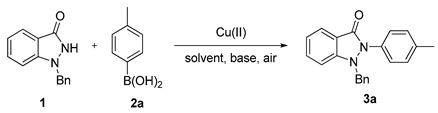
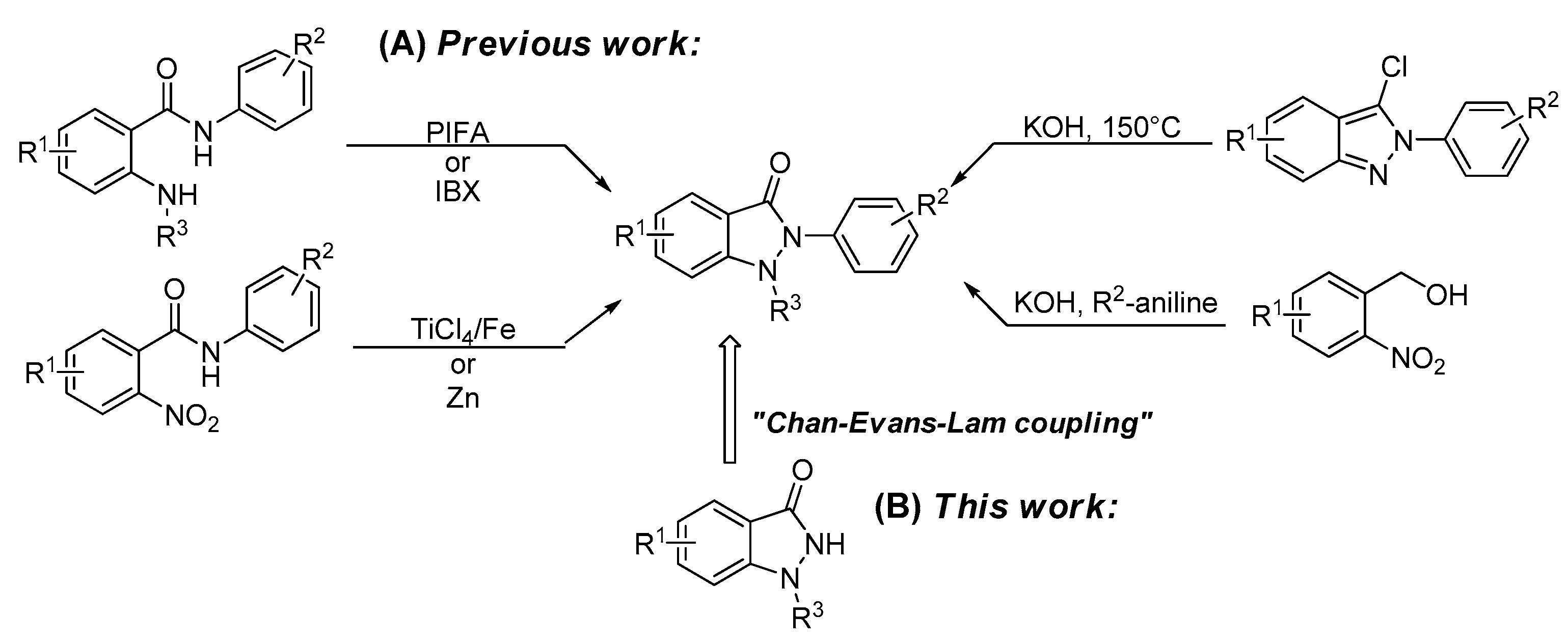
3.2. Chemistry
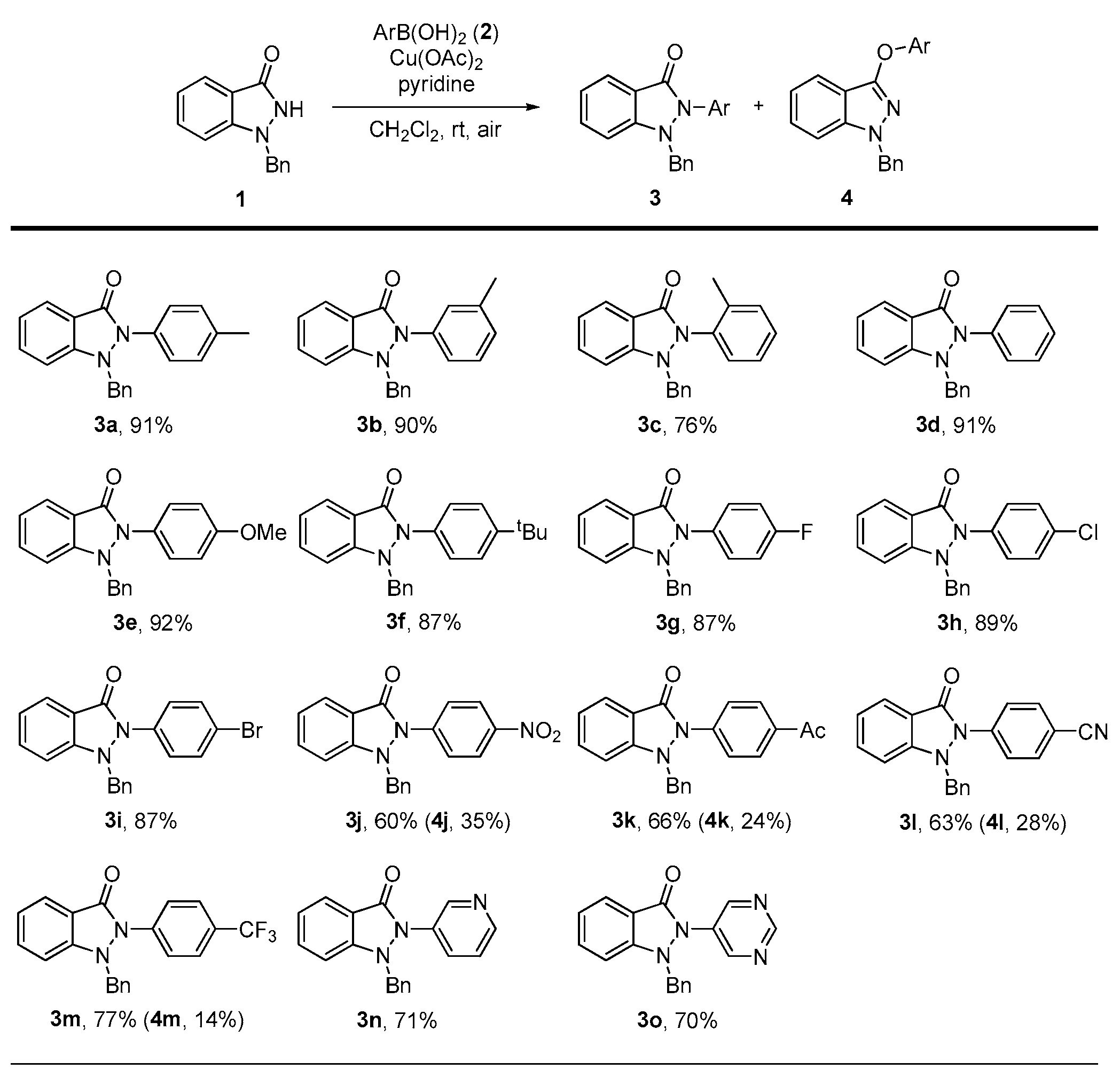
3.2.1. General Procedure for the Synthesis of Compounds (3a–3o, 4j–4m)
![Molecules 28 06706 i002]()
1-Benzyl-1H-indazol-3(2H)-one (1, 0.36 mmol), Cu(OAc)2 (0.18 mmol) and arylboronic acid (0.54 mmol) were dissolved in CH2Cl2 (0.7 mL) and pyridine (7.2 mmol). The mixture was stirred under air at ambient temperature for 24 h. Upon completion of the reaction, the mixture was quenched with 1 M HCl at 0 °C, followed by extraction with ethyl acetate. The organic layer was separated, washed with brine, and dried over Na2SO4. After filtration, the mixture was concentrated in vacuo and subsequently purified by silica gel flash column chromatography to yield pure solid products (3a–3o, 4j–4m).
3a, Yield = 91%, white solid, 1-benzyl-2-
p-tolyl-1
H-indazol-3(2
H)-one, m.p.: 164–166 °C. IR (cm
−1): υ
max 1665, 1509, 1319, 1011.
1H NMR (300 MHz, DMSO-
d6) δ 7.85 (d,
J = 8.3 Hz, 1H), 7.67 (t,
J = 8.0 Hz, 1H), 7.63 (d,
J = 8.1 Hz, 1H), 7.45–7.38 (m, 4H), 7.20 (t,
J = 7.5 Hz, 1H), 7.17–7.13 (m, 3H), 6.84 (d,
J = 5.5 Hz, 2H), 4.91 (s, 2H).
13C NMR (75 MHz, CDCl
3) δ 162.36, 149.81, 136.38, 133.45, 132.57, 132.21, 129.81, 128.61, 128.28, 128.21, 124.50, 124.03, 122.77, 119.82, 113.10, 54.59, 21.11. HRMS (EI) calcd for C
21H
18N
2O (M)
+ 314.1419, found 314.1418. [CAS RN: 1182783-69-2] [
28].
3b, Yield = 90%, white solid, 1-benzyl-2-
m-tolyl-1
H-indazol-3(2
H)-one, m.p.: 124–125 °C.
1H NMR (300 MHz, DMSO-
d6) δ 7.84 (d,
J = 8.4 Hz, 1H), 7.67 (t,
J = 8.1 Hz, 1H), 7.62 (d,
J = 8.3 Hz, 1H), 7.46 (t,
J = 7.6 Hz, 1H), 7.36–7.33 (m, 2H), 7.19–7.16 (m, 2H), 7.14–7.12 (m, 3H), 6.84 (d,
J = 7.3 Hz, 2H), 4.90 (s, 2H), 2.41 (s, 3H).
13C NMR (75 MHz, CDCl
3) δ 162.42, 149.92, 139.16, 135.09, 133.43, 132.29, 128.99, 128.65, 128.27, 128.22, 127.28, 124.62, 124.53, 122.82, 121.09, 119.84, 113.15. 54.78, 21.53. HRMS (EI) calcd for C
21H
18N
2O (M)
+ 314.1419, found 314.1417. [CAS RN: 2906200-90-0] [
28].
3c, Yield = 76%, white solid, 1-benzyl-2-o-tolyl-1H-indazol-3(2H)-one, m.p.: 112–113 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.76 (t, J = 8.3 Hz, 1H), 7.73 (d, J = 7.7 Hz, 1H), 7.70 (t, J = 7.3 Hz, 1H), 7.38–7.35 (m, 3H), 7.32–7.29 (m, 1H), 7.23 (t, J = 7.7 Hz, 1H), 7.20–7.14 (m, 3H), 6.75 (d, J = 6.2 Hz, 2H), 5.16 (d, J = 15.4 Hz, 1H), 4.59 (d, J = 15.8 Hz, 1H), 1.85 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 162.09, 149.63, 137.03, 134.72, 133.67, 132.34, 131.40, 128.56, 128.52, 128.20, 127.96, 127.47, 126.49, 124.59, 122.19, 118.31, 111.92, 53.58, 17.97. HRMS (EI) calcd for C21H18N2O (M)+ 314.1419, found 314.1417.
3d, Yield = 91%, white solid, 1-benzyl-2-phenyl-1
H-indazol-3(2
H)-one, m.p.: 131–132 °C.
1H NMR (300 MHz, DMSO-
d6) δ 7.87 (d,
J = 8.2 Hz, 1H), 7.69 (t,
J = 8.2 Hz, 1H), 7.65 (d,
J = 8.4 Hz, 1H), 7.60–7.55 (m, 4H), 7.39 (t,
J = 6.8 Hz, 1H), 7.21 (t,
J = 7.4 Hz, 1H), 7.19–7.13 (m, 3H), 6.84 (d,
J = 6.9 Hz, 2H), 4.93 (s, 2H).
13C NMR (75 MHz, CDCl
3) δ 162.43, 150.00, 135.21, 133.27, 132.38, 129.19, 128.63, 128.29, 126.33, 124.55, 123.84, 122.91, 119.83, 113.23, 54.82. (EI) calcd for C
20H
16N
2O (M)
+ 300.1263, found 300.1259. [CAS RN: 1182783-55-6] [
29].
3e, Yield = 92%, white solid, 1-benzyl-2-(4-methoxyphenyl)-1
H-indazol-3(2
H)-one, m.p.: 152–154 °C.
1H NMR (300 MHz, DMSO-
d6) δ 7.81 (d,
J = 8.4 Hz, 1H), 7.65 (t,
J = 7.1 Hz, 1H), 7.62 (d,
J = 7.9 Hz, 1H), 7.42 (d, 9.0 Hz, 2H), 7.18 (t,
J = 9.0 Hz, 1H), 7.15–7.11 (m, 5H), 6.85 (d,
J = 7.7 Hz, 2H), 4.88 (s, 2H), 3.82 (s, 3H).
13C NMR (75 MHz, CDCl
3) δ 162.38, 158.25, 149.60, 133.60, 132.17, 128.49, 128.31, 128.21, 127.93, 125.97, 124.48, 122.70, 119.63, 114.52, 112.92, 55.53, 54.41. HRMS (EI) calcd for C
21H
18N
2O
2 (M)
+ 330.1368, found 330.1367. [CAS RN: 886974-93-2] [
29].
3f, Yield = 87%, white solid, 1-benzyl-2-(4-tert-butylphenyl)-1H-indazol-3(2H)-one, m.p.: 117–120 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.82 (d, J = 8.3 Hz, 1H), 7.66 (t, J = 7.1 Hz, 1H), 7.62 (d, J = 7.0 Hz, 1H), 7.60 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 8.6 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.15–7.13 (m, 3H), 6.86 (d, J = 7.7 Hz, 1H), 4.89 (s, 2H), 1.34 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 162.38, 149.90, 149.49, 133.59, 132.46, 132.19, 128.62, 128.31, 128.19, 126.16, 124.50, 123.64, 122.80, 119.86, 113.16, 54.73, 34.62, 31.35. HRMS (EI) calcd for C24H24N2O (M)+ 356.1889, found 356.1889.
3g, Yield = 87%, pink solid, 1-benzyl-2-(4-fluorophenyl)-1
H-indazol-3(2
H)-one, m.p.: 133–134 °C.
1H NMR (300 MHz, DMSO-
d6) δ 7.85 (d,
J = 8.2 Hz, 2H), 7.69 (t,
J = 7.5 Hz, 1H), 7.64 (d,
J = 8.1 Hz, 1H), 7.57–7.52 (m, 2H), 7.45–7.39 (m, 2H), 7.21 (t,
J = 7.3 Hz, 1H), 7.15–7.13 (m, 3H), 6.82 (d,
J = 6.8 Hz, 2H), 4.90 (s, 2H).
13C NMR (75 MHz, CDCl
3) δ 162.59, 162.50, 159.24, 150.06, 133.19, 132.57, 131.31, 131.27, 128.57, 128.40, 128.38, 125.80, 125.68, 124.61, 123.06, 119.64, 116.31, 116.00, 113.20, 54.89. HRMS (EI) calcd for C
20H
15FN
2O (M)
+ 318.1168, found 318.1167. [CAS RN: 2902600-89-7] [
28].
3h, Yield = 89%, white solid, 1-benzyl-2-(4-chlorophenyl)-1
H-indazol-3(2
H)-one, m.p.: 118–122 °C.
1H NMR (300 MHz, DMSO-
d6) δ 7.87 (d,
J = 8.4 Hz, 2H), 7.70 (t,
J = 8.2 Hz, 1H), 7.66 (d,
J = 8.1 Hz, 2H), 7.64 (d,
J = 9.0 Hz, 2H), 7.56 (d,
J = 8.8 Hz, 2H), 7.21 (t,
J = 7.4 Hz, 1H), 7.17–7.10 (m, 3H), 6.81 (d,
J = 7.5 Hz, 2H), 4.92 (s, 2H).
13C NMR (75 MHz, CDCl
3) δ 162.55, 150.24, 133.89, 132.93, 132.68, 131.77, 12937, 128.63, 128.43, 128.36, 124.79, 124.63, 123.19, 119.72, 113.35, 55.09. HRMS (EI) calcd for C
20H
15ClN
2O (M)
+ 334.0873, found 334.0873. [CAS RN: 1182783-74-9] [
28].
3i, Yield = 87%, white solid, 1-benzyl-2-(4-bromophenyl)-1
H-indazol-3(2
H)-one, m.p.: 114–116 °C.
1H NMR (300 MHz, DMSO-
d6) δ 7.88 (d,
J = 8.4 Hz, 1H), 7.77 (d,
J = 8.8 Hz, 2H), 7.70 (t,
J = 7.1 Hz, 1H), 7.64 (d,
J = 7.9 Hz, 1H), 7.51 (d,
J = 8.8 Hz, 2H), 7.21 (t,
J = 7.1 Hz, 1H), 7.16–7.09 (m, 3H), 6.80 (d,
J = 7.9 Hz, 2H), 4.92 (s, 2H).
13C NMR (75 MHz, CDCl
3) δ 162.52, 150.27, 134.43, 132.89, 132.71, 132.33, 128.64, 128.44, 128.36, 125.04, 124.64, 123.21, 119.75, 119.62, 113.39, 55.13. HRMS (EI) calcd for C
20H
15BrN
2O (M)
+ 378.0368, found 378.0364. [CAS RN: 1182783-76-1] [
29].
3j, Yield = 60%, yellow solid, 1-benzyl-2-(4-nitrophenyl)-1
H-indazol-3(2
H)-one, m.p.: 166–168 °C.
1H NMR (300 MHz, DMSO-
d6) δ 8.45 (d,
J = 9.2 Hz, 2H), 7.93 (d,
J = 8.4 Hz, 1H), 7.86 (d,
J = 9.0 Hz, 2H), 7.76 (t,
J = 7.3 Hz, 1H), 7.68 (d,
J = 7.7 Hz, 1H), 7.25 (t,
J = 7.5 Hz, 1H), 7.17–7.09 (m, 3H), 6.77 (d,
J = 6.8 Hz, 2H), 4.97 (s, 2H).
13C NMR (75 MHz, CDCl
3) δ 162.99, 151.10, 144.72, 141.11, 133.57, 132.23, 128.73, 128.50, 124.97, 124.93, 123.88, 122.42, 119.64, 115.64, 113.87, 56.15. HRMS (EI) calcd for C
20H
15N
3O
3 (M)
+ 345.1113, found 345.1110. [CAS RN: 1182783-81-8] [
29].
4j, Yield = 35%, white solid, 1-benzyl-3-(4-nitrophenoxy)-1H-indazole, m.p.: 118–120 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.27 (d, J = 9.2 Hz, 2H), 7.77 (d, J = 8.6 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.47 (t, J = 8.6 Hz, 1H), 7.35–7.23 (m, 7H), 7.15 (t, J = 7.1 Hz, 1H), 5.60 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 161.81, 151.00, 143.24, 141.32, 136.46, 128.80, 127.97, 127.80, 127.23, 125.77, 120.97, 119.49, 117.27, 113.71, 109.78, 53.01. HRMS (EI) calcd for C20H15N3O3 (M)+ 345.1113, found 345.1111.
3k, Yield = 66%, white solid, 2-(4-acetylphenyl)-1-benzyl-1H-indazol-3(2H)-one, m.p.: 130–132 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.18 (d, J = 8.6 Hz, 2H), 7.91 (d, J = 8.3 Hz, 1H), 7.73 (d, J = 8.6 Hz, 2H), 7.72 (t, J = 7.3 Hz, 1H), 7.65 (d, J = 7.5 Hz, 1H), 7.23 (t, J = 7.3 Hz, 1H), 7.16–7.09 (m, 3H), 6.78 (d, J = 6.4 Hz, 2H), 4.95 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 197.06, 162.70, 150.62, 139.51, 134.20, 133.01, 132.61, 129.58, 128.72, 128.52, 128.37, 124.74, 123.44, 122.52, 119.82, 113.63, 55.57, 26.64. HRMS (EI) calcd for C22H18N2O2 (M)+ 342.1368, found 342.1365.
4k, Yield = 24%, white solid, 1-(4-(1-benzyl-1H-indazol-3-yloxy)phenyl)ethanone, m.p.: 95–98 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.93 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 8.8 Hz, 1H), 7.41–7.37 (m, 2H), 7.29–7.14 (m, 7H), 7.06 (t, J = 7.5 Hz, 1H), 5.53 (s, 2H), 2.44 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 196.69, 160.77, 151.79, 141.27, 136.60, 132.38, 130.41, 128.71, 127.82, 127.54, 127.18, 120.61, 119.72, 116.91, 113.85, 109.64, 52.88, 26.45. HRMS (EI) calcd for C22H18N2O2 (M)+ 342.1368, found 342.1365.
3l, Yield = 63%, white solid, 4-(1-benzyl-3-oxo-1
H-indazol-2(3
H)-yl)benzonitrile, m.p.: 96–99 °C. IR (cm
−1): υ
max 2221, 1677, 1595, 1495, 1351.
1H NMR (300 MHz, DMSO-
d6) δ 8.05 (d,
J = 8.4 Hz, 2H), 7.91 (d,
J = 8.2 Hz, 1H), 7.77 (d,
J = 8.4 Hz, 2H), 7.74 (t,
J = 8.0 Hz, 1H), 7.66 (d,
J = 7.9 Hz, 1H), 7.24 (t,
J = 7.5 Hz, 1H), 7.19–7.09 (m, 3H), 6.77 (d,
J = 7.0 Hz, 2H), 4.94 (s, 2H).
13C NMR (75 MHz, CDCl
3) δ 162.85, 150.93, 139.43, 133.36, 133.24, 132.33, 128.68, 128.65, 128.44, 124.83, 123.72, 122.82, 119.63, 118.55, 113.77, 108.94, 55.93. HRMS (EI) calcd for C
21H
15N
3O (M)
+ 325.1215, found 325.1212. [CAS RN: 1182783-78-3] [
29].
4l, Yield = 28%, white solid, 4-(1-benzyl-1H-indazol-3-yloxy)benzonitrile, m.p.: 79–81 °C. IR (cm−1): υmax 2237, 1605, 1481, 1365, 1232, 1167. 1H NMR (300 MHz, DMSO-d6) δ 7.88 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 8.6 Hz, 1H), 7.50–7.44 (m, 2H), 7.33–7.24 (m, 7H), 7.15 (t, J = 7.0 Hz, 1H), 5.60 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 160.22, 151.01, 141.27, 136.47, 133.97, 128.74, 127.88, 127.70, 127.18, 120.83, 119.49, 118.69, 117.83, 113.72, 109.71, 106.60, 52.93. HRMS (EI) calcd for C21H15N3O (M)+ 325.1215, found 325.1212.
3m, Yield = 77%, white solid, 1-benzyl-2-(4-(trifluoromethyl)phenyl)-1H-indazol-3(2H)-one, m.p.: 119–121 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.96 (d, J = 8.6 Hz, 2H), 7.91 (d, J = 8.3 Hz, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.73 (t, J = 8.1 Hz, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.24 (t, J = 7.5 Hz, 1H), 7.17–7.12 (m, 3H), 6.79 (d, J = 7.5 Hz, 2H), 4.95 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 162.78, 150.65, 138.52, 133.03, 132.62, 128.69, 128.53, 128.39, 127.93, 127.50, 126.48, 126.46, 126.43, 126.38, 126.33, 125.76, 124.74, 123.46, 122.95, 122.16, 119.73, 113.61, 55.51. HRMS (EI) calcd for C21H15F3N2O (M)+ 368.1136, found 368.1132.
4m, Yield = 14%, colorless oil, 1-benzyl-3-(4-(trifluoromethyl)phenoxy)-1H-indazole, 1H NMR (300 MHz, DMSO-d6) δ 7.77–7.74 (m, 3H), 7.47 (d, J = 8.3 Hz, 1H), 7.45 (t, J = 9.2 Hz, 1H), 7.33–7.27 (m, 5H), 7.23 (d, J = 8.0 Hz, 2H), 7.13 (t, J = 7.9 Hz, 1H), 5.58 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 159.46, 151.65, 141.32, 136.64, 128.75, 127.85, 127.60, 127.21, 127.01, 126.99, 126.96, 126.93, 126.91, 125.92, 125.60, 125.16, 122.34, 120.64, 119.75, 117.38, 113.84, 109.66, 52.92. HRMS (EI) calcd for C21H15F3N2O (M)+ 368.1136, found 368.1136.
3n, Yield = 71%, white solid, 1-benzyl-2-(pyridin-3-yl)-1H-indazol-3(2H)-one, m.p.: 153–156 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.76 (s, 1H), 8.56 (d, J = 4.8 Hz, 1H), 7.94 (d, J = 6.8 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.72 (t, J = 7.1 Hz, 1H), 7.67 (d, J = 7.9 Hz, 1H), 7.62 (dd, J = 8.2, 4.8 Hz, 1H), 7.23 (t, J = 7.3 Hz, 1H), 7.17–7.10 (m, 3H), 6.81 (d, J = 7.7 Hz, 2H), 4.94 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 162.88, 150.68, 147.05, 144.28, 133.00, 132.62, 132.33, 130.56, 128.58, 128.53, 128.41, 124.66, 123.75, 123.39, 119.49, 113.54, 55.36. HRMS (EI) calcd for C19H15N3O (M)+ 301.1215, found 301.1214.
3o, Yield = 70%, white solid, 1-benzyl-2-(pyrimidin-5-yl)-1H-indazol-3(2H)-one, m.p.: 213–216 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.99 (s, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.75 (t, J = 7.1 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.26 (t, J = 7.1 Hz, 1H), 7.20–7.10 (m, 3H), 6.86 (d, J = 7.7 Hz, 2H), 4.97 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 162.43, 150.00, 135.21, 133.27, 132.38, 129.19, 128.63, 128.29, 126.33, 124.55, 123.84, 122.91, 119.83, 113.23, 54.82. HRMS (EI) calcd for C18H14N4O (M)+ 302.1168, found 302.1168.
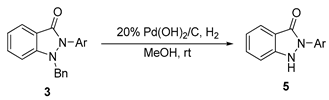
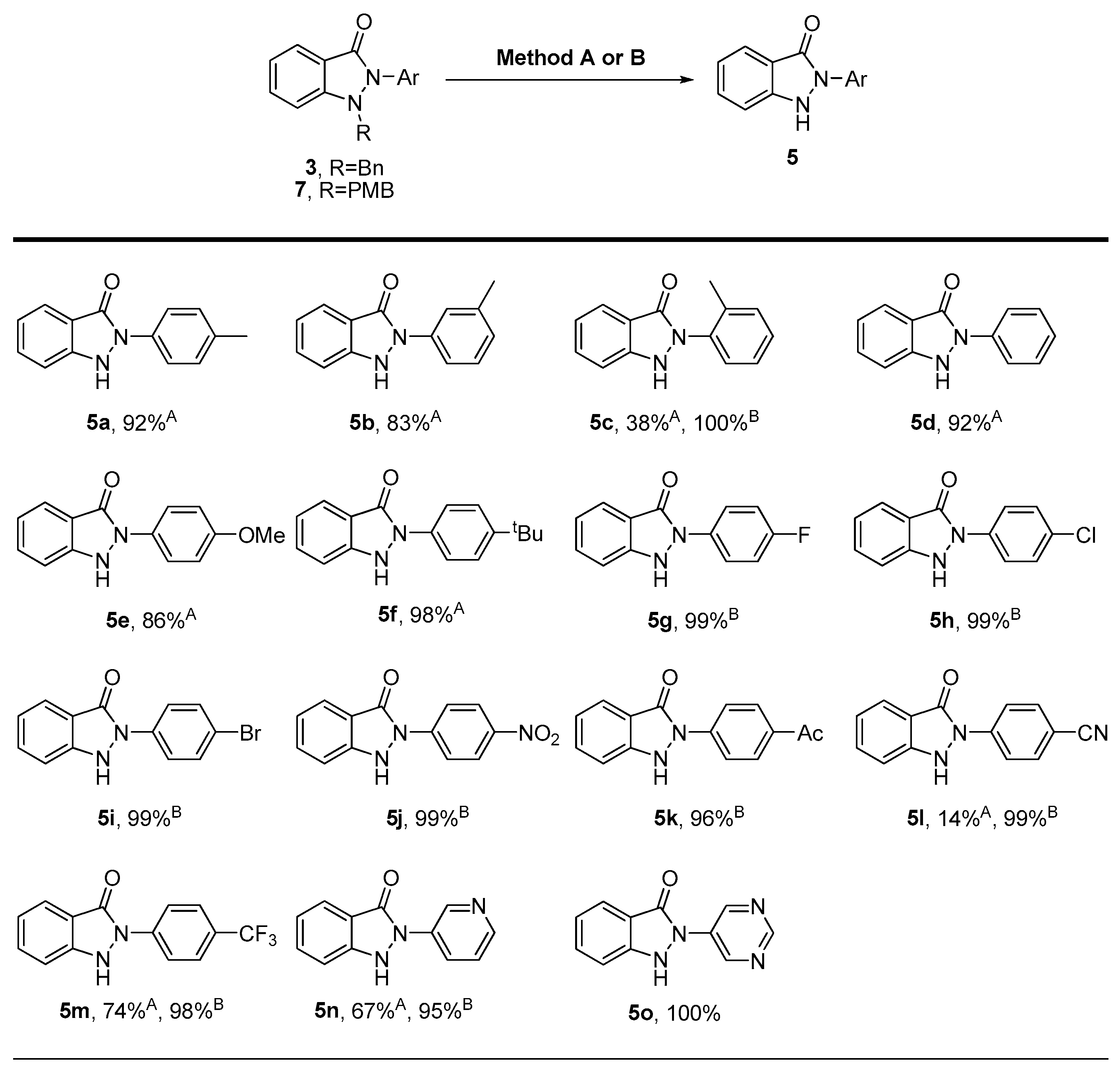
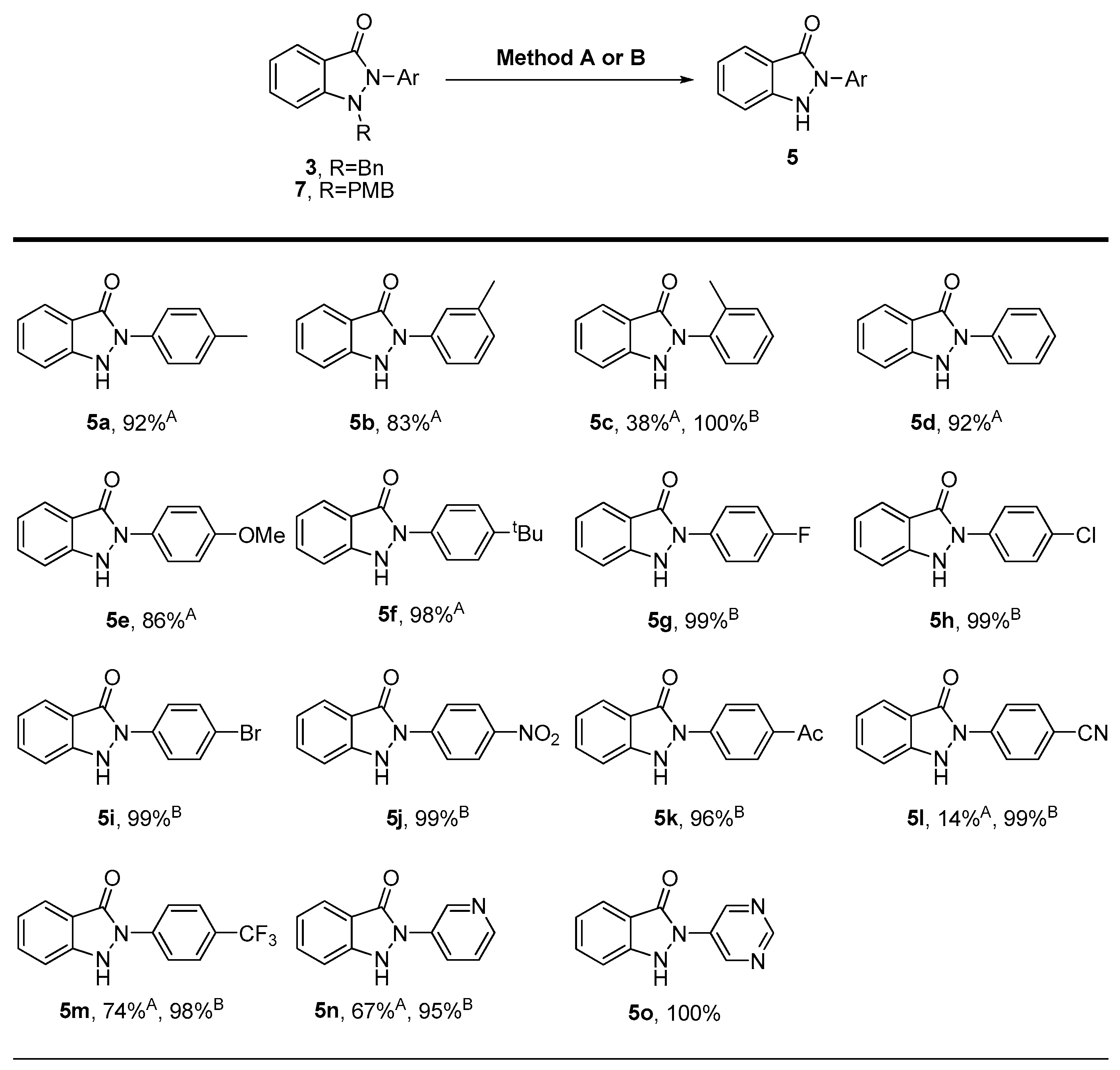
3.2.2. General Procedure for the Synthesis of Compounds (5a–5f, 5l–5n)
![Molecules 28 06706 i003]()
N(2)-aryl-substituted-N(1)-benzyl-1H-indazol-3(3H)-one (3, 0.16 mmol), 20% Pd(OH)2/C (0.08 mmol) were dissolved in MeOH (6.4 mL). The mixture was stirred under a hydrogen gas balloon at room temperature for 4 h. The progress of the reaction was monitored using TLC chromatography. Upon completion, the mixture was filtered through celite and washed with CH2Cl2. The volatile components were evaporated, and the resulting residues subsequently purified through silica gel flash column chromatography to yield pure solid products (5a–5f, 5l–5n).
5a, Yield = 92%, white solid, 2-
p-tolyl-1
H-indazol-3(2
H)-one, m.p.: 209–211 °C.
1H NMR (300 MHz, DMSO-
d6) δ 10.64 (s, 1H), 7.80 (d,
J = 8.4 Hz, 2H), 7.73 (d,
J = 7.9 Hz, 1H), 7.59 (t,
J = 7.7 Hz, 1H), 7.36–7.29 (m, 3H), 7.18 (t,
J = 7.5 Hz, 2H), 2.33 (s, 3H).
13C NMR (75 MHz, DMSO-
d6) δ 159.96, 146.41, 135.19, 134.11, 132.32, 129.41, 123.32, 121.75, 118.91, 118.13, 112.55, 20.49. HRMS (EI) calcd for C
14H
12N
2O (M)
+ 224.0950, found 224.0949. [CAS RN: 74152-88-8] [
30].
5b, Yield = 83%, white solid, 2-m-tolyl-1H-indazol-3(2H)-one, m.p.: 192–195 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 7.75–7.73 (m, 3H), 7.60 (t, J = 7.8 Hz, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.19 (t, J = 7.4 Hz, 1H), 7.07 (d, J = 8.0 Hz, 1H), 2.38 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 160.15, 146.53, 138.43, 137.55, 132.49, 128.92, 125.57, 123.41, 121.83, 119.33, 118.10, 116.13, 112.61, 21.24. HRMS (EI) calcd for C14H12N2O (M)+ 224.0950, found 224.0948.
5c, Yield = 38%, white solid, 2-
o-tolyl-1
H-indazol-3(2
H)-one, m.p.: 142–144 °C.
1H NMR (300 MHz, DMSO-
d6) δ 10.64 (s, 1H), 7.80 (d,
J = 8.4 Hz, 2H), 7.73 (d,
J = 7.9 Hz, 1H), 7.59 (t,
J = 7.7 Hz, 1H), 7.36–7.29 (m, 3H), 7.18 (t,
J = 7.5 Hz, 2H), 2.33 (s, 3H).
13C NMR (75 MHz, DMSO-
d6) δ 159.96, 146.41, 135.19, 134.11, 132.32, 129.41, 123.32, 121.75, 118.91, 118.13, 112.55, 20.49. HRMS (EI) calcd for C
14H
12N
2O (M)
+ 224.0950, found 224.0950. [CAS RN: 74152-87-7] [
14].
5d, Yield = 92%, white solid, 2-phenyl-1
H-indazol-3(2
H)-one, m.p.: 202–205 °C.
1H NMR (300 MHz, DMSO-
d6) δ 10.66 (s, 1H), 7.93 (d,
J = 8.6 Hz, 2H), 7.75 (d,
J = 7.7 Hz, 1H), 7.61 (t,
J = 7.6 Hz, 1H), 7.51 (t,
J = 8.0 Hz, 2H), 7.37 (d,
J = 8.3 Hz, 1H), 7.22 (p,
J = 7.3 Hz, 2H).
13C NMR (75 MHz, DMSO-
d6) δ 160.20, 146.58, 137.57, 132.52, 129.05, 124.85, 123.41, 121.85, 118.86, 118.08, 112.63. HRMS (EI) calcd for C
13H
10N
2O (M)
+ 210.0793, found 210.0792. [CAS RN: 17049-65-9] [
14].
5e, Yield = 86%, white solid, 2-(4-methoxyphenyl)-1
H-indazol-3(2
H)-one, m.p.: 177–178 °C.
1H NMR (300 MHz, DMSO-
d6) δ 10.62 (s, 1H), 7.79 (d,
J = 9.0 Hz, 2H), 7.71 (d,
J = 7.7 Hz, 1H), 7.57 (t,
J = 7.4 Hz, 1H), 7.33 (d,
J = 8.1 Hz, 1H), 7.16 (t,
J = 7.4 Hz, 1H), 7.06 (d,
J = 9.0 Hz, 2H), 3.78 (s, 3H).
13C NMR (75 MHz, DMSO-
d6) δ 159.73, 156.59, 146.26, 132.12, 130.74, 123.25, 121.69, 121.02, 118.08, 114.20, 112.50, 55.33. HRMS (EI) calcd for C
14H
12N
2O
2 (M)
+ 240.0899, found 240.0897. [CAS RN: 74152-89-9] [
14].
5f, Yield = 98%, white solid, 2-(4-tert-butylphenyl)-1H-indazol-3(2H)-one, m.p.: 218–220 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1H), 7.82 (d, J = 7.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.52 (d, J = 7.0 Hz, 2H), 7.36 (d, J = 8.4 Hz, 1H), 7.19 (t, J = 7.6 Hz, 1H), 1.32 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ 159.98, 147.46, 146.47, 135.08, 132.39, 125.77, 123.39, 121.82, 118.88, 118.14, 112.63, 34.25, 31.14. HRMS (EI) calcd for C17H18N2O (M)+ 266.1419, found 266.1418.
5l, Yield = 14%, white solid, 4-(3-oxo-1H-indazol-2(3H)-yl)benzonitrile, m.p.: 152–154 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 8.14 (d, J = 6.8 Hz, 2H), 7.98 (d, J = 6.8 Hz, 2H), 7.79 (d, J = 7.6 Hz, 1H), 7.66 (t, J = 7.2 Hz, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.23 (t, J = 6.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 161.11, 147.26, 141.10, 133.49, 133.45, 123.74, 122.35, 118.78, 118.23, 117.71, 112.87, 106.36. HRMS (EI) calcd for C14H12N2O2 (M)+ 235.0746, found 235.0747.
5m, Yield = 74%, white solid, 2-(4-(trifluoromethyl)phenyl)-1
H-indazol-3(2
H)-one, m.p.: 233–235 °C.
1H NMR (400 MHz, DMSO-
d6) δ 10.75 (s, 1H), 8.17 (d,
J = 8.8 Hz, 2H), 7.89 (d,
J = 8.8 Hz, 2H), 7.79 (d,
J = 8.0 Hz, 1H), 7.66 (t,
J = 7.6 Hz, 1H), 7.41 (d,
J = 8.4 Hz, 1H), 7.23 (t,
J = 7.6 Hz, 1H).
13C NMR (100 MHz, DMSO-
d6) δ 160.97, 147.15, 140.76, 133.26, 126.41, 126.37, 125.59, 124.69, 124.37, 123.69, 122.89, 122.28, 118.35, 117.85, 112.86. HRMS (EI) calcd for C
14H
9N
3O (M)
+ 278.0667, found 278.0663. [CAS RN: 889359-36-8] [
30].
5n, Yield = 67%, white solid, 2-(pyridin-3-yl)-1
H-indazol-3(2
H)-one, m.p.: 183–185 °C.
1H NMR (400 MHz, DMSO-
d6) δ 10.73 (s, 1H), 9.14 (s, 1H), 8.45 (d,
J = 4.8 Hz, 1H), 8.31 (d,
J = 8.4 Hz, 1H), 7.78 (d,
J = 8.0 Hz, 1H), 7.65 (t,
J = 8.0 Hz, 1H), 7.55 (dd,
J = 8.5, 4.8 Hz, 1H), 7.41 (d,
J = 8.4 Hz, 1H), 7.23 (t,
J = 8.0 Hz, 1H).
13C NMR (100 MHz, DMSO-
d6) δ 160.83, 147.23, 145.65, 139.91, 134.36, 133.03, 125.76, 123.91, 123.58, 122.20, 117.64, 112.86. HRMS (EI) calcd for C
12H
9N
3O (M)
+ 211.0746, found 211.0743. [CAS RN: 175653-67-5] [
31].
3.2.3. Preparation of N(1)-PMB-1H-indazol-3(2H)-one 6 [32]
![Molecules 28 06706 i004]()
1H-indazol-3(2H)-one (1 mmol) and NaOH (1 mmol) were dissolved in water (1 mL) at 35 °C, followed by addition of 4-methoxybenzyl chloride (PMB-Cl,1 mmol). The resulting mixture was stirred at 70 °C for 3 h. After the reaction was complete, the solution was cooled to room temperature. The mixture was then quenched with water and extracted with dichloromethane. The organic layer was separated, washed with brine, and dried over anhydrous Na2SO4. After filtration, the mixture was concentrated under vacuum and subsequently purified through silica gel flash column chromatography to obtain a pure solid product 6.
White solid, 1-(4-methoxybenzyl)-1
H-indazol-3(2
H)-one, m.p.: 158–159 °C.
1H NMR (400 MHz, DMSO-
d6) δ 10.66 (br s, 1H), 7.60 (d,
J = 8.4 Hz, 1H), 7.52 (d,
J = 8.4 Hz, 1H), 7.31 (t,
J = 8.0 Hz, 1H), 7.16 (d,
J = 8.8 Hz, 2H), 6.97 (t,
J = 8.0 Hz, 1H), 6.84 (d,
J = 8.8 Hz, 2H), 5.27 (s, 2H), 3.69 (s, 3H).
13C NMR (100 MHz, CDCl
3) δ 159.30, 156.94, 141.89, 128.99, 128.77, 128.69, 121.48, 119.64, 114.17, 113.49, 109.35, 55.32, 52.13. [CAS RN: 1029-30-7] [
32].
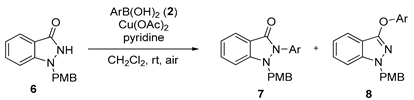
3.2.4. General Procedure for the Synthesis of Compounds (7c, 7g–7o, 8j–8m)
![Molecules 28 06706 i005]()
N(1)-PMB-1H-indazol-3(2H)-one (6, 0.36 mmol), Cu(OAc)2 (0.18 mmol) and arylboronic acid (0.54 mmol) were dissolved in CH2Cl2 (0.7 mL) and pyridine (7.2 mmol). The reaction was conducted at ambient temperature while being exposed to air. After the reaction was complete, the mixture was quenched with 1 M HCl at 0 °C and then extracted with ethyl acetate. The organic layer was separated, washed with brine, and dried over anhydrous Na2SO4. Following filtration, the mixture was concentrated under vacuum and subsequently purified through silica gel flash column chromatography to yield pure solid products. (7c, 7g–7o, 8j–8m).
7c, Yield = 82%, white solid, 1-(4-methoxybenzyl)-2-o-tolyl-1H-indazol-3(2H)-one, m.p.: 103–104 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 8.0 Hz, 1H), 7.72–7.68 (m, 2H), 7.37–7.31 (m, 4H), 7.22 (t, J = 8.0 Hz, 1H), 6.74–6.68 (m, 4H), 5.05 (d, J = 15.6 Hz, 1H), 4.52 (d, J = 15.6 Hz, 1H), 3.65 (s, 3H) 1.92 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.09, 159.46, 149.61, 136.89, 133.77, 132.24, 131.41, 129.37, 128.44, 127.40, 126.59, 126.47, 124.56, 122.14, 118.45, 113.83, 112.08, 55.20, 53.08, 18.10.
7g, Yield = 88%, white solid, 2-(4-fluorophenyl)-1-(4-methoxybenzyl)-1H-indazol-3(2H)-one, m.p.: 110–112 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (d, J = 8.2 Hz, 1H), 7.71 (t, J = 7.5 Hz, 1H), 7.67–7.64 (m, 3H), 7.60–7.57 (d, J = 8.8 Hz, 2H), 7.22 (t, J = 7.5 Hz, 1H), 6.73 (d, J = 8.7 Hz, 2H), 6.68 (d, J = 8.7 Hz, 2H), 4.85 (s, 2H), 3.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.65, 162.03, 159.55, 150.03, 132.49, 131.37, 129.97, 125.67, 125.59, 124.98, 124.60, 123.05, 119.84, 116.26, 116.04, 113.65, 113.36, 55.15, 54.35.
7h, Yield = 88%, white solid, 2-(4-chlorophenyl)-1-(4-methoxybenzyl)-1H-indazol-3(2H)-one, m.p.: 129–130 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.86 (d, J = 8.5 Hz, 1H), 7.70 (t, J = 8.4 Hz, 1H), 7.65 (d, J = 7.5 Hz, 1H), 7.59–7.56 (m, 2H), 7.46–7.41 (m, 2H), 7.22 (t, J = 8.0 Hz, 1H), 6.75 (d, J = 8.4 Hz, 2H), 6.69 (d, J = 8.8 Hz, 2H), 4.84 (s, 2H), 3.64 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.63, 159.57, 150.23, 133.99, 132.63, 131.67, 130.06, 129.39, 124.74, 124.64, 123.19, 119.95, 119.94, 113.64, 113.53, 55.15, 54.57.
7i, Yield = 90%, white solid, 2-(4-bromophenyl)-1-(4-methoxybenzyl)-1H-indazol-3(2H)-one, m.p.: 122–123 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (d, J = 8.5 Hz, 1H), 7.78 (d, J = 8.8 Hz, 2H), 7.71 (t, J = 7.4 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.53 (d, J = 8.8 Hz, 2H), 7.22 (t, J = 7.4 Hz, 1H), 6.73 (d, J = 8.4 Hz, 2H), 6.68 (d, J = 8.8 Hz, 2H), 4.85 (s, 2H), 3.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.63, 159.57, 150.23, 133.99, 132.63, 131.67, 130.06, 129.39, 124.74, 124.64, 123.19, 119.95, 119.94, 113.64, 113.53, 55.15, 54.57.
7j, Yield = 55%, yellow solid, 1-(4-methoxybenzyl)-2-(4-nitrophenyl)-1H-indazol-3(2H)-one, m.p.: 179–180 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.46 (d, J = 8.8 Hz, 2H), 7.93 (d, J = 8.5 Hz, 1H), 7.87 (d, J = 8.8 Hz, 2H), 7.77 (t, J = 7.4 Hz, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.27 (t, J = 7.4 Hz, 1H), 6.71–6.65 (m, 4H), 4.91 (s, 2H), 3.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.02, 159.76, 151.07, 144.61, 141.19, 133.45, 130.11, 124.95, 124.90, 124.00, 123.80, 122.32, 119.83, 113.97, 113.74, 55.56, 55.14.
8j, Yield = 27%, yellow oil, 1-(4-methoxybenzyl)-3-(4-nitrophenoxy)-1H-indazole. 1H NMR (400 MHz, DMSO-d6) δ 8.29 (d, J = 9.1 Hz, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.46 (t, J = 7.4 Hz, 1H), 7.33 (d, J = 9.4 Hz, 2H), 7.24 (d, J = 8.5 Hz, 2H), 7.15 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.7 Hz, 2H), 5.52 (s, 2H), 3.71 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.87, 159.33, 150.85, 143.21, 141.15, 128.72, 128.51, 127.71, 125.77, 120.90, 119.45, 117.22, 114.15, 113.70, 109.84, 55.27, 52.58.
7k, Yield = 69%, white solid, 2-(4-acetylphenyl)-1-(4-methoxybenzyl)-1H-indazol-3(2H)-one, m.p.: 132–134 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 8.2 Hz, 1H), 7.76–7.71 (m, 3H), 7.66 (d, J = 7.8 Hz, 1H), 7.24 (t, J = 7.4 Hz, 1H), 6.72–6.66 (m, 4H), 4.89 (s, 2H), 3.63 (s, 3H), 2.65 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 197.12, 162.80, 159.65, 150.65, 139.63, 134.16, 132.96, 130.13, 129.61, 124.75, 124.47, 123.43, 122.50, 120.04, 113.78, 113.67, 55.15, 55.07, 26.64.
8k, Yield = 27%, white solid, 1-(4-(1-(4-methoxybenzyl)-1H-indazol-3-yloxy)phenyl)ethanone, m.p.: 100–101 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 8.5 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.24–7.20 (m, 4H), 7.11 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 5.50 (s, 2H), 3.70 (s, 3H), 2.55 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 196.78, 160.89, 159.28, 151.41, 141.17, 141.16, 132.41, 130.47, 128.72, 127.50, 120.59, 119.78, 116.94, 114.13, 113.91, 109.75, 55.28, 52.52, 26.50.
7l, Yield = 67%, white solid, 4-(1-(4-methoxybenzyl)-3-oxo-1H-indazol-2(3H)-yl)benzonitrile, m.p.: 148–149 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.06 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 8.2 Hz, 1H), 7.79 (d, J = 8.8 Hz, 2H), 7.75 (t, J = 8.4 Hz, 1H), 7.67 (d, J = 7.5 Hz, 1H), 7.25 (t, J = 8.4 Hz, 1H), 6.71–6.65 (m, 4H), 4.88 (s, 2H), 3.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.93, 159.72, 150.92, 139.52, 133.28, 133.26, 130.09, 124.83, 124.15, 123.69, 122.76, 119.85, 118.60, 113.90, 113.72, 108.83, 55.38, 55.14.
8l, Yield = 32%, white solid, 4-(1-(4-methoxybenzyl)-1H-indazol-3-yloxy)benzonitrile, m.p.: 107–108 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (d, J = 8.7 Hz, 2H), 7.77 (d, J = 8.5 Hz, 1H), 7.47 (d, J = 8.5 Hz, 1H), 7.46 (t, J = 7.5 Hz, 1H), 7.29 (d, J = 8.8 Hz, 2H), 7.23 (d, J = 8.8 Hz, 2H), 7.13 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 8.7 Hz, 2H), 5.51 (s, 2H), 3.71 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 160.32, 159.28, 150.86, 141.12, 134.02, 128.69, 128.55, 127.65, 120.79, 119.51, 118.74, 117.79, 114.11, 113.73, 109.79, 106.57, 55.26, 52.54.
7m, Yield = 80%, white solid, 1-(4-methoxybenzyl)-2-(4-(trifluoromethyl)phenyl)-1H-indazol-3(2H)-one, m.p.: 103–104 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J = 8.7 Hz, 2H), 7.92 (d, J = 8.5 Hz, 1H), 7.82 (d, J = 8.5 Hz, 2H), 7.74 (t, J = 7.5 Hz, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.25 (t, J = 7.5 Hz, 1H), 6.73–6.66 (m, 4H), 4.89 (s, 2H), 3.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.89, 159.66, 150.68, 138.62, 132.98, 130.12, 127.80, 127.50, 126.67, 126.42, 124.77, 124.48, 123.45, 122.93, 122.67, 119.96, 113.70, 55.15, 55.03.
8m, Yield = 16%, colorless oil, 1-(4-methoxybenzyl)-3-(4-(trifluoromethyl)phenoxy)-1H-indazole. 1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.31 (d, J = 8.7 Hz, 2H), 7.23 (d, J = 8.7 Hz, 2H), 7.13 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 5.50 (s, 2H), 3.70 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.52, 159.28, 151.53, 141.17, 128.71, 127.52, 127.02, 126.98, 125.52, 125.20, 122.80, 120.59, 119.75, 117.36, 114.13, 113.86, 109.74, 55.27, 52.51.
7n, Yield = 71%, white solid, 1-(4-methoxybenzyl)-2-(pyridin-3-yl)-1H-indazol-3(2H)-one, m.p.: 144–145 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (d, J = 2.3 Hz, 1H), 8.57 (dd, J = 4.8, 1.4 Hz, 1H), 7.97 (dt, J = 8.4, 1.9 Hz, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.73 (t, J = 7.4 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.63 (dd, J = 8.2, 4.8 Hz, 1H), 7.25 (t, J = 7.4 Hz, 1H), 6.75–6.68 (m, 4H), 4.88 (s, 2H), 3.63 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 13C-NMR (101 MHz, CHLOROFORM-D) δ 162.97, 159.67, 150.78, 146.98, 144.30, 132.93, 132.47, 130.49, 130.00, 124.69, 124.54, 123.75, 123.38, 119.77, 113.75, 113.70, 55.14, 54.94.
7o, Yield = 91%, white solid, 1-(4-methoxybenzyl)-2-(pyrimidin-5-yl)-1H-indazol-3(2H)-one, m.p.: 165–166 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 9.01 (s, 2H), 7.92 (d, J = 8.5 Hz, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.27 (t, J = 7.5 Hz, 1H), 6.78 (d, J = 8.5 Hz, 2H), 6.69 (d, J = 8.5 Hz, 2H), 4.91 (s, 2H), 3.64 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.24, 159.87, 155.35, 151.64, 150.25, 133.56, 131.54, 130.02, 124.90, 124.03, 123.91, 119.42, 114.04, 113.94, 55.70, 55.19.
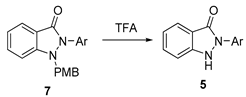
3.2.5. General Procedure for the Synthesis of Compounds (5c, 5g–5o)
![Molecules 28 06706 i006]()
N(1)-PMB-protected N(2)-aryl-substituted indazol-3-ones (7, 0.15 mmol) were dissolved in trifluoroacetic acid (TFA, 2.3 mL). The mixture was stirred at 60 °C for 1 h. After the reaction was complete, the mixture was concentrated under vacuum and subsequently purified through silica gel flash column chromatography to obtain pure solid products. (5c, 5g–5o).
5c, Yield = 100%, white solid, 2-o-tolyl-1H-indazol-3(2H)-one.
5g, Yield = 99%, white solid, 2-(4-fluorophenyl)-1
H-indazol-3(2
H)-one, m.p.: 208–210 °C.
1H NMR (400 MHz, DMSO-
d6) δ 10.66 (s, 1H), 7.96–7.92 (m, 2H), 7.75 (d,
J = 7.6 Hz, 1H), 7.62 (t,
J = 6.8 Hz, 1H), 7.39–7.34 (m, 3H), 7.20 (t,
J = 7.2 Hz, 1H).
13C NMR (100 MHz, DMSO-
d6) δ 159.73, 156.59, 146.26, 132.12, 130.74, 123.25, 121.69, 121.02, 118.08, 114.20, 112.50, 55.33. HRMS (EI) calcd for C
13H
9FN
2O (M)
+ 228.0699, found 228.0697. [CAS RN: 135066-29-4] [
19].
5h, Yield = 99%, white solid, 2-(4-chlorophenyl)-1
H-indazol-3(2
H)-one, m.p.: 210–212 °C.
1H NMR (400 MHz, DMSO-
d6) δ 10.68 (s, 1H), 7.97 (d,
J = 6.8 Hz, 2H), 7.76 (d,
J = 7.6 Hz, 1H), 7.64 (t,
J = 8.0 Hz, 1H), 7.58 (d,
J = 6.8 Hz, 1H), 7.38 (d,
J = 8.4 Hz, 1H), 7.22 (t,
J = 8.0 Hz, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 160.42, 146.76, 136.47, 132.82, 129.00, 128.64, 123.50, 122.07, 120.20, 117.94, 112.72. HRMS (EI) calcd for C
13H
9ClN
2O (M)
+ 244.0403, found 244.0404. [CAS RN: 17049-63-7] [
30].
5i, Yield = 99%, white solid, 2-(4-bromophenyl)-1
H-indazol-3(2
H)-one, m.p.: 217–219 °C.
1H NMR (400 MHz, DMSO-
d6) δ 10.67 (s, 1H), 7.91 (d,
J = 6.8 Hz, 2H), 7.76 (d,
J = 8.0 Hz, 1H), 7.70 (d,
J = 7.2 Hz, 2H), 7.63 (t,
J = 7.2 Hz, 1H), 7.38 (d,
J = 8.0 Hz, 1H), 7.23 (t,
J = 7.2 Hz, 1H).
13C NMR (100 MHz, DMSO-
d6) δ 160.43, 146.76, 136.88, 132.83, 131.90, 123.49, 122.07, 120.47, 117.94, 116.77, 112.72. HRMS (EI) calcd for C
13H
9BrN
2O (M)
+ 287.9898, found 287.9896. [CAS RN: 135066-31-8] [
19].
5j, Yield = 99%, white solid, 2-(4-nitrophenyl)-1
H-indazol-3(2
H)-one, m.p.: 243–245 °C.
1H NMR (400 MHz, DMSO-
d6) δ 10.85 (s, 1H), 8.41 (d,
J = 8.8 Hz, 2H), 8.21 (d,
J = 9.2 Hz, 2H), 7.80 (d,
J = 7.6 Hz, 1H), 7.68 (t,
J = 7.6 Hz, 1H), 7.42 (d,
J = 8.0 Hz, 1H), 7.24 (t,
J = 8.2 Hz, 1H).
13C NMR (100 MHz, DMSO-
d6) δ 161.32, 147.46, 143.07, 142.74, 133.68, 125.10, 123.84, 122.48, 117.95, 117.61, 112.93. HRMS (EI) calcd for C
13H
9N
3O
3 (M)
+ 255.0644, found 255.0645. [CAS RN: 120274-01-3] [
33].
5k, Yield = 96%, white solid, 2-(4-acetylphenyl)-1H-indazol-3(2H)-one, m.p.: 233–235 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H), 8.12–8.08 (m, 4H), 7.78 (d, J = 8.0 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.22 (t, J = 7.2 Hz, 1H), 2.60 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 196.67, 160.88, 147.10, 141.24, 133.21, 132.67, 129.57, 123.66, 122.22, 117.87, 117.62, 112.82, 26.60. HRMS (EI) calcd for C15H12N2O2 (M)+ 252.0899, found 252.0894.
5l, Yield = 99%, white solid, 4-(3-oxo-1H-indazol-2(3H)-yl)benzonitrile.
5m, Yield = 98%, white solid, 2-(4-(trifluoromethyl)phenyl)-1H-indazol-3(2H)-one.
5n, Yield = 95%, white solid, 2-(pyridin-3-yl)-1H-indazol-3(2H)-one.
5o, Yield = 100%, white solid, 2-(pyrimidin-5-yl)-1H-indazol-3(2H)-one, m.p.: 228–230 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 9.34 (s, 2H), 9.06 (s, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.68 (t, J = 7.2 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.25 (t, J = 7.2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 161.27, 154.03, 147.80, 146.05, 133.48, 133.00, 123.68, 122.46, 117.15, 113.01. HRMS (EI) calcd for C11H8N4O (M)+ 212.0698, found 212.0697.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}