
A Simple Screening and Optimization Bioprocess for Long-Chain Peptide Catalysts Applied to Asymmetric Aldol Reaction

 ,
, 
Abstract
:1. Introduction
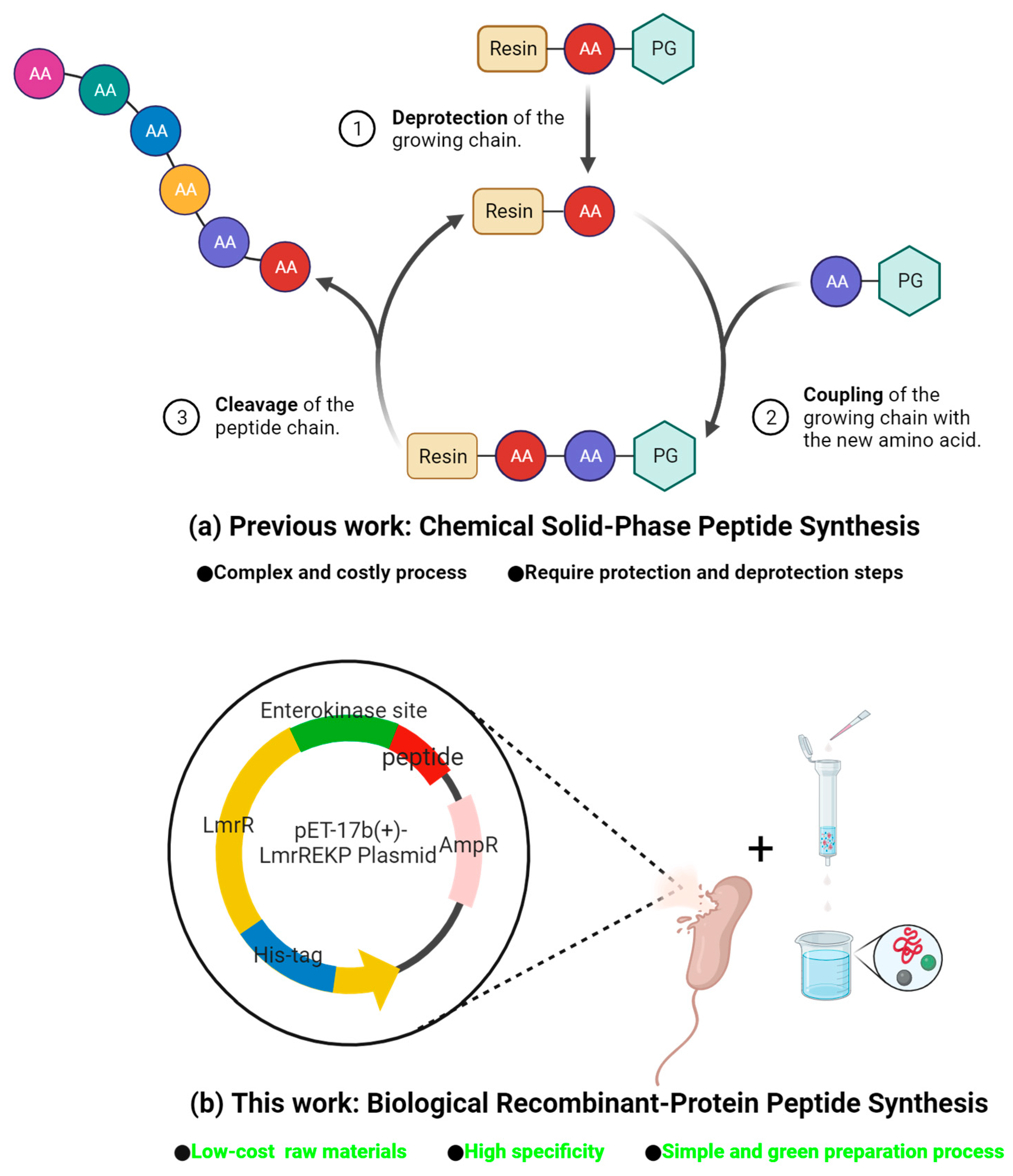
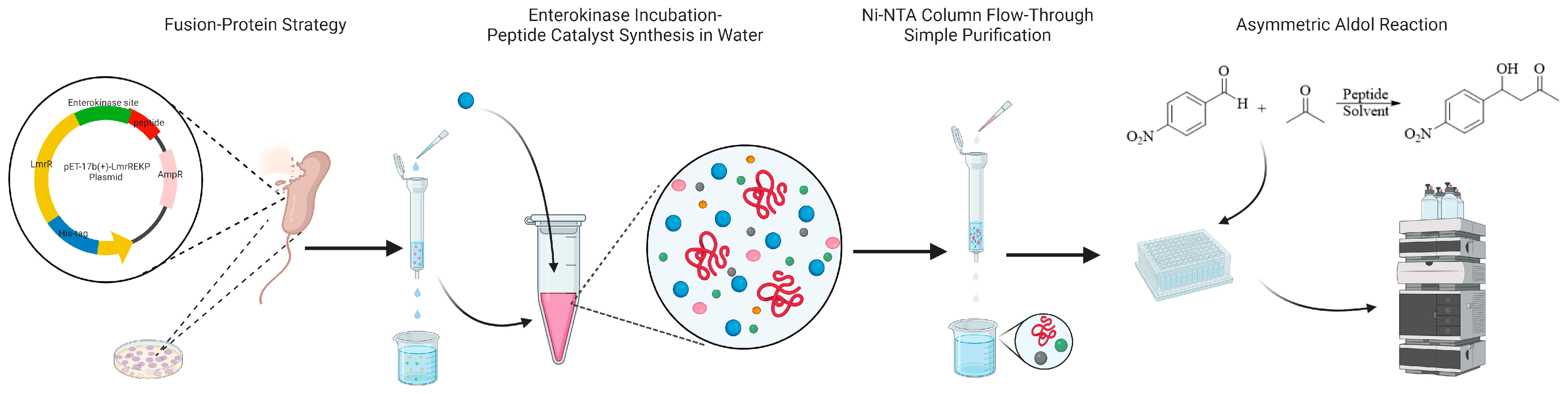
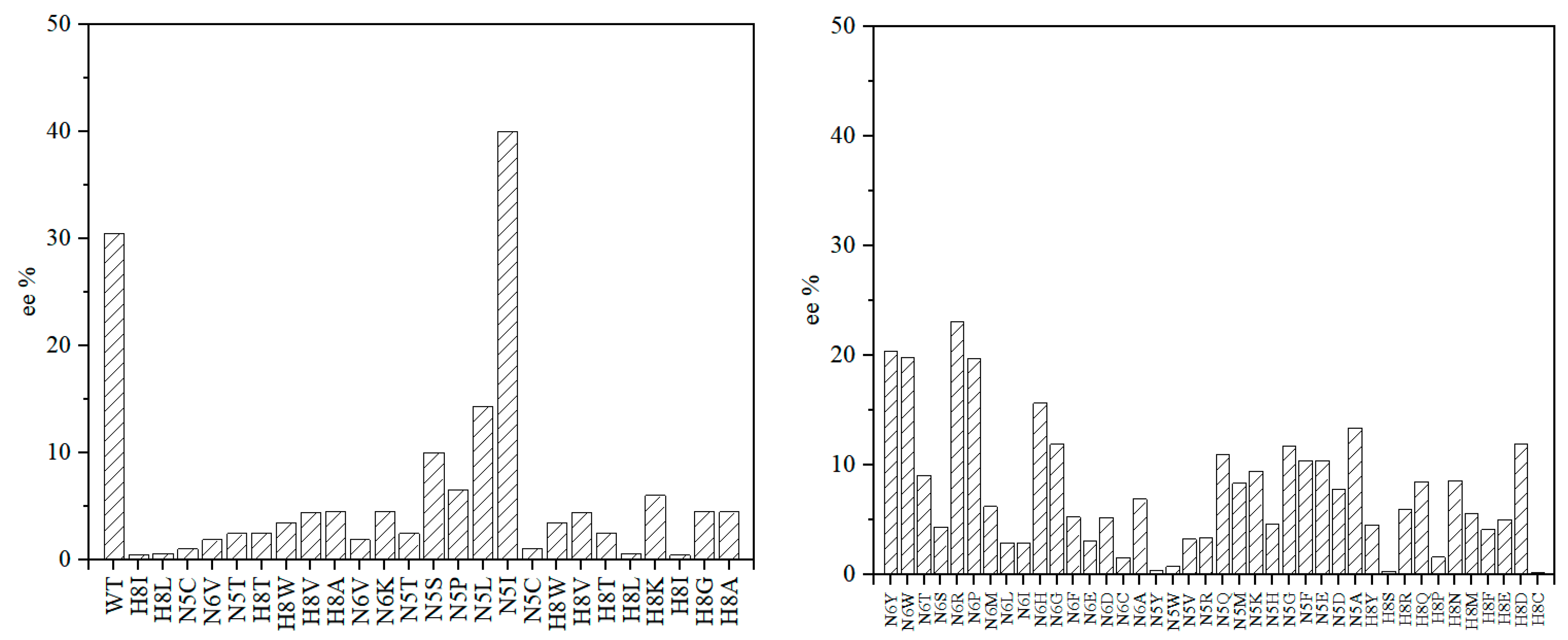
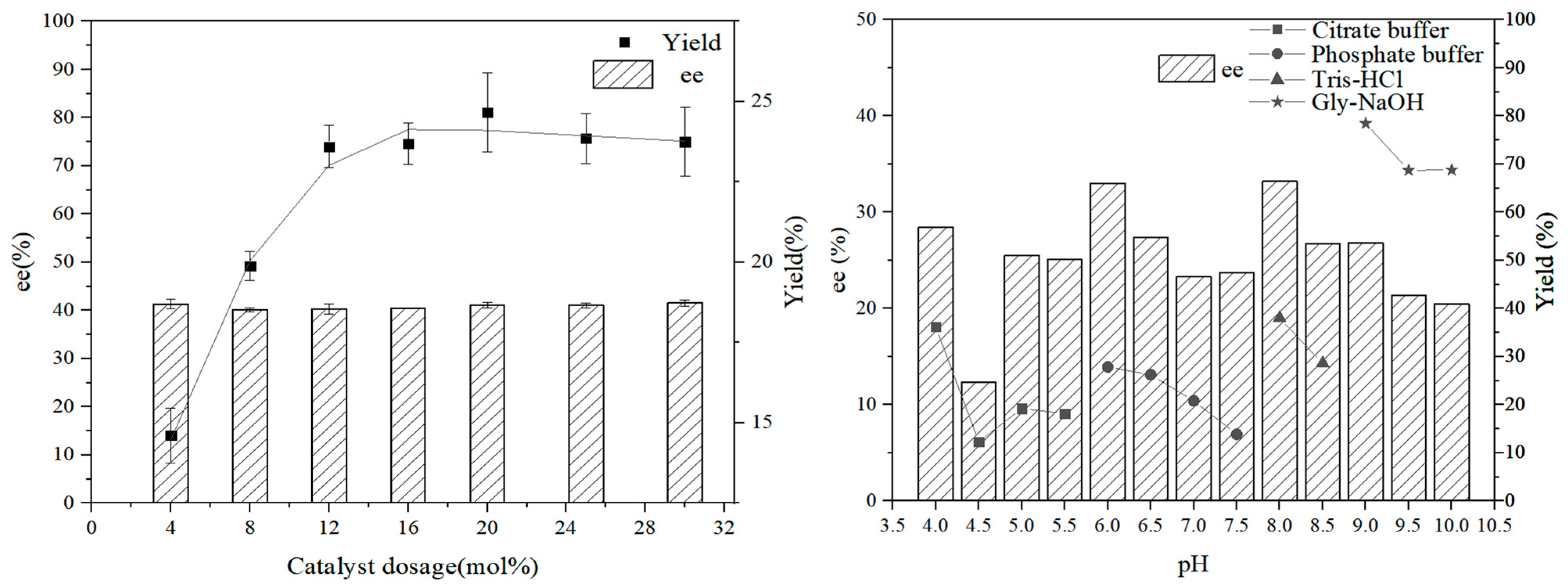
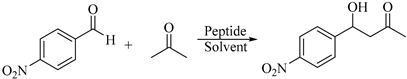
2. Results and Discussion
3. Materials and Methods
3.1. General Materials
3.2. Construction of LmrR Fusion Protein Plasmid Containing Peptide and Enterokinase Sites
3.3. Preparation and Purification of Peptides and Establishment of Peptide Library
3.4. Agarose Gel Electrophoresis Analysis
3.5. SDS-PAGE Analysis
3.6. Chemical Synthesis of Peptides
3.7. Chiral HPLC Analysis
3.8. Preparation of Aldol Reaction Standard Product Racemer and NMR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| Amp | ampicillin |
| Arg | arginine |
| D | aspartic acid |
| DCE | 1,2-dichloroethane |
| DCM | dichloromethane |
| DMSO | dimethyl sulfoxide |
| E. coli | Escherichia coli |
| EK | enterokinase |
| Gly | glycine |
| His | histidine |
| IPTG | isopropyl-β-d-thiogalactoside |
| K | lysine |
| LB | Luria–Bertani |
| Ni-NTA | Nickel-nitrilotriacetic acid |
| P | peptide |
| PCR | Polymerase Chain Reaction |
| SDS-PAGE | sodium dodecyl sulfate−polyacrylamide gel electrophoresis |
References
- Soai, K.; Niwa, S. Enantioselective Addition of Organozinc Reagents to Aldehydes. Chem. Rev. 1992, 92, 833–856. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Yu, S.-L.; Shen, W.-Y.; Gao, J.-X. Iron-, Cobalt-, and Nickel-Catalyzed Asymmetric Transfer Hydrogenation and Asymmetric Hydrogenation of Ketones. Acc. Chem. Res. 2015, 48, 2587–2598. [Google Scholar] [CrossRef]
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic Enantioselective Aldol Reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. [Google Scholar] [CrossRef]
- Trost, B.M.; Brindle, C.S. The Direct Catalytic Asymmetric Aldol Reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. [Google Scholar] [CrossRef]
- He, Y.-H.; Li, H.-H.; Chen, Y.-L.; Xue, Y.; Yuan, Y.; Guan, Z. Chymopapain-Catalyzed Direct Asymmetric Aldol Reaction. Adv. Synth. Catal. 2012, 354, 712–719. [Google Scholar] [CrossRef]
- Haoran, W.; Zhi, W.; Hong, Z.; Ge, C.; Hong, Y.; Lei, W. Enzyme Catalytic Promiscuity: Asymmetric Aldol Addition Reaction Catalyzed by a Novel Thermophilic Esterase in Organic Solvent. Green Chem. Lett. Rev. 2014, 7, 145–149. [Google Scholar] [CrossRef]
- Finn, M.G.; Lerner, R.A.; Barbas, C.F. Cofactor-Induced Refinement of Catalytic Antibody Activity: A Metal-Specific Allosteric Effect. J. Am. Chem. Soc. 1998, 120, 2963–2964. [Google Scholar] [CrossRef]
- Xu, Y.; Yamamoto, N.; Janda, K.D. Catalytic Antibodies: Hapten Design Strategies and Screening Methods. Bioorganic Med. Chem. 2004, 12, 5247–5268. [Google Scholar] [CrossRef]
- List, B. Introduction: Organocatalysis. Chem. Rev. 2007, 107, 5413–5415. [Google Scholar] [CrossRef]
- Dondoni, A.; Massi, A. Asymmetric Organocatalysis: From Infancy to Adolescence. Angew. Chem. Int. Ed. 2008, 47, 4638–4660. [Google Scholar] [CrossRef]
- Ashokkumar, V.; Chithiraikumar, C.; Siva, A. Binaphthyl-Based Chiral Bifunctional Organocatalysts for Water Mediated Asymmetric List–Lerner–Barbas Aldol Reactions. Org. Biomol. Chem. 2016, 14, 9021–9032. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.L. Highlights of the Recent Patent Literature: Focus on Asymmetric Organocatalysis. Org. Process Res. Dev. 2022, 26, 2224–2239. [Google Scholar] [CrossRef]
- Inoue, H.; Kikuchi, M.; Ito, J.; Nishiyama, H. Chiral Phebox–Rhodium Complexes as Catalysts for Asymmetric Direct Aldol Reaction. Tetrahedron 2008, 64, 493–499. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Li, W.; Liu, Y.; Guan, Z.; He, Y.-H. Trypsin-Catalyzed Direct Asymmetric Aldol Reaction. J. Mol. Catal. B Enzym. 2013, 87, 83–87. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Enantioselective Aldol Cyclodehydrations Catalyzed by Antibody 38C2. Org. Lett. 1999, 1, 59–62. [Google Scholar] [CrossRef]
- Zheng, B.-L.; Liu, Q.-Z.; Guo, C.-S.; Wang, X.-L.; He, L. Highly Enantioselective Direct Aldol Reaction Catalyzed by Cinchona Derived Primary Amines. Org. Biomol. Chem. 2007, 5, 2913–2915. [Google Scholar] [CrossRef]
- Bastida, D.; Liu, Y.; Tian, X.; Escudero-Adán, E.; Melchiorre, P. Asymmetric Vinylogous Aldol Reaction via H-Bond-Directing Dienamine Catalysis. Org. Lett. 2013, 15, 220–223. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Córdova, A.; Zou, W.; Dziedzic, P.; Ibrahem, I.; Reyes, E.; Xu, Y. Direct Asymmetric Intermolecular Aldol Reactions Catalyzed by Amino Acids and Small Peptides. Chem. A Eur. J. 2006, 12, 5383–5397. [Google Scholar] [CrossRef]
- Kofoed, J.; Nielsen, J.; Reymond, J.-L. Discovery of New Peptide-Based Catalysts for the Direct Asymmetric Aldol Reaction. Bioorganic Med. Chem. Lett. 2003, 13, 2445–2447. [Google Scholar] [CrossRef]
- Wennemers, H. Asymmetric Catalysis with Peptides. Chem. Commun. 2011, 47, 12036–12041. [Google Scholar] [CrossRef] [PubMed]
- Córdova, A.; Notz, W.; Barbas, C.F., III. Direct Organocatalytic Aldol Reactions in Buffered Aqueous Media. Chem. Commun. 2002, 24, 3024–3025. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Yang, Z.-H.; Chen, X.-H.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. A Highly Efficient Organocatalyst for Direct Aldol Reactions of Ketones with Aldedydes. J. Am. Chem. Soc. 2005, 127, 9285–9289. [Google Scholar] [CrossRef] [PubMed]
- Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C.F. Organocatalytic Direct Asymmetric Aldol Reactions in Water. J. Am. Chem. Soc. 2006, 128, 734–735. [Google Scholar] [CrossRef]
- Huang, W.; Tian, H.; Xu, H.; Zheng, L.; Liu, Q.; Zhang, S. L-Valine Dipeptide Organocatalysts with Two Amide Units for the Direct Asymmetric Aldol Reaction in Brine. Catal. Lett. 2011, 141, 872–876. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Liu, L.; Sang, K.; Zhang, C.; Satoh, T. Asymmetric Aldol Reaction Catalyzed by Amino Acid Tetrapeptides (L-Pro-L-Pro-L-Phe-L-Phe-OMe). React. Chem. Eng. 2023, 8, 2022–2028. [Google Scholar] [CrossRef]
- Al-Momani, L.A.; Lang, H.; Lüdeke, S. A Pathway for Aldol Additions Catalyzed by L-Hydroxyproline-Peptides via a β-Hydroxyketone Hemiaminal Intermediate. Chemistry 2023, 5, 1203–1219. [Google Scholar] [CrossRef]
- Bayat, S.; Tejo, B.A.; Abdulmalek, E.; Salleh, A.B.; Normi, Y.M.; Abdul Rahman, M.B. Rational Design of Mimetic Peptides Based on Aldo-Ketoreductase Enzyme as Asymmetric Organocatalysts in Aldol Reactions. RSC Adv. 2014, 4, 38859–38868. [Google Scholar] [CrossRef]
- Peme, T.; Brady, D.; Juma, W.; Makatini, M. Development of Fructose-1,6-Bisphosphate Aldolase Enzyme Peptide Mimics as Biocatalysts in Direct Asymmetric Aldol Reactions. RSC Adv. 2021, 11, 36670–36681. [Google Scholar] [CrossRef]
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef]
- Metrano, A.J.; Chinn, A.J.; Shugrue, C.R.; Stone, E.A.; Kim, B.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms. Chem. Rev. 2020, 120, 11479–11615. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic Amide Formation from Non-Activated Carboxylic Acids and Amines. Chem. Soc. Rev. 2014, 43, 2714–2742. [Google Scholar] [CrossRef] [PubMed]
- Krattiger, P.; Kovasy, R.; Revell, J.D.; Ivan, S.; Wennemers, H. Increased Structural Complexity Leads to Higher Activity: Peptides as Efficient and Versatile Catalysts for Asymmetric Aldol Reactions. Org. Lett. 2005, 7, 1101–1103. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, R.; Adamala, K.; Gasperi, T.; Polticelli, F.; Stano, P. Small and Random Peptides: An Unexplored Reservoir of Potentially Functional Primitive Organocatalysts. The Case of Seryl-Histidine. Life 2017, 7, 19. [Google Scholar] [CrossRef]
- Carvalho, S.; Peralta Reis, D.Q.; Pereira, S.V.; Kalafatovic, D.; Pina, A.S. Catalytic Peptides: The Challenge between Simplicity and Functionality. Isr. J. Chem. 2022, 62, e202200029. [Google Scholar] [CrossRef]
- Schnitzer, T.; Wiesner, M.; Krattiger, P.; Revell, J.D.; Wennemers, H. Is More Better? A Comparison of Tri- and Tetrapeptidic Catalysts. Org. Biomol. Chem. 2017, 15, 5877–5881. [Google Scholar] [CrossRef] [PubMed]
- Dryland, A.; Sheppard, R.C. Peptide Synthesis. Part 8. A System for Solid-Phase Synthesis under Low Pressure Continuous Flow Conditions. J. Chem. Soc. Perkin Trans. 1986, 1, 125–137. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, F.; Wang, Q.; Zhang, Y.; Xu, B. Small Peptide Nanofibers as the Matrices of Molecular Hydrogels for Mimicking Enzymes and Enhancing the Activity of Enzymes. Chem. Soc. Rev. 2010, 39, 3425–3433. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar]
- Deng, T.; Ge, H.; He, H.; Liu, Y.; Zhai, C.; Feng, L.; Yi, L. The Heterologous Expression Strategies of Antimicrobial Peptides in Microbial Systems. Protein Expr. Purif. 2017, 140, 52–59. [Google Scholar] [CrossRef]
- Issaq, H.J.; Conrads, T.P.; Janini, G.M.; Veenstra, T.D. Methods for Fractionation, Separation and Profiling of Proteins and Peptides. Electrophoresis 2002, 23, 3048–3061. [Google Scholar] [CrossRef] [PubMed]
- Spinck, M.; Piedrafita, C.; Robertson, W.E.; Elliott, T.S.; Cervettini, D.; De La Torre, D.; Chin, J.W. Genetically Programmed Cell-Based Synthesis of Non-Natural Peptide and Depsipeptide Macrocycles. Nat. Chem. 2023, 15, 61–69. [Google Scholar] [CrossRef]
- Kaguchi, R.; Katsuyama, A.; Sato, T.; Takahashi, S.; Horiuchi, M.; Yokota, S.; Ichikawa, S. Discovery of Biologically Optimized Polymyxin Derivatives Facilitated by Peptide Scanning and In Situ Screening Chemistry. J. Am. Chem. Soc. 2023, 145, 3665–3681. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Pu, Z.; Xiao, L.; Xu, G.; Yang, L.; Yu, H.; Wu, J. Mutability-Landscape-Guided Engineering of l-Threonine Aldolase Revealing the Prelog Rule in Mediating Diastereoselectivity of C-C Bond Formation. Angew. Chem. Int. Ed. 2022, 62, e202213855. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Peptide Catalyst | ee% | Yield% |
| 1 | LVYISNSTER | 7(R) | 2.4 |
| 2 | MYSFNNDHSEGAHPR | 31(R) | 5.9 |
| 3 | GTIYSK | 3(S) | 39.2 |
| 4 | GAMLAK | 13(S) | 55.6 |
| 5 | GDNDLVLSDFPK | 19(S) | 9.4 |
| 6 | GNYQFR | 17(S) | 13.0 |
| 7 | GDHSAIR | 25(S) | 18.2 |
| Entry | Peptide Catalyst | ee% | Yield% |
|---|---|---|---|
| 1 | MYSFNNDHSEGAHPA | 5(R) | trace |
| 2 | MYSFINDHSEGAHPR | 40(R) | 13.6 |
| 3 | GTIYSR | 53(S) | 1.5 |
| 4 | GAMLAR | 39(R) | 16.8 |
| Entry | Solvent | ee% | Yield% |
|---|---|---|---|
| 1 | Acetone | 18 | 7.1 |
| 2 | Acetone/H2O (3:1) | 40 | 13.6 |
| 3 | Acetone/H2O (2:3) | 34 | 1.9 |
| 4 | MeOH | 44 | 9.0 |
| 5 | EtOH | 38 | 5.7 |
| 6 | i-PrOH | 29 | 6.0 |
| 7 | DMSO/H2O (100:1) | 73 | 1.8 |
| 8 | DMF/H2O (100:1) | 58 | 8.0 |
| 9 | CH3CN | 22 | 1.7 |
| 10 | toluene | 11 | 3.0 |
| 11 | DCM | 34 | 0.8 |
| 12 | DCE | 41 | 0.6 |
| 13 | Et2O | 23 | 9.2 |
| 14 | n-hexane | 22 | 5.8 |
| 15 | pyridine | 51 | 2.5 |
| 16 | ethyl acetate | 18 | 4.3 |
| Entry | Temperature °C | ee% | Yield% |
|---|---|---|---|
| 1 | 4 | 13 | 21.2 |
| 2 | 25 | 27 | 78.5 |
| 3 | 30 | 28 | 81.2 |
| 4 | 37 | 34 | 87.2 |
| 5 | 45 | 33 | 79.4 |
| Entry | Solvent | Peptide Dosage% | Temperature (°C) | ee% | Yield% | |
|---|---|---|---|---|---|---|
| H2O or Buffer Solution (μL) | Acetone (μL) | |||||
| 1 | 3 | 200 | 4 | 25 | 73 | 1.8 |
| 2 | 3 | 200 | 4 | 37 | 69 | 14.6 |
| 3 | 3 | 200 | 4 | 45 | 61 | 17.2 |
| 4 | 10 | 200 | 4 | 25 | 68 | 9.5 |
| 5 | 20 | 200 | 4 | 25 | 58 | 8.7 |
| 6 | 25 | 200 | 4 | 25 | 56 | 8.4 |
| 7 | 3 | 150 | 4 | 25 | 69 | 6.9 |
| 8 | 3 | 250 | 4 | 25 | 72 | 10.1 |
| 9 | 3 | 300 | 4 | 25 | 73 | 13.0 |
| 10 | 10 | 300 | 12 | 25 | 65 | 20.7 |
| 11 | 10 | 300 | 20 | 25 | 68 | 31.6 |
| 12 | 10 | 300 | 30 | 25 | 66 | 29.9 |
| 13 | 10 | 300 | 20 | 37 | 63 | 80.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Teng, H.; Wang, L.; Li, P.; Yuan, X.; Sang, X.; Wu, J.; Yang, L.; Xu, G. A Simple Screening and Optimization Bioprocess for Long-Chain Peptide Catalysts Applied to Asymmetric Aldol Reaction. Molecules 2023, 28, 6985. https://doi.org/10.3390/molecules28196985
Wang S, Teng H, Wang L, Li P, Yuan X, Sang X, Wu J, Yang L, Xu G. A Simple Screening and Optimization Bioprocess for Long-Chain Peptide Catalysts Applied to Asymmetric Aldol Reaction. Molecules. 2023; 28(19):6985. https://doi.org/10.3390/molecules28196985
Chicago/Turabian StyleWang, Shulin, Haidong Teng, Lan Wang, Pengcheng Li, Xinghao Yuan, Xi Sang, Jianping Wu, Lirong Yang, and Gang Xu. 2023. "A Simple Screening and Optimization Bioprocess for Long-Chain Peptide Catalysts Applied to Asymmetric Aldol Reaction" Molecules 28, no. 19: 6985. https://doi.org/10.3390/molecules28196985
APA StyleWang, S., Teng, H., Wang, L., Li, P., Yuan, X., Sang, X., Wu, J., Yang, L., & Xu, G. (2023). A Simple Screening and Optimization Bioprocess for Long-Chain Peptide Catalysts Applied to Asymmetric Aldol Reaction. Molecules, 28(19), 6985. https://doi.org/10.3390/molecules28196985