
Detection of Aromatic Hydrocarbons in Aqueous Solutions Using Quartz Tuning Fork Sensors Modified with Calix[4]arene Methoxy Ester Self-Assembled Monolayers: Experimental and Density Functional Theory Study

 ,
,  and
and
Abstract
:1. Introduction
2. Results
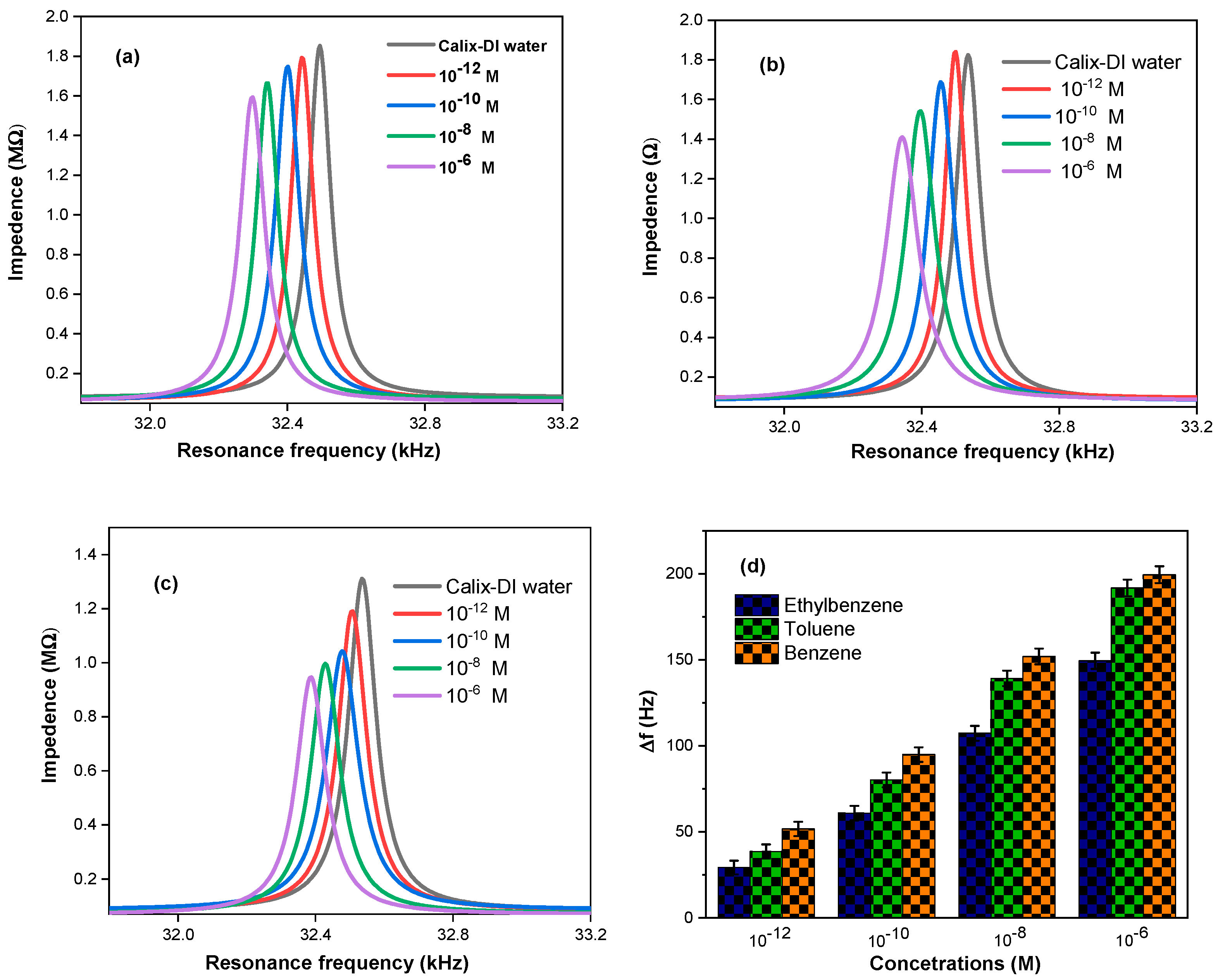
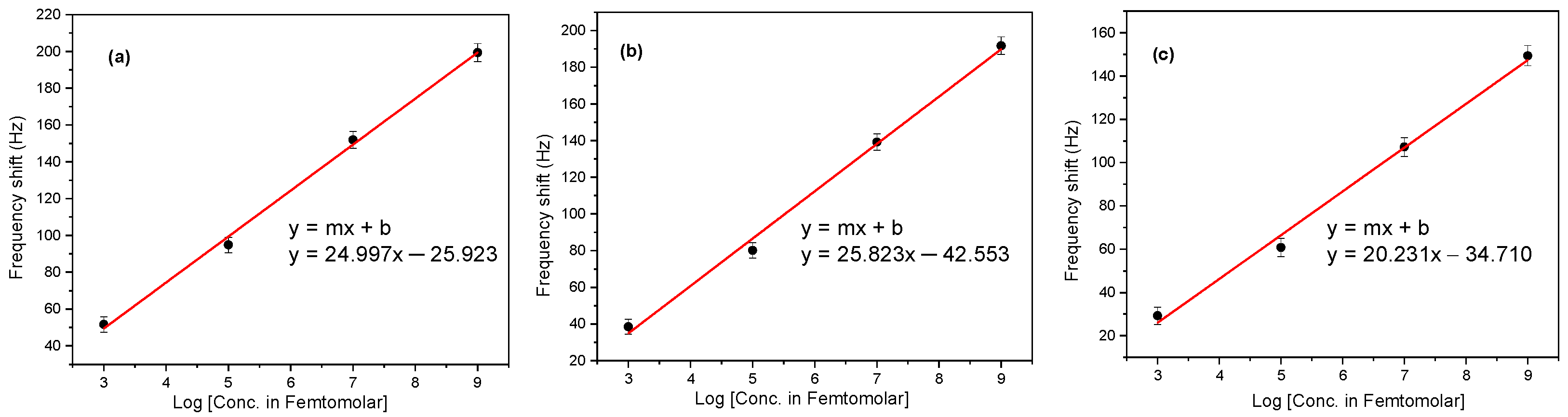
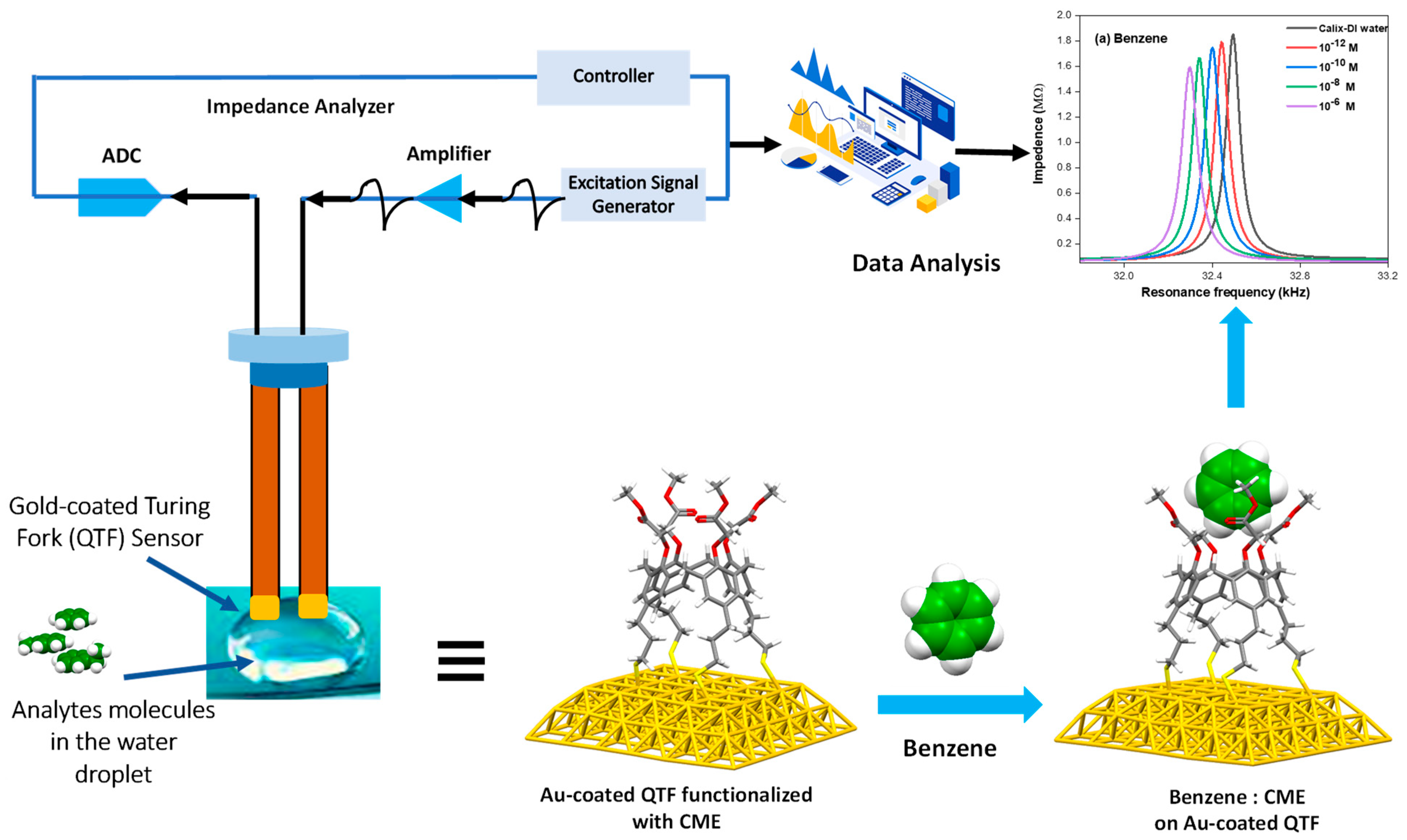
2.1. Resonance Frequency Measurements
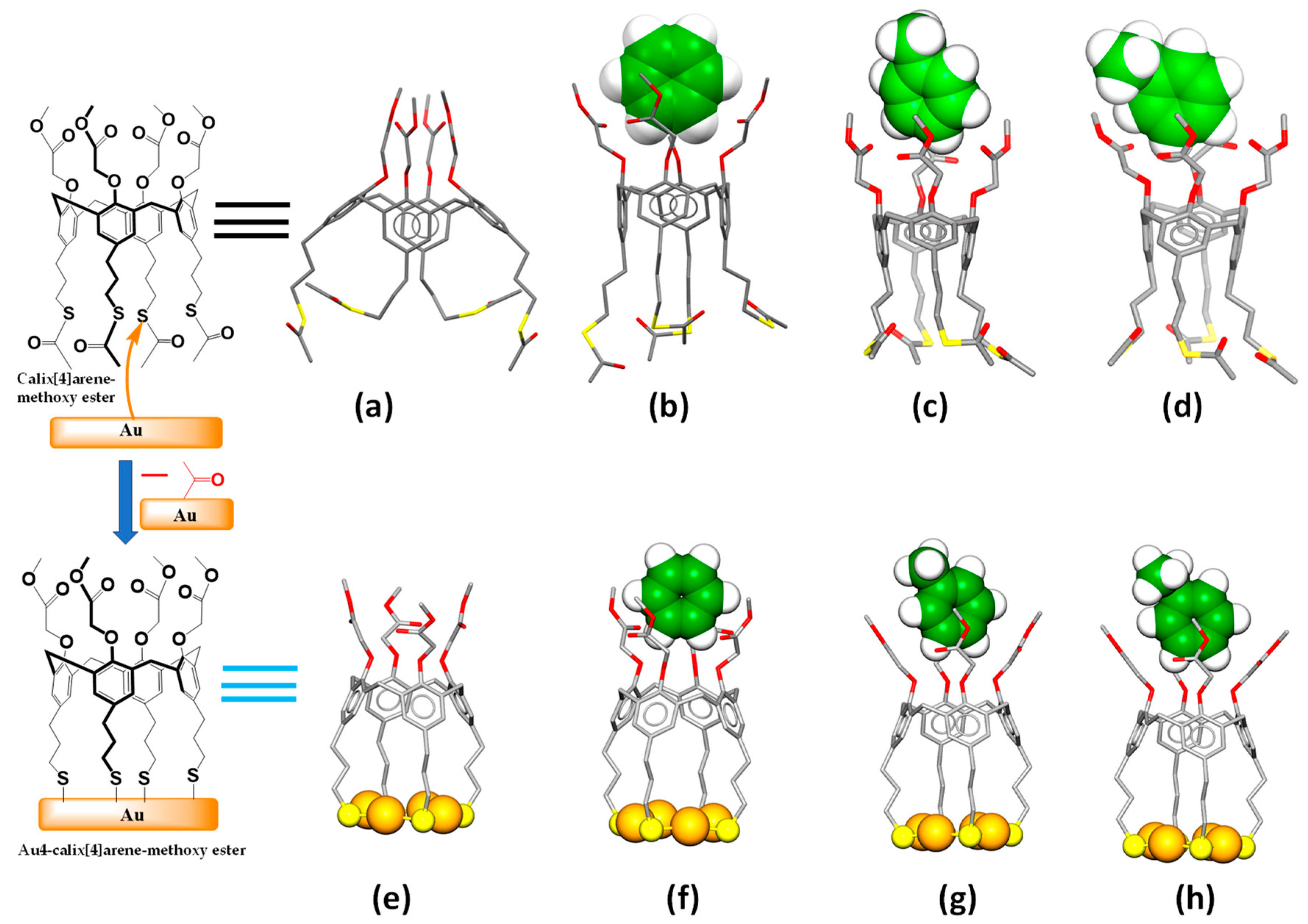
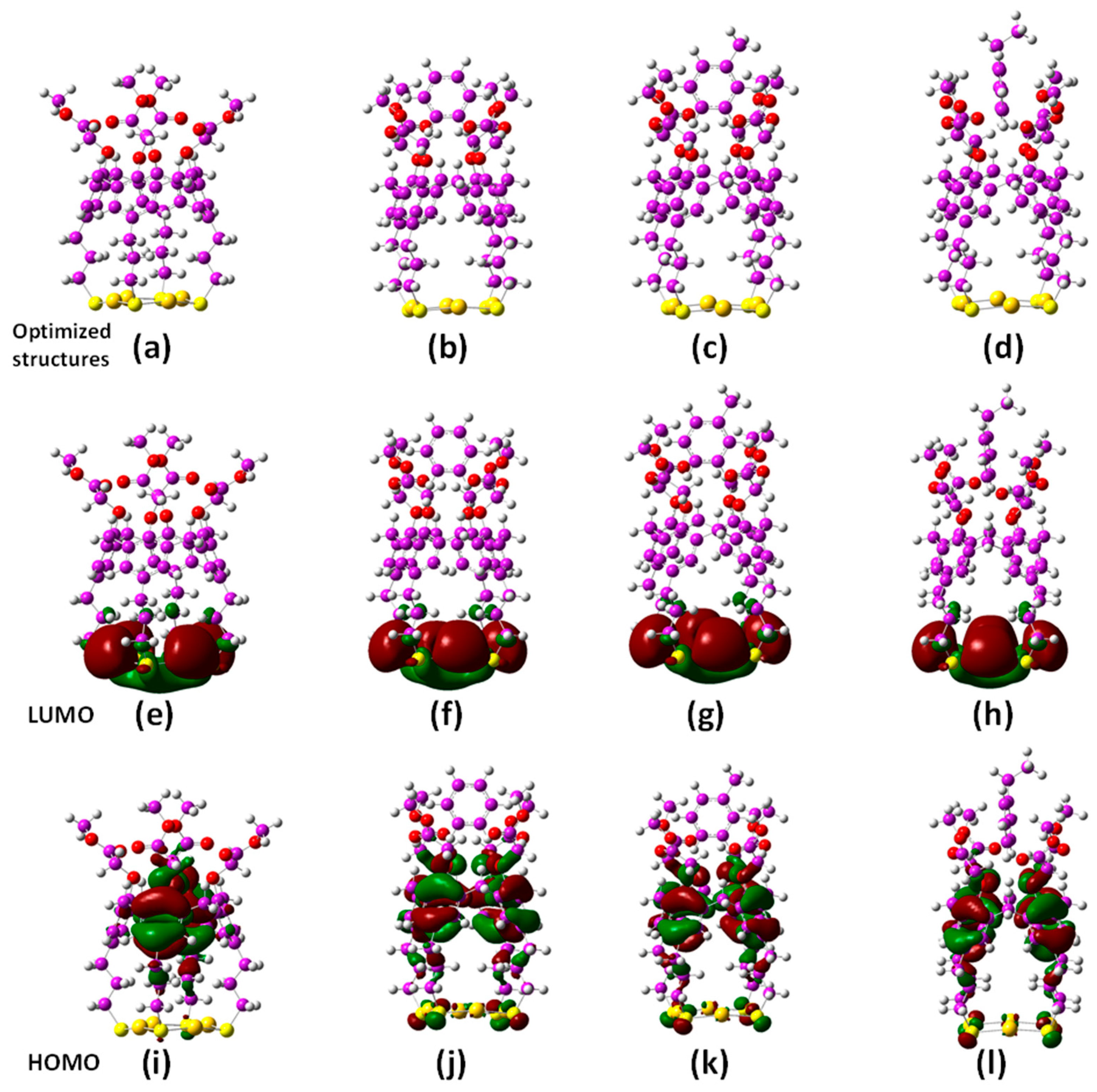
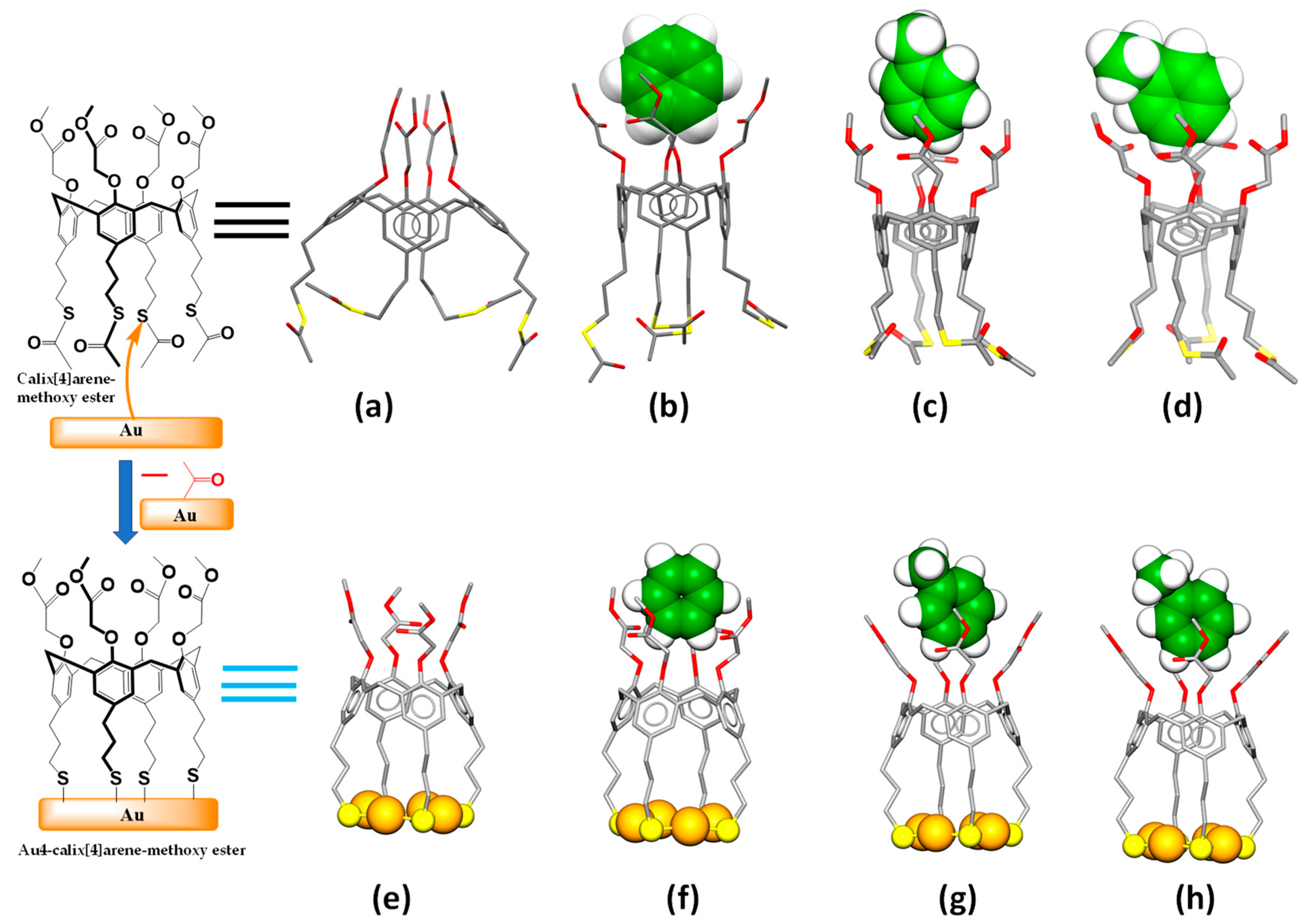
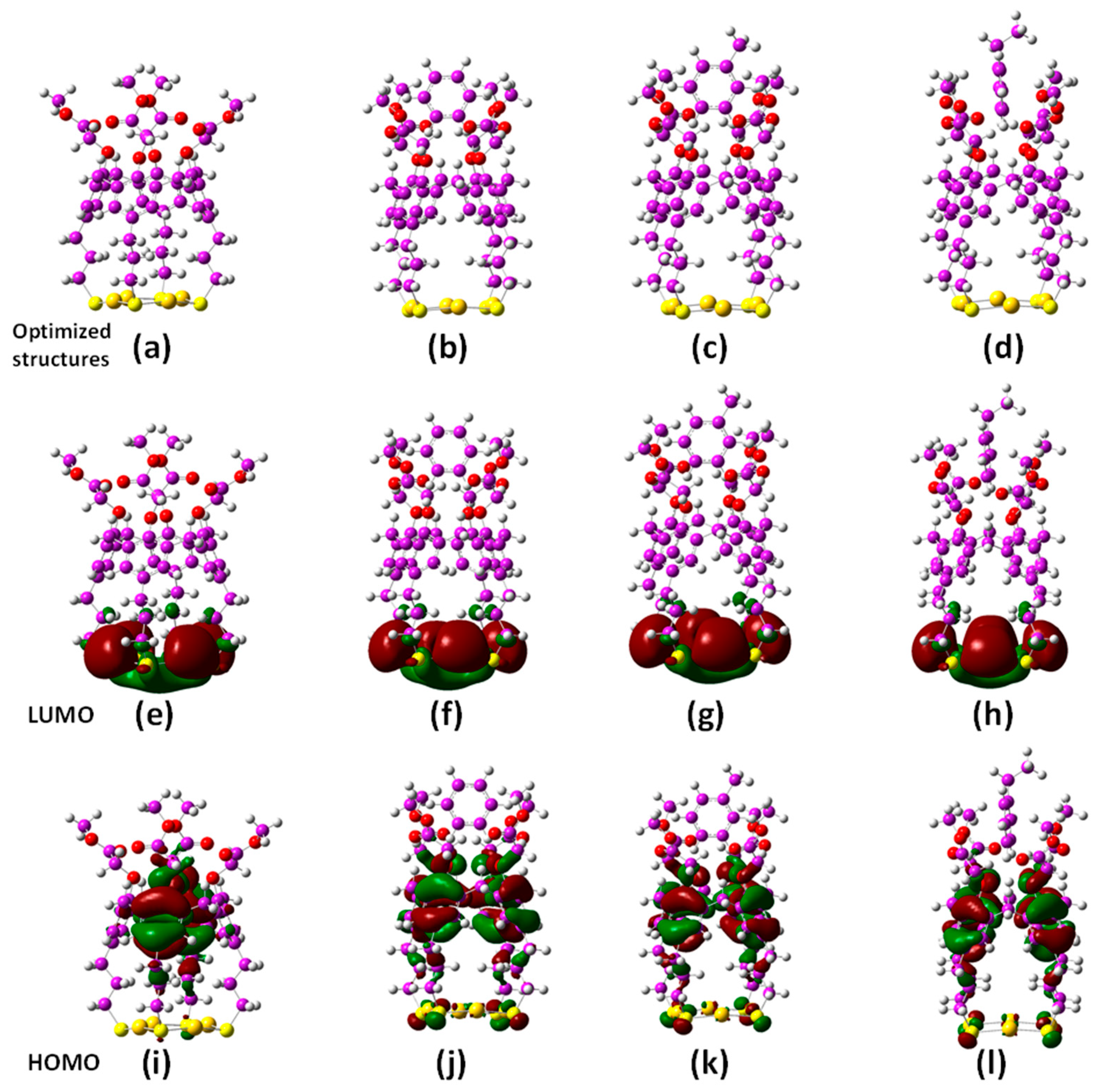
2.2. Quantum Chemical DFT Calculations
3. Discussion
3.1. Analysis of the Resonance Frequency Measurements
3.2. DFT Calculations
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Experimental Setup and Instrumentation
4.3. Experimental Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chary, N.S.; Fernandez-Alba, A.R. Determination of volatile organic compounds in drinking and environmental waters. Trends Anal. Chem. 2012, 32, 60–75. [Google Scholar] [CrossRef]
- Yu, B.; Yuan, Z.; Yu, Z.; Xue-song, F. BTEX in the environment: An update on sources, fate, distribution, pretreatment, analysis, and removal techniques. Chem. Eng. J. 2022, 435, 134825. [Google Scholar] [CrossRef]
- Chen, D.; Lawrence, K.G.; Sandler, D.P. Nontraditional occupational exposures to crude oil combustion disasters and respiratory disease risk: A narrative review of the literature. Curr. Allergy Asthma Rep. 2023, 23, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Nriagu, J.; Udofia, E.A.; Ekong, I.; Ebuk, G. Health risks associated with oil pollution in the Niger Delta, Nigeria. Int. J. Environ. Res. Public Health 2016, 13, 346. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, N.; Cuadras, A.; Rovira, E.; Borrull, F.; Marcé, R.M. Chronic Risk Assessment of Exposure to Volatile Organic Compounds In The Atmosphere Near The Largest Mediterranean Industrial Site. Environ. Int. 2012, 39, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Suzuki, N. Toxicities of Polycyclic Aromatic Hydrocarbons for Aquatic Animals. Int. J. Environ. Res. Public Health. 2020, 17, 1363. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, H.; Mosleh, M.H.; Mandal, P.; Lea-Langton, A.; Sedighi, M. Emissions of volatile organic compounds from crude oil processing–Global emission inventory and environmental release. Sci. Total Environ. 2020, 727, 138654. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://pubchem.ncbi.nlm.nih.gov/compound/241 (accessed on 15 September 2023).
- Yang, Y.; Miller, D.J.; Hawthorne, S.B. Toluene solubility in water and organic partitioning from gasoline and diesel fuel into water at elevated temperatures and pressures. J. Chem. Eng. Data 1997, 42, 908–913. [Google Scholar] [CrossRef]
- Available online: https://pubchem.ncbi.nlm.nih.gov/compound/7500 (accessed on 15 September 2023).
- US Environmental Protection Agency. National Primary Drinking Water Regulations. Available online: https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations (accessed on 29 August 2023).
- Adeniran, M.A.; Oladunjoye, M.A.; Doro, K.O. Soil and groundwater contamination by crude oil spillage: A review and implications for remediation projects in Nigeria. Front. Environ. Sci. 2023, 11, 1137496. [Google Scholar] [CrossRef]
- Zogorski, J.S.; Carter, J.M.; Ivahnenko, T.; Lapham, W.W.; Moran, M.J.; Rowe, B.L.; Squillace, P.J.; Toccalino, P.L. Volatile organic compounds in the nation’s ground water and drinking-water supply wells. US Geol. Surv. Circ. 2006, 1292, 101. [Google Scholar]
- Smith, M.T. Advances in understanding benzene health effects and susceptibility. Annu. Rev. Public Health 2010, 31, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H. Iron oxide catalysts for dehydrogenation of ethylbenzene in the presence of steam. Catal. Rev. Sci. Eng. 1974, 8, 285–305. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Zhang, M. Source Apportionment of Personal Exposure to Carbonyl Compounds and BTEX at Homes in Beijing, China. Aerosol Air Qual. Res. 2014, 14, 330–337. [Google Scholar] [CrossRef]
- Jurdáková, H.; Kubinec, R.; Jurčišinová, M.; Krkošová, Ž.; Blaško, J.; Ostrovský, I.; Soják, L.; Berezkin, V.G. Gas chromatography analysis of benzene, toluene, ethylbenzene and xylenes using newly designed needle trap device in aqueous samples. J. Chromatogr. A 2008, 1194, 61–164. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.; Bianco, G.; Calace, S.; Masi, S.; Mancini, I.M.; Mazzone, G.; Caniani, D. Method development and optimization for the determination of benzene, toluene, ethylbenzene and xylenes in water at trace levels by static headspace extraction coupled to gas chromatography–barrier ionization discharge detection. J. Chromatogr. A 2018, 1548, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.; Ghamari, F.; Yamini, Y.; Ghorbani Shahna, F.; Koolivand, A. Ion-pair-based hollow-fiber liquid-phase microextraction combined with high-performance liquid chromatography for the simultaneous determination of urinary benzene, toluene, and styrene metabolites. J. Sep. Sci. 2018, 41, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Karlitschek, P.; Lewitzka, F.; Bünting, U.; Niederkrüger, M.; Marowsky, G. Detection of aromatic pollutants in the environment by using UV-laser-induced fluorescence. Appl. Phys. B 1998, 67, 497–504. [Google Scholar] [CrossRef]
- Iyer, A.; Mitevska, V.; Samuelson, J.; Campbell, S.; Bhethanabotla, V.R. Polymer–plasticizer coatings for BTEX detection using quartz crystal microbalance. Sensors 2021, 21, 5667. [Google Scholar] [CrossRef]
- Rahman, S.; Al-Gawati, M.A.; Alfaifi, F.S.; Muthuramamoorthy, M.; Alanazi, A.F.; Albrithen, H.; Alzahrani, K.E.; Assaifan, A.K.; Alodhayb, A.N.; Georghiou, P.E. The Effect of Counterions on the Detection of Cu2+ Ions in Aqueous Solutions Using Quartz Tuning Fork (QTF) Sensors Modified with L-Cysteine Self-Assembled Monolayers: Experimental and Quantum Chemical DFT Study. Chemosensors 2023, 11, 88. [Google Scholar] [CrossRef]
- Alshammari, A.; Aldosari, F.; Qarmalah, N.B.; Lsloum, A.; Muthuramamoorthy, M.; Alodhayb, A. Detection of chemical host-guest interactions using a quartz tuning fork sensing system. IEEE Sens. J. 2020, 20, 12543–12551. [Google Scholar] [CrossRef]
- Neri, P.; Sessler, J.L.; Wang, M.-X. (Eds.) Calixarenes and Beyond; Springer International Publishing AG: Cham, Switzerland, 2016. [Google Scholar]
- Zeybek, N.; Acikbas, Y.; Bozkurt, S.; Sirit, A.; Çapan, R.; Erdoğan, M.; Özkaya, C. Developing of the calixarene based diamide chemical sensor chip for detection of aromatic hydrocarbons’ vapors. J. BAUN Inst. Sci. Technol. 2021, 23, 291–300. [Google Scholar] [CrossRef]
- Georghiou, P.E.; Rahman, S.; Assiri, Y.; Valluru, G.K.; Menelaou, M.; Alodhayb, A.N.; Braim, M.; Beaulieu, L.Y. Development of calix [4]arenes modified at their narrow- and wide-rims as potential metal ions sensor layers for microcantilever sensors: Further studies. Can. J. Chem. 2022, 100, 144–149. [Google Scholar] [CrossRef]
- Georghiou, P.E.; Rahman, S.; Valluru, G.; Dawe, L.N.; Rahman, S.S.; Alodhayb, A.N.; Beaulieu, L.Y. Synthesis of an upper-and lower-rim functionalized calix [4] arene for detecting calcium ions using a microcantilever sensor. New J. Chem. 2013, 37, 298–1301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of polarization and diffuse functions to the LANL2DZ basis set for p-block elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Armbruster, D.A.; Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29 (Suppl. 1), S49–S52. [Google Scholar]
- Theodorsson, E. Limit of Blank, Limit of Detection and Limit of Quantitation. Available online: https://www.eflm.eu/files/efcc/Zagreb-Theodorsson_2.pdf (accessed on 15 September 2023).
- Shrivastava, A.; Gupta, V.B. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J. GaussView, Version 6.0.16; Semichem Inc.: Shawnee Mission, KS, USA, 2019. [Google Scholar]
- Mandler, D.; Kraus-Ophir, S. Self-assembled monolayers(SAMs) for electrochemical sensing. J. Solid State Electrochem. 2011, 15, 1535−1558. [Google Scholar] [CrossRef]
- Valluru, G. Synthesis and Applications of Some Upper and Lower Rim Functionalized Calix[4] Arenes and Calix [4] Naphthalene Derivatives. Ph.D. Dissertation, Memorial University of Newfoundland, St. John’s, NL, Canada, August 2015. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Kestner, N.R.; Combariza, J.E. Basis set superposition errors: Theory and practice. In Reviews in Computational Chemistry; Wiley-VCH, John Wiley and Sons, Inc.: New York, NY, USA, 1999; Volume 13, p. 99. [Google Scholar]
- Muhammad, S.; Irfan, A.; Al-Sehemi, A.G.; Al-Assiri, M.S.; Kalam, A.; Chaudhry, A.R. Quantum chemical investigation of spectroscopic studies and hydrogen bonding interactions between water and methoxybenzeylidene-based humidity sensor. J. Theor. Comput. Chem. 2015, 14, 1550029. [Google Scholar] [CrossRef]
- Muhammad, S.; Liu, C.; Zhao, L.; Wu, S.; Su, Z. A theoretical investigation of intermolecular interaction of a phthalimide based “on–off” sensor with different halide ions: Tuning its efficiency and electro-optical properties. Theor. Chem. Acc. 2009, 122, 77–86. [Google Scholar] [CrossRef]
- Uddin, K.M.; Ralph, D.; Henry, D.J. Mechanistic investigation of halopentaaquachromium (III) complexes: Comparison of computational and experimental results. Comput. Theor. Chem. 2015, 1070, 152–161. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Luo, J.; Xue, Z.Q.; Liu, W.M.; Wu, J.L.; Yang, Z.Q. Koopmans’ theorem for large molecular systems within density functional theory. J. Phys. Chem. A 2006, 110, 12005–12009. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔIEs (kJ mol−1) of the Hypothetical 1:1 Supramolecular Structures of CME with Benzene, Toluene, and Ethylbenzene | |||
|---|---|---|---|---|
| CAM-B3LYP/LANL2DZ | PBE/LANL2DZ | |||
| Uncorrected BSSE | Corrected BSSE | Uncorrected BSSE | Corrected BSSE | |
| [CME]⊃[Benzene] | −51.99 | −34.19 | −48.18 | −31.08 |
| [CME]⊃[Toluene] | −45.66 | −30.66 | −44.91 | −29.95 |
| [CME]⊃[Ethylbenzene] | −41.92 | −25.06 | −43.08 | −26.13 |
| [Au4-CME]⊃[Benzene] | −71.42 | −48.60 | −61.53 | −44.18 |
| [Au4-CME]⊃[Toluene] | −65.40 | −44.85 | −59.40 | −42.57 |
| [Au4-CME]⊃[Ethylbenzene] | −54.39 | −42.38 | −51.57 | −41.14 |
| Complex | ΔIEs (kJ mol−1) of the Hypothetical 1:1 Supramolecular Structures of CME with Benzene, Toluene, and Ethylbenzene | |
|---|---|---|
| CAM-B3LYP/LANL2DZ | PBE/LANL2DZ | |
| Uncorrected BSSE | Uncorrected BSSE | |
| [CME]⊃[Benzene] | −32.12 | −29.77 |
| [CME]⊃[Toluene] | −31.28 | −28.80 |
| [CME]⊃[Ethylbenzene] | −30.55 | −28.34 |
| [Au4-CME]⊃[Benzene] | −60.45 | −53.31 |
| [Au4-CME]⊃[Toluene] | −58.06 | −50.93 |
| [Au4-CME]⊃[Ethylbenzene] | −55.47 | −47.19 |
| Complex | ΔIEs (kJ mol−1) of the Hypothetical 1:1 Supramolecular Structures of CME with Benzene, Toluene, and Ethylbenzene | ||
|---|---|---|---|
| Gas Phase | Water Solvent | ||
| Uncorrected BSSE | Corrected BSSE | Uncorrected BSSE | |
| [Au4-CME]⊃[Benzene] | −96.86 | −86.39 | −84.98 |
| [Au4-CME]⊃[Toluene] | −87.80 | −77.23 | −75.25 |
| [Au4-CME]⊃[Ethylbenzene] | −79.33 | −67.63 | −66.28 |
| HOMO Energy eV | LUMO Energy eV | H–L Gap eV | Ionization Potential (IP) eV | Electron Affinity (EA) eV | Electronegativity (χ) eV | Chemical Potential (μ) eV | Hardness (η) eV | Softness (S) eV | Electrophilicity Index (ω) eV | |
|---|---|---|---|---|---|---|---|---|---|---|
| Gas phase | ||||||||||
| Au4-Calix[4]arene | −7.230 | −0.181 | 7.050 | 7.230 | 0.181 | 3.706 | −3.706 | 3.525 | 0.284 | 1.948 |
| Au4-CME⊃benzene | −7.337 | −0.202 | 7.135 | 7.337 | 0.202 | 3.770 | −3.770 | 3.568 | 0.280 | 1.992 |
| Au4-CME⊃toluene | −7.325 | −0.196 | 7.129 | 7.325 | 0.196 | 3.761 | −3.761 | 3.564 | 0.281 | 1.984 |
| Au4-CME⊃ethylbenzene | −7.321 | −0.195 | 7.126 | 7.321 | 0.195 | 3.758 | −3.758 | 3.563 | 0.281 | 1.982 |
| Water solvent system | ||||||||||
| Au4-CME | −7.526 | −0.206 | 7.319 | 7.526 | 0.206 | 3.866 | −3.866 | 3.660 | 0.273 | 2.042 |
| Au4-CME⊃benzene | −7.625 | −0.160 | 7.465 | 7.625 | 0.160 | 3.892 | −3.892 | 3.732 | 0.268 | 2.030 |
| Au4-CME⊃toluene | −7.622 | −0.159 | 7.463 | 7.622 | 0.159 | 3.890 | −3.890 | 3.732 | 0.268 | 2.028 |
| Au4-CME⊃ethylbenzene | −7.605 | −0.162 | 7.443 | 7.605 | 0.162 | 3.883 | −3.883 | 3.722 | 0.269 | 2.026 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.; Al-Gawati, M.A.; Alfaifi, F.S.; Alenazi, W.K.; Alarifi, N.; Albrithen, H.; Alodhayb, A.N.; Georghiou, P.E. Detection of Aromatic Hydrocarbons in Aqueous Solutions Using Quartz Tuning Fork Sensors Modified with Calix[4]arene Methoxy Ester Self-Assembled Monolayers: Experimental and Density Functional Theory Study. Molecules 2023, 28, 6808. https://doi.org/10.3390/molecules28196808
Rahman S, Al-Gawati MA, Alfaifi FS, Alenazi WK, Alarifi N, Albrithen H, Alodhayb AN, Georghiou PE. Detection of Aromatic Hydrocarbons in Aqueous Solutions Using Quartz Tuning Fork Sensors Modified with Calix[4]arene Methoxy Ester Self-Assembled Monolayers: Experimental and Density Functional Theory Study. Molecules. 2023; 28(19):6808. https://doi.org/10.3390/molecules28196808
Chicago/Turabian StyleRahman, Shofiur, Mahmoud A. Al-Gawati, Fatimah S. Alfaifi, Wadha Khalaf Alenazi, Nahed Alarifi, Hamad Albrithen, Abdullah N. Alodhayb, and Paris E. Georghiou. 2023. "Detection of Aromatic Hydrocarbons in Aqueous Solutions Using Quartz Tuning Fork Sensors Modified with Calix[4]arene Methoxy Ester Self-Assembled Monolayers: Experimental and Density Functional Theory Study" Molecules 28, no. 19: 6808. https://doi.org/10.3390/molecules28196808
APA StyleRahman, S., Al-Gawati, M. A., Alfaifi, F. S., Alenazi, W. K., Alarifi, N., Albrithen, H., Alodhayb, A. N., & Georghiou, P. E. (2023). Detection of Aromatic Hydrocarbons in Aqueous Solutions Using Quartz Tuning Fork Sensors Modified with Calix[4]arene Methoxy Ester Self-Assembled Monolayers: Experimental and Density Functional Theory Study. Molecules, 28(19), 6808. https://doi.org/10.3390/molecules28196808