
Neuronal Nitric Oxide Synthase and Post-Translational Modifications in the Development of Central Nervous System Diseases: Implications and Regulation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Overstimulation of nNOS in CNS Disease Development
2.1. NO-Mediated PTM and Alzheimer Disease
2.2. NO-Mediated PTM and Parkinson Disease
2.3. NO-Mediated PTM and Neurological Disorders
3. Inhibitors of nNOS as Modulators of CNS Disease Development
3.1. nNOS Inhibitors
3.2. PSD95-nNOS Interaction Inhibitors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Murad, F. Discovery of some of the biological effects of nitric oxide and its role in cell signaling. Biosci. Rep. 2004, 24, 452–474. [Google Scholar] [CrossRef]
- Knott, A.B.; Bossy-Wetzel, E. Nitric oxide in health and disease of the nervous system. Antioxid. Redox Signal. 2009, 11, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.A.; da Silva, R.S.; Miranda, K.M.; Switzer, C.H.; Wink, D.A.; Fukuto, J.M. Biological nitric oxide signalling: Chemistry and terminology. Br. J. Pharmacol. 2013, 169, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef]
- Hobbs, A.J. Soluble guanylate cyclase: The forgotten sibling. Trends Pharmacol. Sci. 1997, 18, 484–491. [Google Scholar] [CrossRef]
- Kass, D.A.; Takimoto, E.; Nagayama, T.; Champion, H.C. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc. Res. 2007, 75, 303–314. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Bradley, S.A.; Steinert, J.R. Nitric Oxide-Mediated Posttranslational Modifications: Impacts at the Synapse. Oxid. Med. Cell. Longev. 2016, 2016, 5681036. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, D.Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef]
- Zhu, L.J.; Li, F.; Zhu, D.Y. nNOS and Neurological, Neuropsychiatric Disorders: A 20-Year Story. Neurosci. Bull. 2023, 39, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; Austin, S.A. Neurovascular Protective Function of Endothelial Nitric Oxide—Recent Advances. Circ. J. 2016, 80, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Bechade, C.; Colasse, S.; Diana, M.A.; Rouault, M.; Bessis, A. NOS2 expression is restricted to neurons in the healthy brain but is triggered in microglia upon inflammation. Glia 2014, 62, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Bechade, C.; Pascual, O.; Triller, A.; Bessis, A. Nitric oxide regulates astrocyte maturation in the hippocampus: Involvement of NOS2. Mol. Cell. Neurosci. 2011, 46, 762–769. [Google Scholar] [CrossRef]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef]
- Eissa, N.; Sadeq, A.; Sasse, A.; Sadek, B. Role of Neuroinflammation in Autism Spectrum Disorder and the Emergence of Brain Histaminergic System. Lessons Also for BPSD? Front. Pharmacol. 2020, 11, 886. [Google Scholar] [CrossRef]
- Thiel, V.E.; Audus, K.L. Nitric oxide and blood-brain barrier integrity. Antioxid. Redox Signal. 2001, 3, 273–278. [Google Scholar] [CrossRef]
- Kim, H.G.; Moon, M.; Choi, J.G.; Park, G.; Kim, A.J.; Hur, J.; Lee, K.T.; Oh, M.S. Donepezil inhibits the amyloid-beta oligomer-induced microglial activation in vitro and in vivo. Neurotoxicology 2014, 40, 23–32. [Google Scholar] [CrossRef]
- Caruso, G.; Grasso, M.; Fidilio, A.; Torrisi, S.A.; Musso, N.; Geraci, F.; Tropea, M.R.; Privitera, A.; Tascedda, F.; Puzzo, D.; et al. Antioxidant Activity of Fluoxetine and Vortioxetine in a Non-Transgenic Animal Model of Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 809541. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef]
- Justo, A.F.O.; Suemoto, C.K. The modulation of neuroinflammation by inducible nitric oxide synthase. J. Cell. Commun. Signal. 2022, 2, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Merino-Gracia, J.; Costas-Insua, C.; Canales, M.Á.; Rodríguez-Crespo, I. Insights into the c-terminal peptide binding specificity of the pdz domain of neuronal nitric-oxide synthase: Characterization of the interaction with the tight junction protein claudin-3. J. Biol. Chem. 2016, 291, 11581–11595. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Rothe, F.; Canzler, U.; Wolf, G. Subcellular localization of the neuronal isoform of nitric oxide synthase in the rat brain: A critical evaluation. Neuroscience 1998, 83, 259–269. [Google Scholar] [CrossRef]
- Park, J.H.; Straub, V.A.; O’Shea, M. Anterograde signaling by nitric oxide: Characterization and in vitro reconstitution of an identified nitrergic synapse. J. Neurosci. 1998, 18, 5463–5476. [Google Scholar] [CrossRef]
- Hardingham, N.; Dachtler, J.; Fox, K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell. Neurosci. 2013, 7, 190. [Google Scholar] [CrossRef]
- Paul, V.; Ekambaram, P. Involvement of nitric oxide in learning & memory processes. Indian J. Med. Res. 2011, 133, 471–478. [Google Scholar]
- Gallo, E.F.; Iadecola, C. Neuronal nitric oxide contributes to neuroplasticity-associated protein expression through cGMP, protein kinase G, and extracellular signal-regulated kinase. J. Neurosci. 2011, 31, 6947–6955. [Google Scholar] [CrossRef]
- Moosavi, M.; Abbasi, L.; Zarifkar, A.; Rastegar, K. The role of nitric oxide in spatial memory stages, hippocampal ERK and CaMKII phosphorylation. Pharmacol. Biochem. Behav. 2014, 122, 164–172. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Sandín, S.; Alonso, M.T.; García-Sancho, J. Calcium homoeostasis modulator 1 (CALHM1) reduces the calcium content of the endoplasmic reticulum (ER) and triggers ER stress. Biochem. J. 2011, 437, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. Current and future treatments in Alzheimer’s disease. Semin. Neurol. 2019, 39, 227–240. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, M.; Gopinath, P.; Govindaraju, T. Role of Post-translational Modifications in Alzheimer’s Disease. Chembiochem 2020, 21, 1052–1079. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical Analysis of Protein Nitrotyrosine and Dityrosine in the Alzheimer Brain Indicates Region-Specific Accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [CrossRef]
- Quinn, J.; Davis, F.; Woodward, W.R.; Eckenstein, F. Beta-amyloid plaques induce neuritic dystrophy of nitric oxide-producing neurons in a transgenic mouse model of Alzheimer’s disease. Exp. Neurol. 2001, 168, 203–212. [Google Scholar] [CrossRef]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef]
- Guix, F.X.; Wahle, T.; Vennekens, K.; Snellinx, A.; Chávez-Gutiérrez, L.; Ill-Raga, G.; Ramos-Fernandez, E.; Guardia-Laguarta, C.; Lleó, A.; Arimon, M.; et al. Modification of γ-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer’s disease. EMBO Mol. Med. 2012, 4, 660–673. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Guivernau, B.; Bonet, J.; Valls-Comamala, V.; Bosch-Morató, M.; Godoy, J.A.; Inestrosa, N.C.; Perálvarez-Marín, A.; Fernández-Busquets, X.; Andreu, D.; Oliva, B.; et al. Amyloid-β peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J. Neurosci. 2016, 36, 11693–11703. [Google Scholar] [CrossRef] [PubMed]
- Guix, F.X.; Ill-Raga, G.; Bravo, R.; Nakaya, T.; de Fabritiis, G.; Coma, M.; Pietro Miscione, G.; Villà-Freixa, J.; Suzuki, T.; Fernàndez-Busquets, X.; et al. Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain 2009, 132, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Thongboonkerd, V.; Klein, J.B.; Lynn, B.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J. Neurochem. 2003, 85, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.-K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.-i.; Lipton, S.A. Aberrant protein Snitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef]
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, G.; Rizza, S.; Montagna, C.; Filomeni, G. Established principles and emerging concepts on the interplay between mitochondrial physiology and S-(De) nitrosylation: Implications in cancer and neurodegeneration. Int. J. Cell Biol. 2012, 2012, 361872. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Nakamura, T.; Cao, G.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by β-amyloid peptide. Proc. Natl. Acad. Sci. USA 2011, 108, 14330–14335. [Google Scholar] [CrossRef]
- Qu, J.; Nakamura, T.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-nitrosylation of Cdk5. Prion 2012, 6, 364–370. [Google Scholar] [CrossRef]
- Cordes, C.M.; Bennett, R.G.; Siford, G.L.; Hamel, F.G. Redox regulation of insulin degradation by insulin-degrading enzyme. PLoS ONE 2011, 6, e18138. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Z.; Tan, H.; Zhu, S.; Sun, Y.; Li, M.X.; Wang, J.F. Nitrosylation of vesicular transporters in brain of amyloid precursor protein/presenilin 1 double transgenic mice. J. Alzheimer’s Dis. 2017, 55, 1683–1692. [Google Scholar] [CrossRef]
- Lipton, S.A. Hidden networks of aberrant protein transnitrosylation contribute to synapse loss in Alzheimer’s disease. Free Radic. Biol. Med. 2022, 193, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P. Understanding the molecular basis of Parkinson’s disease, identification of biomarkers and routes to therapy. Expert. Rev. Proteom. 2010, 2010, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A. Levodopa Therapy for Parkinson’s Disease: Pharmacokinetics and Pharmacodynamics. Mov. Disord. 2015, 30, 64–72. [Google Scholar] [CrossRef]
- Dhanawat, M.; Mehta, D.K.; Gupta, S.; Das, R. Understanding the Pathogenesis Involved in Parkinson’s Disease and Potential Therapeutic Treatment Strategies. Cent. Nerv. Syst. Agents Med. Chem. 2020, 20, 88–102. [Google Scholar] [CrossRef]
- Gatto, E.M.; Riob’o, N.A.; Carreras, M.C.; Chernavsky, A.; Rubio, A.; Satz, M.L.; Poderoso, J.J. Overexpression of neutrophil neuronal nitric oxide synthase in Parkinson’s disease. Nitric Oxide 2000, 4, 534–539. [Google Scholar] [CrossRef]
- Eve, D.J.; Nisbet, A.P.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Joniec, I.; Ciesielska, A.; Kurkowska-Jastrzebska, I.; Przybylkowski, A.; Czlonkowska, A.; Czlonkowski, A. Age- and sex-differences in the nitric oxide synthase expression and dopamine concentration in the murine model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 2009, 1261, 7–19. [Google Scholar] [CrossRef]
- Burai, R.; Ait Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef]
- Blanchard-Fillion, B.; Souza, J.M.; Friel, T.; Jiang, G.C.T.; Vrana, K.; Sharov, V.; Barrón, L.; Schöneich, C.; Quijano, C.; Alvarez, B.; et al. Nitration and Inactivation of Tyrosine Hydroxylase by Peroxynitrite. J. Biol. Chem. 2001, 276, 46017–46023. [Google Scholar] [CrossRef]
- Wang, Y.; Sung, C.C.; Chung, K.K. Novel enhancement mechanism of tyrosine hydroxylase enzymatic activity by nitric oxide through S-nitrosylation. Sci. Rep. 2017, 7, 44154. [Google Scholar] [CrossRef] [PubMed]
- Stykel, M.G.; Ryan, S.D. Nitrosative stress in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Zoupa, E.; Pitsikas, N. The Nitric Oxide (NO) Donor Sodium Nitroprusside (SNP) and Its Potential for the Schizophrenia Therapy: Lights and Shadows. Molecules 2021, 26, 3196. [Google Scholar] [CrossRef] [PubMed]
- Kartawy, M.; Khaliulin, I.; Amal, H. Systems biology reveals S-nitrosylation dependent regulation of mitochondrial functions in mice with Shank3 mutation associated with autism spectrum disorder. Brain Sci. 2021, 11, 677. [Google Scholar] [CrossRef]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Amal, H.; Barak, B.; Bhat, V.; Gong, G.; Joughin, B.A.; Wang, X.; Wishnok, J.S.; Feng, G.; Tannenbaum, S.R. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol. Psychiatr. 2018, 25, 1835–1848. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Ojha, S.K.; Kartawy, M.; Hamoudi, W.; Choudhary, A.; Stern, S.; Aran, A.; Amal, H. The NO Answer for Autism Spectrum Disorder. Adv. Sci. 2023, 10, 2205783. [Google Scholar] [CrossRef]
- Zhu, X.; Dong, J.; Han, B.; Huang, R.; Zhang, A.; Xia, Z.; Chang, H.; Chao, J.; Yao, H. Neuronal nitric oxide synthase contributes to PTZ kindling epilepsy-induced hippocampal endoplasmic reticulum stress and oxidative damage. Front. Cell. Neurosci. 2017, 11, 377. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, Y.; Yang, J.; Zhang, C.; He, Y.; Qi, H.; Zeng, Y.; Zhang, A.; Liu, X.; Zhu, X. Inhibition of neuronal nitric oxide synthase protects against hippocampal neuronal injuries by increasing neuropeptide Y expression in temporal lobe epilepsy mice. Free Radic. Biol. Med. 2022, 188, 45–61. [Google Scholar] [CrossRef]
- Bashkatova, V.; Narkevich, V.; Vitskova, G.; Vanin, A. The influence of anticonvulsant and antioxidant drugs on nitric oxide level and lipid peroxidation in the rat brain during penthylenetetrazole-induced epileptiform model seizures. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 487–492. [Google Scholar] [CrossRef]
- Mikami, Y.; Kanemaru, K.; Okubo, Y.; Nakaune, T.; Suzuki, J.; Shibata, K.; Sugiyama, H.; Koyama, R.; Murayama, T.; Ito, A.; et al. Nitric Oxide-induced Activation of the Type 1 Ryanodine Receptor Is Critical for Epileptic Seizure-induced Neuronal Cell Death. eBioMedicine 2016, 11, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Lu, R.; Dong, G.; Chen, X.; Yun, W.; Zhou, X. The role of S-nitrosylation of kainate-type of ionotropic glutamate receptor 2 in epilepsy induced by kainic acid. J. Neurochem. 2017, 144, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Maccallini, C.; Amoroso, R. Targeting neuronal nitric oxide synthase as a valuable strategy for the therapy of neurological disorders. Neural Regen. Res. 2016, 11, 1731–1734. [Google Scholar] [CrossRef]
- Annedi, S.C. Cell-Permeable Inhibitors of Neuronal Nitric Oxide Synthase Open New Prospects for the Treatment of Neurological Disorders. J. Med. Chem. 2015, 58, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, H.; Flinspach, M.; Poulos, T.L.; Silverman, R.B. Computer modeling of selective regions in the active site of nitric oxide synthases: Implication for the design of isoform-selective inhibitors. J. Med. Chem. 2003, 46, 5700–5711. [Google Scholar] [CrossRef]
- Carrión, M.D.; Rubio-Ruiz, B.; Franco-Montalban, F.; Amoia, P.; Zuccarini, M.C.; De Simone, C.; Camacho, M.E.; Amoroso, R.; Maccallini, C. New amidine-benzenesulfonamides as iNOS inhibitors for the therapy of the triple negative breast cancer. Eur. J. Med. Chem. 2023, 248, 115112. [Google Scholar] [CrossRef]
- Maccallini, C.; Montagnani, M.; Paciotti, R.; Ammazzalorso, A.; De Filippis, B.; Di Matteo, M.; Di Silvestre, S.; Fantacuzzi, M.; Giampietro, L.; Potenza, M.A.; et al. Selective Acetamidine-Based Nitric Oxide Synthase Inhibitors: Synthesis, Docking, and Biological Studies. ACS Med. Chem. Lett. 2015, 6, 635–640. [Google Scholar] [CrossRef]
- Maccallini, C.; Marinelli, L.; Indorf, P.; Cacciatore, I.; Fantacuzzi, M.; Clement, B.; Di Stefano, A.; Amoroso, R. A Novel Prodrug of a nNOS Inhibitor with Improved Pharmacokinetic Potential. ChemMedChem 2020, 15, 2157–2163. [Google Scholar] [CrossRef]
- Annedi, S.C.; Ramnauth, J.; Maddaford, S.P.; Renton, P.; Rakhit, S.; Mladenova, G.; Dove, P.; Silverman, S.; Andrews, J.S.; Felice, M.D.; et al. Discovery of cis-N-(1-(4-(methylamino)cyclohexyl)indolin-6-yl)thiophene-2-carboximidamide: A 1,6-disubstituted indoline derivative as a highly selective inhibitor of human neuronal nitric oxide synthase (nNOS) without any cardiovascular liabilities. J. Med. Chem. 2012, 55, 943–955. [Google Scholar] [CrossRef]
- Moore, P.K.; Bland-Ward, P.A. 7-Nitroindazole: An inhibitor of nitric oxide synthase. Methods Enzymol. 1996, 268, 393–398. [Google Scholar] [CrossRef]
- Maccallini, C.; Di Matteo, M.; Vullo, D.; Ammazzalorso, A.; Carradori, S.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Pandolfi, A.; Supuran, C.T.; et al. Indazole, Pyrazole, and Oxazole Derivatives Targeting Nitric Oxide Synthases and Carbonic Anhydrases. ChemMedChem 2016, 11, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Maccallini, C.; Gallorini, M.; Sisto, F.; Akdemir, A.; Ammazzalorso, A.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Carradori, S.; Cataldi, A.; et al. New azolyl-derivatives as multitargeting agents against breast cancer and fungal infections: Synthesis, biological evaluation and docking study. J. Enzyme Inhib. Med. Chem. 2021, 36, 1632–1645. [Google Scholar] [CrossRef]
- Vasu, D.; Li, H.; Hardy, C.D.; Poulos, T.L.; Silverman, R.B. 2-Aminopyridines with a shortened amino sidechain as potent, selective, and highly permeable human neuronal nitric oxide synthase inhibitors. Bioorg. Med. Chem. 2022, 69, 116878. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Reidl, C.T.; Li, H.; Chreifi, G.; Poulos, T.L.; Silverman, R.B. First Contact: 7-Phenyl-2-Aminoquinolines, Potent and Selective Neuronal Nitric Oxide Synthase Inhibitors That Target an Isoform Specific Aspartate. J. Med. Chem. 2020, 63, 4528–4554. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Yokoyama, R.; Shibata, T.; Dawson, V.L.; Dawson, T.M. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridin (MPTP)-induced dopaminergic neurotoxicity. Proc. Natl. Acad. Sci. USA 1996, 93, 4565–4571. [Google Scholar] [CrossRef]
- Yuste, J.E.; Echeverry, M.B.; Ros-Bernal, F.; Gomez, A.; Ros, C.M.; Campuzano, C.M.; Fernandez-Villalba, E.; Herrero, M.T. 7-Nitroindazole down-regulates dopamine/DARPP-32 signaling in neostriatal neurons in a rat model of Parkinson’s disease. Neuropharmacology 2012, 63, 1258–1267. [Google Scholar] [CrossRef]
- Ehara, A.; Nakadate, K.; Sugimoto, H.; Yoshimoto, K.; Ueda, S. Role of neuronal nitric oxide synthase in slowly progressive dopaminergic neurodegeneration in the Zitter rat. Nitric Oxide 2018, 78, 41–50. [Google Scholar] [CrossRef]
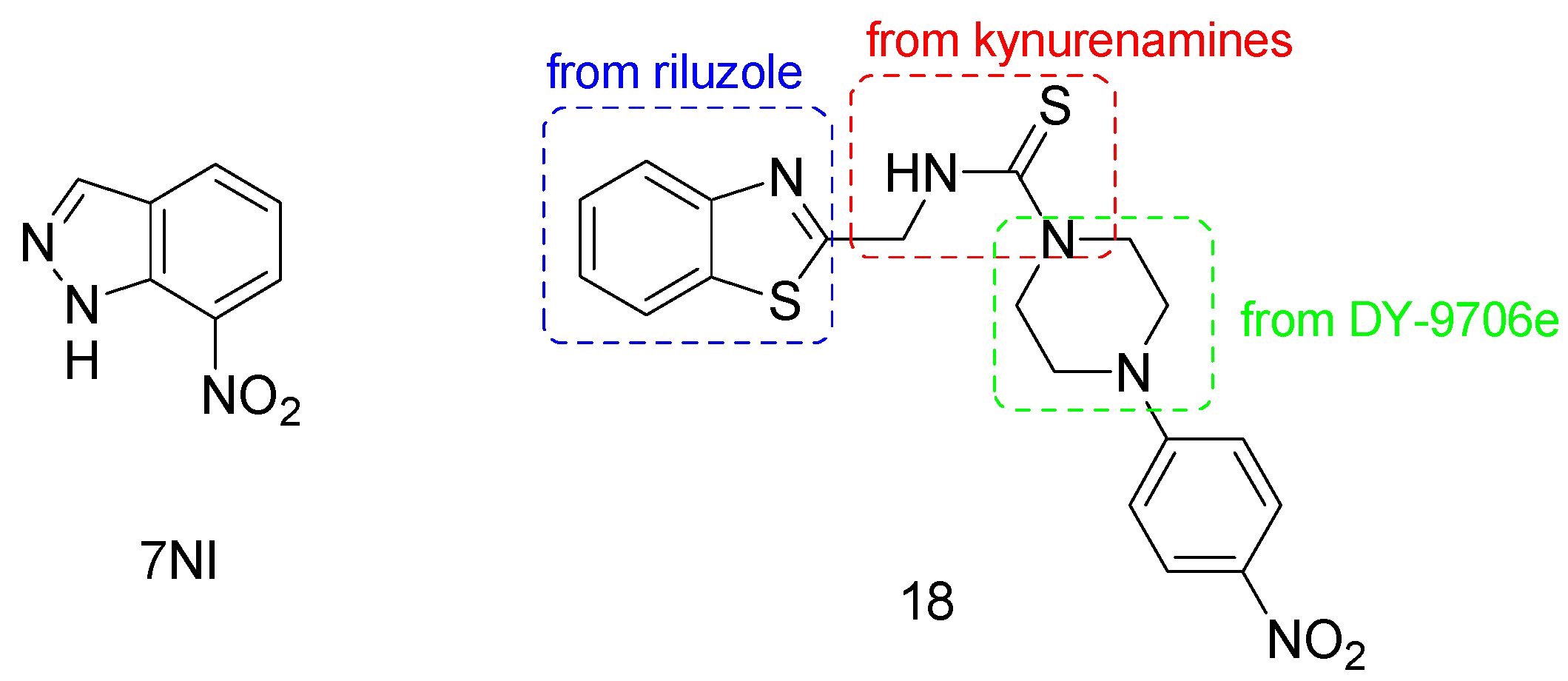
- Agrawal, S.; Kumari, R.; Sophronea, T.; Kumari, N.; Luthra, P.M. Design and synthesis of benzo[d]thiazol-2-yl-methyl-4-(substituted)-piperazine-1-carbothioamide as novel neuronal nitric oxide inhibitors and evaluation of their neuroprotecting effect in 6-OHDA-induced unilateral lesioned rat model of Parkinson’s disease. Biomed. Pharmacother. 2022, 156, 113838. [Google Scholar] [CrossRef]
- Chayah, M.; Carrion, M.C.; Gallo, M.A.; Jimenez, R.; Duarte, J.; Camacho, M.E. Development of urea and thiourea kynurenamine derivatives: Synthesis, molecular modeling, and biological evaluation as nitric oxide synthase inhibitors. ChemMedChem 2015, 10, 874–882. [Google Scholar] [CrossRef]
- Hashiguchi, A.; Kawano, T.; Yano, S.; Morioka, M.; Hamada, J.; Sato, T.; Shirasaki, Y.; Ushio, Y.; Fukunaga, K. The neuroprotective effect of a novel calmodulin antagonist, 3-[2-[4-(3-chloro-2-methylphenyl)-1-piperazinyl]ethyl]-5,6-dimethoxy-1-(4-imidazolylmethyl)-1H-indazole dihydrochloride 3.5 hydrate, in transient forebrain ischemia. Neuroscience 2003, 121, 379–386. [Google Scholar] [CrossRef]
- Gu, Y.; Zhu, D. nNOS-mediated protein-protein interactions: Promising targets for treating neurological and neuropsychiatric disorders. J. Biomed. Res. 2021, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Hillier, B.J.; Lim, W.A.; Bredt, D.S. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999, 274, 27467–27473. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.; Clausen, B.H.; Møller, M.; Vestergaard, B.; Chi, C.N.; Round, A.; Sørensen, P.L.; Nissen, K.B.; Kastrup, J.S.; Gajhede, M.; et al. A high-affinity, dimeric inhibitor of PSD-95 bivalently interacts with PDZ1-2 and protects against ischemic brain damage. Proc. Natl. Acad. Sci. USA 2012, 109, 3317–3322. [Google Scholar] [CrossRef]
- Kucharz, K.; Søndergaard Rasmussen, I.; Bach, A.; Strømgaard, K.; Lauritzen, M. PSD-95 uncoupling from NMDA receptors by Tat- N-dimer ameliorates neuronal depolarization in cortical spreading depression. J. Cereb. Blood Flow Metab. 2017, 37, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Nobili, P.; Cipelletti, B.; Cagnoli, C.; Zambon, S.; Locatelli, D.; de Curtis, M.; Battaglia, G.S. Targeting PSD95-nNOS interaction by Tat-N-dimer peptide during status epilepticus is neuroprotective in MAM-pilocarpine rat model. Neuropharmacology 2019, 153, 82–97. [Google Scholar] [CrossRef]
- Balboa, J.R.; Essig, D.J.; Ma, S.; Karer, N.; Clemmensen, L.S.; Pedersen, S.W.; Joerger, A.C.; Knapp, S.; Østergaard, S.; Strømgaard, K. Development of a Potent Cyclic Peptide Inhibitor of the nNOS/PSD-95 Interaction. J. Med. Chem. 2023, 66, 976–990. [Google Scholar] [CrossRef]
- Zhou, L.; Li, F.; Xu, H.B.; Luo, C.X.; Wu, H.Y.; Zhu, M.M.; Lu, W.; Ji, X.; Zhou, Q.G.; Zhu, D.Y. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat. Med. 2010, 16, 1439–1443. [Google Scholar] [CrossRef]
- Cai, W.H.; Wu, S.G.; Pan, Z.; Xiao, J.; Li, F.; Cao, J.; Zang, W.; Tao, Y. Disrupting interaction of PSD-95 with nNOS attenuates hemorrhage-induced thalamic pain. Neuropharmacology 2018, 141, 238–248. [Google Scholar] [CrossRef]
- Tao, W.Y.; Yu, L.J.; Jiang, S.; Cao, X.; Chen, J.; Bao, X.Y.; Li, F.; Xu, Y.; Zhu, X.L. Neuroprotective effects of ZL006 in Aβ1-42-treated neuronal cells. Neural Regen. Res. 2020, 15, 2296–2305. [Google Scholar] [CrossRef]
- Hu, W.; Guan, L.S.; Dang, X.B.; Ren, P.Y.; Zhang, Y.L. Small-molecule inhibitors at the PSD-95/nNOS interface attenuate MPP+-induced neuronal injury through Sirt3 mediated inhibition of mitochondrial dysfunction. Neurochem. Int. 2014, 79, 57–64. [Google Scholar] [CrossRef]
- Florio, S.K.; Loh, C.; Huang, S.M.; Iwamaye, A.E.; Kitto, K.F.; Fowler, K.W.; Treiberg, J.A.; Hayflick, J.S.; Walker, J.M.; Fairbanks, C.A.; et al. Disruption of nNOS-PSD95 protein-protein interaction inhibits acute thermal hyperalgesia and chronic mechanical allodynia in rodents. Br. J. Pharmacol. 2009, 158, 494–506. [Google Scholar] [CrossRef]
- Bach, A.; Pedersen, S.W.; Dorr, L.A.; Vallon, G.; Ripoche, I.; Ducki, S.; Lian, L.Y. Biochemical investigations of the mechanism of action of small molecules ZL006 and IC87201 as potential inhibitors of the nNOS-PDZ/PSD-95-PDZ interactions. Sci. Rep. 2015, 5, 12157. [Google Scholar] [CrossRef]
- Doucet, M.V.; Levine, H.; Dev, K.K.; Harkin, A. Small-molecule inhibitors at the PSD-95/nNOS interface have antidepressant like properties in mice. Neuropsychopharmacology 2013, 38, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.X.; Lin, Y.H.; Qian, X.D.; Tang, Y.; Zhou, H.H.; Jin, X.; Ni, H.Y.; Zhang, F.Y.; Qin, C.; Li, F.; et al. Interaction of nNOS with PSD-95 negatively controls regenerative repair after stroke. J. Neurosci. 2014, 34, 13535–13548. [Google Scholar] [CrossRef]
- Luo, C.X.; Zhu, X.J.; Zhou, Q.G.; Wang, B.; Wang, W.; Cai, H.H.; Sun, Y.J.; Hu, M.; Jiang, J.; Hua, Y.; et al. Reduced neuronal nitric oxide synthase is involved in ischemia-induced hippocampal neurogenesis by up-regulating inducible nitric oxide synthase expression. J. Neurochem. 2007, 103, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Dao, V.T.; Elbatreek, M.H.; Fuchß, T.; Grädler, U.; Schmidt, H.H.H.W.; Shah, A.M.; Wallace, A.; Knowles, R. Nitric Oxide Syn-thase Inhibitors into the Clinic at Last. Handb. Exp. Pharmacol. 2021, 264, 169–204. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maccallini, C.; Amoroso, R. Neuronal Nitric Oxide Synthase and Post-Translational Modifications in the Development of Central Nervous System Diseases: Implications and Regulation. Molecules 2023, 28, 6691. https://doi.org/10.3390/molecules28186691
Maccallini C, Amoroso R. Neuronal Nitric Oxide Synthase and Post-Translational Modifications in the Development of Central Nervous System Diseases: Implications and Regulation. Molecules. 2023; 28(18):6691. https://doi.org/10.3390/molecules28186691
Chicago/Turabian StyleMaccallini, Cristina, and Rosa Amoroso. 2023. "Neuronal Nitric Oxide Synthase and Post-Translational Modifications in the Development of Central Nervous System Diseases: Implications and Regulation" Molecules 28, no. 18: 6691. https://doi.org/10.3390/molecules28186691
APA StyleMaccallini, C., & Amoroso, R. (2023). Neuronal Nitric Oxide Synthase and Post-Translational Modifications in the Development of Central Nervous System Diseases: Implications and Regulation. Molecules, 28(18), 6691. https://doi.org/10.3390/molecules28186691