Abstract
Molecular switches which can be triggered by light to interconvert between two or more well-defined conformation differing in their chemical or physical properties are fundamental for the development of materials with on-demand functionalities. Recently, a novel molecular switch based on a the azodicarboxamide core has been reported. It exhibits a volume-conserving conformational change upon excitation, making it a promising candidate for embedding in confined environments. In order to rationally implement and efficiently utilize the azodicarboxamide molecular switch, detailed insight into the coordinates governing the excited-state dynamics is needed. Here, we report a detailed comparative picture of the molecular motion at the atomic level in the presence and absence of explicit solvent. Our hybrid quantum mechanics/molecular mechanics (QM/MM) excited state simulations reveal that, although the energy landscape is slightly modulated by the solvation, the light-induced motion is dominated by a bending-assisted pedalo-type motion independent of the solvation. To support the predicted mechanism, we simulate time-resolved IR spectroscopy from first principles, thereby resolving fingerprints of the light-induced switching process. Our calculated time-resolved data are in good agreement with previously reported measured spectra.
1. Introduction
In recent decades, light-triggered molecular switches have been intensively studied not only because of their fundamental role in biological processes, such as vision [1], but also as promising candidates for the development of stimuli-responsive [2,3,4,5] functional materials, such as molecular devices [6,7,8,9], photo-responsive drug delivery systems [10], porous materials [11], liquid crystals [12] and optical memories [13], to name a few. Recently, a novel molecular photo-switch based on the azodicarboxamide chromophore unit (Figure 1) stepped into the spotlight thanks to a unique feature: an ultrafast photoinduced volume-conserving motion. Time-resolved UV-pump/IR-probe spectroscopy [14] supplemented with QM(TDDFT)/MM non-adiabatic molecular dynamics simulation on the solvated model [15] demonstrated that the chromophore, in contrast to other azo-compounds, does not undergo the usual trans-cis photoisomerization but rather a volume-conserving pedalo-type motion, rendering possible the application of this new class of chromophores under constrained conditions such as in solid state [16], on surfaces [17,18] and inside polymers [19].
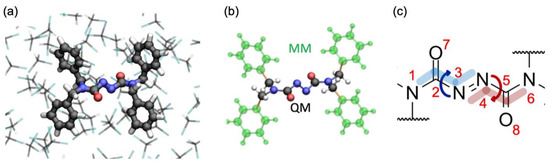
Figure 1.
(a) QM/MM set up of the system (balls and sticks representation) in a chloroform solvent (stick representation). (b) Partitioning scheme of the solute; the QM region contains the azodicarboxamide unit, while the MM region contains the phenyl rings; bonds across which the QM/MM cut is applied are highlighted in orange. (c) Lewis structure of the photo-active azodicarboxamide unit with atom labeling; note that the GS geometry is not planar, showing a 75° distortion of the N1-C2-N3-N4 and N3-N4-C5-N6 dihedrals.
The operation mechanism of the azodicarboxamide Is, however, a matter of debate. Based on microsolvation non-adiabatic molecular dynamics simulations at the CASSCF level, Liu and co-workers reported the usual trans-cis isomerization as the dominant decay mechanism [20]. Thus, a key factor for the exploitation of azodicarboxamide in different environments is to understand whether the pedalo-type motion is favored only in a medium which hinders the volume-demanding trans-cis process or whether it is an intrinsic property of the photo-switch. To elucidate the operation mechanism in detail and to evaluate in particular the coupling of the photochromic unit with the molecular environment, we report non-adiabatic molecular dynamics simulations on the isolated system and compare them with the dynamics on the solvated model. We find that, while the solvent affects the topology of the excited state (ES) potential energy surface (PES), the dynamical behavior is essentially unaltered. Moreover, to validate the finding and to allow a direct comparison with the experiments, we developed a protocol to simulate transient IR spectra based on the non-adiabatic molecular dynamics, which is applicable beyond the azodicarboxamide switching unit. Our results emphasize the importance of dynamical treatment of the photodynamic process.
2. Results and Discussion
2.1. Static Calculation: Critical Points and MEPs
GS optimization in gas-phase at the DFT and MP2 level of theory show, in line with calculations on the solvated model [15], that the GS equilibrium of the photochromic unit is characterized by 75° distortion from planarity around the N1-C2-N3-N4 () and N3-N4-C5-N6 () dihedrals (Table 1, see Figure 1 for atom notation). It is worth noting that the two carbonyl groups are decoupled from the central C2-N3-N4-C5 () fragment, i.e., the molecule does not exhibit a conjugated π-system in the GS. This is most clearly demonstrated by the frontier orbitals, and , being essentially localized on the N=N fragment and decoupled from the remaining π-orbitals (see Figure 2).

Table 1.
Structural parameters obtained at several critical points along the S0 and S1 potential energy surfaces of the gas-phase and solvated chromophore. The S0 minimum was obtained at the MP2/6-31G* level, whereas the remaining points at the SS-CASPT2/SA-2-CASSCF(18,12) level.
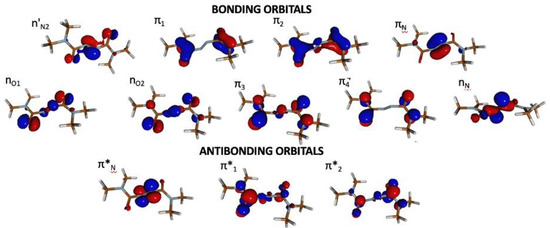
Figure 2.
Active space (18,12) employed for CASPT2 calculations.
The electronic structure analysis, carried out at the GS equilibrium geometry at the CASPT2 and TDDFT levels of theory (Table 2), shows that the lowest ES (S1), which appears in the visible range (below 3 eV), is characterized by a transition involving orbitals localized on the N=N fragment (Figure 2) with a vanishingly small oscillator strength at equilibrium. A triad of ES appear near degenerate around 4.5 eV described by a transition (S2) and a pair of degenerate transitions from and to (S3 or S4). In passing, we note the different energy order of the aforementioned transitions at the CASPT2 and TD-DFT levels. In all three transitions, there is a net charge transfer from the carbonyl groups to the central fragment. Due to the decoupling of the carbonyl and N=N fragments, the oscillator strengths associated with these transitions are rather weak (Table 2). Overall, the electronic structure of isolated azodicarboxamide resembles closely that in chloroform [15].
Table 2.
QM/MM vertical excitation energies (in eV/nm) and oscillator strengths (o.s.), where either CASPT2(18,12)/&-31G* or CAM-B3LYP/6-31G* are used for the QM calculation. CASPT2 and TD-DFT calculations are performed on the respective MP2 and DFT GS minimum. Electronic transitions refer to orbitals shown in Figure 2.
Through analysis of the PES, we could trace the possible deactivation pathways which connect the FC region of the lowest electronic ES S1() to the CI seam with the GS (Figure 3). Optimization at both TDDFT/CAM-B3LYP and SS-CASPT2/SA-2-CASSCF(18,12) levels shows that the driving force for the vibrational relaxation from the FC point is the pedalo-type motion characterized by the simultaneous torsion around and , which leads to the planarization of the azodicarboxamide unit (both dihedrals acquire values close to 180°, Table 1), accompanied by the opening of the bending angles C2-N3-N4 () and N3-N4-C5 () from ca. 110° to 125°. Those modes drive the system toward a plateau on the PES of the S1 state (labeled S1 Plateau in Figure 3). The geometrical changes can be rationalized in the following way: in the closed shell GS the oxygen and nitrogen lone pairs (labeled , , and ) are all doubly occupied. Their spatial proximity would cause an increased steric repulsion in a planar azodicarboxamide, which would outweigh the gain from the conjugation in the π-system. Therefore, the chromophore adopts a highly twisted conformation. Upon S1 excitation, the electron density in the nitrogen lone pair is depleted. The reduced steric repulsion facilitates the planarization thereby increasing the conjugation strength, evidenced by the constructive interference in the C2-N3 and N4-C5 segments of the anti-bonding orbital in the S1 plateau (see Figure S1 in the Supplementary Information, SI). To better accommodate the electron density in the plane, the angles and simultaneously widen to 125°.
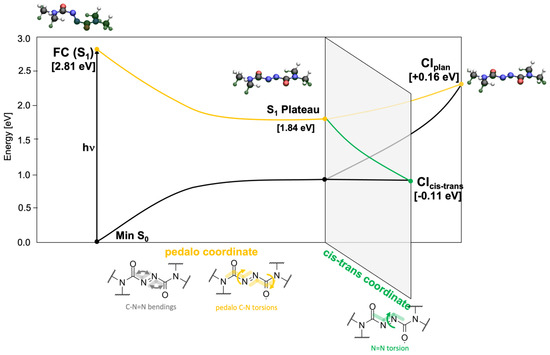
Figure 3.
Scheme of the potential energy profile connecting the FC region of the S1 state to the S1/GS CI seam, obtained at QM(CASPT2)/MM level in gas phase based on relaxed scans along torsion and bending coordinates reported in Table S1 in the SI section.
We located two minimum energy CI along the S1()/S0 CI seam—labeled CIcis-trans and CIplan—potentially relevant for the internal conversion to the GS, as shown schematically in Figure 3. While being almost isoenergetic, they are characterized by different structural parameters, documented in Table 1. CIcis-trans exhibits torsion around the N-N bond ( acquires a value of 112°) and lies about 0.1 eV below the S1 Plateau. In fact, it could be reached without an energetic barrier both at CAST2 and TDDFT level (green path in Figure 3). Conversely, CIplan exhibits an overall planarization of the three dihedral angles, , and accompanied by a further widening of the angles and to ca. 135° (yellow path in Figure 3). CIplan is located at about 0.15 eV above the S1 Plateau. We note the close resemblance of the two CIs with the planar and twisted S1()/S0 CIs found in azobenzene [21], which bears the same photoactive CNNC unit. The qualitative scheme of Figure 3 is based on relaxed scans along the cis-trans and pedalo coordinates carried out at the TDDFT and CASPT2 levels, documented in Figure S2 of the SI.
The S1() PES of the isolated azodicarboxamide resembles the one reported for the solvated model [15], exhibiting CIs with comparable geometrical features. Contrarily to what was reported in chloroform (CIcis-trans and CIplan lying ca. 0.2 eV above the S1 minimum), in gas-phase, no energy is present along the trans-cis coordinate. Thus, at least from an energetical point of view, this would suggest a higher propensity for the chromophore to undergo trans-cis isomerization in gas-phase instead of the pedalo-type motion.
2.2. Dynamics
Based on the static calculation we recognize two energetically competitive decay paths. Even though our previous dynamical study in chloroform demonstrated a clear preference for the pedalo-motion, it is not obvious the same dynamical behavior can be expected in gas-phase, in particular taking into account the stabilization of the CI associated with the cis-trans path. Static PES analysis has only a limited predictive power, as it neglects the role of the momentum conservation, which can divert the system from the minimum energy path. To obtain a more realistic picture of the mechanism, we run trajectory-based mixed quantum classical dynamics simulation.
Figure 4 shows a comparison between the experimental (green) and simulated (violet) linear absorption spectra in chloroform. The UV-vis spectrum shows a weak band above 400 nm, which is attributed to the transition (Table 2), and a more intense UV band due to the triad of states S2–S4. On the basis of this analysis, it can be deduced that the experimental pump pulse utilized to initiate the photoreaction in chloroform, centered at 355 nm (3.5 eV, indicated with a red arrow in Figure 4), addresses the manifold of states S2–S4. Mixed quantum classical dynamics reported previously in solvent were initiated in the brightest state of that manifold. Hence, the gas-phase simulations reported in this work follow the same selection criteria in order to allow a direct comparison.
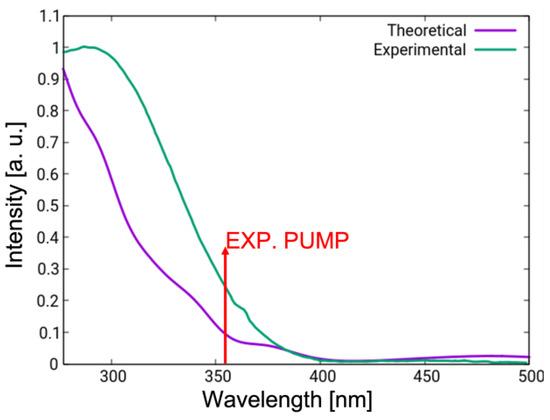
Figure 4.
Linear absorption spectrum in chloroform simulated at the TD-DFT/CAM-B3LYP level on the basis of vertical excitation energies of 200 geometries obtained by means of Wigner sampling (violet) compared to the experimental spectrum (green). Red arrow indicates the central wavelength of the experimental pump pulse.
Analysis of the statistics of the 50 trajectories shows that the internal conversion to the lowest electronic ES S1 occurs on a ~100 fs time scale. Repopulation of the GS is initiated shortly after and concluded within 400 fs (Figure 5a).
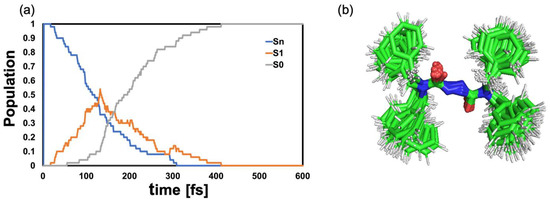
Figure 5.
(a) Population dynamics in gas-phase. Sn comprises states S2–S4; the choice of the initial state is based on the oscillator strengths. (b) Superimposed geometries of the S1 S0 hopping events for all trajectories.
To shed light into the geometrical deformations along the dynamics, we analyzed the variation of the torsional angles and , which act as markers for the pedalo mechanism, and of the torsional angle , representative of the trans-cis mechanism. The distribution of the values of those dihedrals in the equilibrium demonstrated the greater flexibility of and , peaking at 115° and 245°, respectively (i.e., 65° with respect to the plane), with standard deviation of ca. 10°, with respect to , which barely deviates from planarity (Figure 6, top row). The rigidity is further evidenced by the analysis of the evolution of the average values of the three dihedrals. It demonstrates a propensity for the pedalo-type motion. While does not deviate from 180° along the dynamics, / exhibit a rapid concerted increase/decrease associated with the planarization of the azodicarboxamide unit on the time scale of 100 fs (Figure 6, middle and bottom rows). Moreover, planarization is initiated already in the manifold of the higher states S2–S4. Indeed, and reach average values of 168° and 187°, respectively, at the S2/S1 hopping geometries and approach 180° (, ) at the S1/S0 hopping geometries (shown superimposed in Figure 5b). As anticipated in the previous section, the planarization is accompanied by alteration of the angles and . After a rapid decrease from their GS values of 112° to 107°, the average values of and progressively increase to 120° modulated by coherent oscillations with ca. 35 fs period (Figure 6, middle and bottom rows). After decay to S0, the angles return to the equilibrium values of 112°. The initial decrease is associated with the dynamics in the S2–S4 manifold, as confirmed by the average values at the S2/S1 hopping geometries (/). Conversely, at the S1/S0 hopping geometries, and show values around 134°, as anticipated by the static analysis (Table 1). In fact, the / opening is necessary condition to reach CIplan, and at least one of the two angles and is found to have a value above 132° (see / distribution at the hopping geometries, Figure S3 in the SI).
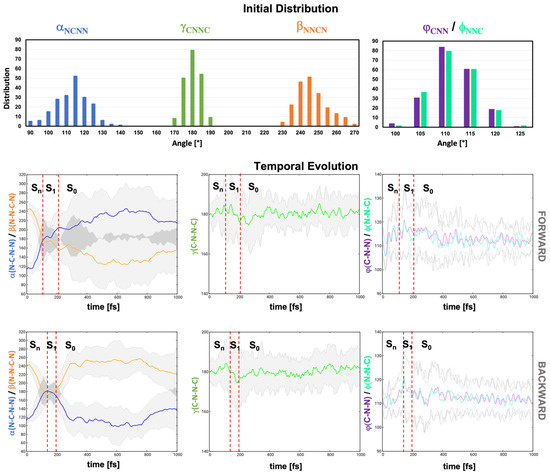
Figure 6.
Initial distribution od dihedrals (N1-C2-N3-N4) (blue), (N3-N4-C5--N6) (orange), (C2-N3-N4-C5) (green) and angles (C2-N3-N4) (violet) and (N3-N4-C5) (top). Average evolution of , , , and undergoing forward (middle) and backward (bottom) pedalo-type motion. Gray areas represent the standard deviation with respect to the average value. Red dashed lines indicate average S2/S1 and S1/S0 hopping times.
The fact that the operation mechanism is dominated by the pedalo-type motion rather than the trans-cis isomerization, whose conical intersection CIcis-trans is energetically more favored, can be rationalized by the conservation of momentum along the pedalo-type motion and the opening of and , activated upon decay to S1. By the time the / widening kicks in, torsion around / has already leads to the near planarization of the azodicarboxamide unit. As a consequence, the system reaches the S1/S0 CI seam, the torsional / modes are distributed around , which facilitated the forward pedalo-type motion upon crossing CIplan and the completion the pedalo-cycle in the GS.
As noted earlier, the azodicarboxamide switch bears a certain resemblance to azobenzene. Azobenzene, whose lowest ES is of character, exhibits minimum energy S1/S0 Cis reminiscent of CIplan and CIcis-trans. There is, however, a striking difference in their energetics. Whereas in azodicarboxamide, the two CIs are 0.2 eV apart and easily accessible out of the S1 Plateau, CIplan is considerably destabilized on the S1 PES of azobenzene (ca. 0.8 eV above CIcis-trans). This makes it accessible only with sufficient kinetic energy at disposal. Indeed, in azobenzene, CIplan acts as an efficient GS decay funnel only upon exciting the second ES (of nature). As in azodicarboxamide, it is the widening of the and angles, activated upon S2/S1 decay, which conserves enough momentum to drive azobenzene to CIplan [21,22]. The relative stabilization of CIplan in azodicarboxamide is a consequence of the smaller widening (i.e., smaller distortion) required to reach an S1/S0 degeneracy (/ compared to 145° in azobenzene). This is a consequence of the overall smaller S1–S0 energy gap in the S1 Plateau (0.95 eV compared to 1.85 eV in azobenzene) caused by the much stronger destabilization of the GS due to the increased in-plane electrostatic repulsion between the doubly occupied oxygen and nitrogen lone pairs. The increased accessibility of CIplan makes the pedalo-type motion the exclusive decay mechanism in azodicarboxamide.
In conclusion, the presence, adjacent to the CNNC fragment, of a group bearing a lone pair (i.e., the carbonyl) increase the in-plane electrostatic repulsion and is responsible both for the non-planar GS conformation and for the dominance of the pedalo-type decay mechanism over the trans-cis isomerization. Consequently, the volume-conserving decay mechanism is an intrinsic feature of azodicarboxamide and is not enforced by the environment.
2.3. Spectroscopy
To support atomistic interpretation, we simulated the transient IR spectra both in gas-phase and in chloroform (using the dynamics reported in our previous study [15]), following the procedure illustrated in the Methods section. In the following, we compare the simulated spectrum in chloroform with the experiment (the gas-phase spectrum is reported in Figures S4 and S5 in the SI) [14]. In particular, we focus on the carbonyl stretching absorbing in the spectral window 1600–1800 cm−1. Figure 7b shows the evolution of the instantaneous frequency along a representative trajectory shown in Figure 7a. It can be noted that immediately after excitation, when the system is in a higher lying ES (labeled as Sn), the carbonyl stretching frequency red-shifts by 200 cm−1 to 1600 cm−1. A gradual blue-shift toward 1800 cm−1 is observed in the following 200 fs upon internal conversion before the trajectory returns to the GS after ca. 350 fs (indicated by a red vertical line in Figure 7b). Subsequently, the value of the carbonyl stretching continues to oscillate around 1800 cm−1. Vibrational analysis in the minima of the ground and excited electronic states at DFT/TDDFT level supports the observed trend. While in the GS, the symmetric and anti-symmetric stretching have a frequency of 1817 cm−1 and 1841 cm−1, in S1, we note red shifts to 1788 cm−1 and 1795 cm−1, respectively. Even more pronounced red shifts are observed in S2 with values of 1688 and 1753 cm−1. These red shifts can be understood by examining the frontier orbitals of the initially excited pair of degenerate electronic states S2 and S3, described through and transitions, respectively: (a) and show bonding contribution on the carbonyl fragment (Figure 2), which is weakened upon exciting an electron; (b) upon planarization, the carbonyl and diazo fragment align and the orbital delocalizes over the azodicarboxamide unit, thereby exhibiting anti-bonding contribution on the carbonyl fragment. Thus, upon excitation and planarization, the C=O bond is weakened which is resembles in the pronounced red shift of the associated stretching modes. A more modest red shift is observed in S1() as has no contribution on the carbonyl group and the hole created therein upon excitation does not weaken the C=O bond.
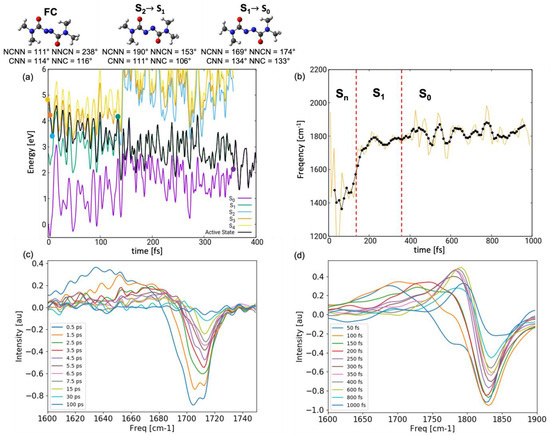
Figure 7.
(a) Energetics of a representative trajectory together with conformational changes from the FC region toward CIplan: dynamics initiated in brightest state S4 (yellow), hopping events indicated by dots. (b) Time evolution of the frequency of the anti-symmetric carbonyl stretching along the representative trajectory show in (a); red dashed lines indicate the time of the S2/S1 and S1/S0 hopping events. (c) Experimental and (d) simulated UV-pump/IR-probe spectra in chloroform at various delay times.
The transient UV-pump/IR-probe spectrum resolves the red shift of the carbonyl stretching mode in the ES as a positive signal peaking at 1700 cm−1 at early times (50 fs in Figure 7d) and progressively blue-shifting toward 1780 cm−1 in the next 300 fs. From there on, the red (blue) shift is slowed down but is clearly visible in the diminishing intensity of the overall signal indicating a destructive interference with the bleach around 1825 cm−1 present in the spectra due to the depletion of the GS. The persisting res-shift after the return to the GS is due to the cooling of the vibrationally hot wave packet, a process which takes several picoseconds and is, thus, not concluded within the simulation window.
The simulated spectrum reproduces qualitatively the experimental one (Figure 7c), though we note several quantitative discrepancies. The simulated spectrum is blue-shifted (e.g., the bleach has a 100 cm−1 blue-shift). This is due to a well-known tendency of DFT to overestimate normal mode frequencies. In fact, functional-specific scaling factors have been derived for GS frequency calculations aimed at correcting this error. As no scaling factors are available for electronic ES, we chose not to apply such a correction protocol. More striking is the difference in the time scales of the simulated and experimentally observed dynamics. The simulations predict a rapid blue-shift within 0.4 ps associated with ultrafast decay to the GS through CIplan. Thus, one could argue that the entire ES dynamics may remain concealed to the experiment, with the first measurement reported only after 0.5 ps. Here, we argue that the disagreement is a consequence of the strong overestimation of the decay rate in the simulations. First, the simplified hopping routine relying on the diminishing energy gap equates the hopping probability to unity every time the two surfaces come close. In reality, hopping will be promoted via vibronic coupling along selected modes, thus slowing down the decay. Second, the energy at which the dynamics is initiated in S2–S4 (both in gas-phase and in solution) is ca. 1 eV higher compared to the wavelength (355 nm/3.50 eV) at which the system is pumped experimentally (indicate with a red arrow in Figure 4). Upon internal conversion to S1, this energy in converted into kinetic energy, which facilitates the approach to the S1/GS CI region and accelerates the decay. This hypothesis is supported by the outcome of a trajectory initiated without initial kinetic energy in the S1 state (comparable to pumping at 500 nm/2.50 eV, Figure S4 in the SI). We observe an increase of ES lifetime by almost an order of magnitude both in gas-phase (hop to the GS after 679 fs) and in solvent (hop after 888 fs). We note that in both cases, the system follows the bending-assisted pedalo-type mechanism, i.e., the few hundred femtoseconds spent roaming in the S1 Plateau are not enough to activate modes toward the energetically more favored CIcis-trans.
In light of this arguments, we can be quite confident that at delay time fs, the experiment resolves the later dynamics in S1 missing out the motion occurring in the initially excited higher-lying states. This explains why the excitation-induced red shift in the simulated spectrum (>100 cm−1 difference between maximum of the positive and negative peak at fs) is more pronounced compared to the experiments (ca. 50 cm−1 difference between maximum of the positive and negative peak at fs).
Overall, the resemblance between the theoretical and experimental transient IR spectra validates the molecular dynamics protocol a strongly supports the dominance of the pedalo-type mechanism over trans-cis isomerization.
3. Methods
All the calculations reported in this work were performed with the COBRAMM [23] QM/MM interface, which links the QM packages Gaussian09 [24] and OpenMolcas [25], used to describe the photo-responsive core of the system with the MM suite AMBER [26], used for describing the environment.
3.1. QM/MM Setup
For the solvated system, a representative geometry embedded in a droplet of chloroform with radius 15 Å was obtained by sampling the conformational freedom by means of a classical MM simulation as described in Ref. [15]. A High-Medium-Low layer partitioning was employed in which the central photoactive moiety of the chromophore is described at QM level (High layer), while the four phenyls substituents, as well as chloroform, are treated at the MM level (Figure 1a). In the MM calculation, the azodicarboxamide is parametrized using the GAFF [27] force field [15], while chloroform is described following the work of Cieplak et al. [28]. The High layer-Medium layer cut was placed between the sp3- and sp2-hybridized carbons of the linker group and of the phenyl ring, respectively, as indicated in Figure 1b. The phenyl rings and solvent molecules within 9 Å of the solute were allowed to move (Medium layer), while the remaining solvent molecules were kept fixed. Concerning the gas-phase model, the initial setup has been obtained by stripping the solvent and re-optimizing the chromophore employing a High-Medium layer partitioning.
3.2. Electronic Structure Calculation
For both the gas-phase and solvated model, the basis set employed is 6-31G*. Electronic ground state (GS) calculations were performed at the density functional theory (DFT) level using the functional CAM-B3LYP [29] and at the Møller Plesset second-order perturbation theory (MP2) [30] level. ES energies in the Frank–Condon region were calculated using a time-dependent (TD) version of DFT, again using the functional CAM-B3LYP, and with complete active state self-consistent field (CASSCF), averaging over seven states, corrected by second–order multi-reference perturbation theory (CASPT2) in its single state (SS) flavor [31]. The active space includes the valence π-orbitals and the oxygen and nitrogen lone-pairs of the chromophore, leading to a total of 18 electrons in 12 orbitals (Figure 2). We will refer to the level of theory as SS-CASPT2/SA-7-CASSCF (18,12). Critical geometries along the lowest ES were optimized at the same computational level; however, only the lowest two state were used in the state-averaging to reduce the computational cost. Conical intersection optimizations were performed at the QM(CASPT2)/MM level with the gradient projection algorithm of Bearpark et al. [31,32] implemented in COBRAMM.
3.3. Linear Absorption Spectra in Chloroform
In total, 200 geometries were generated around the GS equilibrium using the Wigner sampling technique through an interface with a stand-alone script, part of the quantum molecular dynamics program JADE [32]. The sampling was performed at room temperature (300 K) and high-frequency modes belonging to C-H and N-H vibrations were excluded from the sampling. Each of the sampled geometries was subjected to a 10-ps long MM equilibration run, in which the QM region was kept fixed through the harmonic constraints, allowing the MM region to adapt at each sampled geometry. The linear absorption spectrum was simulated according to Equation (1):
where and denote the geometry dependent oscillator strength and excitation energy at given geometry coordinates and is the standard deviation used to account phenomenologically for the spectral line shape. The sums run over the number of snapshots (n) and over the manifold of excited states I. The single point calculations were performed at the QM(TD-DFT)/MM level of theory using a uniform value for eV.
3.4. Mixed Quantum Classical Dynamics Gas-Phase
Mixed quantum-classical dynamics simulations in gas-phase were performed at the QM(TD-DFT)/MM level of theory considering four ES. In total, 50 initial conditions were generated using the protocol outlined in Section 3.3. Thereby, the initial velocities were obtained by combining the velocities of the QM part (from the Wigner sampling) with those of the MM atoms (the four phenyl substituents) from the last snapshot of the MM equilibration. Each trajectory was initiated in the brightest of the triad state below 400 nm (labeled S2–S4, see the discussion of the electronic structure in Section 2.1. for details) and propagated for 2 ps using a time step of 1 fs. Nonadiabatic events were evaluated with a simplified scheme relying on the energy gap as a criterium for changing the electronic state with a threshold of 2 kcal/mol. Back hopping was always allowed between excited states, while it was forbidden once the trajectory hopped to the electronic GS.
3.5. Theory and Implementation of the Time-Resolved IR Spectroscopy Simulations
We have simulated the transient UV-pump/IR-probe spectra both in gas-phase and in solvent. To this aim, we made use of the procedure employed by Stock et al. [33]. Working in the weak regime in which light–matter interactions can be treated perturbatively, we consider a system described by the Hamiltonian , which at time t = 0 is excited by an ultrashort optical pump pulse to an electronic ES. In the dipole approximation, the time-dependent interaction with the electric field is described by a time-dependent operator , where is the transition dipole moment operator and is the electric field. The excitation is assumed to be impulsive, implying that the nuclear wave packet, which before the arrival of the pump pulse is at equilibrium in the GS, is projected vertically on the ES where it starts to evolve. The field-free evolution is governed by the Schrödinger equation (Equation (2)):
While the exact solution can be obtained by means of explicit quantum dynamics, the Schrödinger equation can be solved with some approximations by means of semi-classical and mixed quantum-classical approaches. At time after the interaction with the pump pulse, the system interacts with an IR probe pulse, which promotes it to a higher vibrational level on the same electronic state. The interaction with the pulse perturbs the system creating an oscillating electric dipole which generates a macroscopic polarization , itself the source of an electric field emitted by the system during the time interval after the interaction with the probe pulse [34]. Mathematically, the polarization is described by the following equation (Equation (3)):
Which is the expectation value of the dipole operator. denotes the system, initially in its electronic GS. Following the interaction with the pump pulse (formally two wave-matter interactions, one with the bra and one with the ket), the system evolves on the ES surface during the time interval . Subsequently, an interaction with an IR pulse brings the (ket) wave packet to a vibrational ES. During the time interval after the IR pulse, the left- and right-hand side are propagated under the influence of a different vibrational Hamiltonian, a phenomenon termed coherence. The electronic density oscillating between two quantized vibrational levels during the time is the source of an emitted field with a frequency proportional to the energy difference between the vibrational levels. Although the interaction of the pump and probe pulses with the system induces a third-order nonlinear response, replacing the zero-order stationary initial state by a first-order nonstationary (excited) state , which is time-dependent due to the initial preparation of the system, it is possible to describe the polarization by following linear expression (Equation (4)) [35]:
In the semi-classical line shape theory [36,37], the coherence dynamics can be approximated by the fluctuation of the energy gap between the two vibrational levels along the trajectory (Equation (5)):
The time dependence of the energy gap is related to the evolution of the coordinates . In the harmonic approximation, the energy difference between two vibrational levels is , thus equal to the frequency of the vibrational mode.
In practice, the trajectory is propagated during the interval subject to the ES Hamiltonian as if there was no interaction with the IR pulse, and at every time step, the instantaneous frequency of the mode(s) of interest is calculated.
The experimental spectrum was recorded in the frequency range associated with the carbonyl stretching [14]. For both types of light-induced mechanisms—pedalo-type motion and trans-cis isomerization—the C=O groups are not directly involved but rather act as a spectator mode of the nuclear dynamics, whose frequencies are modulated in the course of the internal conversion in response to the changing electronic structure. To obtain the instantaneous frequency , we tracked the change of the value of the anti-symmetric (we note that in transient IR spectroscopy only anti-symmetric modes are active) carbonyl stretch along each trajectory defined as , where and are the value of the two CO distances at each time instance . Figure 8a shows the evolution of the anti-symmetric carbonyl stretching value along a representative trajectory. We observe a periodically oscillating function with a period of ca. 20 fs. The instantaneous frequency was then obtained as the inverse of the time interval between two consecutive maxima or minima, as shown in the inset of Figure 8a. The data processing results in a dataset of instantaneous frequencies in ca. 10 fs intervals (yellow profile in Figure 8b). To smoothen the profile and minimize the effects of outliers, we averaged over five oscillation periods .
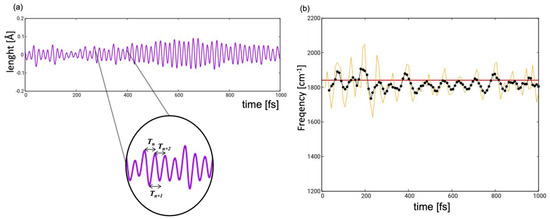
Figure 8.
(a) Oscillating function representing the antisymmetric stretching which originates from subtracting the values of the two carbonyl groups at each time step along a representative trajectory. (b) Time evolution (yellow line) of the frequency of the antisymmetric stretching mode computed as the inverse of the time interval between two consecutive maxima or minima (see inset of (a)); black dotted line indicates the time-evolution of frequency averaged over five periods; red horizontal line represents the frequency of the antisymmetric stretching calculated at the GS equilibrium structure at DFT/CAM-B3LYP level.
In mixed quantum-classical theory, the wave packet is approximated by a swarm of trajectories each one propagated independently, and the total polarization is computed by summing over all trajectories (Equation (6)):
where runs over the total of trajectories and is the magnitude of the IR intensity assumed to be coordinate independent. Fourier transformation of the polarization with respect to the interval gives the transient IR spectrum (Equation (7)):
where we introduce a damping function , with as the phenomenological lifetime of the coherence due to the dephasing of the signal. In this work, was set to 300 fs.
Finally, the transient difference spectrum is calculated as (Equation (8)):
where the reference spectrum represents the steady spectrum of the GS. In practice, is obtained with the same protocol as by running the same set of 50 trajectories in the GS for the duration of 2 ps.
We are aware that there are more accurate protocols to obtain the instantaneous frequencies , a notable example being the mode tracking procedure by Neugebauer et al. [38]. Our protocol is justified by the observation that the carbonyl stretching couples only weakly to other modes. To support these statements, the IR spectrum in the GS gives a steady spectrum peaking at 1825 cm−1, in good agreement with the analytically computed value for the anti-symmetric stretching of 1841 cm−1.
4. Conclusions
The recently reported peculiar behavior of azodicarboxamide to undergo a volume-conserving pedalo-type decay mechanism upon excitation has attracted attention due to the potential exploration as a molecular switch in confined environments [14,15]. In contrast to chemically related dyes containing a central diazo unit, such as azobenzene, azodicarboxamide does not undergo volume-demanding cis-trans isomerization. In the present contribution, by means of mixed quantum classical dynamics simulation at the QM/TD-DFT)/MM level, we demonstrate that the pedalo-type motion is an intrinsic feature of the system determined by its electronic composition, in particular by the presence of carbonyl groups adjacent to the diazo unit. The strong electrostatic repulsion between the electron densities of the spatially proximate oxygen and nitrogen lone pairs to which a planar azodicarboxamide would be exposed to outweigh the potential gain from the conjugation. This forces a 75° twist along the NCNN/NNCN dihedrals in the trans isomer in the electronic GS, contrarily to azobenzene dyes, which are planar in their trans isomer. Instead, the central CNNC fragment is rather stiff and does not deviate from 180°. Photoexcitation with visible to mid-UV light excites a higher lying excited state (S2–S4), depleting the electron density in the lone pairs. This creates a pronounced drive toward planarization (and an increase of the conjugation strength). Notably, a near complete planarization is observed within the first 100 fs, i.e., even before the system decays to S1. The electronic structure S1 () resembles that of the lowest electronic state of azobenzene and, in accordance, induces a widening of the CNN/NNC bending angles which ultimately steers the system towards a region on the S1/S0 CI seam, where the central dihedral CNNC remains planar, thus denoted CIplan. In contrast to azobenzene, where reaching CIplan requires pronounced CNN/NNC bending deformation (ca. 145°) and, thus, a considerable amount of activation energy (ca. 0.7 eV), in azodicarboxamide, the CIplan can be accessed via minor bending deformation (ca. 135°) and, correspondingly, a smaller barrier (ca. 0.2 eV). This makes CIplan the sole deactivation path in azodicarboxamide, independent of the environment. The reason for this mechanism-determining alteration of the S1 topology is that in azodicarboxamide, the ES planarization decreases notably the S0–S1 energy gap, an effect not present in azobenzene. Upon decay to the GS, which is concluded in 400 fs, the electrostatic repulsion between the lone pairs derives the system out of planarity, allowing the switch to conclude the pedalo cycle within less than a picosecond.
The change of the photodynamics due to the different embedding of the azo-moiety within a molecule (no carbonyl groups in azobenzene vs. two carbonyl groups in azodicarboxamide) highlights, on the one hand, the potential of molecular design to modulate the light-responsive properties. On the other, it emphasizes the need for atomistic simulations of the excited-state dynamics that goes beyond the static picture in order to sort out the relevant pathways. This is particularly relevant in the case of diazocarboxamide, where the static description based on exploring the minimum energy path(s) from the FC to the S1/S0 CI seam favors the decay through a region of the seam (CIcis-trans) characterized by the pronounced twisting of the CNNC central fragment connecting the cis and trans isomers of dicarboxamide. The dynamics simulations demonstrate that, while being energetically more favorable, the system does not follow this decay path as momentum conservation along CNN/NNC bending coordinates and accessibility of CIplan drive the system away from the minimum energy path.
The proposed mechanism has been corroborated by transient IR spectroscopy simulations from first principles tracking the dynamics of the carbonyl stretching around 1800 cm−1, an observer mode which is not directly involved in the isomerization but whose instantaneous frequency is affected by the electronic structure of the states visited in the course of the internal conversion. Upon ES planarization the delocalization of the frontier orbitals over the entire dicarboxamide unit leads to a weakening of the carbonyl bonds, which decreases the stretching frequency. Thus, the approx. 50 cm−1 red shift observed experimentally for the carbonyl band and reproduced in our atomistic simulations in solution and in gas-phase is a clear signature of the planarization completed in the ES before the decay to the GS.
In conclusion, we consider the argument regarding the azodicarboxamide decay mechanism as settled. The ascertainment of the dominant role of volume-conserving pedalo motion both in gas and liquid phase opens the door for applications of this novel switch in confined environments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28020816/s1. QM/MM energy profiles along pedalo-type motion and trans-cis coordinates, molecular frontier orbitals in S1 Plateau, energy values along the minimum energy paths, values of bending angles at all S1/S0 hopping geometries, 0K molecular dynamics simulations, time-resolved UV pump/IR probe spectrum in gas phase and coordinates of critical points. Figure S1: Molecular frontier orbitals and obtained at a geometry from the S1 Plateau (see Figure 2 from main text) with N1C2N3N4, N3N4C5N6 and C2N3N4C5 exhibiting values close to 180°, Figure S2: left) QM/MM energy profiles along the pedalo-type coordinate optimizing either S1 (top) or S0 (bottom) at (TD)-DFT/CAM-B3LYP (open squares) and MP2/SS-CASPT2(18,12) (filled squares) level of theory. Right) QM/MM energy profiles along the trans-cis coordinate optimizing either S1 (top) or S0 (bottom) at (TD)-DFT/CAM-B3LYP (open squares) and MP2/SS-CASPT2(18,12) (filled squares), Table S1: Energy values of critical points along the minimum energy paths of the pedalo-type and trans-cis coordinates calculated at CASPT2(18,12) level, Figure S3: Values of the bending angles C2N3N4 () and N3N4C5 () at all S1/S0 hopping geometries along the dynamics, Figure S4: 0K trajectories started from S1 in gas-phase (left) and solvent (right) until the hopping point with S0: 679 fs and 888 fs, respectively, Figure S5: Simulated Transient UV pump/IR probe obtained in gas phase at different delay times.
Author Contributions
Conceptualization, S.A., I.C. and A.N.; investigation, M.T., I.C. and A.N.; resources, M.G. and A.N.; writing—original draft preparation, M.T. and A.N.; writing—review and editing, M.G., S.A. and I.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Polli, D.; Altoè, P.; Weingart, O.; Spillane, K.M.; Manzoni, C.; Brida, D.; Tomasello, G.; Orlandi, G.; Kukura, P.; Mathies, R.A.; et al. Conical Intersection Dynamics of the Primary Photoisomerization Event in Vision. Nature 2010, 467, 440–443. [Google Scholar] [CrossRef]
- Wang, Z.; Müller, K.; Valášek, M.; Grosjean, S.; Bräse, S.; Wöll, C.; Mayor, M.; Heinke, L. Series of Photoswitchable Azobenzene-Containing Metal–Organic Frameworks with Variable Adsorption Switching Effect. J. Phys. Chem. C 2018, 122, 19044–19050. [Google Scholar] [CrossRef]
- Mutruc, D.; Goulet-Hanssens, A.; Fairman, S.; Wahl, S.; Zimathies, A.; Knie, C.; Hecht, S. Modulating Guest Uptake in Core–Shell MOFs with Visible Light. Angew. Chem. Int. Ed. 2019, 58, 12862–12867. [Google Scholar] [CrossRef]
- Danowski, W.; van Leeuwen, T.; Abdolahzadeh, S.; Roke, D.; Browne, W.R.; Wezenberg, S.J.; Feringa, B.L. Unidirectional Rotary Motion in a Metal–Organic Framework. Nat. Nanotechnol. 2019, 14, 488–494. [Google Scholar] [CrossRef]
- Kolodzeiski, E.; Amirjalayer, S. Atomistic Insight Into the Host–Guest Interaction of a Photoresponsive Metal–Organic Framework. Chem. A Eur. J. 2020, 26, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.D.; Moran, M.J.; Aprahamian, I. New Molecular Switch Architectures. Proc. Natl. Acad. Sci. USA 2016, 115, 9414–9422. [Google Scholar] [CrossRef] [PubMed]
- Dattler, D.; Fuks, G.; Heiser, J.; Moulin, E.; Perrot, A.; Yao, X.; Giuseppone, N. Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors. Chem. Rev. 2020, 120, 310–433. [Google Scholar] [CrossRef] [PubMed]
- Kassem, S.; van Leeuwen, T.; Lubbe, A.S.; Wilson, M.R.; Feringa, B.L.; Leigh, D.A. Artificial Molecular Motors. Chem. Soc. Rev. 2017, 46, 2592–2621. [Google Scholar] [CrossRef] [PubMed]
- Pianowski, Z.L. Recent Implementations of Molecular Photoswitches into Smart Materials and Biological Systems. Chem. A Eur. J. 2019, 25, 5128–5144. [Google Scholar] [CrossRef]
- Fitzmaurice, O.; Bartkowski, M.; Giordani, S. Molecular Switches—Tools for Imparting Control in Drug Delivery Systems. Front. Chem. 2022, 10, 859450. [Google Scholar] [CrossRef]
- Amirjalayer, S. Photonic Materials: On the Molecular Mechanism of a Photo-Responsive Phase Change Memory (Adv. Theory Simul. 5/2021). Adv. Theory Simul. 2021, 4, 2170009. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q. Light-Driven Chiral Molecular Switches or Motors in Liquid Crystals. Adv. Mater. 2012, 24, 1926–1945. [Google Scholar] [CrossRef]
- Tsivgoulis, G.M.; Lehn, J.-M. Photonic Molecular Devices: Reversibly Photoswitchable Fluorophores for Nondestructive Readout for Optical Memory. Angew. Chem. Int. Ed. Engl. 1995, 34, 1119–1122. [Google Scholar] [CrossRef]
- Amirjalayer, S.; Martinez-Cuezva, A.; Berna, J.; Woutersen, S.; Buma, W.J. Photoinduced Pedalo-Type Motion in an Azodicarboxamide-Based Molecular Switch. Angew. Chem. 2018, 130, 1810–1814. [Google Scholar] [CrossRef]
- Conti, I.; Buma, W.J.; Buma, W.J.; Garavelli, M.; Amirjalayer, S.; Amirjalayer, S.; Amirjalayer, S. Photoinduced Forward and Backward Pedalo-Type Motion of a Molecular Switch. J. Phys. Chem. Lett. 2020, 11, 4741–4746. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garibay, M.A. Nanoscale Gadgets. Nat. Mater 2008, 7, 431–432. [Google Scholar] [CrossRef]
- Davis, J.J.; Orlowski, G.A.; Rahman, H.; Beer, P.D. Mechanically Interlocked and Switchable Molecules at Surfaces. Chem. Commun. 2010, 46, 54–63. [Google Scholar] [CrossRef]
- Fahrenbach, A.C.; Warren, S.C.; Incorvati, J.T.; Avestro, A.-J.; Barnes, J.C.; Stoddart, J.F.; Grzybowski, B.A. Organic Switches for Surfaces and Devices. Adv. Mater. 2013, 25, 331–348. [Google Scholar] [CrossRef]
- Xu, G.; Li, S.; Liu, C.; Wu, S. Photoswitchable Adhesives Using Azobenzene-Containing Materials. Chem. Asian. J. 2020, 15, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, J. Nonadiabatic Dynamics Simulation of Photoisomerization Mechanism of Photoswitch Azodicarboxamide: Hydrogen Bonding Effects. J. Photochem. Photobiol. A Chem. 2018, 367, 236–239. [Google Scholar] [CrossRef]
- Conti, I.; Garavelli, M.; Orlandi, G. The Different Photoisomerization Efficiency of Azobenzene in the Lowest Nπ* and Ππ* Singlets: The Role of a Phantom State. J. Am. Chem. Soc. 2008, 130, 5216–5230. [Google Scholar] [CrossRef] [PubMed]
- Aleotti, F.; Nenov, A.; Salvigni, L.; Bonfanti, M.; El-Tahawy, M.M.; Giunchi, A.; Gentile, M.; Spallacci, C.; Ventimiglia, A.; Cirillo, G.; et al. Spectral Tuning and Photoisomerization Efficiency in Push–Pull Azobenzenes: Designing Principles. J. Phys. Chem. A 2020, 124, 9513–9523. [Google Scholar] [CrossRef] [PubMed]
- Weingart, O.; Nenov, A.; Altoè, P.; Rivalta, I.; Segarra-Martí, J.; Dokukina, I.; Garavelli, M. COBRAMM 2.0—A Software Interface for Tailoring Molecular Electronic Structure Calculations and Running Nanoscale (QM/MM) Simulations. J. Mol. Model. 2018, 24, 271. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09 (Revision A02); Gaussian Inc.: Wallingford CT, USA, 2009. [Google Scholar]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; de Vico, L.; Fdez Galván, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. Molcas 8: New Capabilities for Multiconfigurational Quantum Chemical Calculations across the Periodic Table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. Amber 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Cieplak, P.; Caldwell, J.; Kollman, P. Molecular Mechanical Models for Organic and Biological Systems Going beyond the Atom Centered Two Body Additive Approximation: Aqueous Solution Free Energies of Methanol and N-Methyl Acetamide, Nucleic Acid Base, and Amide Hydrogen Bonding and Chloroform/Water Partition Coefficients of the Nucleic Acid Bases. J. Comput. Chem. 2001, 22, 1048–1057. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Bearpark, M.J.; Robb, M.A.; Bernhard Schlegel, H. A Direct Method for the Location of the Lowest Energy Point on a Potential Surface Crossing. Chem. Phys. Lett. 1994, 223, 269–274. [Google Scholar] [CrossRef]
- Du, L.; Lan, Z. An On-the-Fly Surface-Hopping Program JADE for Nonadiabatic Molecular Dynamics of Polyatomic Systems: Implementation and Applications. J. Chem. Theory Comput. 2015, 11, 1360–1374. [Google Scholar] [CrossRef]
- Kobus, M.; Lieder, M.; Nguyen, P.H.; Stock, G. Simulation of Transient Infrared Spectra of a Photoswitchable Peptide. J. Chem. Phys. 2011, 135, 225102. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, S. Principles of Nonlinear Optical Spectroscopy; Oxford Univ. Press: New York, NY, USA, 1995. [Google Scholar]
- Pollard, W.T.; Lee, S.; Mathies, R.A. Wave Packet Theory of Dynamic Absorption Spectra in Femtosecond Pump–Probe Experiments. J. Chem. Phys. 1990, 92, 4012–4029. [Google Scholar] [CrossRef]
- Kubo, R. A Stochastic Theory of Line Shape. In Advances in Chemical Physics; Advances in Chemical Physics: Hoboken, NJ, USA, 1969; pp. 101–127. ISBN 9780470143605. [Google Scholar]
- Tanimura, Y. Stochastic Liouville, Langevin, Fokker–Planck, and Master Equation Approaches to Quantum Dissipative Systems. J. Physical. Soc. Jpn. 2006, 75, 082001. [Google Scholar] [CrossRef]
- Reiher, M.; Neugebauer, J. A Mode-Selective Quantum Chemical Method for Tracking Molecular Vibrations Applied to Functionalized Carbon Nanotubes. J. Chem. Phys. 2003, 118, 1634–1641. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).