

Identification of Potential Antitubulin Agents with Anticancer Assets from a Series of Imidazo[1,2-a]quinoxaline Derivatives: In Silico and In Vitro Approaches

 ,
,  ,
,  , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Compound Selection and Virtual Screening
3.2.2. Protein Preparation and Validation
3.2.3. Grid Generation
3.2.4. Ligand Preparation
3.2.5. Molecular Docking Study
3.2.6. Binding Energy Calculation
3.2.7. Molecular Dynamic Studies
3.2.8. Tubulin Inhibitory Potential
3.2.9. Cell Culture and MTT-Based Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Slobodnick, A.; Shah, B.; Krasnokutsky, S.; Pillinger, M.H. Update on colchicine, 2017. Rheumatology 2018, 57, i4–i11. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, R.; Skvortsova, I.; Kumar, V. Mechanisms of tubulin binding ligands to target cancer cells: Updates on their therapeutic potential and clinical trials. Curr. Cancer Drug Targets 2017, 17, 357–375. [Google Scholar] [CrossRef]
- Zich, J.; Hardwick, K.G. Getting down to the phosphorylated ‘nuts and bolts’ of spindle checkpoint signalling. Trends Biochem. Sci. 2010, 35, 18–27. [Google Scholar] [CrossRef]
- Naaz, F.; Haider, M.R.; Shafi, S.; Yar, M.S. Anti-tubulin agents of natural origin: Targeting taxol, vinca, and colchicine binding domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; Kavallaris, M. Microtubules: A dynamic target in cancer therapy. IUBMB Life 2008, 60, 165–170. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Bhutani, P.; Joshi, G.; Raja, N.; Bachhav, N.; Rajanna, P.K.; Bhutani, H.; Paul, A.T.; Kumar, R. US FDA Approved Drugs from 2015-June 2020: A Perspective. J. Med. Chem. 2021, 64, 2339–2381. [Google Scholar] [CrossRef] [PubMed]
- Zghaib, Z.; Guichou, J.-F.; Vappiani, J.; Bec, N.; Hadj-Kaddour, K.; Vincent, L.-A.; Paniagua-Gayraud, S.; Larroque, C.; Moarbess, G.; Cuq, P. New imidazoquinoxaline derivatives: Synthesis, biological evaluation on melanoma, effect on tubulin polymerization and structure–activity relationships. Bioorganic Med. Chem. 2016, 24, 2433–2440. [Google Scholar] [CrossRef]
- Courbet, A.; Bec, N.; Constant, C.; Larroque, C.; Pugniere, M.; El Messaoudi, S.; Zghaib, Z.; Khier, S.; Deleuze-Masquefa, C.; Gattacceca, F. Imidazoquinoxaline anticancer derivatives and imiquimod interact with tubulin: Characterization of molecular microtubule inhibiting mechanisms in correlation with cytotoxicity. PLoS ONE 2017, 12, e0182022. [Google Scholar] [CrossRef] [PubMed]
- Nabbouh, A.I.; Hleihel, R.S.; Saliba, J.L.; Karam, M.M.; Hamie, M.H.; Wu, H.C.J.M.; Berthier, C.P.; Tawil, N.M.; Bonnet, P.A.A.; Deleuze-Masquefa, C. Imidazoquinoxaline derivative EAPB0503: A promising drug targeting mutant nucleophosmin 1 in acute myeloid leukemia. Cancer 2017, 123, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Skayneh, H.; Jishi, B.; Hleihel, R.; Hamie, M.; El Hajj, R.; Deleuze-Masquefa, C.; Bonnet, P.-A.; El Sabban, M.; El Hajj, H. EAPB0503, an Imidazoquinoxaline Derivative Modulates SENP3/ARF Mediated SUMOylation, and Induces NPM1c Degradation in NPM1 Mutant AML. Int. J. Mol. Sci. 2022, 23, 3421. [Google Scholar] [CrossRef]
- Joshi, G.; Chauhan, M.; Kumar, R.; Thakur, A.; Sharma, S.; Singh, R.; Wani, A.A.; Sharon, A.; Bharatam, P.V.; Kumar, R. Cyclocondensation reactions of an electron deactivated 2-aminophenyl tethered imidazole with mono/1,2-biselectrophiles: Synthesis and DFT studies on the rationalisation of imidazo[1,2-a]quinoxaline versus benzo[f]imidazo[1,5-a][1,3,5]triazepine selectivity switches. Org. Chem. Front. 2018, 5, 3526–3533. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Carr, M.; Greene, L.M.; Bergin, O.; Nathwani, S.M.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis and evaluation of azetidinone analogues of combretastatin A-4 as tubulin targeting agents. J. Med. Chem. 2010, 53, 8569–8584. [Google Scholar] [CrossRef]
- Salam, N.K.; Nuti, R.; Sherman, W. Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating molecular docking and molecular dynamics simulations. In Docking Screens for Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2019; pp. 13–34. [Google Scholar]
- Lee, M.R.; Sun, Y. Improving docking accuracy through molecular mechanics generalized born optimization and scoring. J. Chem. Theory Comput. 2007, 3, 1106–1119. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Schrödinger, R. 2: Maestro; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- LigPrep, S. 2: Ligprep; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Release, S. 1: Desmond Molecular Dynamics System, version 3.7; DE Shaw Research: New York, NY, USA, 2014.
- Maestro-Desmond Interoperability Tools, version 2.2; Shaw Research: New York, NY, USA, 2019.
- Beyer, C.F.; Zhang, N.; Hernandez, R.; Vitale, D.; Lucas, J.; Nguyen, T.; Discafani, C.; Ayral-Kaloustian, S.; Gibbons, J.J. TTI-237: A novel microtubule-active compound with in vivo antitumor activity. Cancer Res. 2008, 68, 2292–2300. [Google Scholar] [CrossRef]
- Kumar, B.; Sharma, P.; Gupta, V.P.; Khullar, M.; Singh, S.; Dogra, N.; Kumar, V. Synthesis and biological evaluation of pyrimidine bridged combretastatin derivatives as potential anticancer agents and mechanistic studies. Bioorg. Chem. 2018, 78, 130–140. [Google Scholar] [CrossRef] [PubMed]








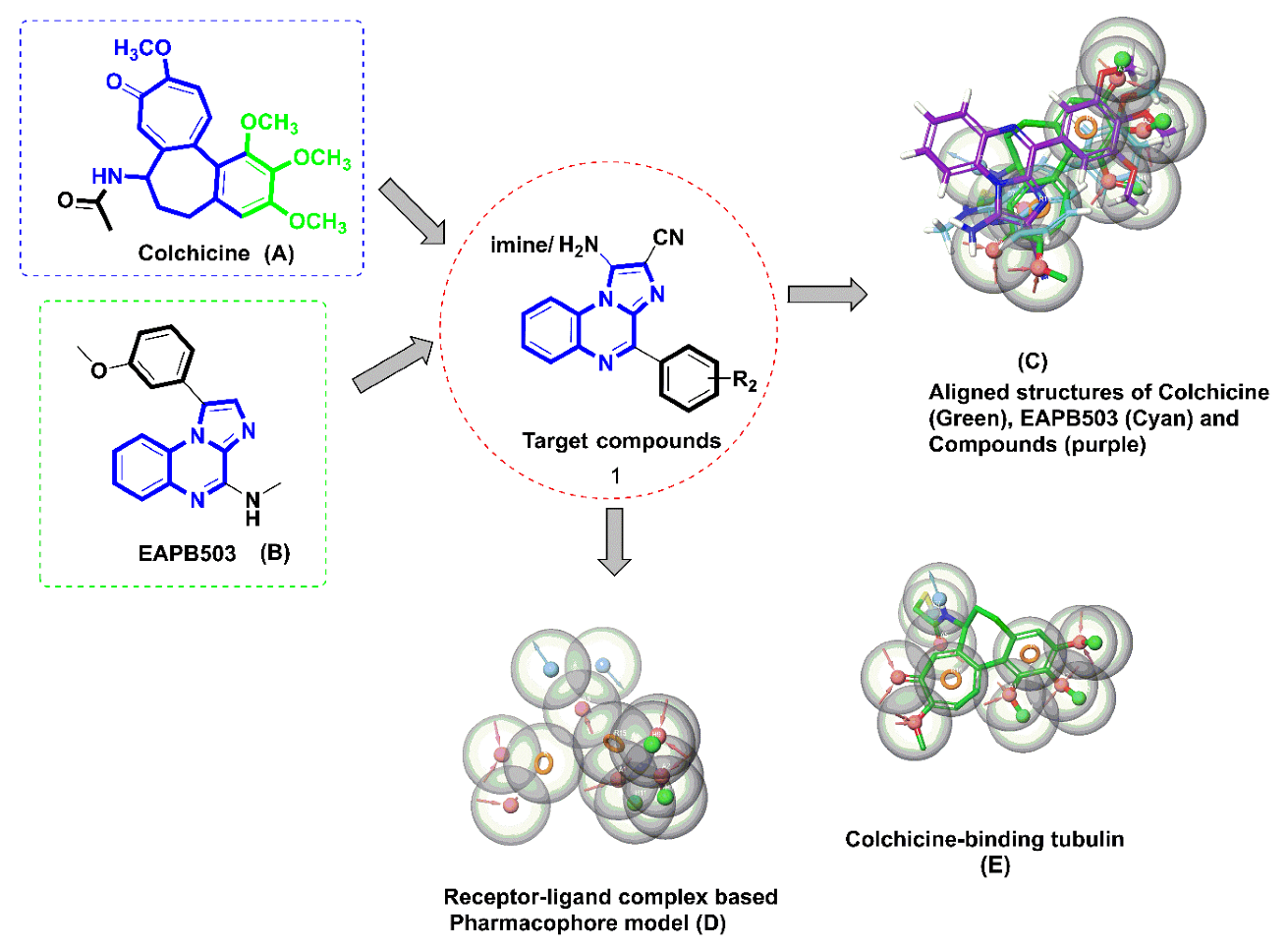{kind=link}
{kind=link}
{kind=link}
{kind=link}
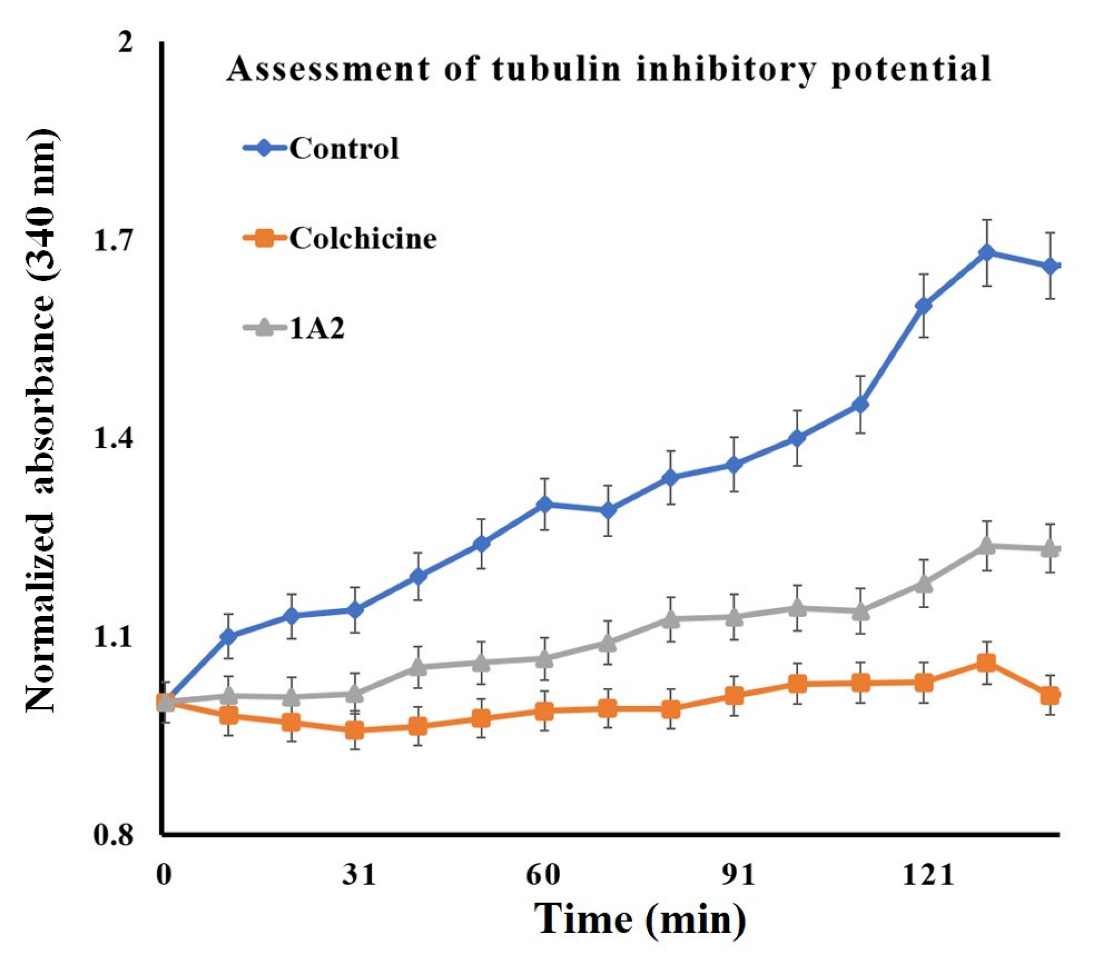{kind=link}
{kind=link}
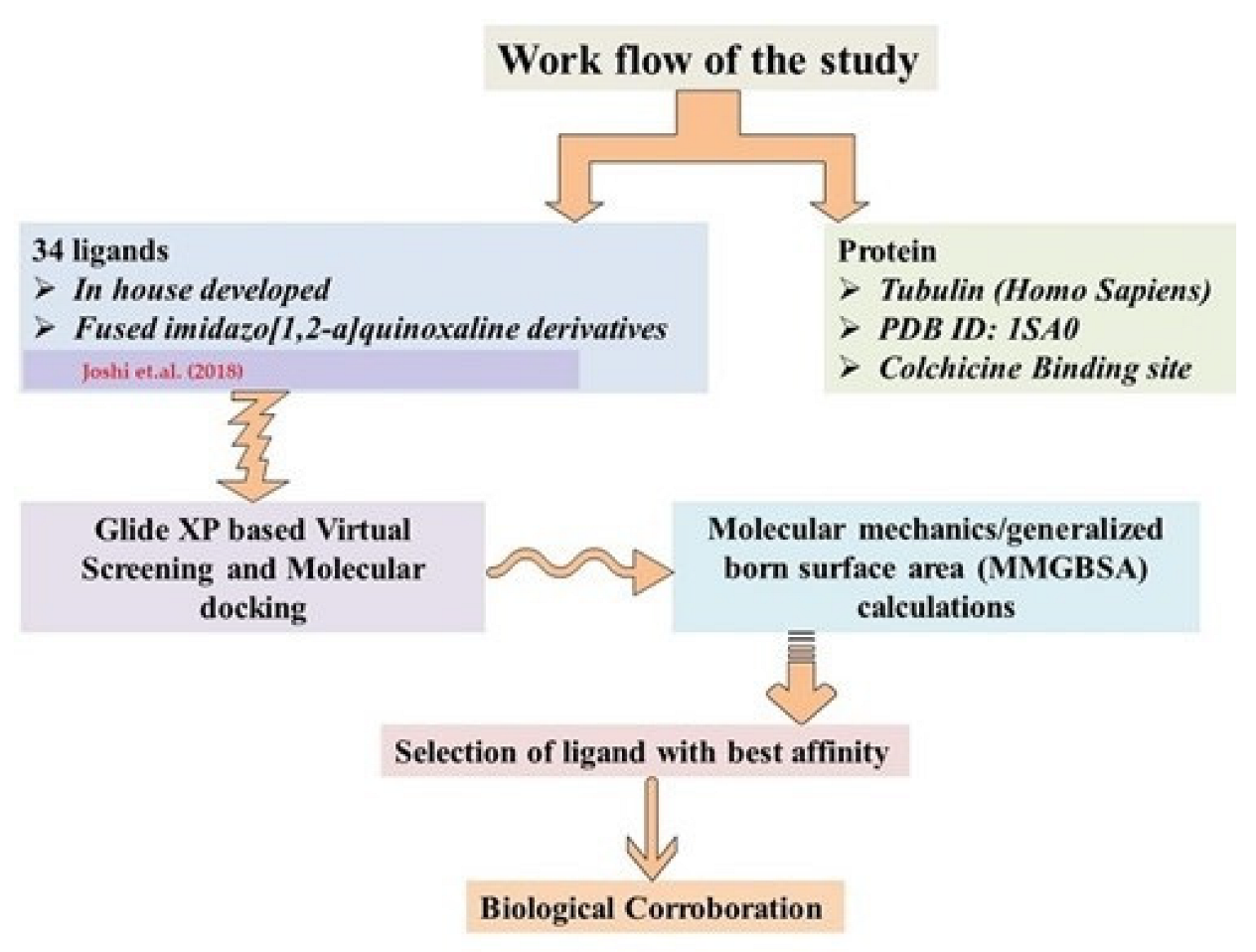{kind=link}
| Sr. No. | Compound Code | G-Score (kcal/mol) | MMGBSA_dG_Bind | MMGBSA_dG_Bind_Coulomb | MMGBSA_dG_Bind_Covalent | MMGBSA_dG_Bind_Hbond | MMGBSA_dG_Bind_Lipo | MMGBSA_dG_Bind_vdW |
|---|---|---|---|---|---|---|---|---|
| 1. | 1A1 | −8.5 | −40.6 | −1.7 | 1.6 | −0.5 | −11.9 | −43.8 |
| 2. | 1A2 | −11.5 | −55.0 | −7.6 | 1.9 | −1.3 | −17.3 | −52.3 |
| 3. | 1B2 | −9.1 | −48.5 | −12.6 | 9.1 | −0.1 | −18.7 | −51.1 |
| 4. | 1B3 | −7.5 | −42.5 | −8.6 | −0.9 | −0.6 | −15.4 | −35.5 |
| 5. | 1B4 | −8.5 | −41.3 | −19.3 | 1.6 | −1.2 | −11.1 | −38.7 |
| 6. | 1B5 | −8.5 | −38.8 | −12.8 | 0.5 | −2.0 | −11.5 | −37.9 |
| 7. | 1B6 | −7.9 | −31.8 | −6.7 | −8.8 | −2.3 | −17.4 | −42.3 |
| 8. | 1C2 | −9.3 | −50.3 | −11.2 | 1.8 | −0.6 | −18.0 | −45.9 |
| 9. | 1C3 | −8.4 | −37.8 | −11.3 | 3.3 | −1.1 | −15.2 | −39.8 |
| 10. | 1C4 | −8.0 | −37.5 | −4.1 | 1.3 | −1.5 | −15.2 | −38.8 |
| 11. | 1C5 | −8.3 | −37.2 | −3.8 | 0.1 | −1.0 | −14.1 | −35.0 |
| 12. | 1C6 | −7.3 | −37.0 | −9.9 | 0.4 | −0.6 | −9.9 | −28.6 |
| 13. | 1C7 | −9.3 | −50.2 | −0.4 | 0.8 | −0.7 | −19.8 | −46.6 |
| 14. | 1C8 | −9.1 | −39.6 | −8.9 | 8.1 | −1.2 | −18.4 | −50.4 |
| 15. | 1C9 | −8.9 | −35.7 | −17.0 | 2.8 | −2.4 | −8.8 | −34.9 |
| 16. | 1C10 | −8.3 | −33.7 | −7.8 | 3.0 | −1.0 | −14.3 | −33.8 |
| 17. | 1C11 | −8.2 | −33.2 | −2.1 | 0.7 | −0.6 | −10.0 | −35.6 |
| 18. | 1D1 | −8.2 | −38.9 | −6.2 | −0.7 | −21.6 | −31.4 | −38.9 |
| 19. | 1D2 | −9.2 | −42.3 | −13.9 | 0.9 | −1.3 | −12.4 | −38.5 |
| 20. | 1E1 | −8.1 | −31.7 | −7.8 | 0.6 | −0.8 | −11.5 | −30.4 |
| 21. | 1E2 | −8.1 | −34.9 | −2.1 | 1.8 | −1.7 | −11.2 | −38.9 |
| 22. | 1E3 | −9.5 | −26.4 | 4.0 | 5.8 | −0.6 | −18.9 | −47.2 |
| 23. | 1E4 | −8.1 | −28.4 | −0.6 | 2.4 | −0.6 | −14.8 | −36.9 |
| 24. | 1E5 | −8.1 | −31.5 | −5.5 | 1.5 | −1.0 | −11.2 | −37.7 |
| 25. | 1E6 | −8.0 | −32.2 | −7.6 | 2.6 | −1.0 | −13.8 | −37.1 |
| 26. | 1E7 | −9.4 | −31.4 | −3.5 | 3.6 | −0.8 | −15.3 | −40.6 |
| 27. | 1E8 | −9.5 | −52.5 | −27.8 | 9.5 | −1.5 | −23.7 | −52.9 |
| 28. | 1E9 | −7.9 | −28.6 | −8.9 | 3.0 | −1.3 | −11.7 | −34.1 |
| 29. | 1E11 | −7.1 | −35.8 | −7.0 | 1.4 | −0.9 | −14.5 | −32.1 |
| 30. | 1E12 | −9.4 | −41.7 | −11.7 | 8.7 | −0.5 | −21.8 | −55.0 |
| 31. | 1E13 | −9.2 | −41.6 | −11.7 | 7.5 | −0.3 | −19.1 | −51.5 |
| 32. | 1E14 | −8.1 | −38.5 | −7.6 | 5.7 | −0.1 | −18.2 | −49.6 |
| 33. | 1E15 | −8.5 | −38.1 | −11.7 | 3.1 | −1.2 | −15.0 | −39.4 |
| 34. | 1E16 | −7.8 | −42.7 | −11.4 | 1.8 | −1.3 | −19.3 | −37.9 |
| 35. | Colchicine | −9.2 | −51.7 | −11.0 | 3.5 | −1.1 | −17.7 | −46.7 |
| Compound | IC50 ± SEM (µM) | |||
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | A549 | HCT-116 | |
| 1A2 | 4.33 ± 0.31 | 6.11 ± 0.23 | 5.87 ± 0.31 | 5.44 ± 0.18 |
| Colchicine | 5.11 ± 0.33 | 5.14 ± 0.35 | 6.55 ± 0.41 | 5.54 ± 0.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goel, K.K.; Hussain, A.; Altamimi, M.A.; Rajput, S.K.; Sharma, P.P.; Kharb, R.; Mahdi, W.A.; Imam, S.S.; Alshehri, S.; Alnemer, O.A.; et al. Identification of Potential Antitubulin Agents with Anticancer Assets from a Series of Imidazo[1,2-a]quinoxaline Derivatives: In Silico and In Vitro Approaches. Molecules 2023, 28, 802. https://doi.org/10.3390/molecules28020802
Goel KK, Hussain A, Altamimi MA, Rajput SK, Sharma PP, Kharb R, Mahdi WA, Imam SS, Alshehri S, Alnemer OA, et al. Identification of Potential Antitubulin Agents with Anticancer Assets from a Series of Imidazo[1,2-a]quinoxaline Derivatives: In Silico and In Vitro Approaches. Molecules. 2023; 28(2):802. https://doi.org/10.3390/molecules28020802
Chicago/Turabian StyleGoel, Kapil Kumar, Afzal Hussain, Mohammad A. Altamimi, Satyendra Kumar Rajput, Prince Prashant Sharma, Rajeev Kharb, Wael A. Mahdi, Syed Sarim Imam, Sultan Alshehri, Osamah Abdulrahman Alnemer, and et al. 2023. "Identification of Potential Antitubulin Agents with Anticancer Assets from a Series of Imidazo[1,2-a]quinoxaline Derivatives: In Silico and In Vitro Approaches" Molecules 28, no. 2: 802. https://doi.org/10.3390/molecules28020802
APA StyleGoel, K. K., Hussain, A., Altamimi, M. A., Rajput, S. K., Sharma, P. P., Kharb, R., Mahdi, W. A., Imam, S. S., Alshehri, S., Alnemer, O. A., & Chaudhary, A. (2023). Identification of Potential Antitubulin Agents with Anticancer Assets from a Series of Imidazo[1,2-a]quinoxaline Derivatives: In Silico and In Vitro Approaches. Molecules, 28(2), 802. https://doi.org/10.3390/molecules28020802