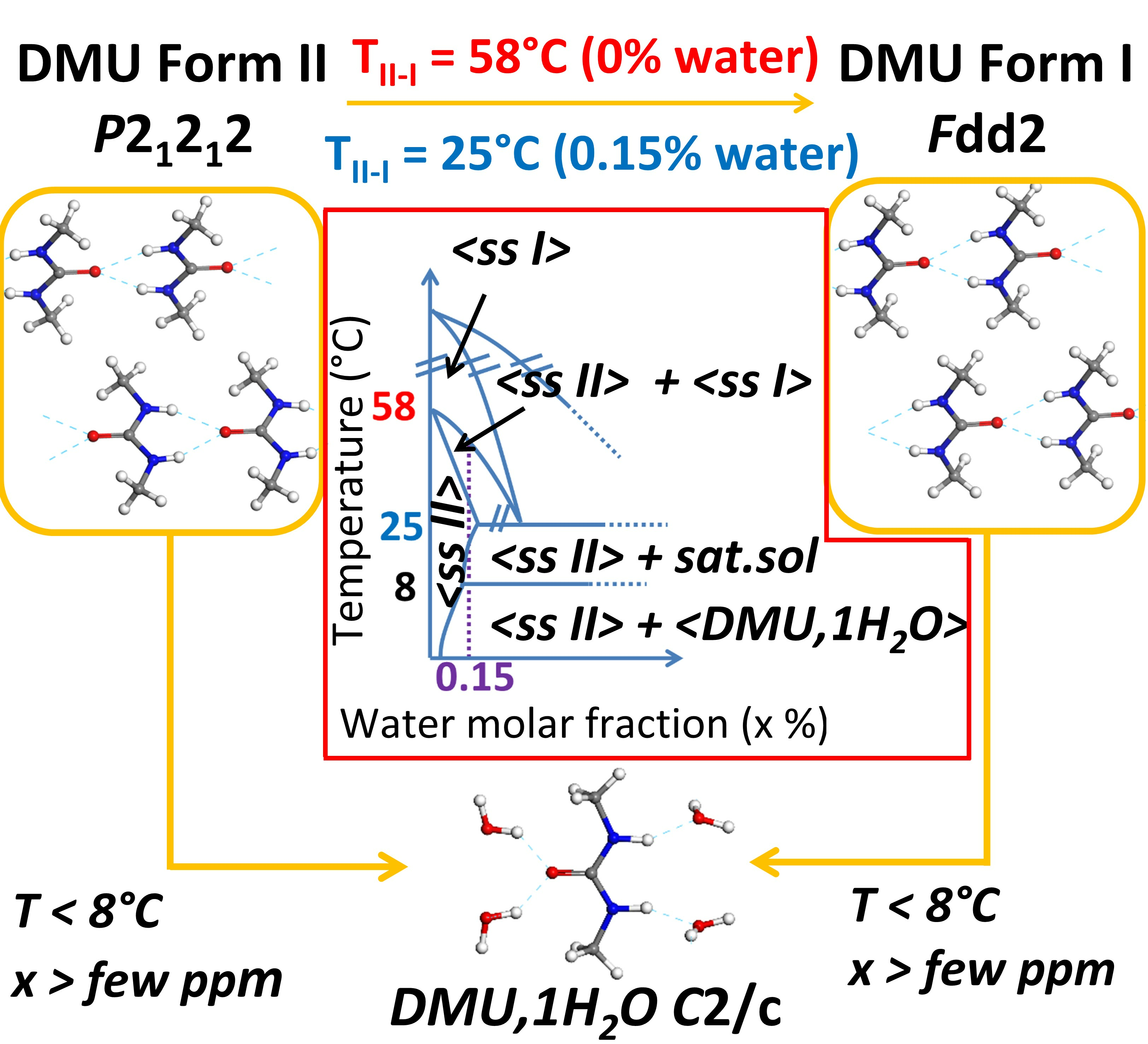
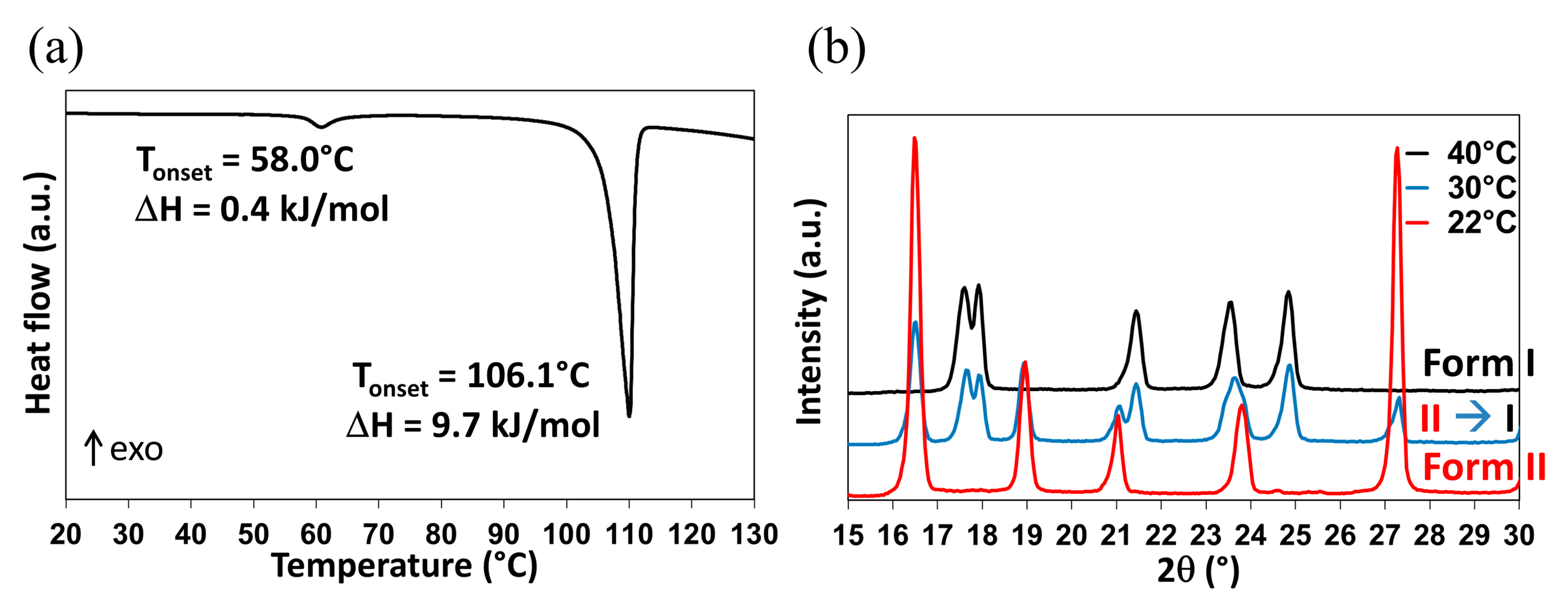
2.1. Influence of Water on DMU Form II—Form I Solid–Solid Transition Temperature
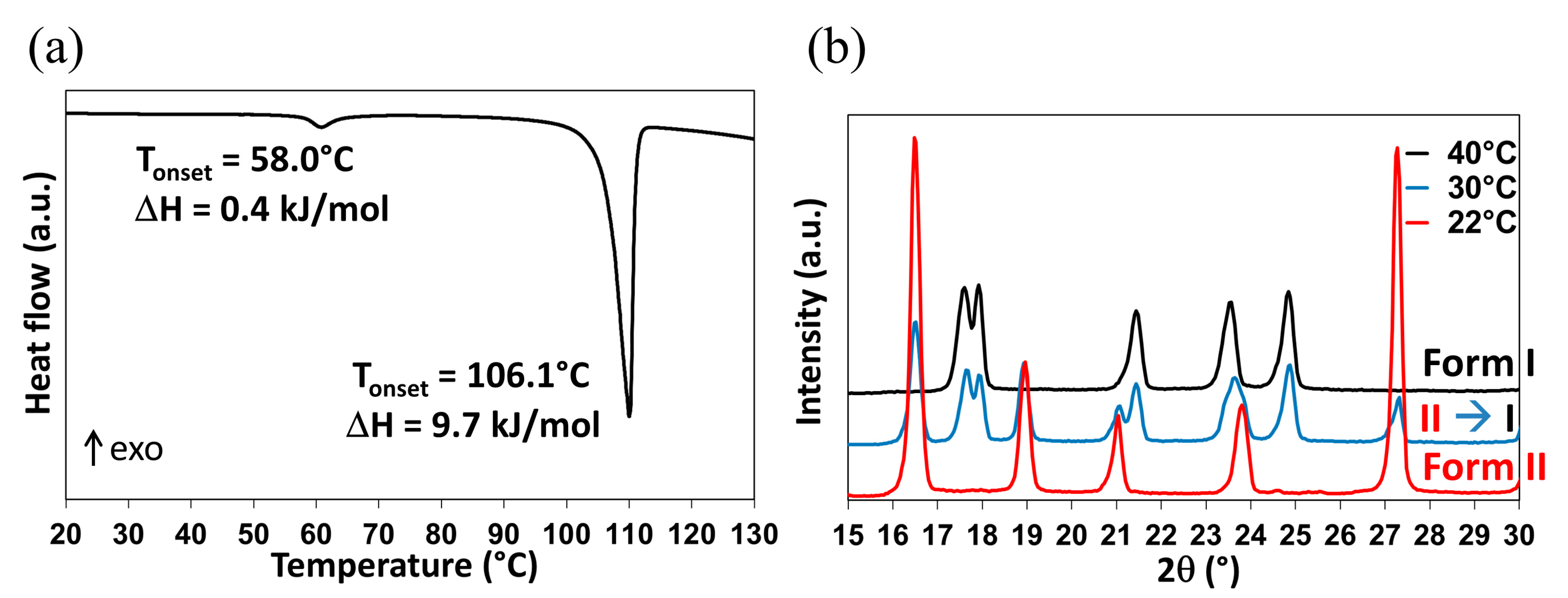
2.1.1. Re-Investigation of the Temperature of Transition
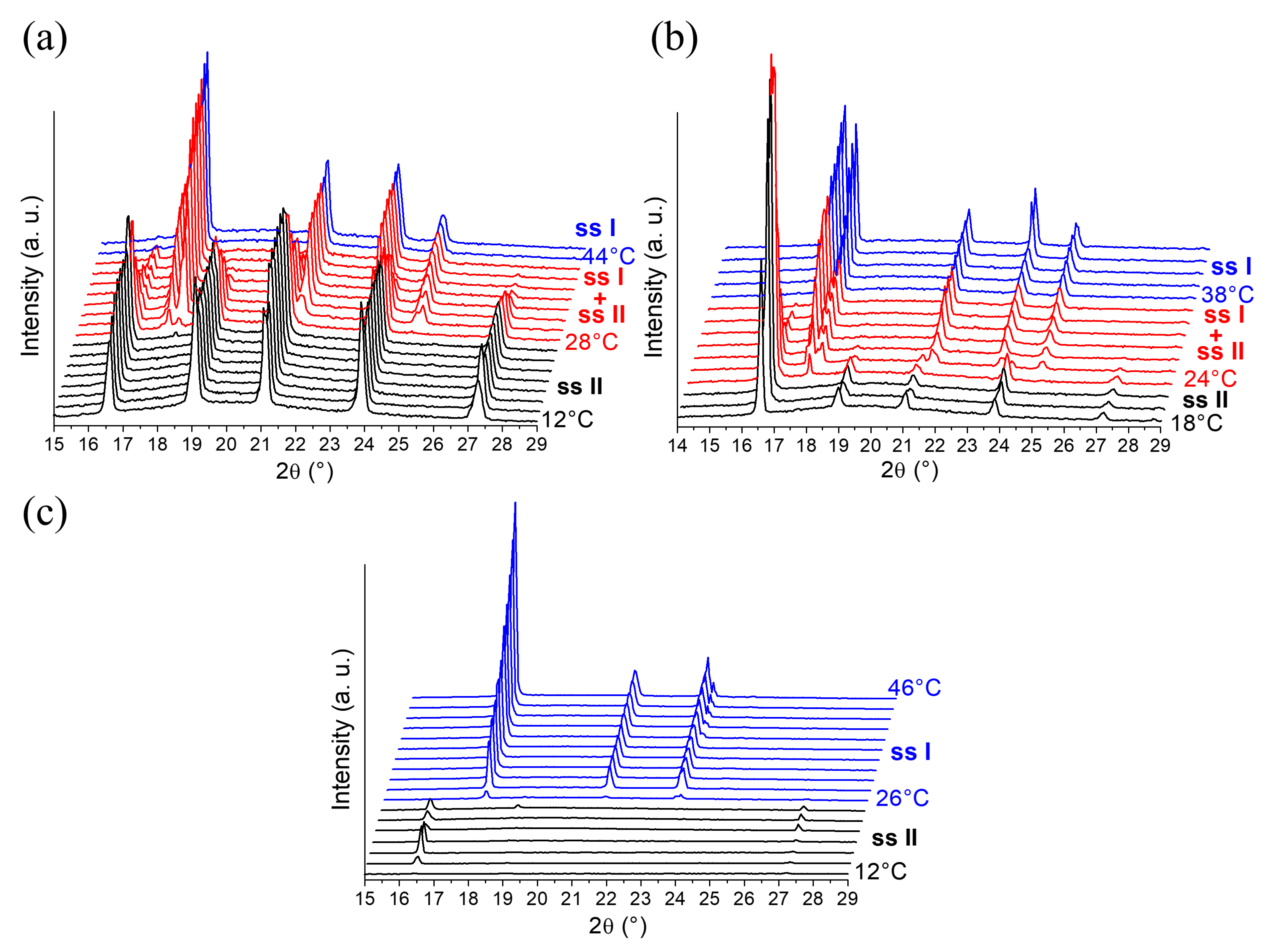
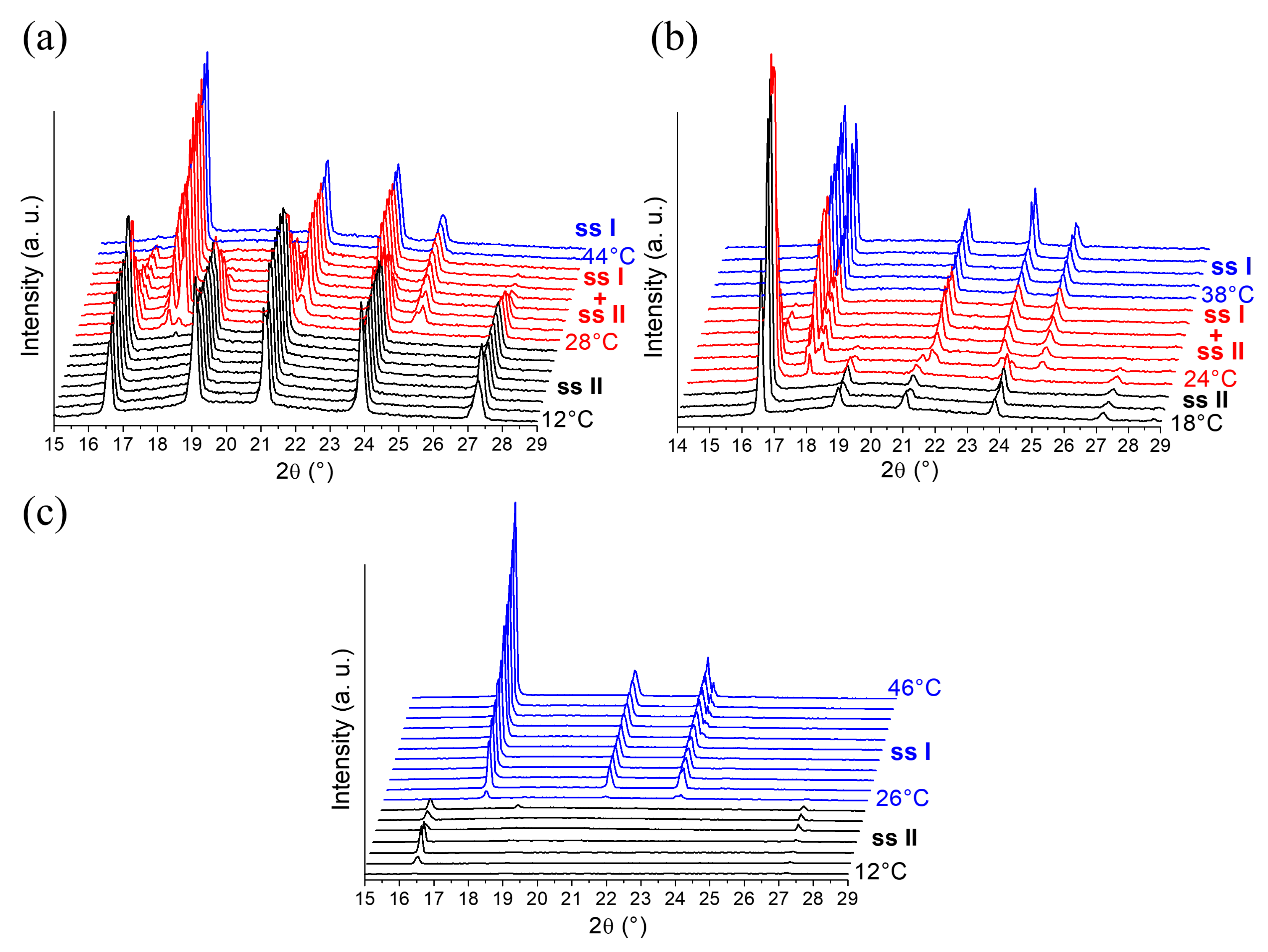
2.1.2. In-situX® Analyses of DMU Samples Crystallized in the Presence of Water
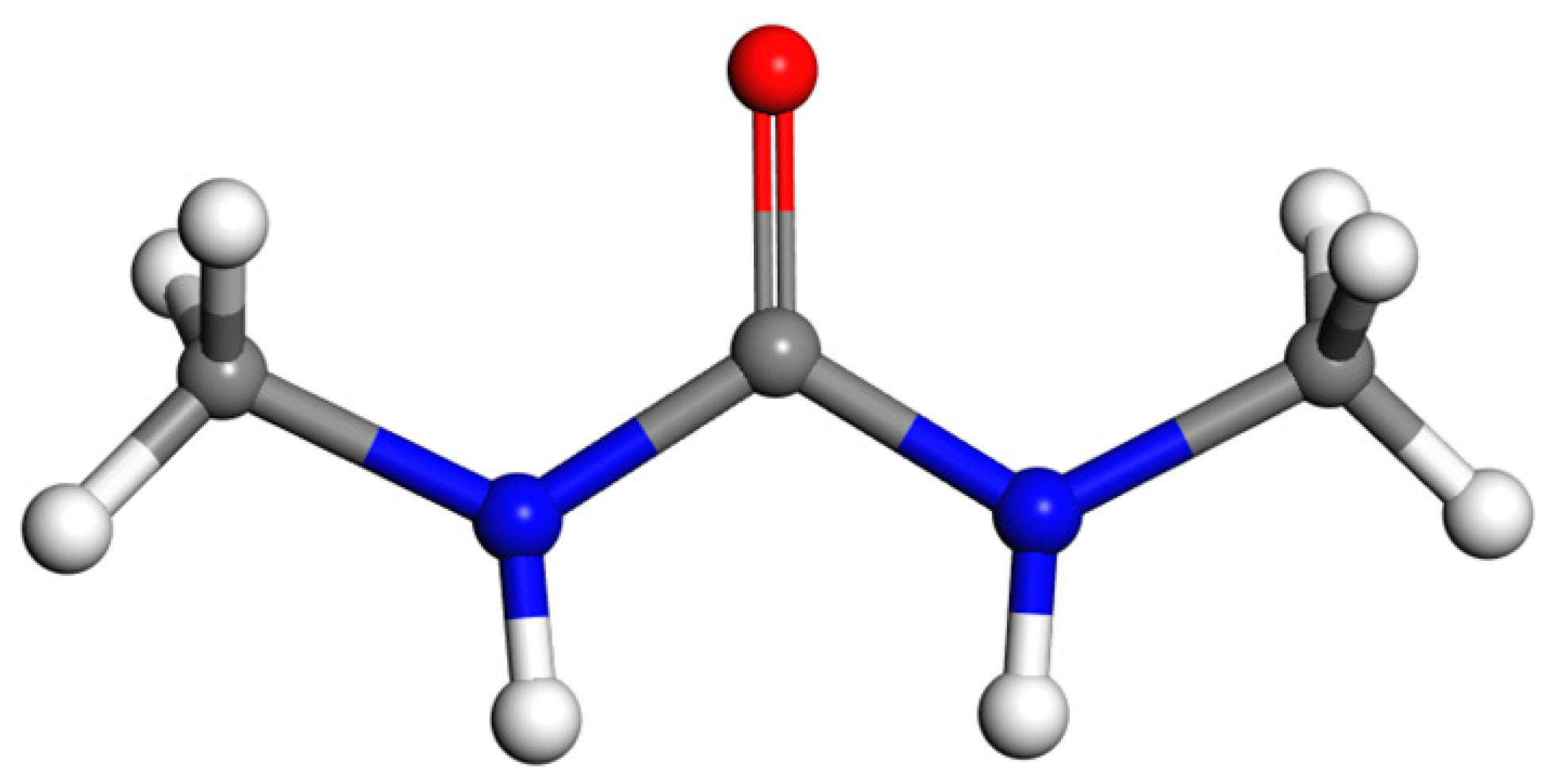
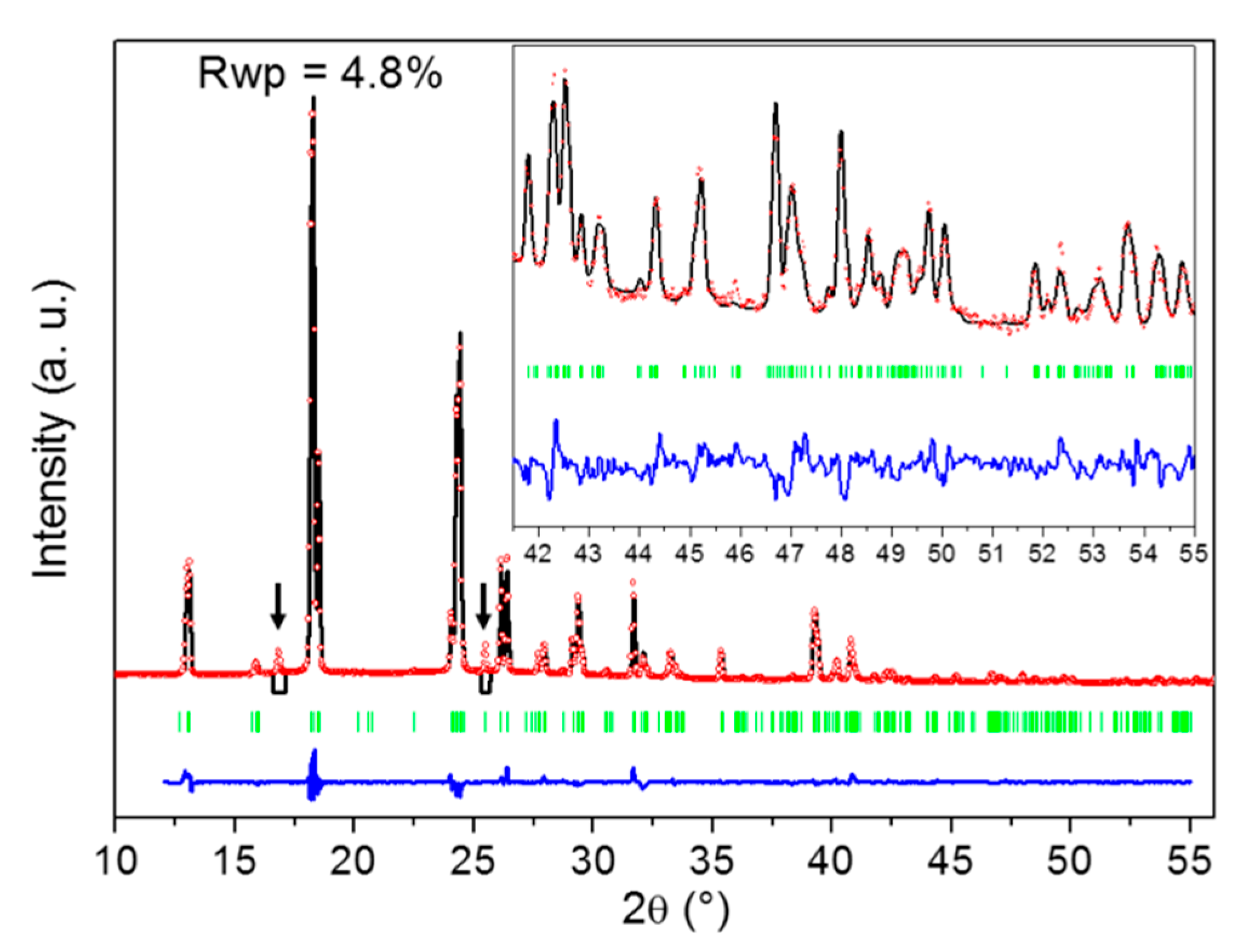
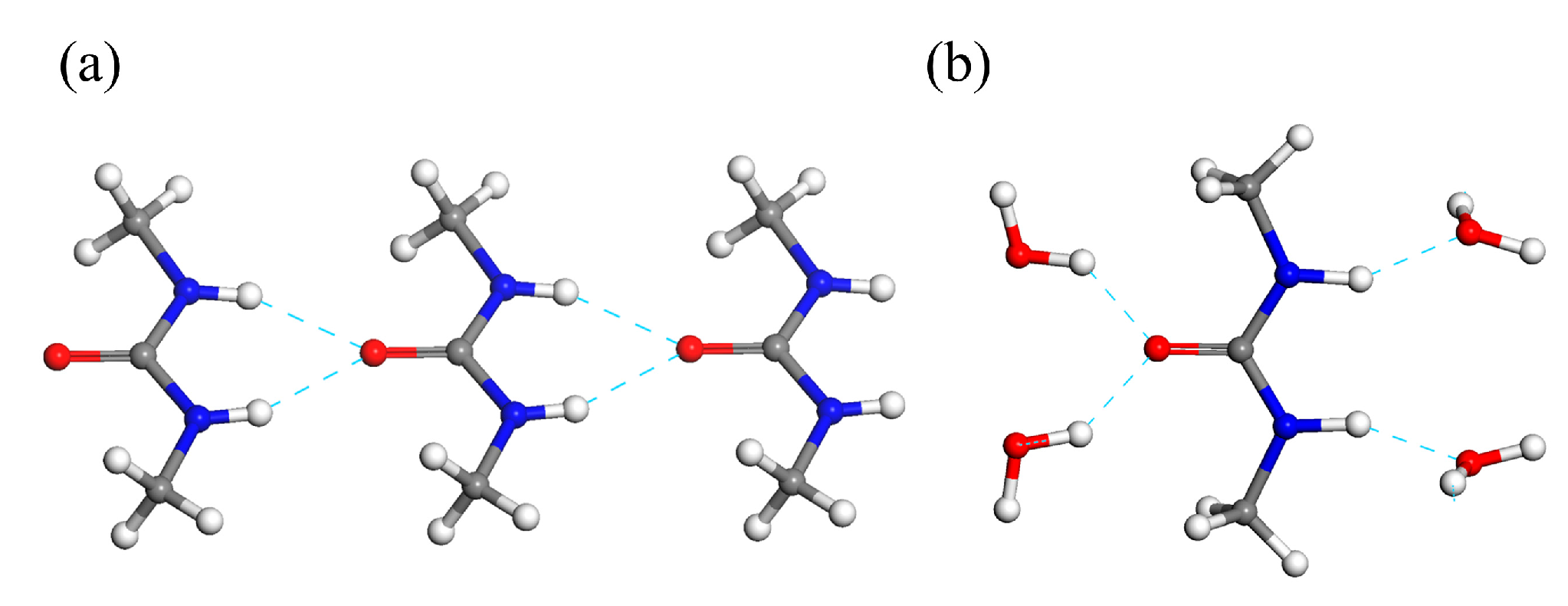
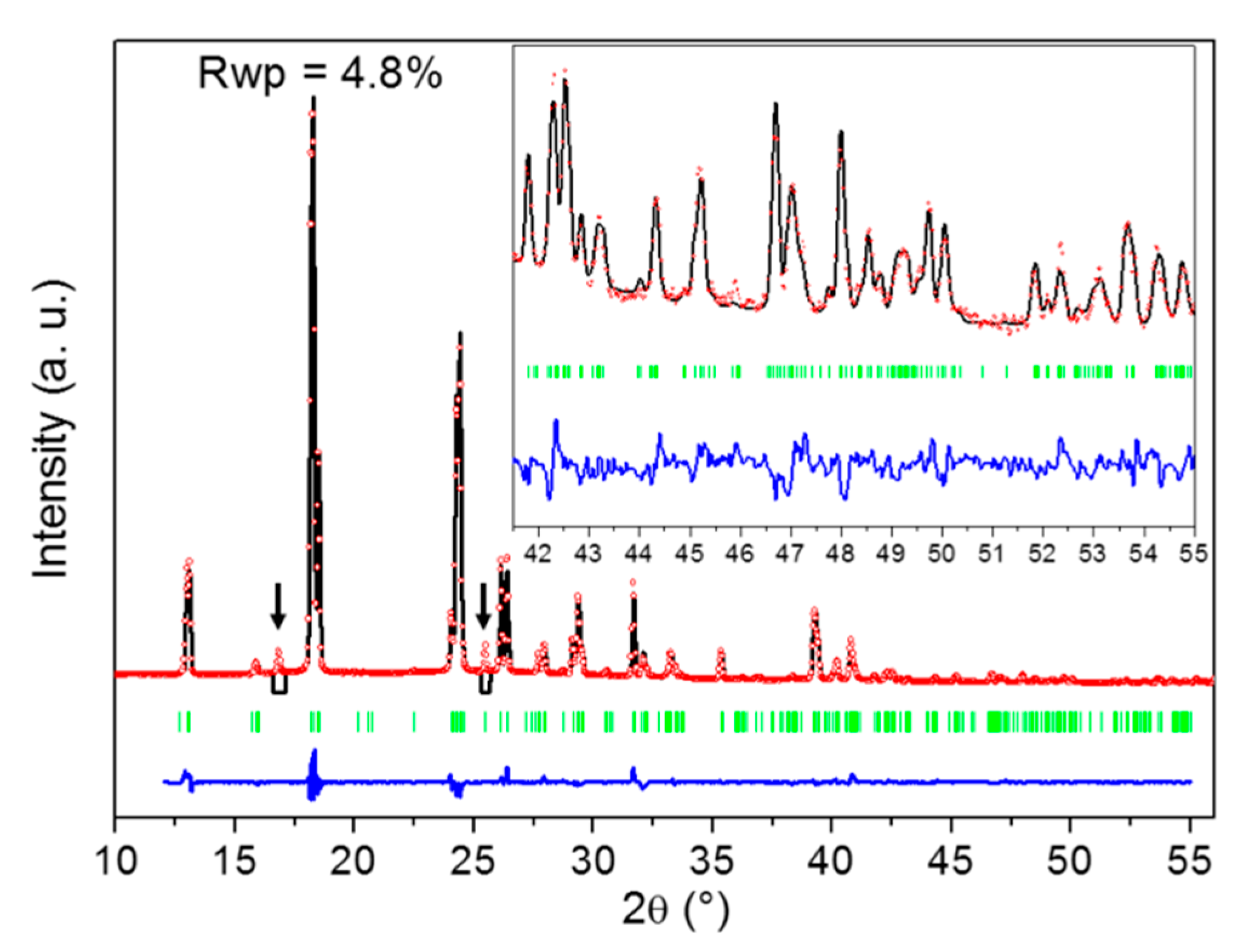
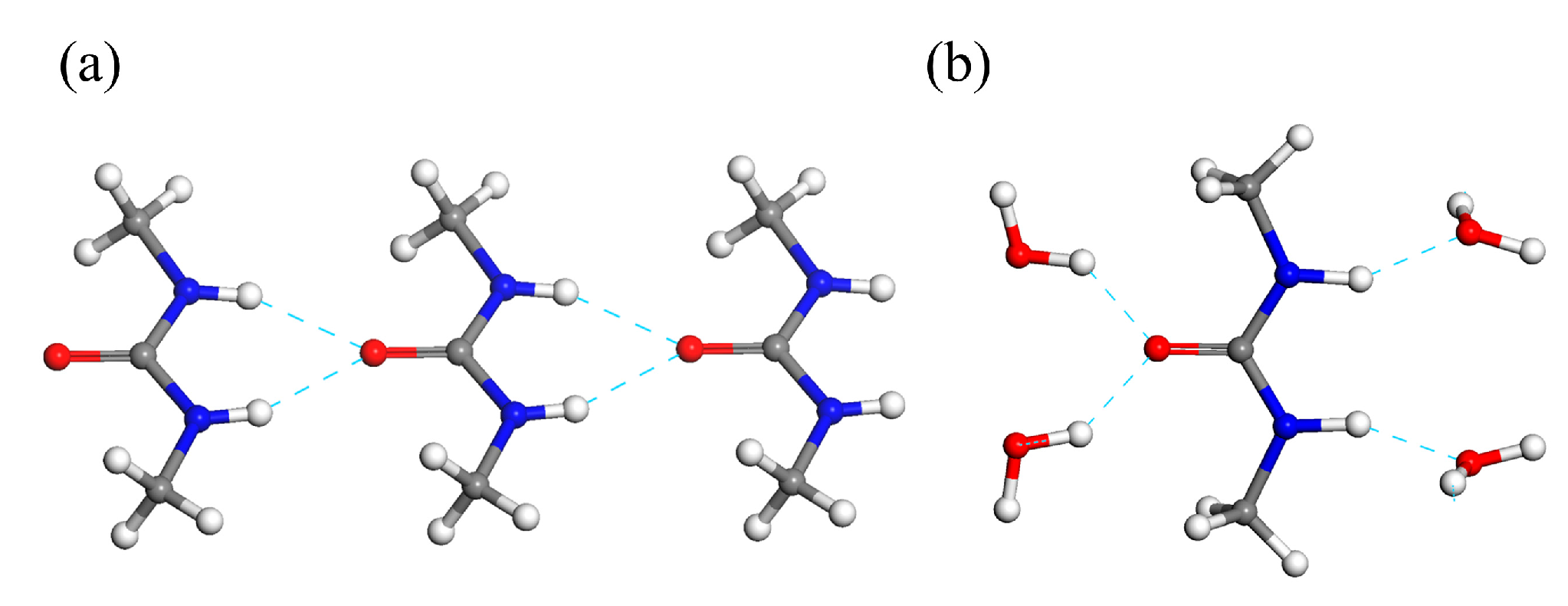
2.2. Identification of the DMU Monohydrate and Resolution of Its Crystal Structure
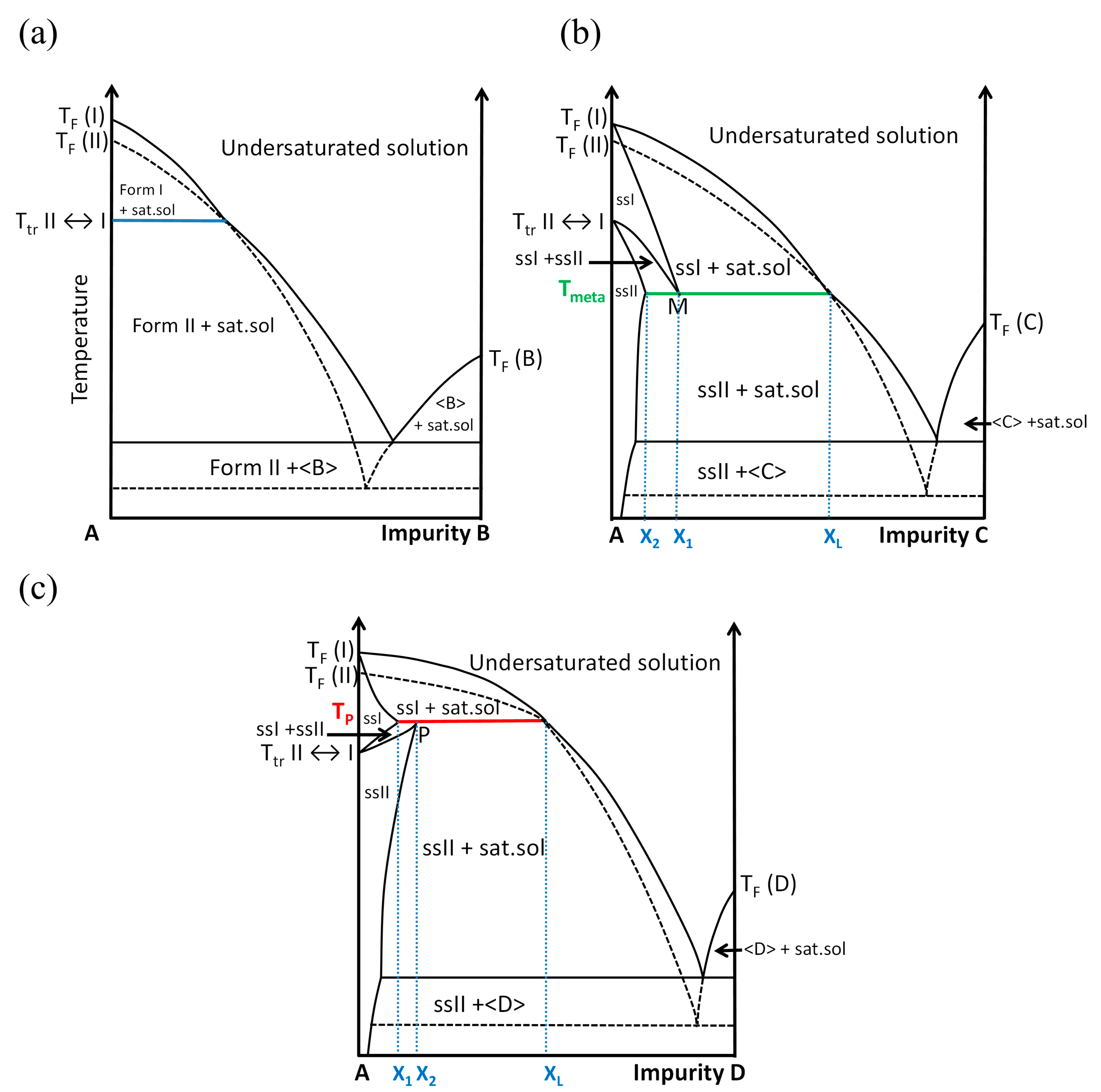
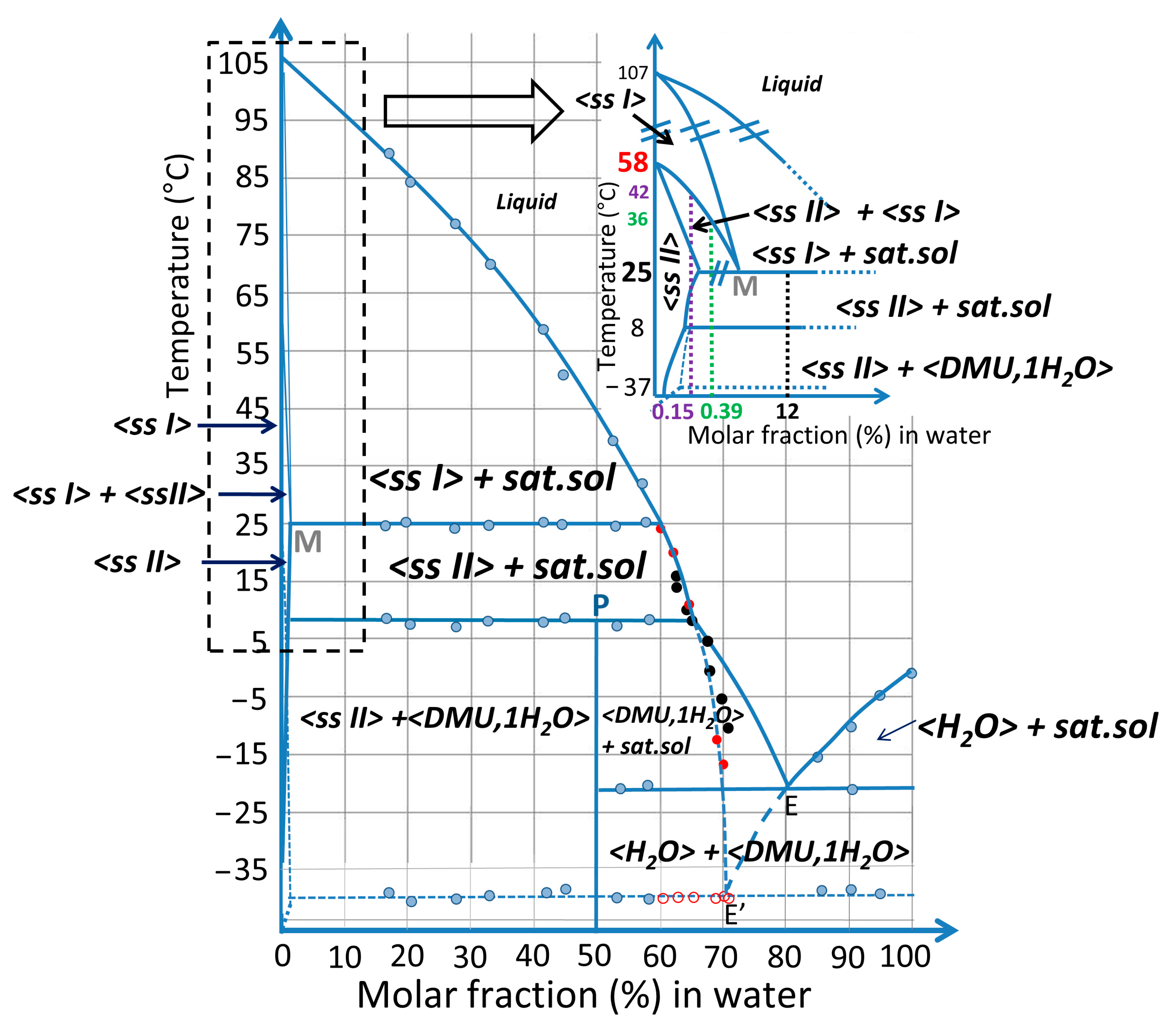
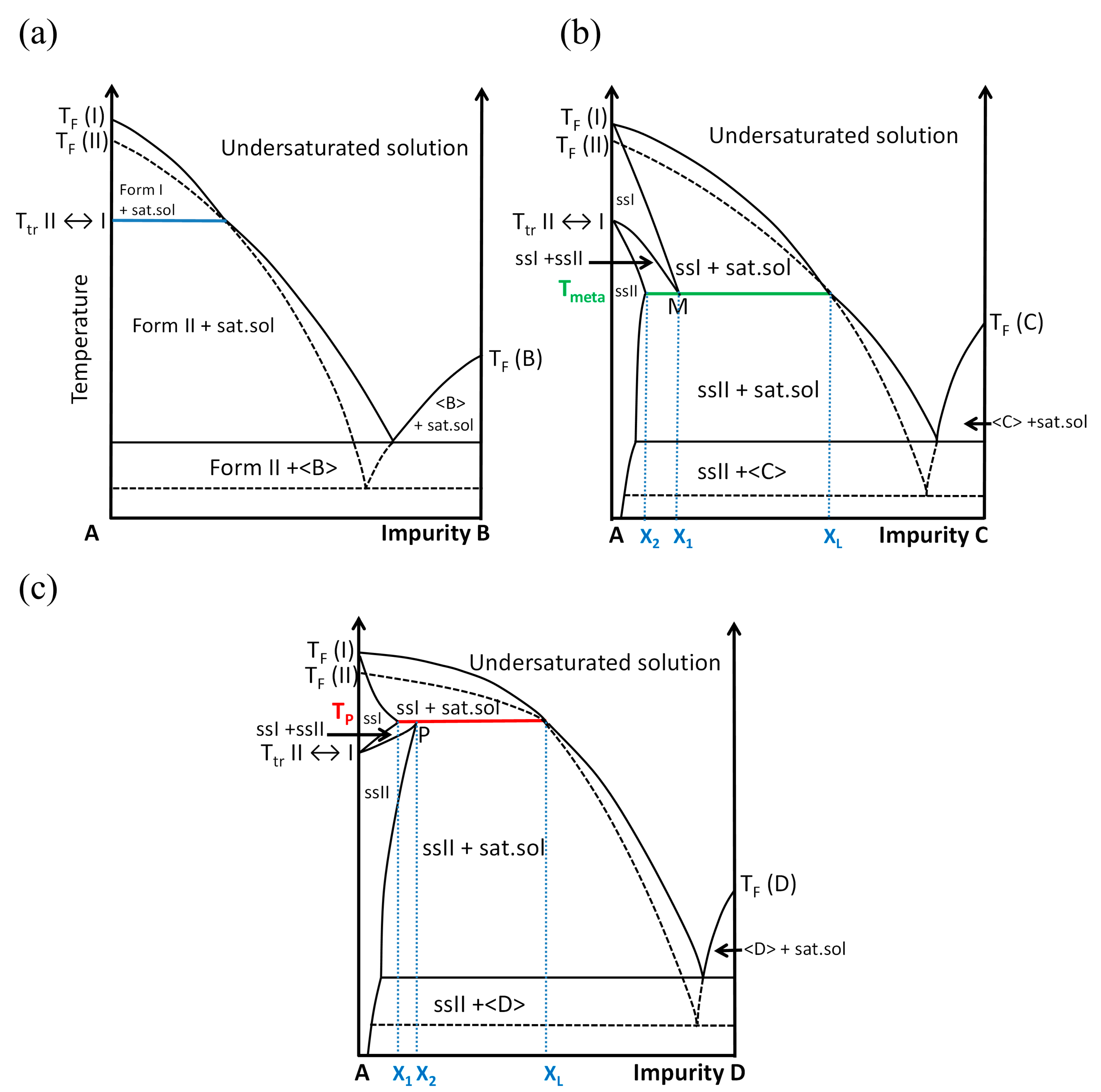
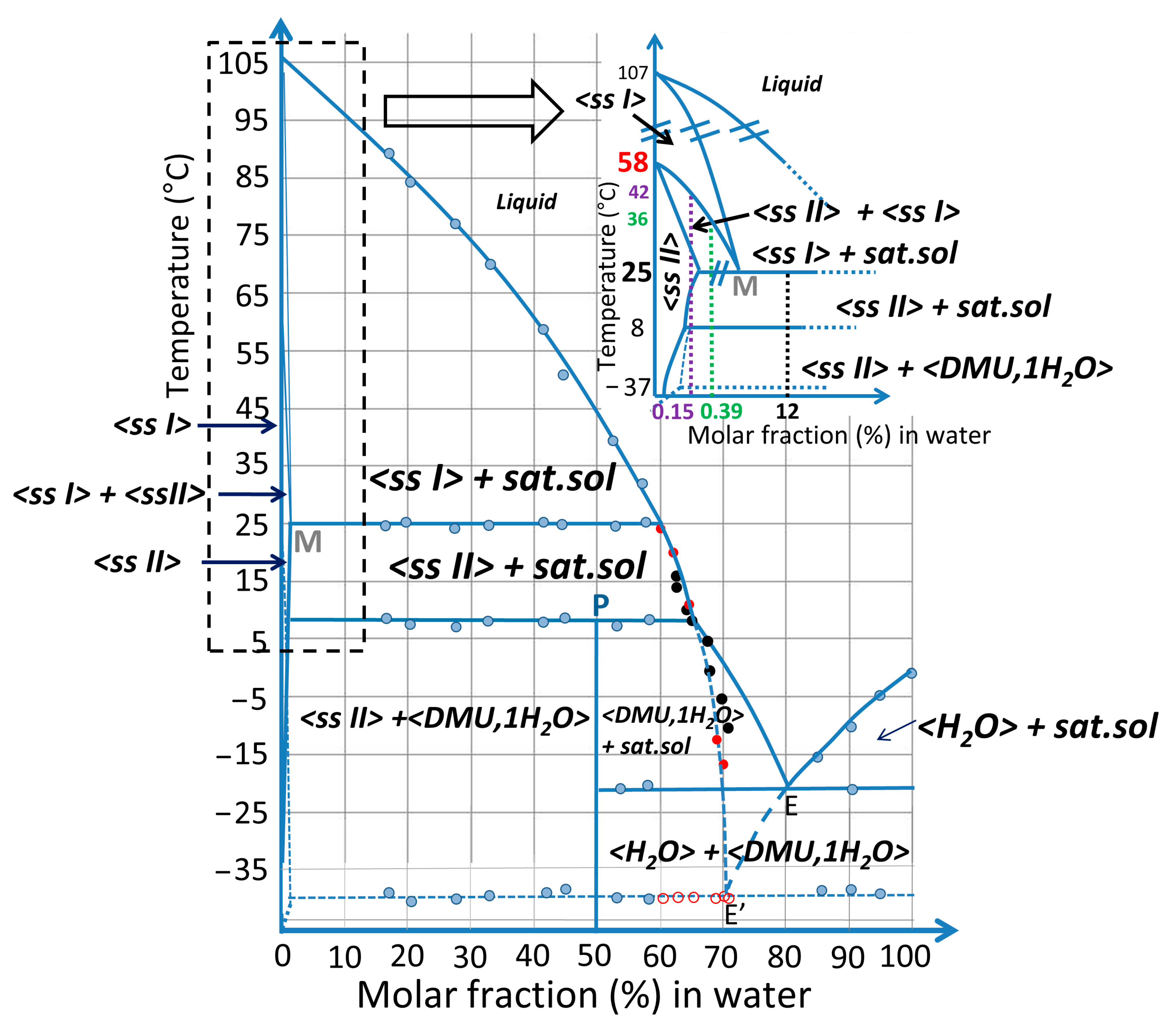
2.3. Exploring the Complete DMU–Water Phase Diagram
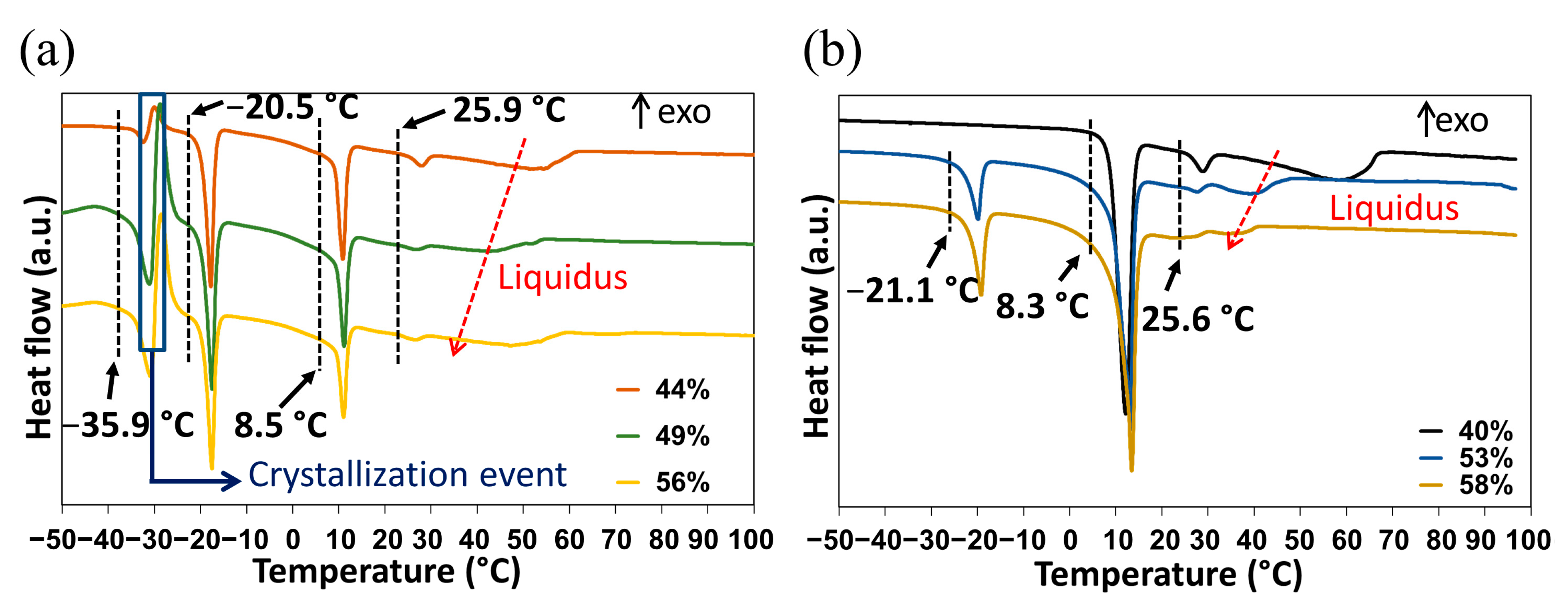
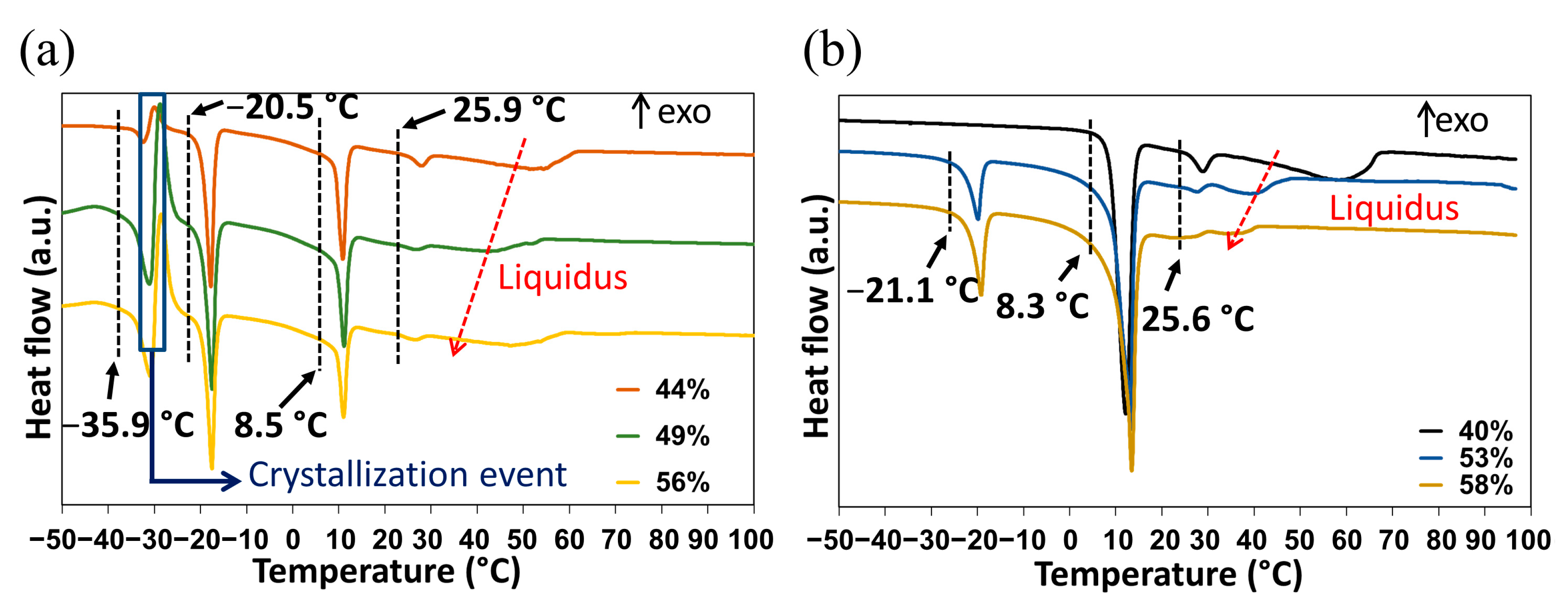
2.3.1. DSC Analyses of Closed Crucibles Enriched with Water
- An endotherm located at circa −36 °C directly followed by an exothermic phenomenon (recrystallization, emphasized by the blue rectangle in Figure 7a). Hence, this endotherm would likely belong to a metastable equilibrium.
- An endotherm at −20 °C can be assigned to the presence of a eutectic equilibrium between the monohydrate and ice.
- A third endotherm located at circa 8 °C, the maximum heat exchange of which is observed for a molar composition in water of 50% (see Supporting Information Figure S8) that can be fairly attributed to the non-congruent fusion of the monohydrate.
- A fourth endotherm at approximately 25 °C which corresponds to the metatectic invariant also previously revealed by In-situX® measurements (see Section 2.1.2).
- A fifth endotherm located at different temperatures corresponding to the end of fusion of the solid solution I (e.g., liquidus line).
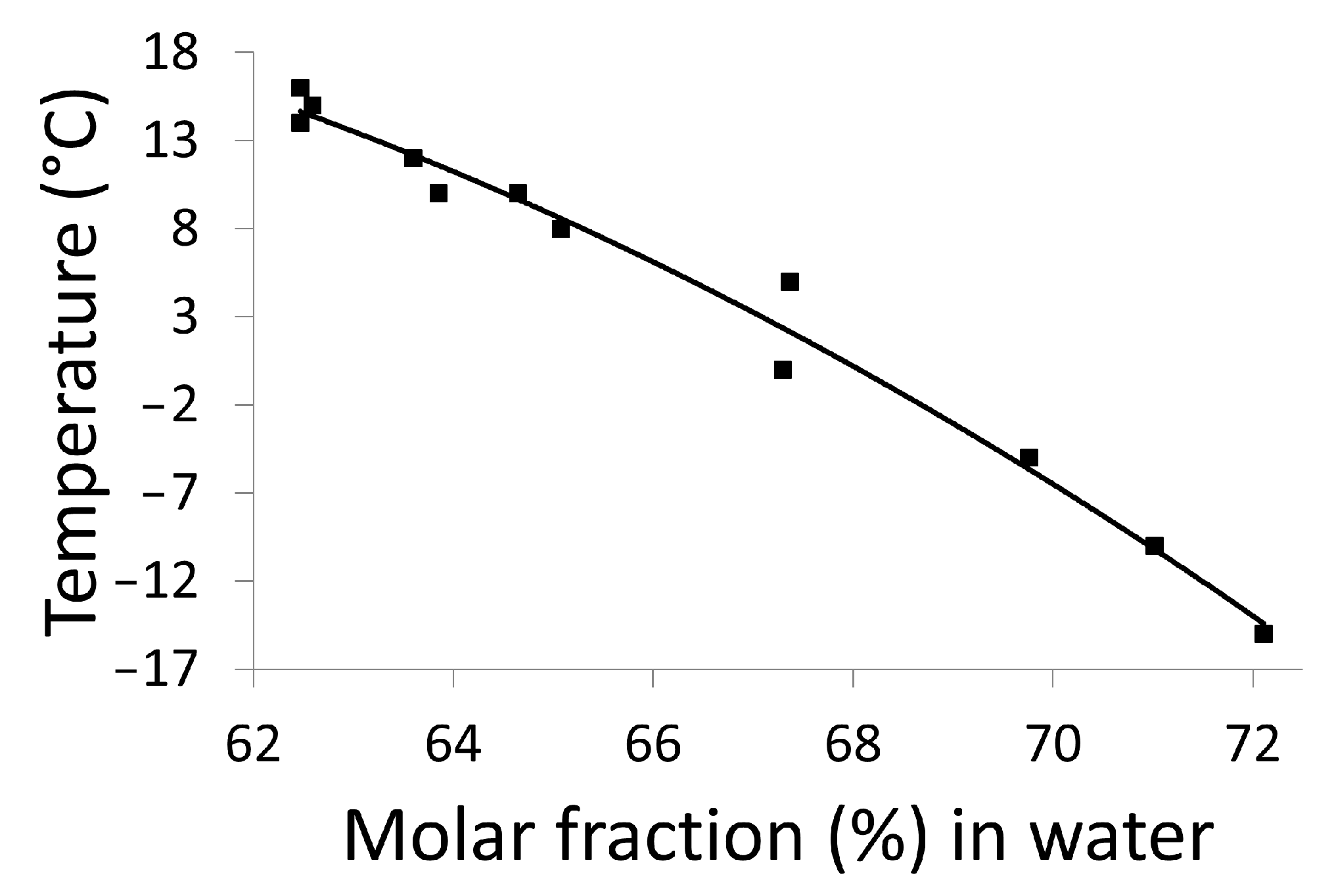
2.3.2. Refractometry Measurements
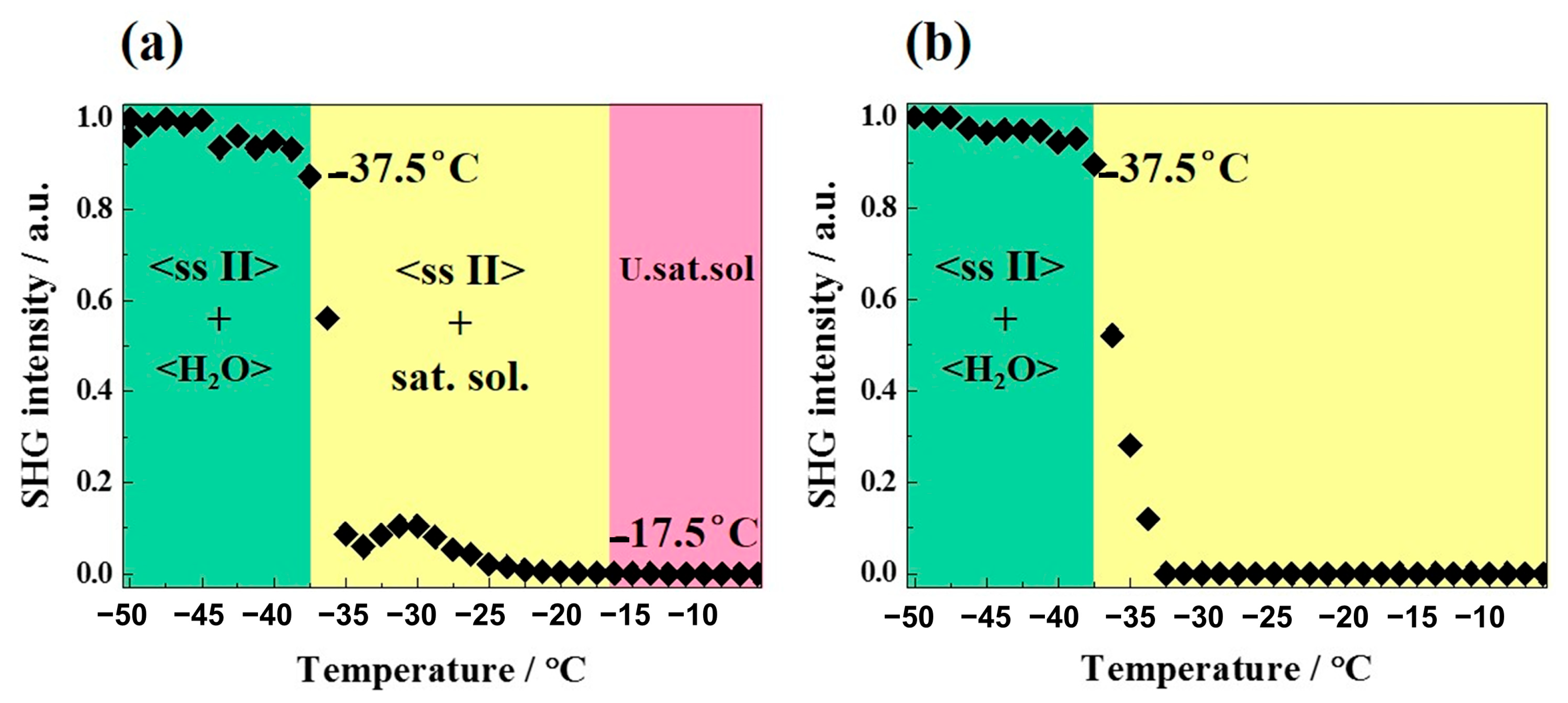
2.3.3. Refinement of the Metastable Eutectic Composition of DMU–Water Binary System by Temperature-Resolved Second Harmonic Generation (TR-SHG)
2.3.4. Plotting the DMU–Water Binary Phase Diagram
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. In-situX® Analyses and Compositions Preparation
4.2.2. DSC Measurements and Compositions Preparation
4.2.3. Temperature-Resolved Second Harmonic Generation (TR-SHG) and Mixtures Preparation
4.2.4. Refractometry Analyses
4.2.5. Temperature-Resolved X-ray Powder Diffraction (TR-XRPD)
4.2.6. Crystal Structure Determination from X-ray Powder Diffraction Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Augsburger, L.L.; Hoag, S.W. (Eds.) Pharmaceutical Dosage Forms: Tablets. Rational Design and Formulation, 3rd ed.; Informa Healthcare: New York, NY, USA, 2008. [Google Scholar]
- Chakravarty, P.; Nagapudi, K. The Importance of Water-Solid Interactions in Small Molecule Drug Development: An Industry Perspective. TrAC Trends Anal. Chem. 2021, 140, 116276. [Google Scholar] [CrossRef]
- Sacchetti, M. Thermodynamics of Water–Solid Interactions in Crystalline and Amorphous Pharmaceutical Materials. J. Pharm. Sci. 2014, 103, 2772–2783. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.W.; Reutzel-Edens, S.M.; Zografi, G. Characterization of the “Hygroscopic” Properties of Active Pharmaceutical Ingredients. J. Pharm. Sci. 2008, 97, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M. The Interaction of Water with Solid Surfaces: Fundamental Aspects Revisited. Surf. Sci. Rep. 2002, 46, 1–308. [Google Scholar] [CrossRef]
- Ahlneck, C.; Zografi, G. The Molecular Basis of Moisture Effects on the Physical and Chemical Stability of Drugs in the Solid State. Int. J. Pharm. 1990, 62, 87–95. [Google Scholar] [CrossRef]
- Brittain, H.G. (Ed.) Polymorphism in Pharmaceutical Solids, 2nd ed.; Drugs and the Pharmaceutical Sciences; Informa Healthcare: New York, NY, USA, 2009. [Google Scholar]
- Reutzel-Edens, S.M.; Braun, D.E.; Newman, A.W. Hygroscopicity and Hydrates in Pharmaceutical Solids. In Polymorphism in the Pharmaceutical Industry; Hilfiker, R., Raumer, M.V., Eds.; Wiley: Hoboken, NJ, USA, 2018; pp. 159–188. [Google Scholar] [CrossRef]
- Morrison, H.; Quan, B.P.; Walker, S.D.; Hansen, K.B.; Nagapudi, K.; Cui, S. Appearance of a New Hydrated Form during Development: A Case Study in Process and Solid-State Optimization. Org. Process Res. Dev. 2015, 19, 1842–1848. [Google Scholar] [CrossRef]
- Khankari, R.K.; Grant, D.J.W. Pharmaceutical Hydrates. Thermochim. Acta 1995, 248, 61–79. [Google Scholar] [CrossRef]
- Holmes, S.T.; Vojvodin, C.S.; Veinberg, N.; Iacobelli, E.M.; Hirsh, D.A.; Schurko, R.W. Hydrates of Active Pharmaceutical Ingredients: A 35Cl and 2H Solid-State NMR and DFT Study. Solid State Nucl. Magn. Reson. 2022, 122, 101837. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, E.; Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M.; Zielińska-Pisklak, M. Pharmaceutical Hydrates Analysis—Overview of Methods and Recent Advances. Pharmaceutics 2020, 12, 959. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, S.; Karjalainen, M.; Shevchenko, A.; Westermarck, S.; Leppänen, E.; Rantanen, J.; Yliruusi, J. Role of Water in the Physical Stability of Solid Dosage Formulations. J. Pharm. Sci. 2005, 94, 2147–2165. [Google Scholar] [CrossRef] [PubMed]
- Steendam, R.; Frijlink, H.W.; Lerk, C.F. Plasticisation of Amylodextrin by Moisture. Consequences for Compaction Behaviour and Tablet Properties. Eur. J. Pharm. Sci. 2001, 14, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Shalaev, E.Y.; Zografi, G. How Does Residual Water Affect the Solid-State Degradation of Drugs in the Amorphous State? J. Pharm. Sci. 1996, 85, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Zelkó, R.; Szakonyi, G. The Effect of Water on the Solid State Characteristics of Pharmaceutical Excipients: Molecular Mechanisms, Measurement Techniques, and Quality Aspects of Final Dosage Form. Int. J. Pharm. Investig. 2012, 2, 18. [Google Scholar] [CrossRef]
- Coquerel, G. Thermodynamic Predictions of Physical Properties—Prediction of Solid Solutions in Molecular Solutes Exhibiting Polymorphism. Chem. Eng. Technol. 2006, 29, 182–186. [Google Scholar] [CrossRef]
- Kochergin, P.M.; Aleksandrova, E.V.; Persanova, L.V. Rational Chemical Schemes for the Synthesis of Medicinal Preparations of the Purine Series (a Review). Pharm. Chem. J. 2001, 35, 388–392. [Google Scholar] [CrossRef]
- Sids Initial Assessment Report for Siam 17, 11–14 November 2003, Arona, Italy. Available online: https://hpvchemicals.oecd.org/ui/handler.axd?id=c3671b32-0e7a-45e9-932c-ee30f71e893d (accessed on 16 January 2017).
- Wei, Y.; Zhao, Q.; Yu, S.; Yin, J. Thermal Stability and Thermodynamics in the Process of Synthesising 1, 3-Dimethylurea. Chem. Eng. Trans. 2020, 81, 667–672. [Google Scholar] [CrossRef]
- Baaklini, G.; Dupray, V.; Coquerel, G. Inhibition of the Spontaneous Polymorphic Transition of Pyrazinamide γ Form at Room Temperature by Co-Spray Drying with 1,3-Dimethylurea. Int. J. Pharm. 2015, 479, 163–170. [Google Scholar] [CrossRef]
- Näther, C.; Döring, C.; Jess, I.; Jones, P.G.; Taouss, C. Thermodynamic and Structural Relationships between the Two Polymorphs of 1,3-Dimethylurea. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 70–76. [Google Scholar] [CrossRef]
- Couvrat, N.; Martins, D.; Lafontaine, A.; Sanselme, M.; Dupray, V.; Taulelle, P.; Lerestif, J.-M.; Lynch, M.; Vaysse-Ludot, L.; Coquerel, G. Partial Blockage of the Reversible Solid-Solid Transition of Strontium Succinate. Chem. Eng. Technol. 2016, 39, 1224–1230. [Google Scholar] [CrossRef]
- Bowlan, P.; Henson, B.F.; Smilowitz, L.; Levitas, V.I.; Suvorova, N.; Oschwald, D. Kinetics of the γ–δ Phase Transition in Energetic Nitramine-Octahydro-1,3,5,7-Tetranitro-1,3,5,7-Tetrazocine. J. Chem. Phys. 2019, 150, 064705. [Google Scholar] [CrossRef]
- Misane, L.; El Allali, S.; Kaddami, M.; Zrineh, A.; Tenu, R.; Berthet, J.; Counioux, J.J. Etude Du Systeme Quasi Ternaire H2O–NH4NO3–Mg(NO3)2·6H2O: Isotherme 60 °C, Coupe Quasi Binaire NH4NO3–Mg(NO3)2·6H2O et Diagramme Polythermique. Thermochim. Acta 2000, 354, 135–144. [Google Scholar] [CrossRef]
- Clow, A. Deliquescence in Urea and Methyl-Ureas. Nature 1940, 146, 26. [Google Scholar] [CrossRef]
- Yuan, L.; Clevers, S.; Couvrat, N.; Cartigny, Y.; Dupray, V.; Coquerel, G. Precise Urea/Water Eutectic Composition by Temperature-Resolved Second Harmonic Generation. Chem. Eng. Technol. 2016, 39, 1326–1332. [Google Scholar] [CrossRef]
- Yuan, L.; Clevers, S.; Burel, A.; Negrier, P.; del Barrio, M.; Ben Hassine, B.; Mondieig, D.; Dupray, V.; Tamarit, J.L.; Coquerel, G. New Intermediate Polymorph of 1-Fluoro-Adamantane and Its Second-Order-like Transition toward the Low Temperature Phase. Cryst. Growth Des. 2017, 17, 3395–3401. [Google Scholar] [CrossRef]
- Xu, J.; Chen, A.; Cai, T. Polymorphism of Purpurin and Low-Level Detection of the Noncentrosymmetric Form by Second Harmonic Generation Microscopy. J. Pharm. Sci. 2023, 112, 282–289. [Google Scholar] [CrossRef]
- Sherman, A.M.; Takanti, N.; Rong, J.; Simpson, G.J. Nonlinear Optical Characterization of Pharmaceutical Formulations. TrAC Trends Anal. Chem. 2021, 140, 116241. [Google Scholar] [CrossRef]
- Dougherty, J.P.; Kurtz, S.K. A Second Harmonic Analyzer for the Detection of Non-Centrosymmetry. J. Appl. Crystallogr. 1976, 9, 145–158. [Google Scholar] [CrossRef]
- Kurtz, S.K.; Perry, T.T. A Powder Technique for the Evaluation of Nonlinear Optical Materials. J. Appl. Phys. 1968, 39, 3798–3813. [Google Scholar] [CrossRef]
- Coquerel, G.; Sanselme, M.; Lafontaine, A. Method and Measuring Scattering of X-Rays, Its Applications and Implementation Device. WO2012/136921A1, 2 April 2012. [Google Scholar]
- Evain, M.; Deniard, P.; Jouanneaux, A.; Brec, R. Potential of the INEL X-Ray Position-Sensitive Detector: A General Study of the Debye–Scherrer Setting. J. Appl. Crystallogr. 1993, 26, 563–569. [Google Scholar] [CrossRef]
- Huang, T.C.; Toraya, H.; Blanton, T.N.; Wu, Y. X-Ray Powder Diffraction Analysis of Silver Behenate, a Possible Low-Angle Diffraction Standard. J. Appl. Crystallogr. 1993, 26, 180–184. [Google Scholar] [CrossRef]
- Neumann, M.A. X-Cell: A Novel Indexing Algorithm for Routine Tasks and Difficult Cases. J. Appl. Crystallogr. 2003, 36, 356–365. [Google Scholar] [CrossRef]
- Pawley, G.S. Unit-Cell Refinement from Powder Diffraction Scans. J. Appl. Crystallogr. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Engel, G.E.; Wilke, S.; König, O.; Harris, K.D.M.; Leusen, F.J.J. PowderSolve—A Complete Package for Crystal Structure Solution from Powder Diffraction Patterns. J. Appl. Crystallogr. 1999, 32, 1169–1179. [Google Scholar] [CrossRef]
- MS Modeling (Materials Studio), Version 5.5. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/ (accessed on 4 October 2016).
- Rietveld, H.M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Toraya, H.; Marumo, F. Preferred Orientation Correction in Powder Pattern-Fitting. Mineral. J. 1981, 10, 211–221. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Form | DMU Monohydrate | DMU Form I [22] | DMU Form II [22] |
|---|---|---|---|
| M (g/mol) | 106.13 | 88.11 | 88.11 |
| Temperature (°C) | −20 | −93 | −173 |
| CCDC number | 1966048 | 924553 | 924554 |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | C2/c | Fdd2 | P21212 |
| a (Å) | 19.1479 (8) | 11.3837 (2) | 10.8522 (6) |
| b (Å) | 13.9557 (5) | 19.6293 (4) | 4.9102 (3) |
| c (Å) | 13.4926 (4) | 4.5608 (1) | 4.5766 (3) |
| β (°) | 134.644 (2) | 90 | 90 |
| Z | 16 | 8 | 2 |
| Z’ | 2 | 0.5 | 0.5 |
| Volume (Å3) | 2565 (1) | 1019 (1) | 244 (1) |
| Density (g/cm3) | 1.099 (1) | 1.149 (1) 1.120 (1) * | 1.200 (1) 1.151 (1) * |
| DMU Monohydrate | DMU Form I [22] | DMU Form II [22] | |||
|---|---|---|---|---|---|
| Type A Chains | Type B Chains | ||||
| O∙∙∙O distances (Å) | ODMU∙∙∙O1w | 2.76 (2) | 2.71 (2) | ||
| ODMU∙∙∙O2w | 2.74 (2) | 2.77 (2) | |||
| O∙∙∙N distances (Å) | O3w∙∙∙N1DMU | 2.83 (2) | 3.00 (2) | 2.85 (2) | 2.86 (2) |
| O4w∙∙∙N2DMU | 2.88 (2) | 2.82 (2) | |||
| N–H∙∙∙O angles (°) | N1DMU–H1DMU∙∙∙O3w | 152 (2) | 152 (2) | 154 (4) | 153 (2) |
| N2DMU–H2DMU∙∙∙O4w | 155 (2) | 155 (1) | |||
| O–H∙∙∙O angles (°) | O1w–H1w∙∙∙ODMU | 142 (2) | 160 (9) | ||
| O2w–H2w∙∙∙ODMU | 138 (4) | 163 (4) | |||
| T Invariant (°C) | Nature of the Invariant | Stability of the Equilibrium | Phases in Equilibrium |
|---|---|---|---|
| −37 | Eutectic | Metastable | <ss II>(<<1%) + <Ice> ↔ doubly saturated liquid (between 70% and 70.5%) |
| −20 | Eutectic | Stable | <monohydrate> + <water> ↔ doubly saturated liquid (≈80%) |
| 8 | Peritectic | Stable | <monohydrate> ↔ <ssII> (<<1%) + doubly saturated liquid(≈65%) |
| 25 | Metatectic | Stable | <ssII> (<<1%) + doubly saturated liquid (≈60%) ↔ <ss I> (<<1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baaklini, G.; Schindler, M.; Yuan, L.; Jores, C.D.S.; Sanselme, M.; Couvrat, N.; Clevers, S.; Négrier, P.; Mondieig, D.; Dupray, V.; et al. Critical Influence of Water on the Polymorphism of 1,3-Dimethylurea and Other Heterogeneous Equilibria. Molecules 2023, 28, 7061. https://doi.org/10.3390/molecules28207061
Baaklini G, Schindler M, Yuan L, Jores CDS, Sanselme M, Couvrat N, Clevers S, Négrier P, Mondieig D, Dupray V, et al. Critical Influence of Water on the Polymorphism of 1,3-Dimethylurea and Other Heterogeneous Equilibria. Molecules. 2023; 28(20):7061. https://doi.org/10.3390/molecules28207061
Chicago/Turabian StyleBaaklini, Grace, Manon Schindler, Lina Yuan, Clément De Saint Jores, Morgane Sanselme, Nicolas Couvrat, Simon Clevers, Philippe Négrier, Denise Mondieig, Valérie Dupray, and et al. 2023. "Critical Influence of Water on the Polymorphism of 1,3-Dimethylurea and Other Heterogeneous Equilibria" Molecules 28, no. 20: 7061. https://doi.org/10.3390/molecules28207061
APA StyleBaaklini, G., Schindler, M., Yuan, L., Jores, C. D. S., Sanselme, M., Couvrat, N., Clevers, S., Négrier, P., Mondieig, D., Dupray, V., Cartigny, Y., Gbabode, G., & Coquerel, G. (2023). Critical Influence of Water on the Polymorphism of 1,3-Dimethylurea and Other Heterogeneous Equilibria. Molecules, 28(20), 7061. https://doi.org/10.3390/molecules28207061