
Fabrication of PEG-PLGA Microparticles with Tunable Sizes for Controlled Drug Release Application

 and
and 
Abstract
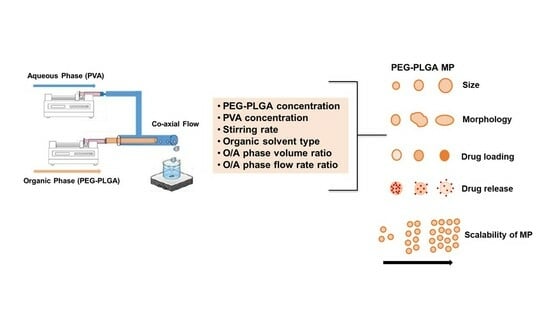
:
1. Introduction
2. Results and Discussion
2.1. Optimization of Experimental Design and Mechanical Parameters
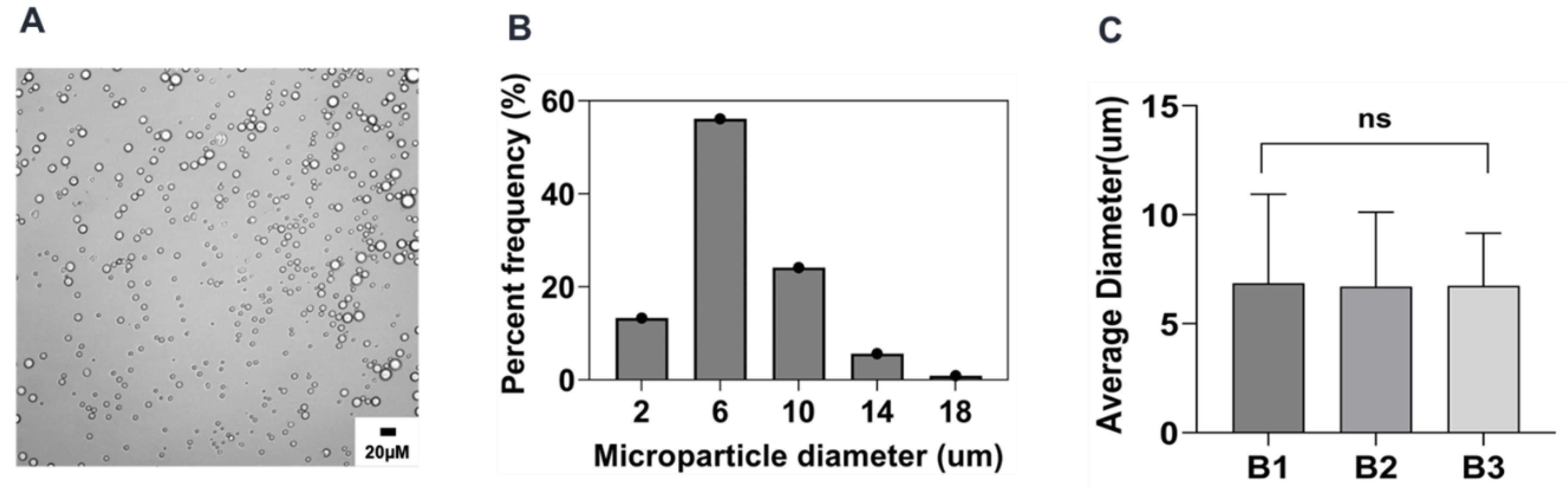
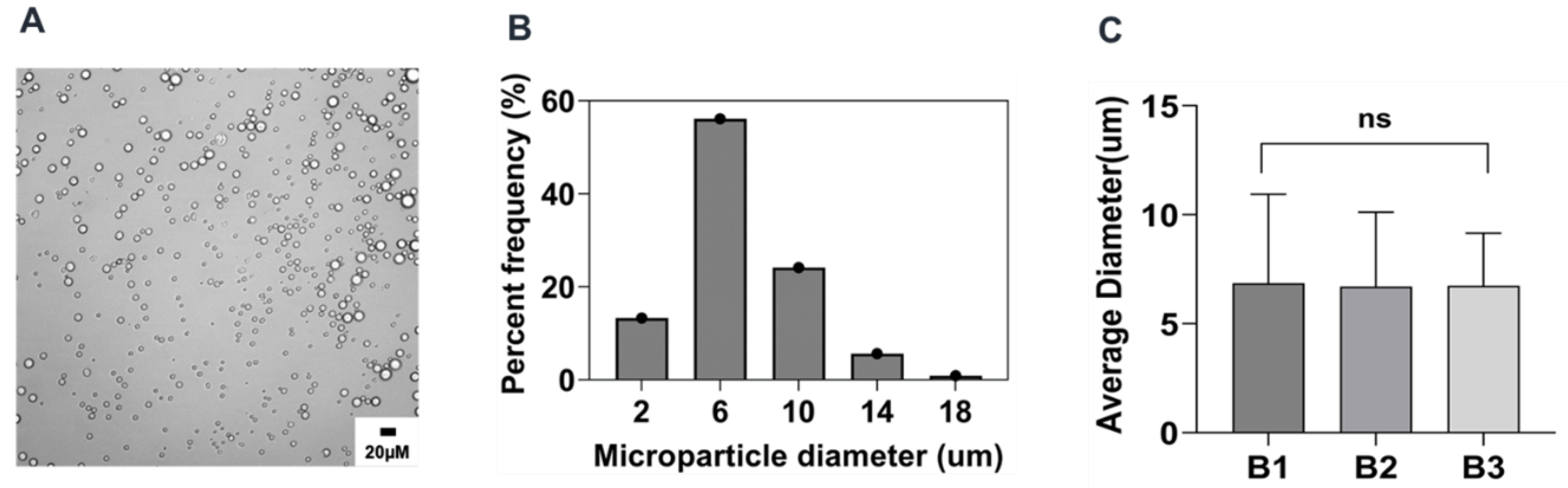
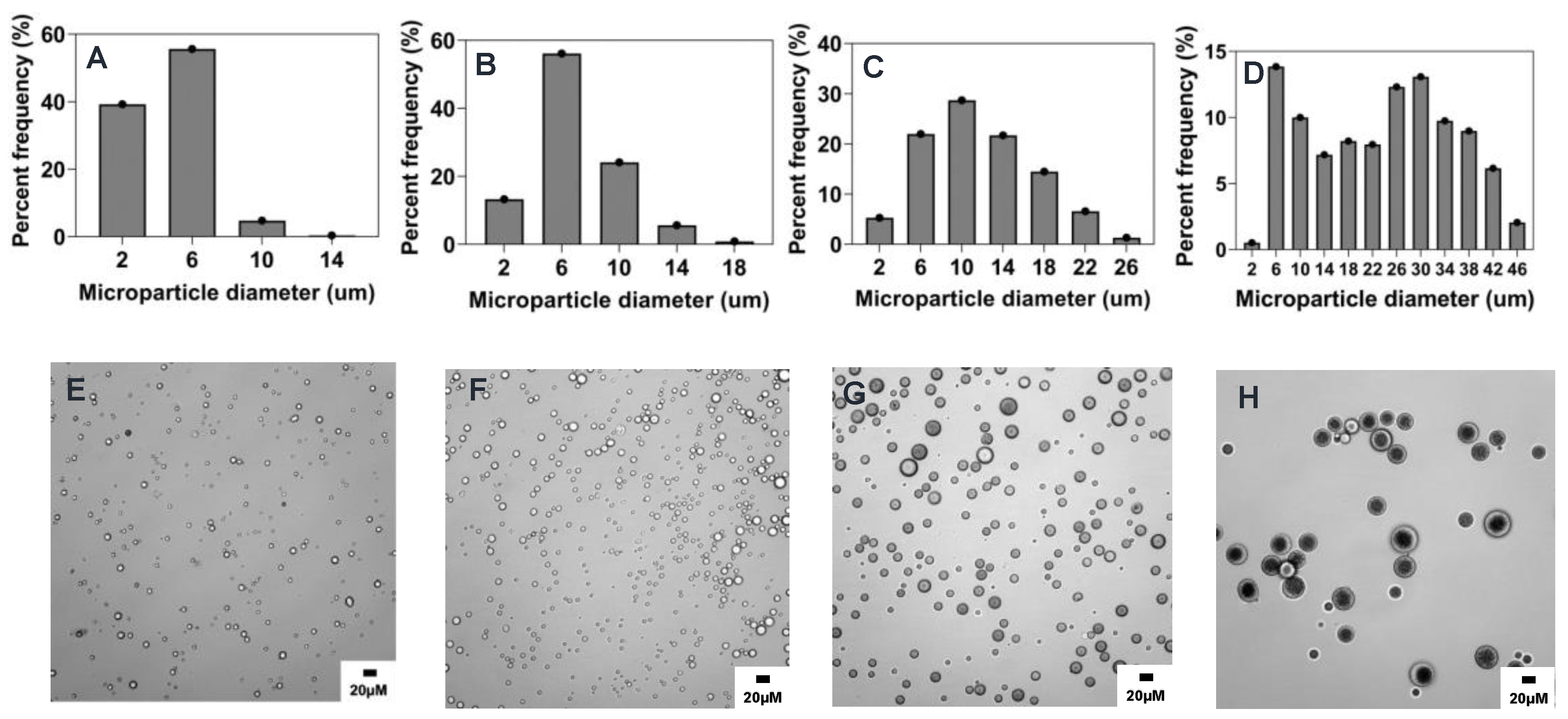
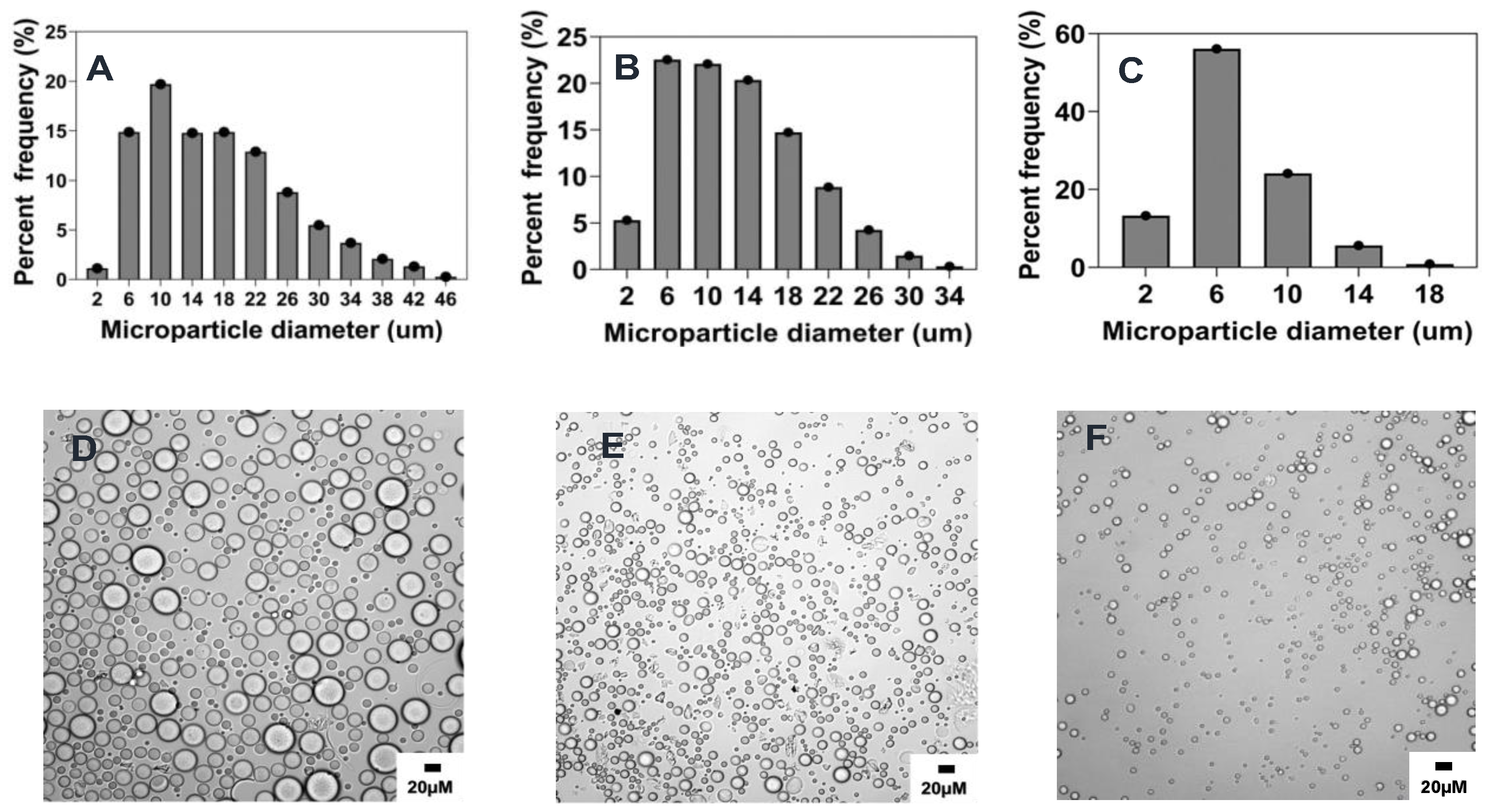
2.2. Preparation and Characterization of Reference Formulation
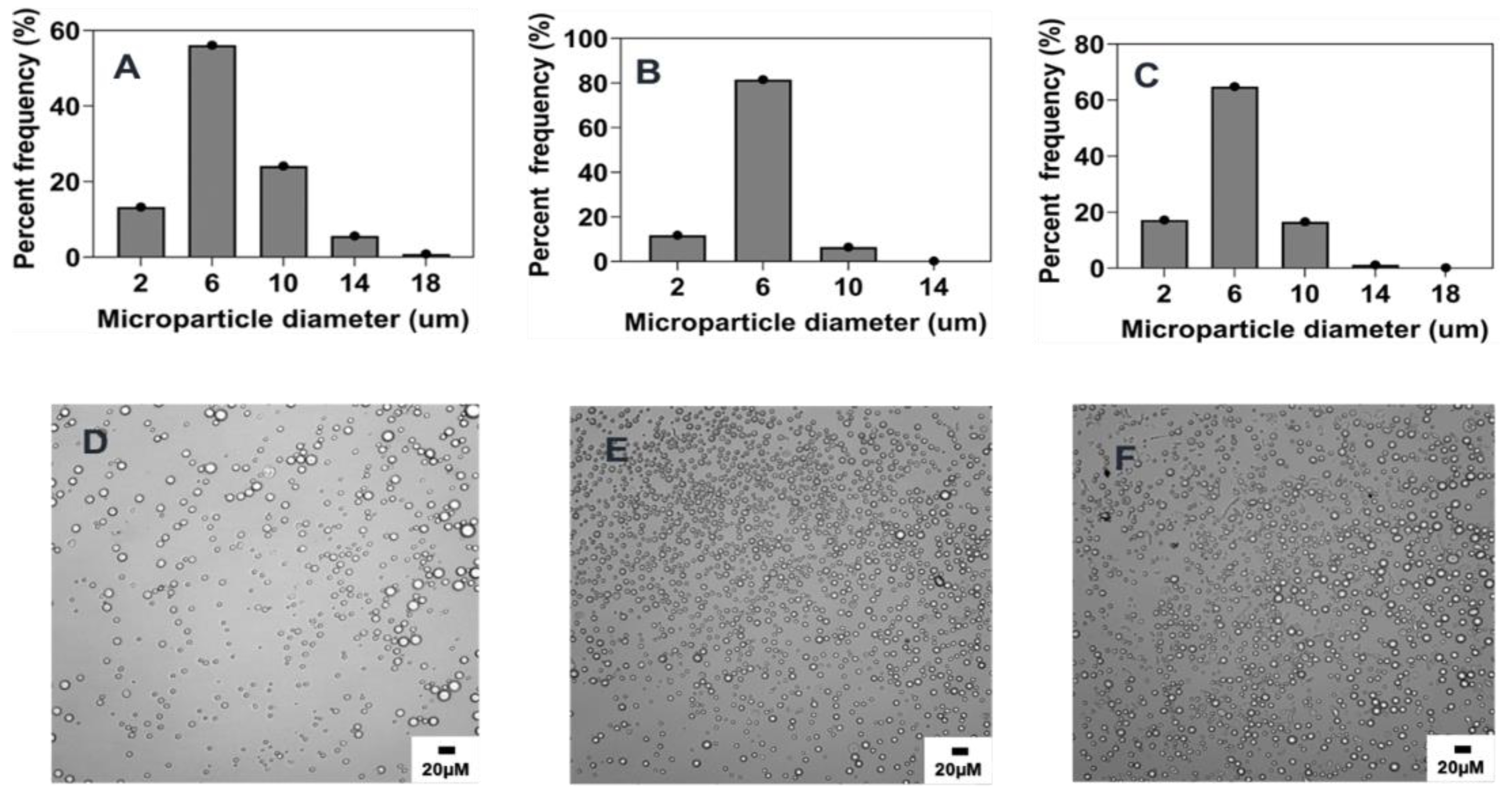
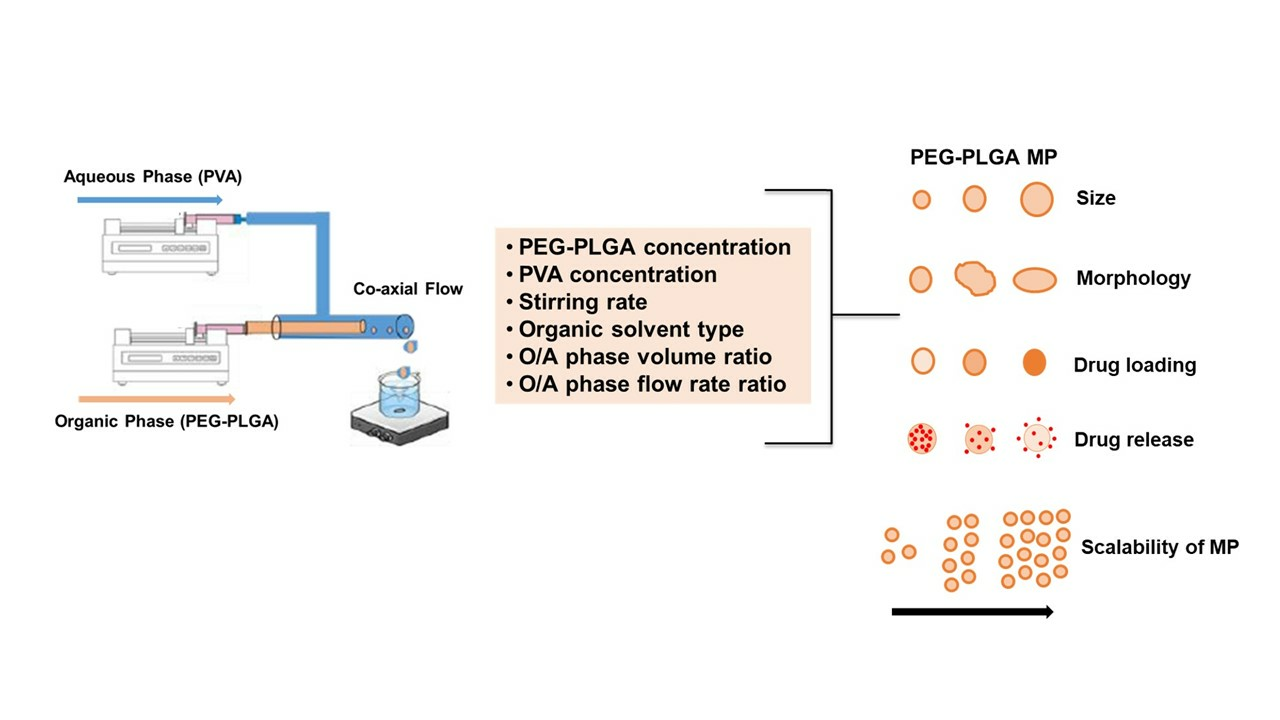
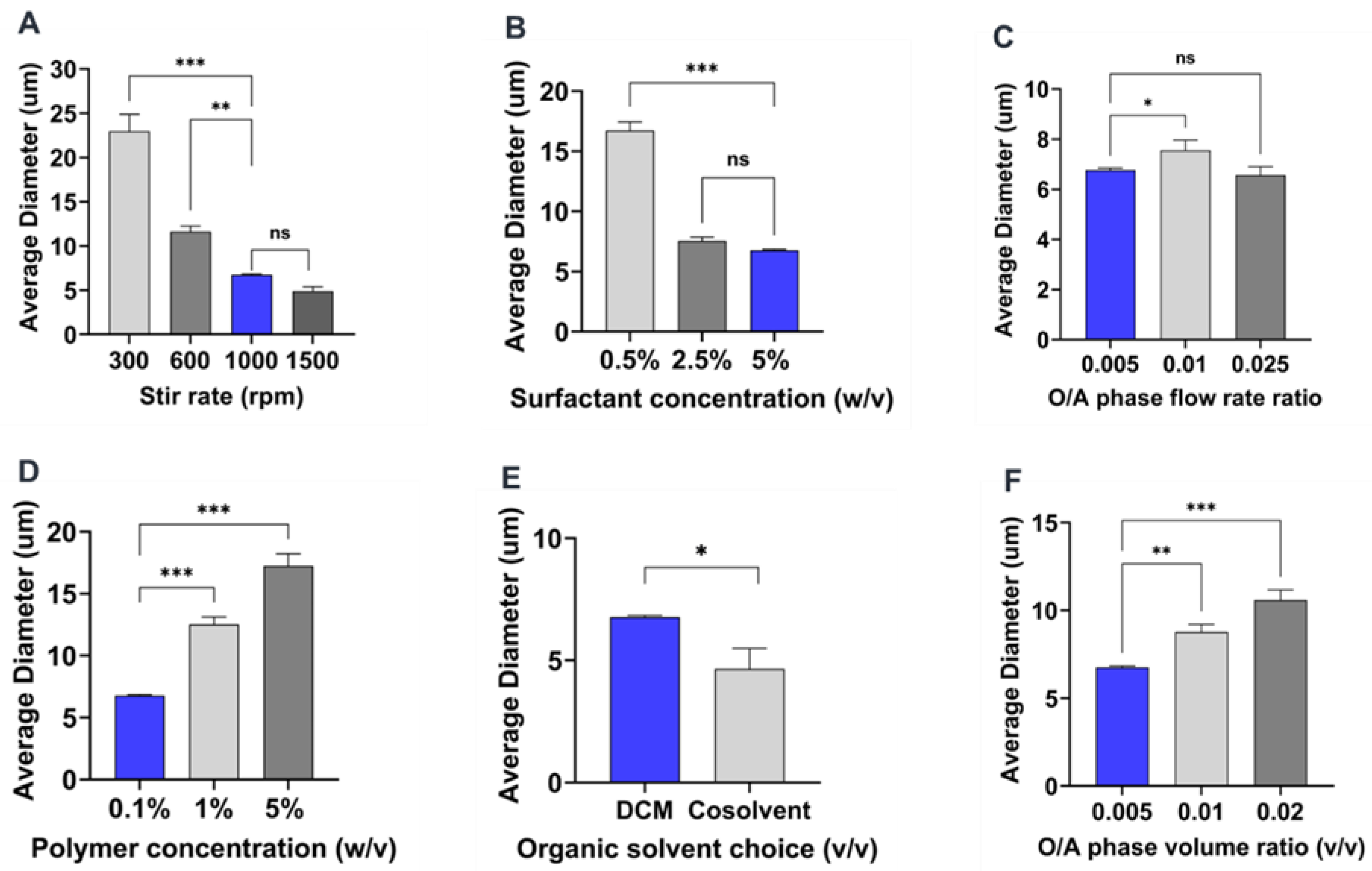
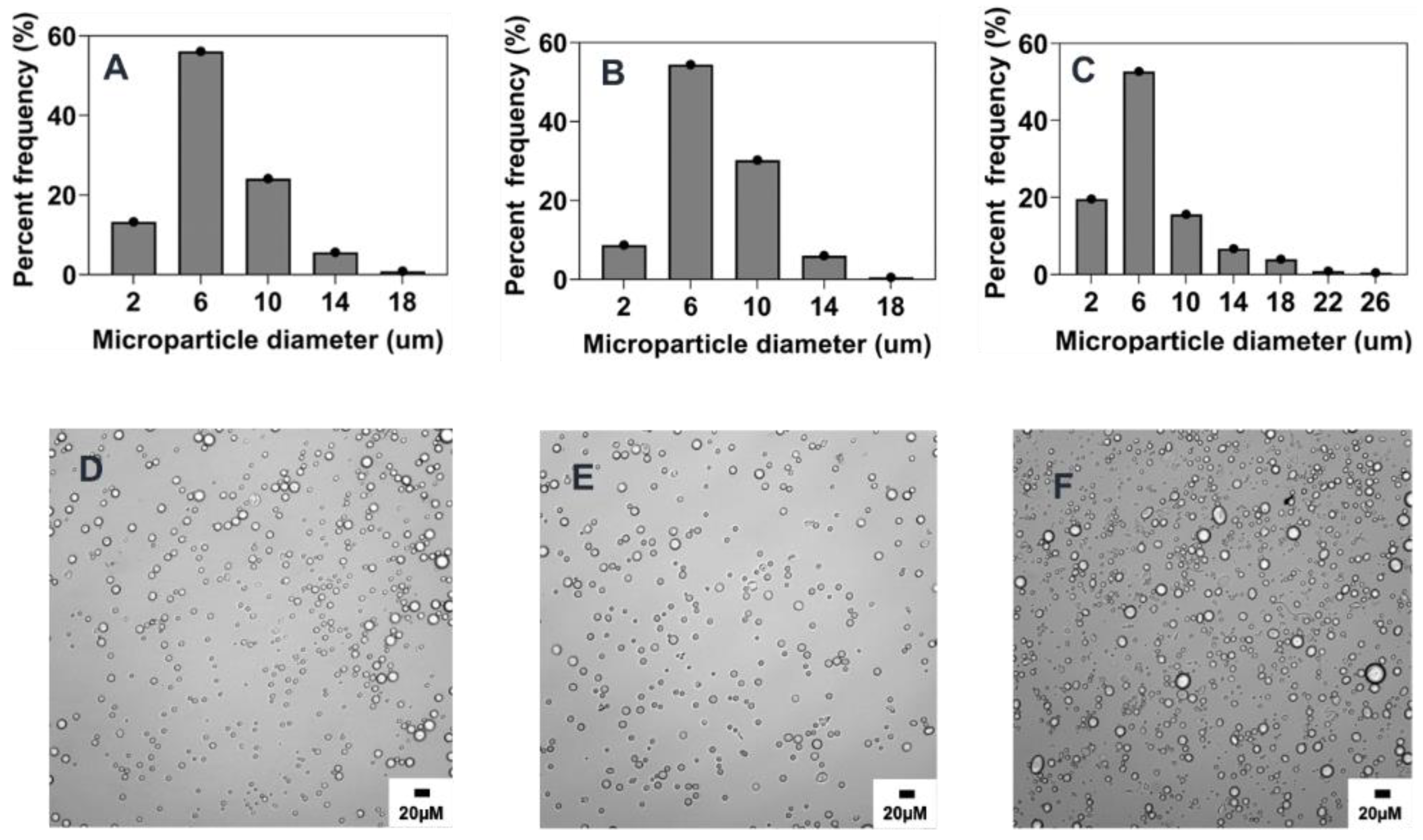
2.3. Summary of the Effect of Process Parameters on Microparticle Mean Size and Size Distributions
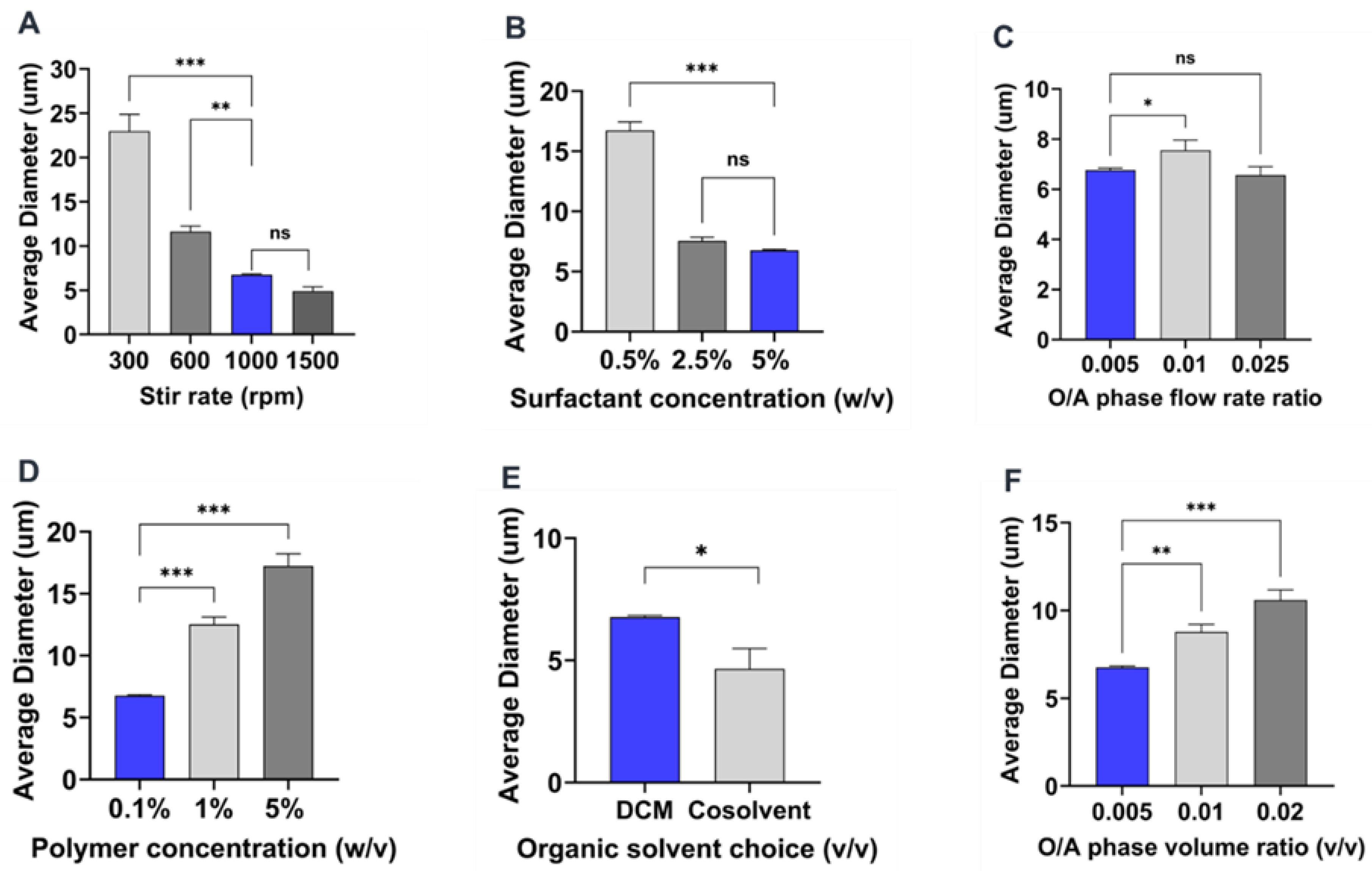
2.4. Influence of Process Parameters on Microparticle Characteristics
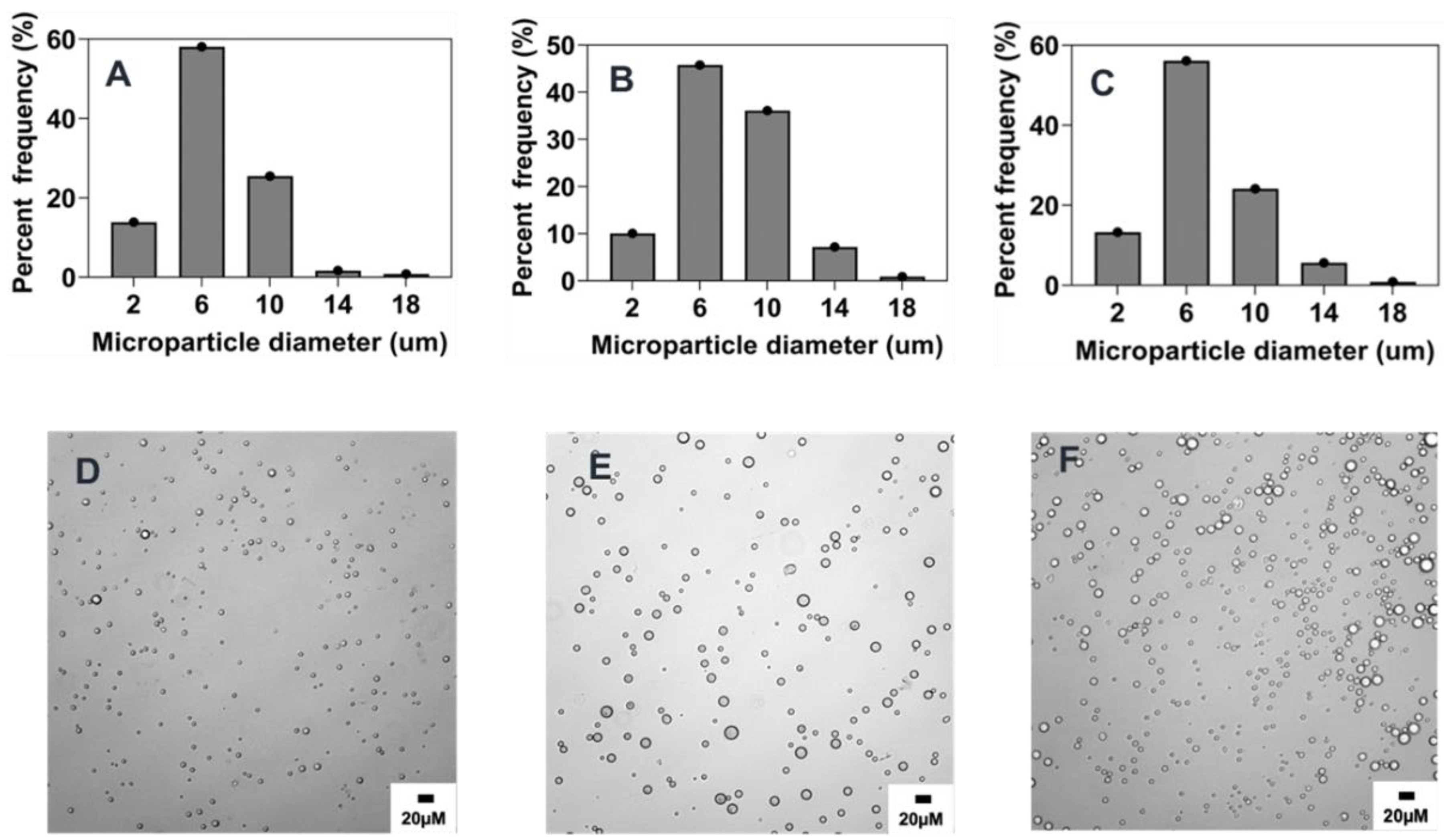
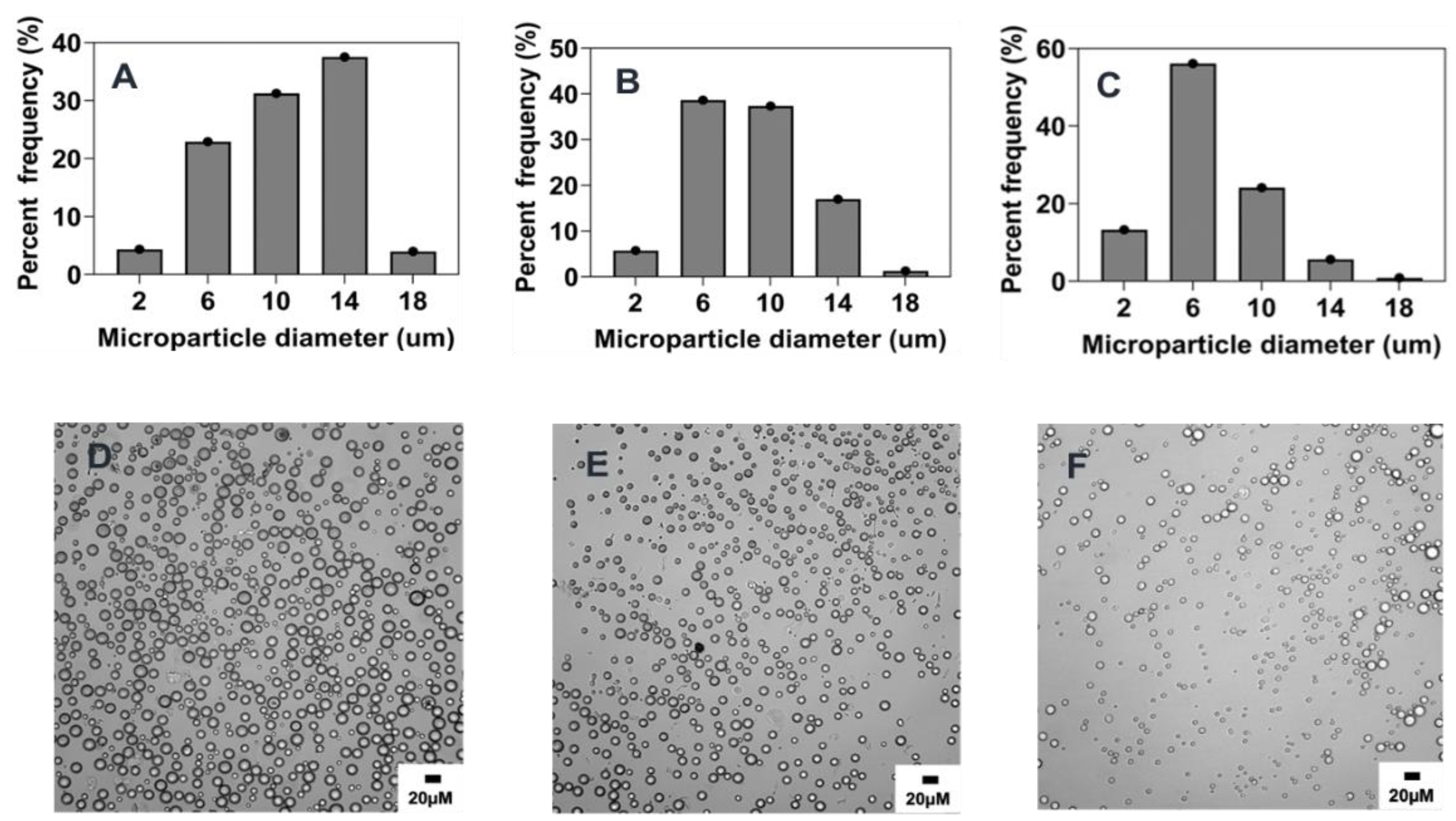
2.4.1. Effect of Stirring Rate
2.4.2. Effect of Surfactant Concentration
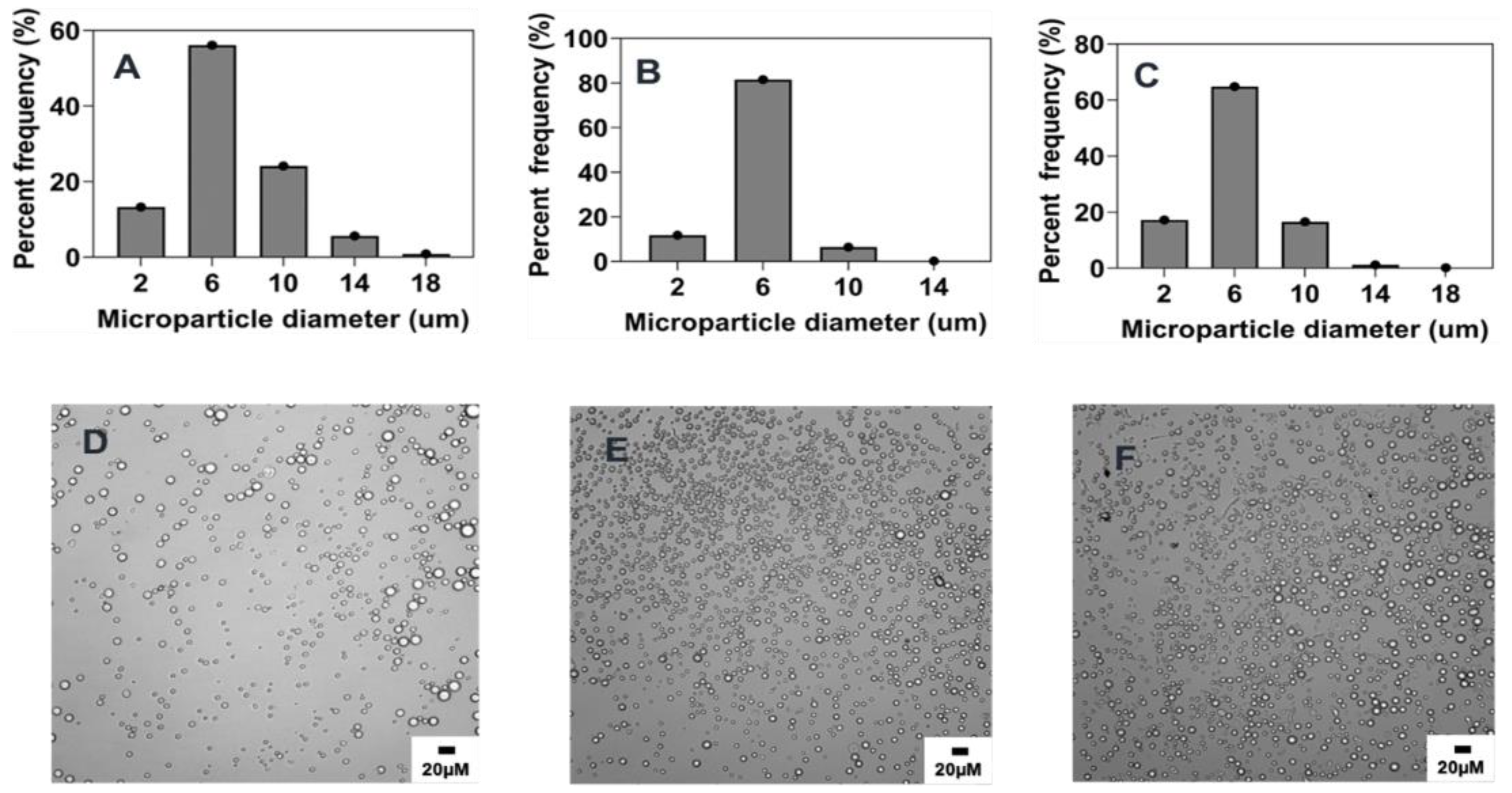
2.4.3. Effect of Organic and Aqueous Phase Flow Rate Ratio
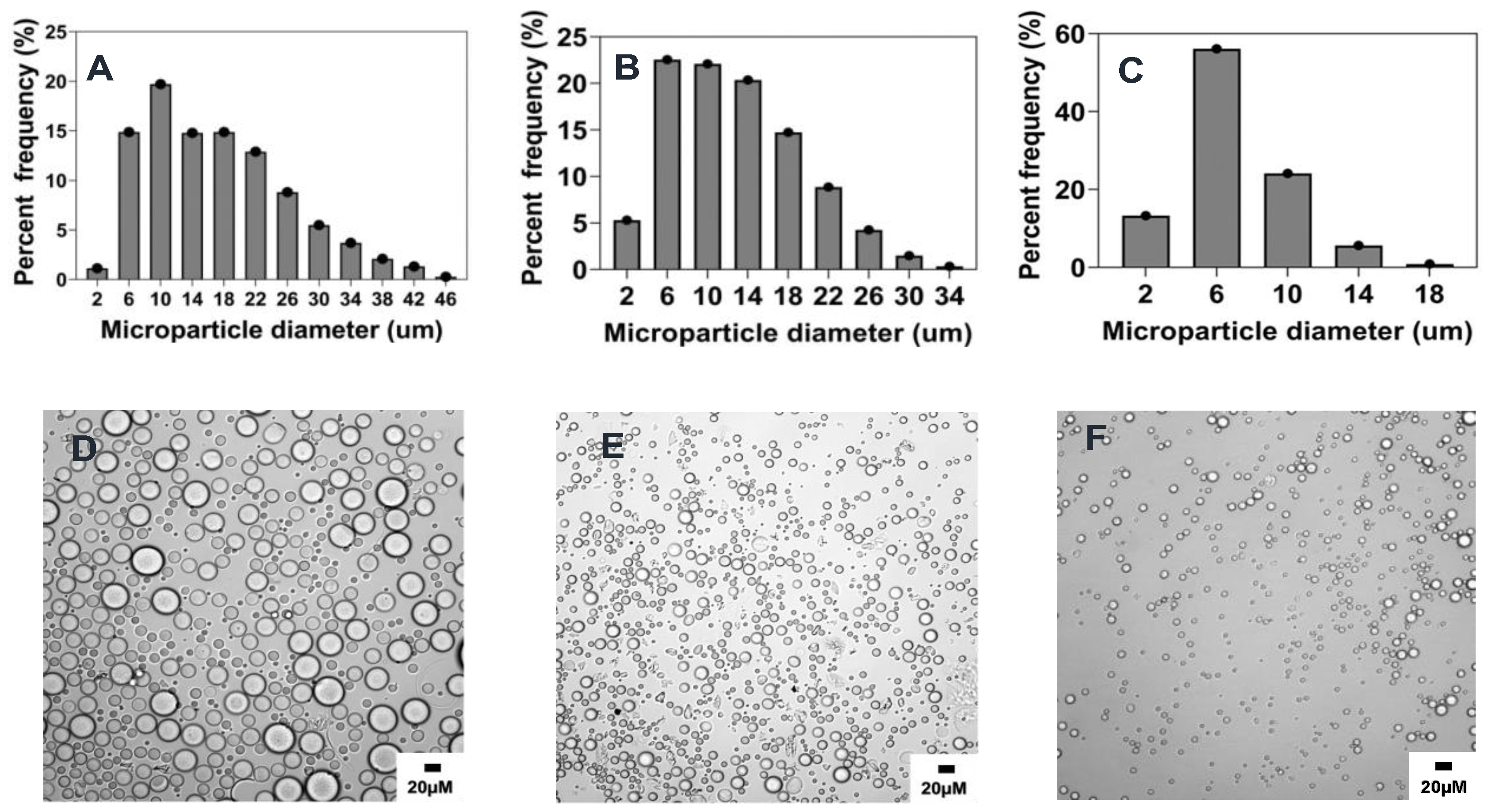
2.4.4. Effect of Polymer Concentration
2.4.5. Effect of Organic Solvent Choice
2.4.6. Effect of Organic to Aqueous Phase Volume Ratio
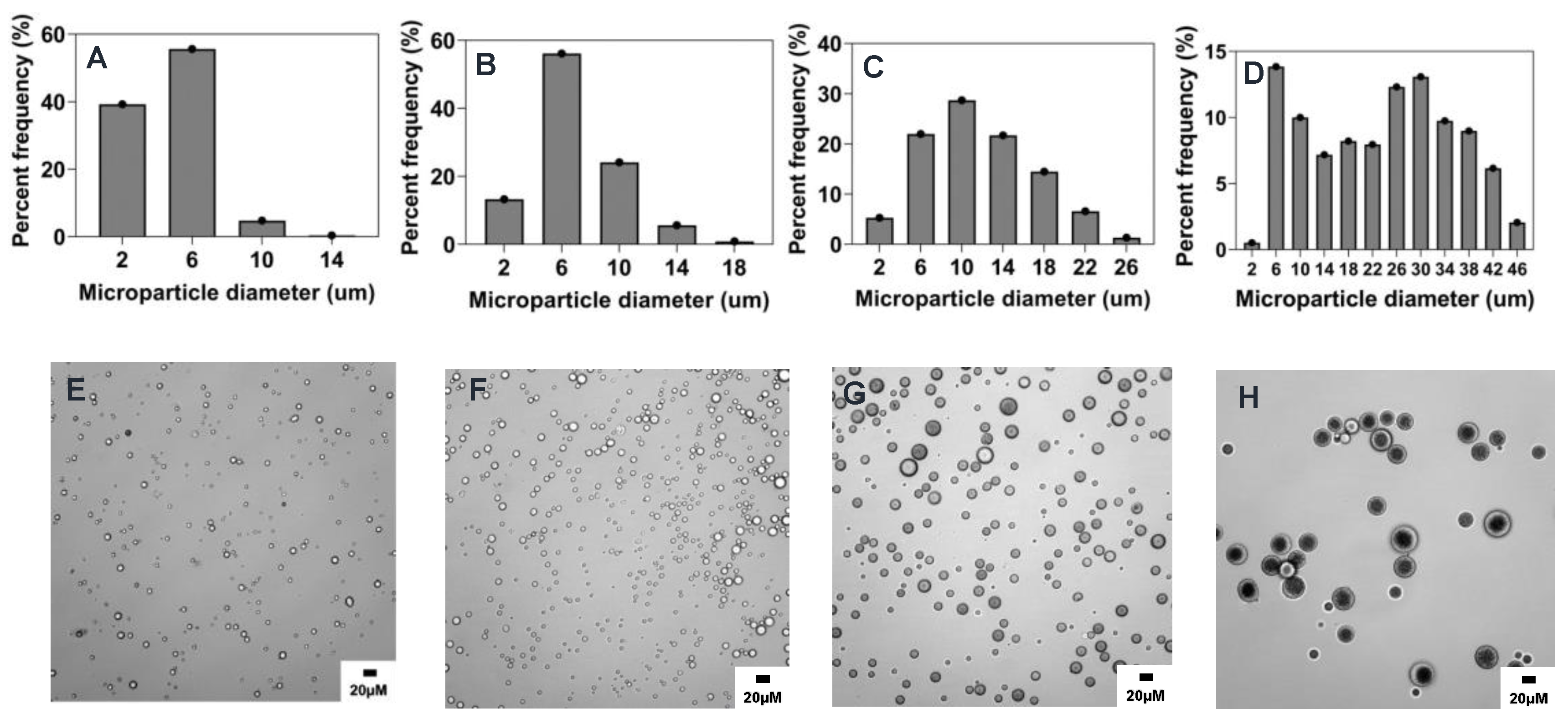
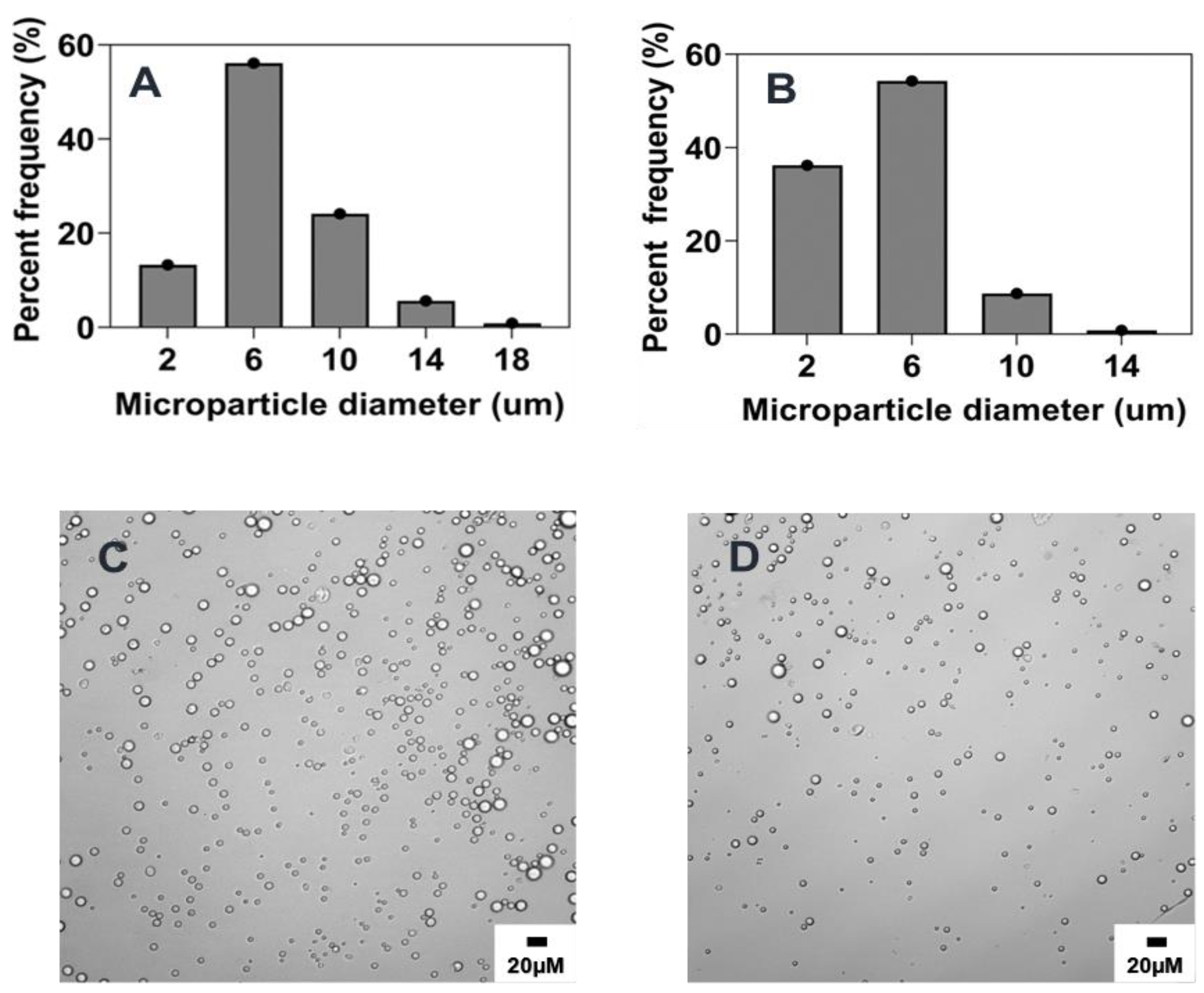
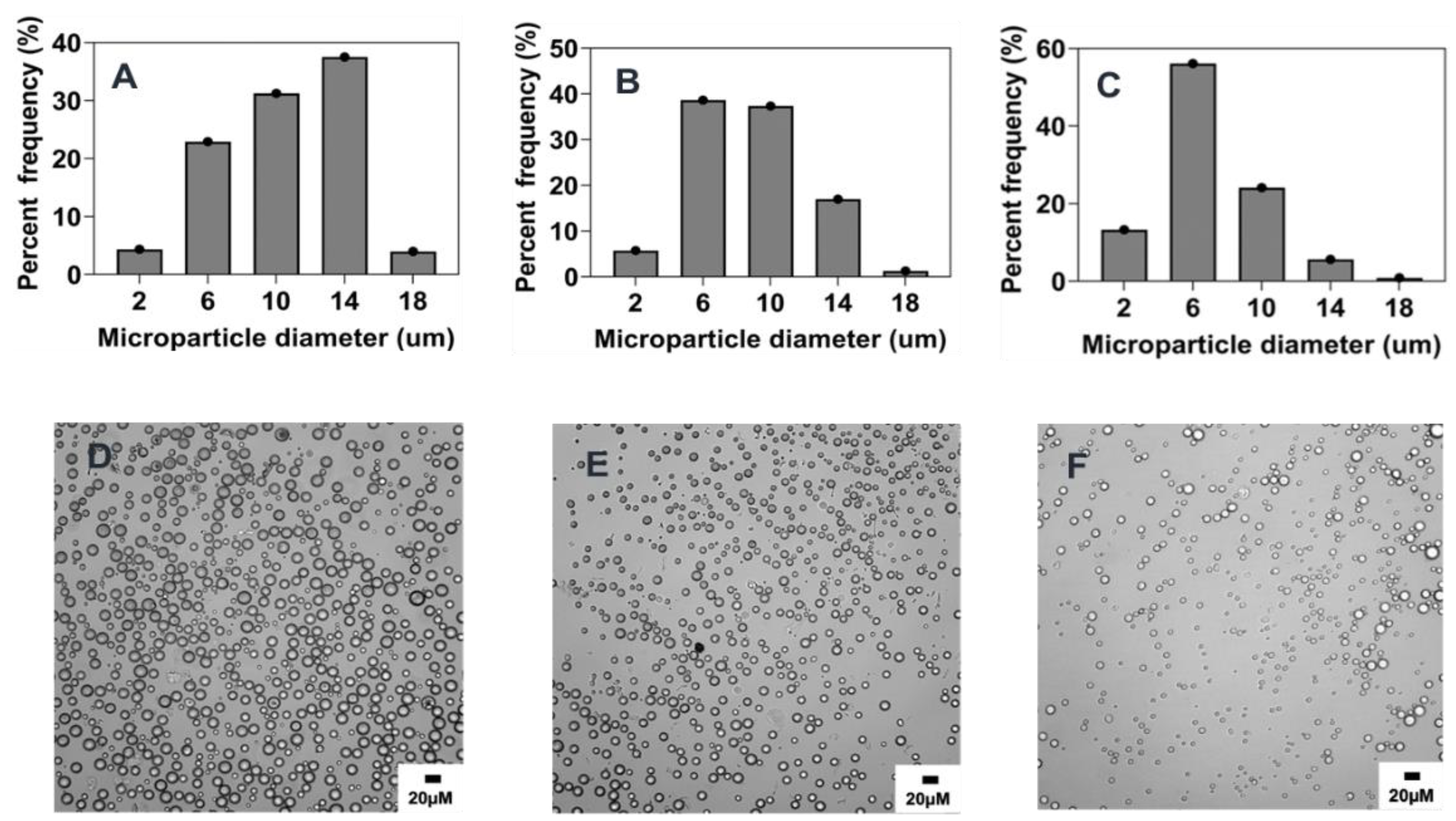
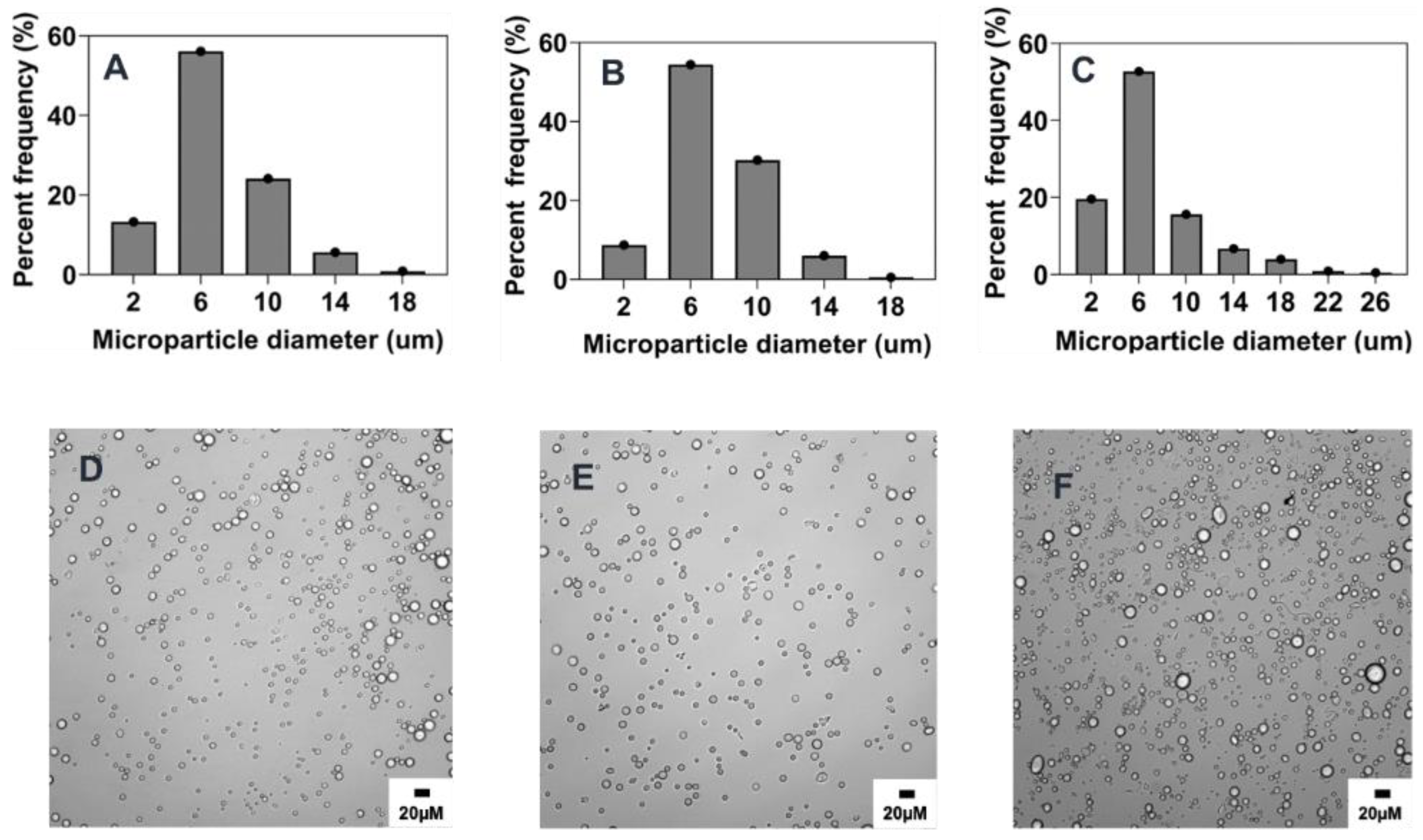
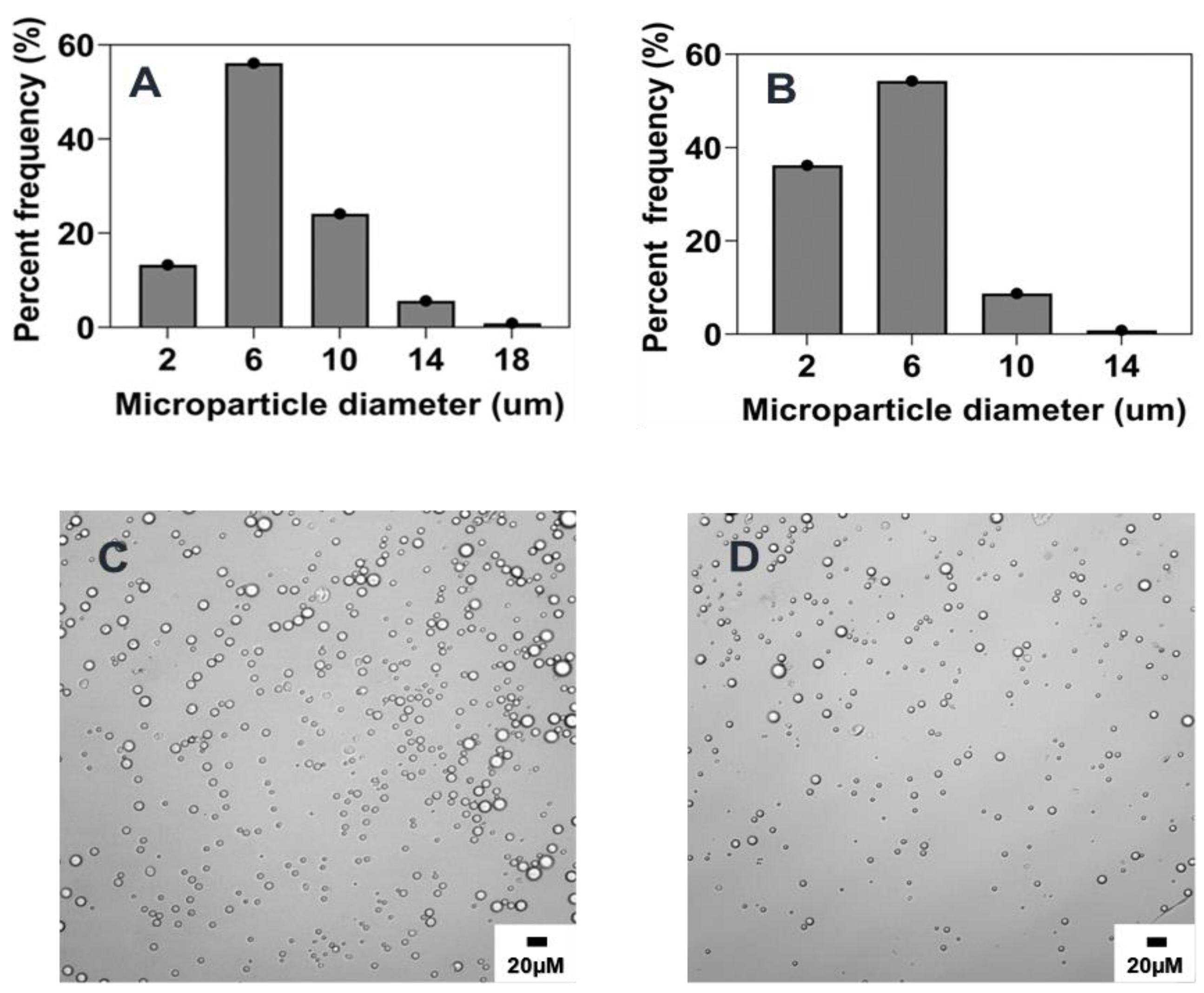
2.5. Scalability and Tunability of Co-Flow Phase Separation Method
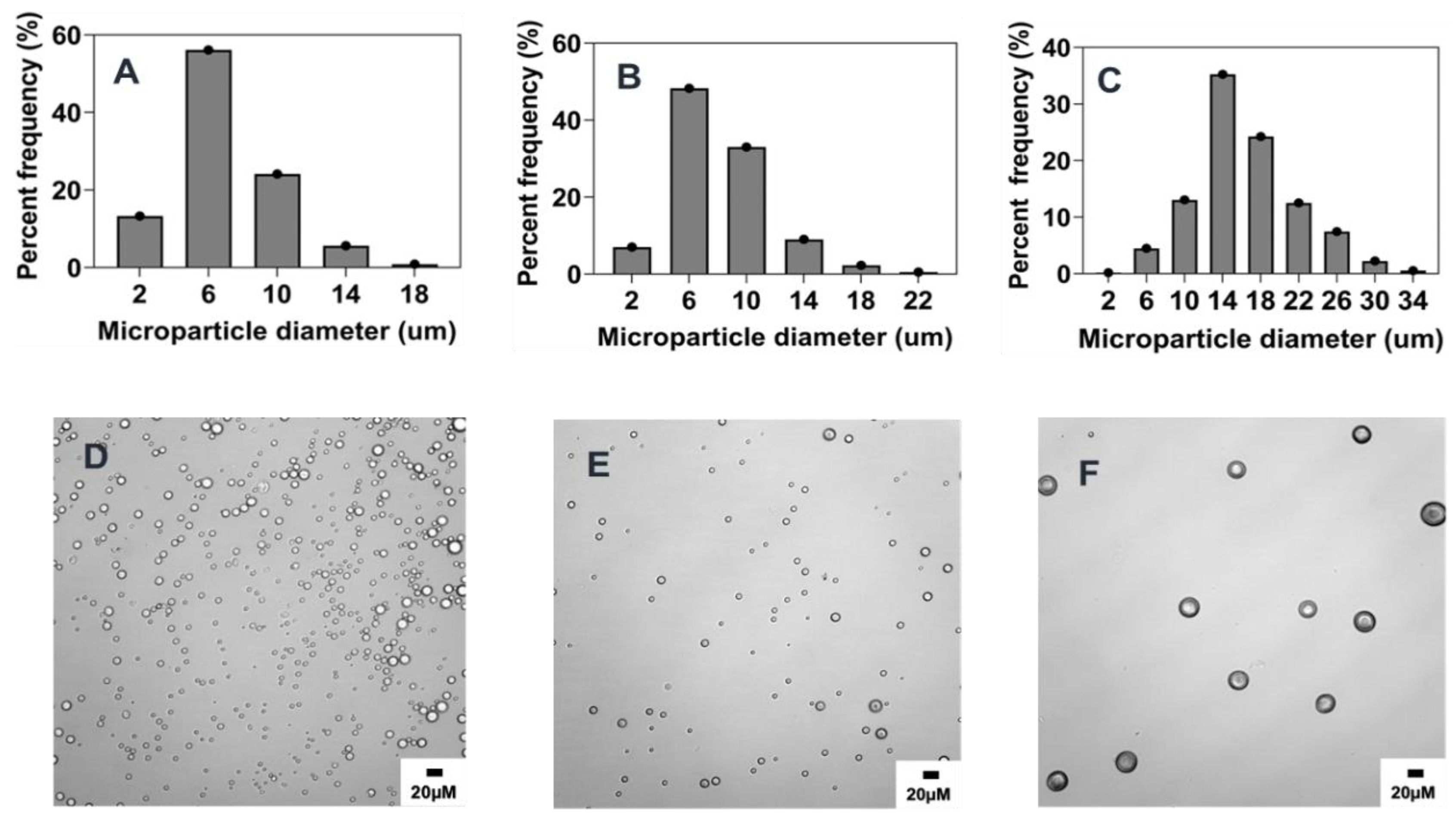
2.5.1. Combined Effect of Increasing Phase Volume Ratio, Surfactant Concentration and Stir Rate
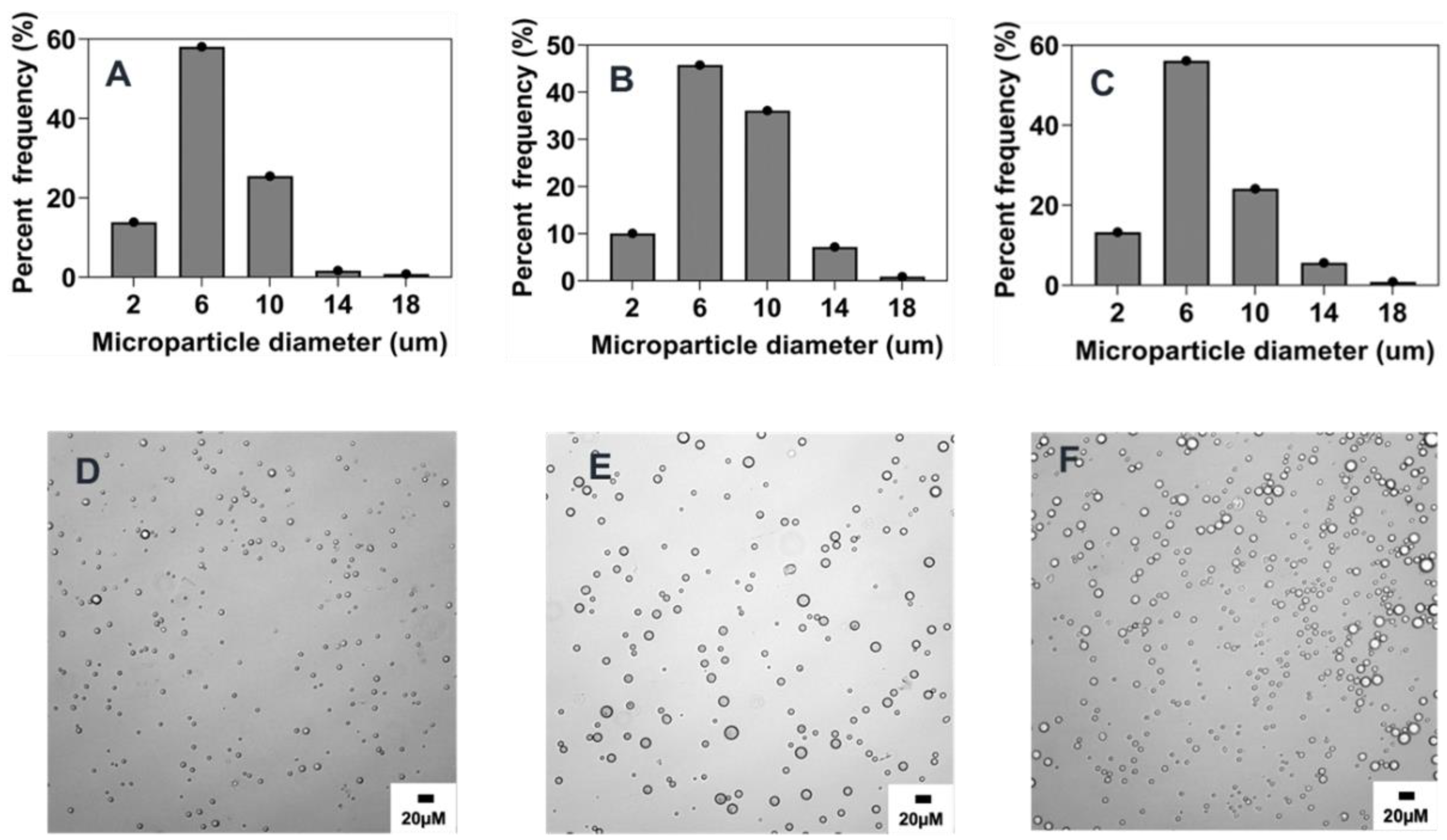
2.5.2. Combined Effect of Increasing Polymer Concentration, Surfactant Concentration, and Stir Rate
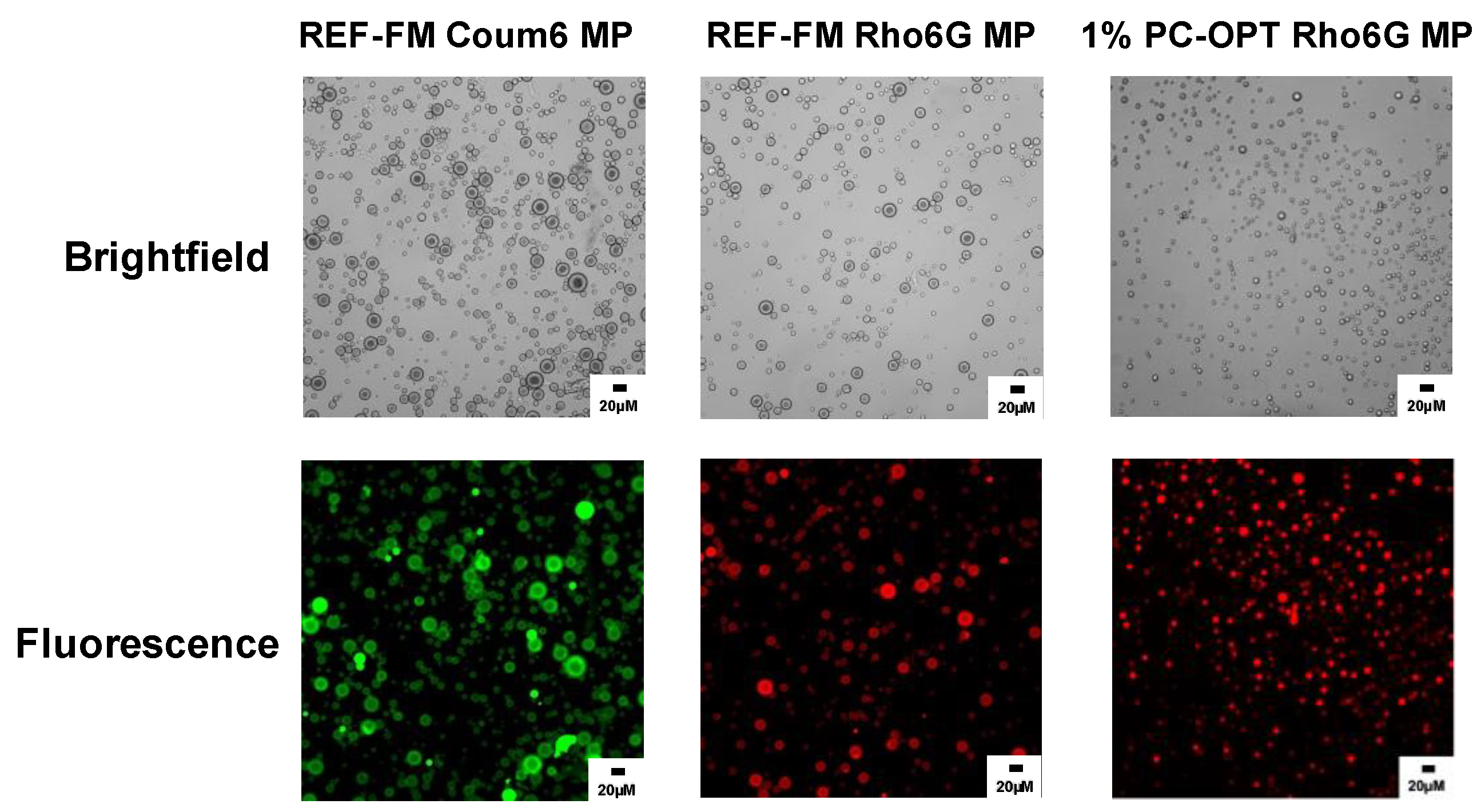
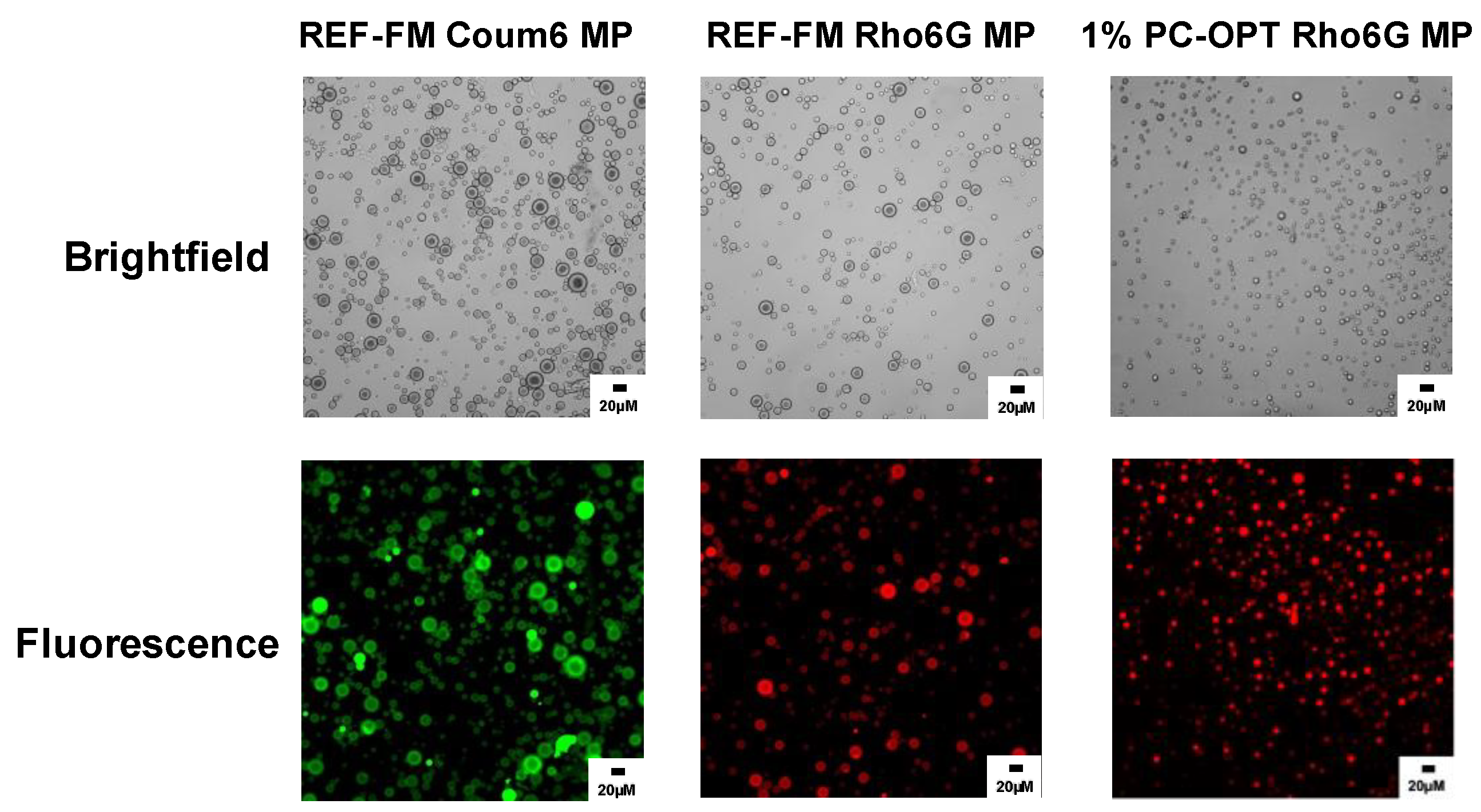
2.6. Encapsulation of Model Dyes in PEG-PLGA Microparticles
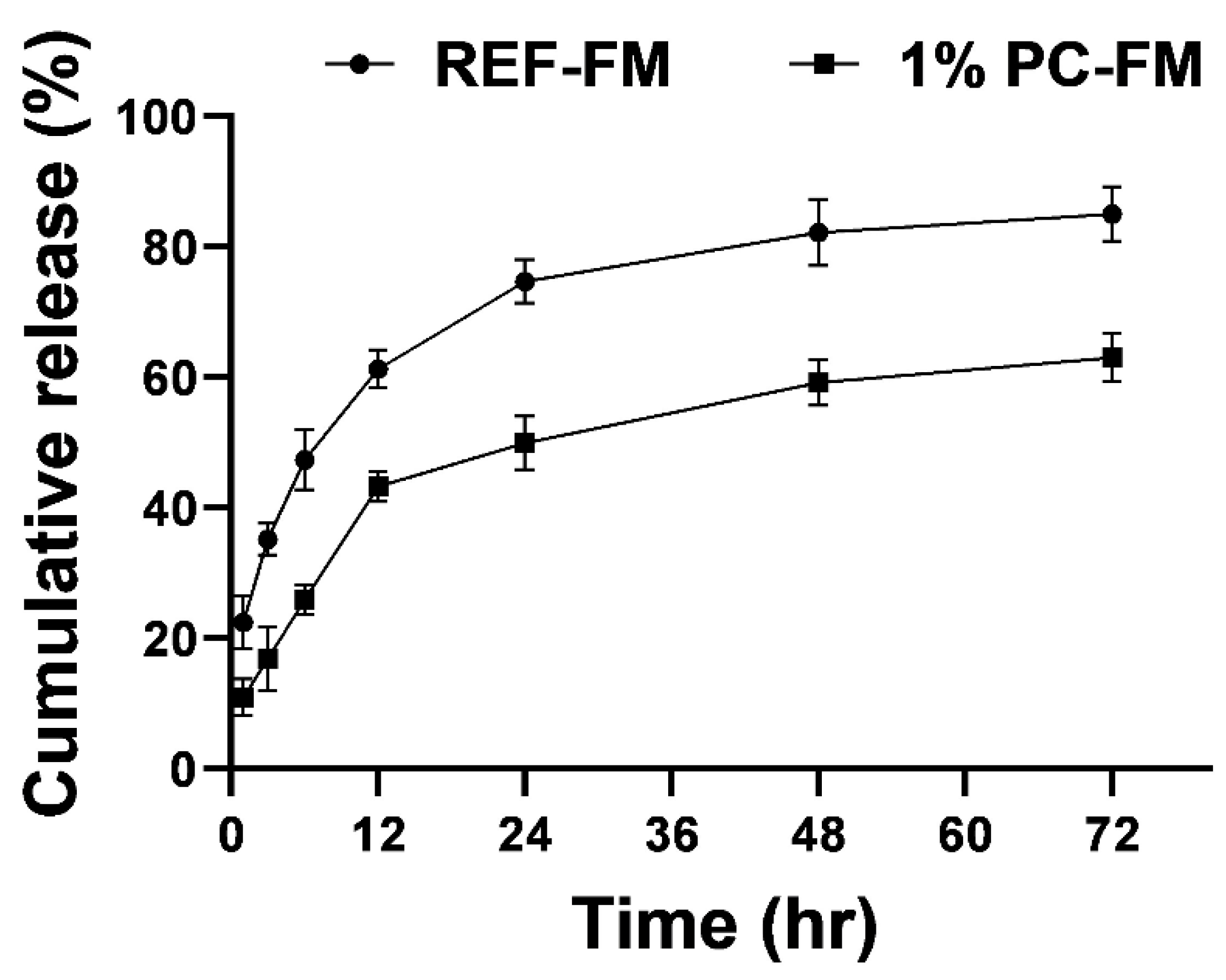
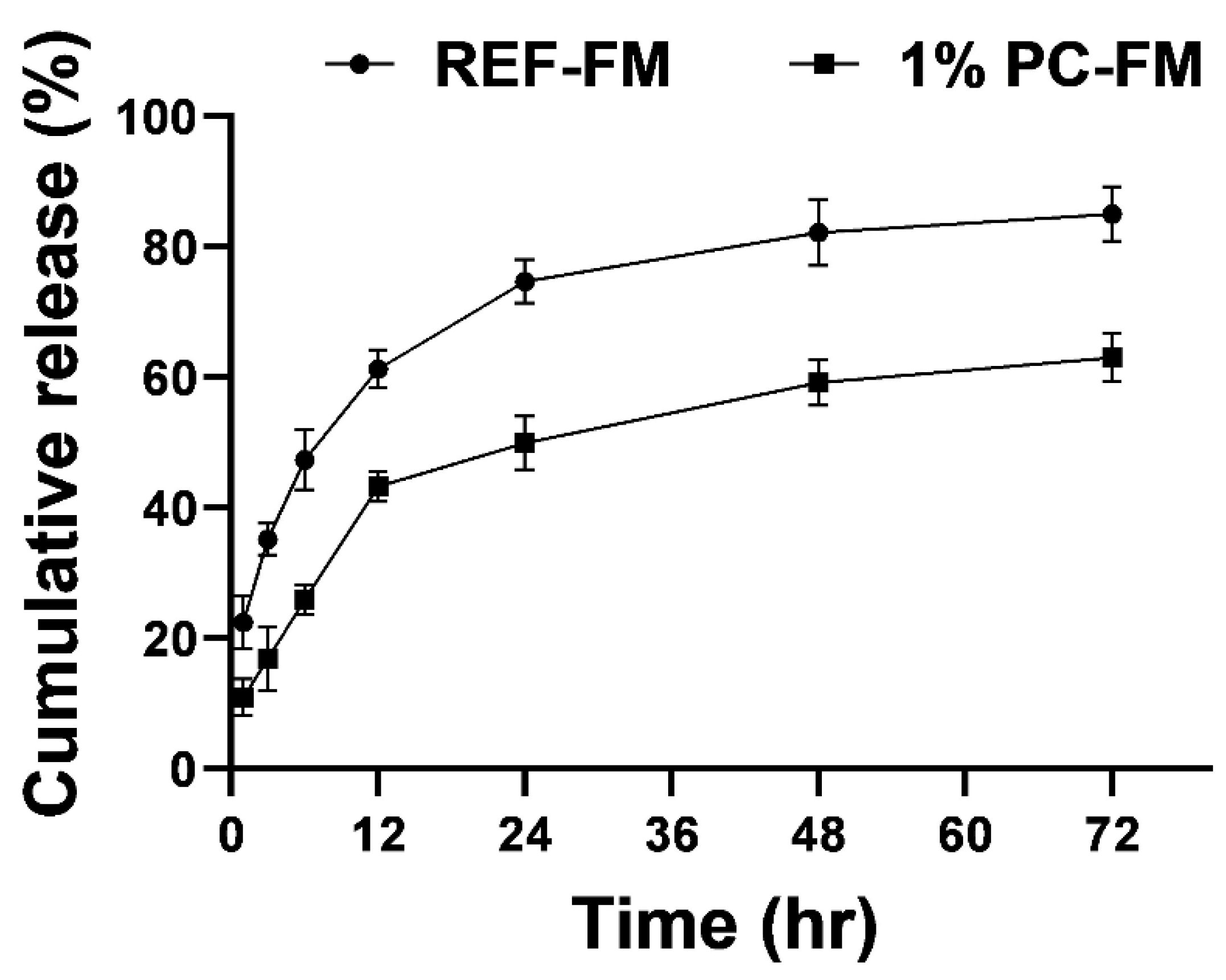
2.7. In-Vitro Release Studies
3. Materials and Method
3.1. Materials
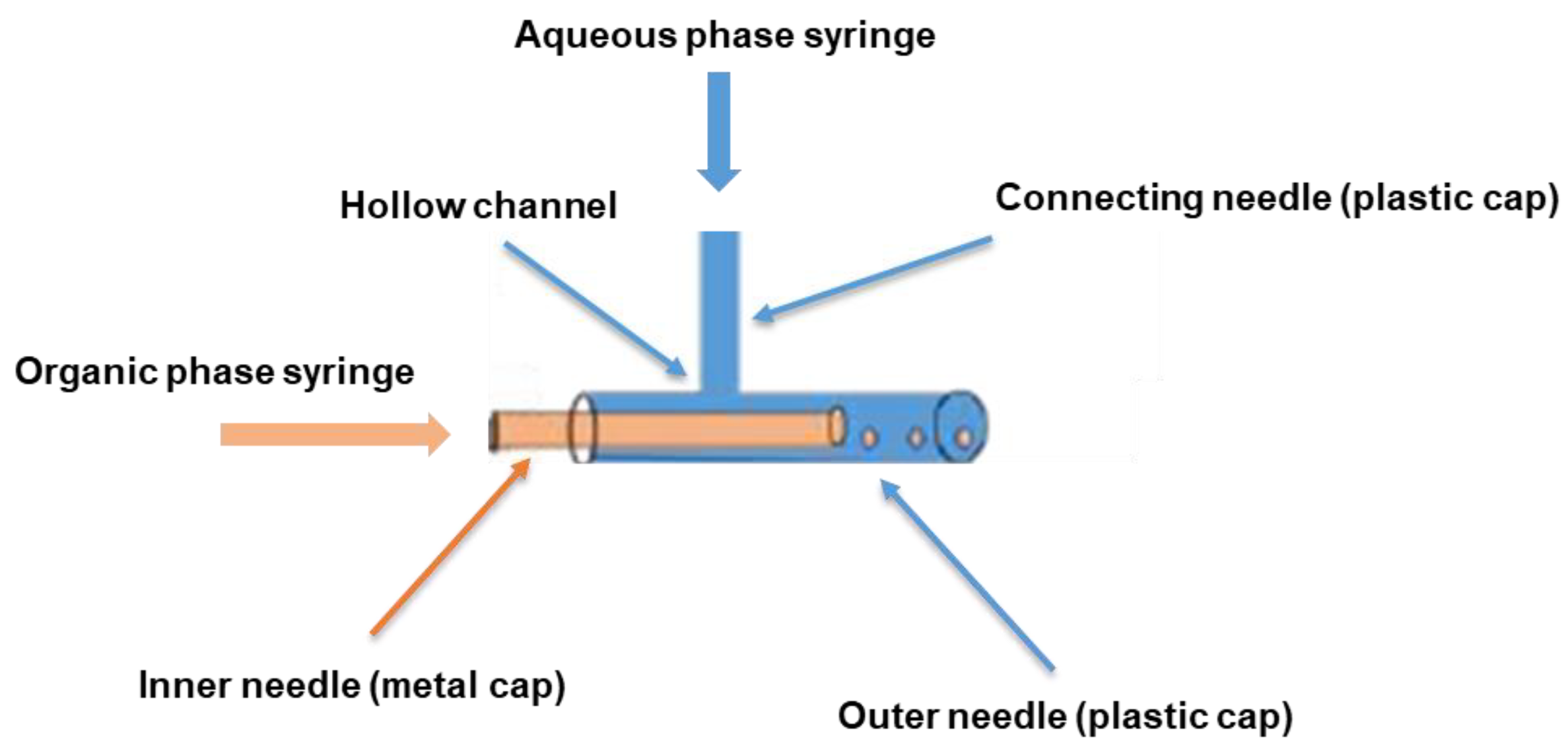
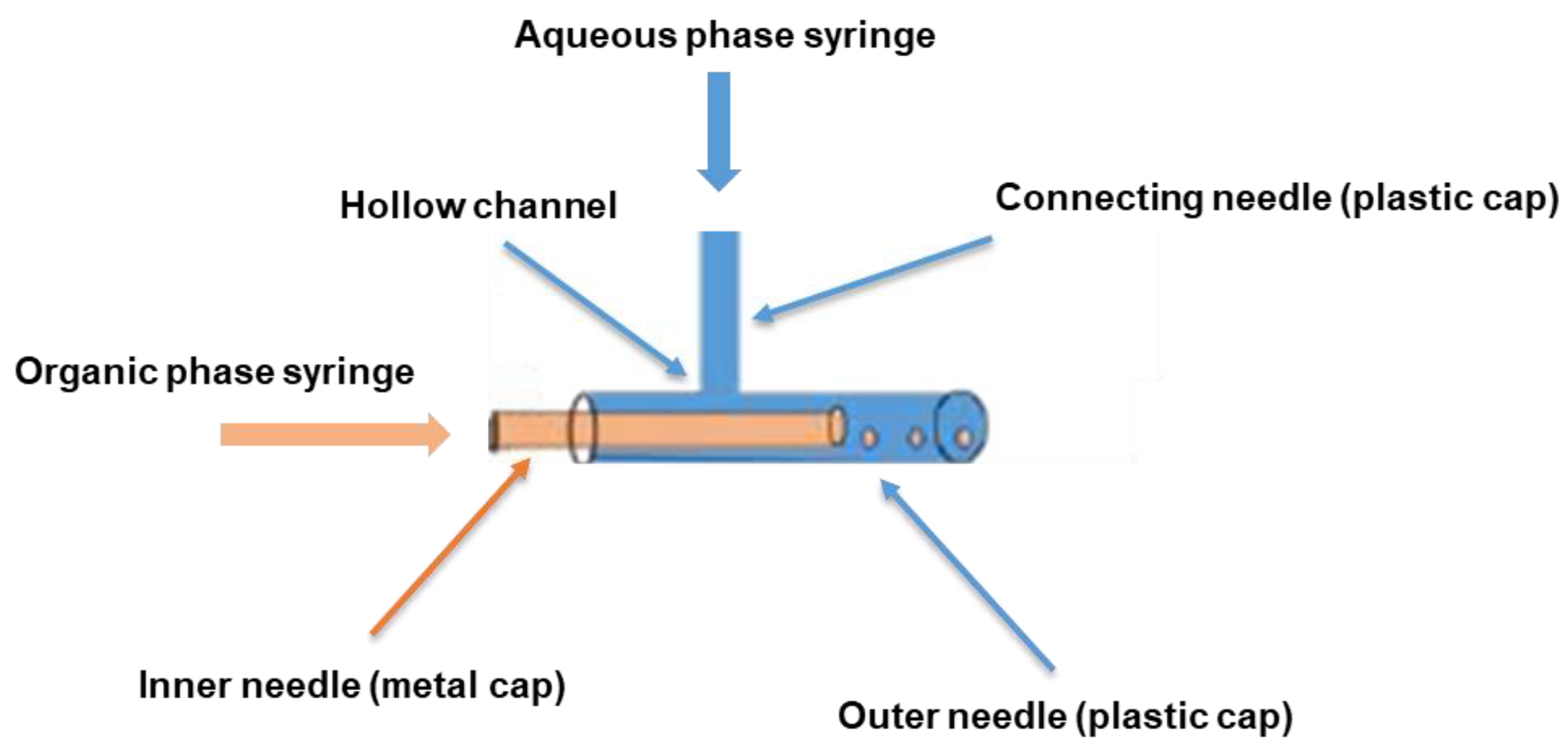
3.2. Assembly and Assessment of the Coaxial Needle Construct
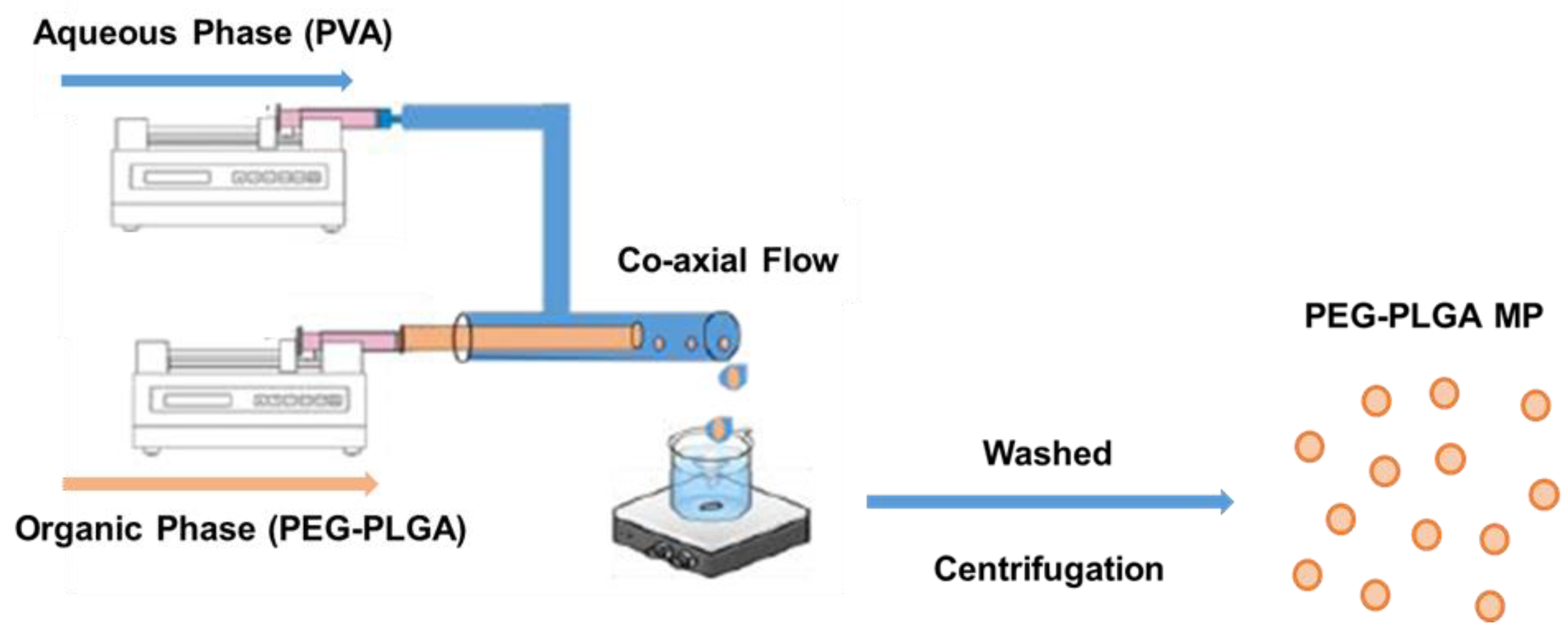
3.3. Preparation of PEG-PLGA Microparticles
3.4. Preparation of Dye Loaded PEG-PLGA Microparticles
3.5. Characterization of PEG-PLGA Microparticles
3.6. In Vitro Release of Rhodamine 6G from Dye-Loaded Microparticles
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blasi, P. Poly(lactic acid)/poly(lactic-co-glycolic acid)-based microparticles: An overview. J. Pharm. Investig. 2019, 49, 337–346. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Prabhuraj, R.S.; Bomb, K.; Srivastava, R.; Bandyopadhyaya, R. Selection of superior targeting ligands using PEGylated PLGA nanoparticles for delivery of curcumin in the treatment of triple-negative breast cancer cells. J. Drug Deliv. Sci. Technol. 2020, 57, 101722. [Google Scholar] [CrossRef]
- Jusu, S.M.; Obayemi, J.D.; Salifu, A.A.; Nwazojie, C.C.; Uzonwanne, V.; Odusanya, O.S.; Soboyejo, W.O. Drug-encapsulated blend of PLGA-PEG microspheres: In vitro and in vivo study of the effects of localized/targeted drug delivery on the treatment of triple-negative breast cancer. Sci. Rep. 2020, 10, 14188. [Google Scholar] [CrossRef]
- Sharma, S.; Parmar, A.; Kori, S.; Sandhir, R. PLGA-based nanoparticles: A new paradigm in biomedical applications. TrAC Trends Anal. Chem. 2016, 80, 30–40. [Google Scholar] [CrossRef]
- Ding, D.; Zhu, Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 1041–1060. [Google Scholar] [CrossRef]
- Lagreca, E.; Onesto, V.; Di Natale, C.; La Manna, S.; Netti, P.A.; Vecchione, R. Recent advances in the formulation of PLGA microparticles for controlled drug delivery. Prog. Biomater. 2020, 9, 153–174. [Google Scholar] [CrossRef]
- Busatto, C.; Pesoa, J.; Helbling, I.; Luna, J.; Estenoz, D. Effect of particle size, polydispersity and polymer degradation on progesterone release from PLGA microparticles: Experimental and mathematical modeling. Int. J. Pharm. 2018, 536, 360–369. [Google Scholar] [CrossRef]
- Bao, T.-Q.; Hiep, N.-T.; Kim, Y.-H.; Yang, H.-M.; Lee, B.-T. Fabrication and characterization of porous poly(lactic-co-glycolic acid) (PLGA) microspheres for use as a drug delivery system. J. Mater. Sci. 2011, 46, 2510–2517. [Google Scholar] [CrossRef]
- Wan, F.; Yang, M. Design of PLGA-based depot delivery systems for biopharmaceuticals prepared by spray drying. Int. J. Pharm. 2016, 498, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Yonet-Tanyeri, N.; Amer, M.; Balmert, S.C.; Korkmaz, E.; Falo, L.D., Jr.; Little, S.R. Microfluidic Systems For Manufacturing of Microparticle-Based Drug-Delivery Systems: Design, Construction, and Operation. ACS Biomater. Sci. Eng. 2022, 8, 2864–2877. [Google Scholar] [CrossRef]
- Nicolete, R.; dos Santos, D.F.; Faccioli, L.H. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int. Immunopharmacol. 2011, 11, 1557–1563. [Google Scholar] [CrossRef]
- Tran, M.K.; Swed, A.; Boury, F. Preparation of polymeric particles in CO(2) medium using non-toxic solvents: Formulation and comparisons with a phase separation method. Eur. J. Pharm. Biopharm. 2012, 82, 498–507. [Google Scholar] [CrossRef]
- Patel, M.; Jha, A.; Patel, R. Potential application of PLGA microsphere for tissue engineering. J. Polym. Res. 2021, 28, 214. [Google Scholar] [CrossRef]
- Vysloužil, J.; Doležel, P.; Kejdušová, M.; Mašková, E.; Mašek, J.; Lukáč, R.; Košťál, V.; Vetchý, D.; Dvořáčková, K. Influence of different formulations and process parameters during the preparation of drug-loaded PLGA microspheres evaluated by multivariate data analysis. Acta Pharm. 2014, 64, 403–417. [Google Scholar] [CrossRef]
- Mark, D.; Haeberle, S.; Roth, G.; Von Stetten, F.; Zengerle, R. Microfluidic Lab-on-a-Chip Platforms: Requirements, Characteristics and Applications. Chem Soc Rev: Dordrecht, The Netherlands, 2010; pp. 305–376. [Google Scholar]
- Matsumoto, A.; Kitazawa, T.; Murata, J.; Horikiri, Y.; Yamahara, H. A novel preparation method for PLGA microspheres using non-halogenated solvents. J. Control. Release 2008, 129, 223–227. [Google Scholar] [CrossRef]
- Bile, J.; Bolzinger, M.A.; Vigne, C.; Boyron, O.; Valour, J.P.; Fessi, H.; Chevalier, Y. The parameters influencing the morphology of poly(varepsilon-caprolactone) microspheres and the resulting release of encapsulated drugs. Int. J. Pharm. 2015, 494, 152–166. [Google Scholar] [CrossRef]
- Ellis, A.; Jacquier, J.C. Manufacture of food grade κ-carrageenan microspheres. J. Food Eng. 2009, 94, 316–320. [Google Scholar] [CrossRef]
- Mahmoodi, N.; Ai, J.; Hassannejad, Z.; Ebrahimi-Barough, S.; Hasanzadeh, E.; Hadi, A.; Nekounam, H.; Rahimi-Movaghar, V. Are reported methods for synthesizing nanoparticles and microparticles by magnetic stirrer reproducible? J. Comput. Appl. Mech. 2020, 51, 498–500. [Google Scholar] [CrossRef]
- Nosrati, Z.; Li, N.; Michaud, F.; Ranamukhaarachchi, S.; Karagiozov, S.; Soulez, G.; Martel, S.; Saatchi, K.; Häfeli, U.O. Development of a Coflowing Device for the Size-Controlled Preparation of Magnetic-Polymeric Microspheres as Embolization Agents in Magnetic Resonance Navigation Technology. ACS Biomater. Sci. Eng. 2018, 4, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Noviendri, D.; Jaswir, I.; Taher, M.; Mohamed, F.; Salleh, H.M.; Noorbatcha, I.A.; Octavianti, F.; Lestari, W.; Hendri, R.; Ahmad, H.; et al. Fabrication of Fucoxanthin-Loaded Microsphere(F-LM) By Two Steps Double-Emulsion Solvent Evaporation Method and Characterization of Fucoxanthin before and after Microencapsulation. J. Oleo Sci. 2016, 65, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Moura, M.J.; Martins, S.P.; Duarte, B.P. Production of chitosan microparticles cross-linked with genipin–Identification of factors influencing size and shape properties. Biochem. Eng. J. 2015, 104, 82–90. [Google Scholar] [CrossRef]
- Valot, P.; Baba, M.; Nedelec, J.M.; Sintes-Zydowicz, N. Effects of process parameters on the properties of biocompatible ibuprofen-loaded microcapsules. Int. J. Pharm. 2009, 369, 53–63. [Google Scholar] [CrossRef]
- Mohanty, S.; Panigrahi, A.K. Multiparticulate drug delivery system for colon targeting. Int. J. Pharm. Pharm. Sci. 2015, 7, 433–436. [Google Scholar]
- Mensah, R.A.; Kirton, S.B.; Cook, M.T.; Styliari, I.D.; Hutter, V.; Chau, D.Y.S. Optimising poly(lactic-co-glycolic acid) microparticle fabrication using a Taguchi orthogonal array design-of-experiment approach. PLoS ONE 2019, 14, e0222858. [Google Scholar] [CrossRef]
- Safari, H.; Adili, R.; Holinstat, M.; Eniola-Adefeso, O. Modified two-step emulsion solvent evaporation technique for fabricating biodegradable rod-shaped particles in the submicron size range. J. Colloid Interface Sci. 2018, 518, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Gupta, I.; Jonnalagadda, U.S.; Kwan, J.J. Complementary Effects of Porosigen and Stabilizer on the Structure of Hollow Porous Poly(lactic-co-glycolic acid) Microparticles. ACS Appl. Polym. Mater. 2020, 2, 3696–3703. [Google Scholar] [CrossRef]
- Otte, A.; Sharifi, F.; Park, K. Interfacial tension effects on the properties of PLGA microparticles. Colloids Surf. B Biointerfaces 2020, 196, 111300. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Qi, F.; Miao, C.; Yue, H.; Gong, F.; Wu, J.; Ma, G.; Su, Z. Direct and controllable preparation of uniform PLGA particles with various shapes and surface morphologies. Colloids Surf. A Physicochem. Eng. Asp. 2016, 500, 177–185. [Google Scholar] [CrossRef]
- Utada, A.S.; Fernandez-Nieves, A.; Stone, H.A.; Weitz, D.A. Dripping to jetting transitions in coflowing liquid streams. Phys. Rev. Lett. 2007, 99, 094502. [Google Scholar] [CrossRef] [PubMed]
- Operti, M.C.; Dolen, Y.; Keulen, J.; van Dinther, E.A.W.; Figdor, C.G.; Tagit, O. Microfluidics-Assisted Size Tuning and Biological Evaluation of PLGA Particles. Pharmaceutics 2019, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Rapier, C.E.; Shea, K.J.; Lee, A.P. Investigating PLGA microparticle swelling behavior reveals an interplay of expansive intermolecular forces. Sci. Rep. 2021, 11, 14512. [Google Scholar] [CrossRef] [PubMed]
- Sheen, S.; Lee, J.; Woo, C.G. Application of coflow premixed flame for generating aggregate silica particles and its limitation. AIP Adv. 2022, 12, 095007. [Google Scholar] [CrossRef]
- Vasiliauskas, R.; Liu, D.; Cito, S.; Zhang, H.; Shahbazi, M.A.; Sikanen, T.; Mazutis, L.; Santos, H.A. Simple Microfluidic Approach to Fabricate Monodisperse Hollow Microparticles for Multidrug Delivery. ACS Appl. Mater. Interfaces 2015, 7, 14822–14832. [Google Scholar] [CrossRef]
- Zhu, K.J.; Jiang, H.L.; Du, X.Y.; Wang, J.; Xu, W.X.; Liu, S.F. Preparation and characterization of hCG-loaded polylactide or poly(lactide-co-glycolide) microspheres using a modified water-in-oil-in-water (w/o/w) emulsion solvent evaporation technique. J. Microencapsul. 2001, 18, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, C. Tuning the Size of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles Fabricated by Nanoprecipitation. Biotechnol. J. 2018, 13, 1700203. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Esnaashari, S.S.; Mujokoro, B.; Dorkoosh, F.; Khosravani, M.; Adabi, M. Investigation of Effective Parameters on Size of Paclitaxel Loaded PLGA Nanoparticles. Adv. Pharm. Bull. 2018, 8, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef]
- Jafari, S.M.; Assadpoor, E.; He, Y.; Bhandari, B. Re-coalescence of emulsion droplets during high-energy emulsification. Food Hydrocoll. 2008, 22, 1191–1202. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, B.; Sun, R.; Liu, W.; Zhu, Q.; Zhang, X.; Wang, R.; Chen, C. PLGA-based biodegradable microspheres in drug delivery: Recent advances in research and application. Drug Deliv. 2021, 28, 1397–1418. [Google Scholar] [CrossRef] [PubMed]
- Heiskanen, H.; Denifl, P.; Hurme, M.; Pitkänen, P.; Oksman, M. Effect of Physical Properties and Emulsification Conditions on the Microsphere Size Prepared Using a Solvent Extraction Process. J. Dispers. Sci. Technol. 2012, 33, 234–244. [Google Scholar] [CrossRef]
- Abulateefeh, S.R.; Spain, S.G.; Thurecht, K.J.; Aylott, J.W.; Chan, W.C.; Garnett, M.C.; Alexander, C. Enhanced uptake of nanoparticle drug carriers via a thermoresponsive shell enhances cytotoxicity in a cancer cell line. Biomater. Sci. 2013, 1, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Lee, Y.B.; Oh, I.J. Cellular uptake of poly(dl-lactide-co-glycolide) nanoparticles: Effects of drugs and surface characteristics of nanoparticles. J. Pharm. Investig. 2015, 45, 659–667. [Google Scholar] [CrossRef]
- O’Donnell, P.B.; McGinity, J.W. Preparation of microspheres by the solvent evaporation technique. Adv. Drug Deliv. Rev. 1997, 28, 25–42. [Google Scholar] [CrossRef]
- Krishnamachari, Y.; Madan, P.; Lin, S. Development of pH- and time-dependent oral microparticles to optimize budesonide delivery to ileum and colon. Int. J. Pharm. 2007, 338, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Shi, Y.; Li, L.; Xu, J.; Schaper, A.; Kissel, T. Effects of process and formulation parameters on characteristics and internal morphology of poly(d,l-lactide-co-glycolide) microspheres formed by the solvent evaporation method. Eur. J. Pharm. Biopharm. 2008, 68, 214–223. [Google Scholar] [CrossRef]
- Sharma, N.; Madan, P.; Lin, S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study. Asian J. Pharm. Sci. 2016, 11, 404–416. [Google Scholar] [CrossRef]
- Rodrigues de Azevedo, C.; von Stosch, M.; Costa, M.S.; Ramos, A.M.; Cardoso, M.M.; Danhier, F.; Préat, V.; Oliveira, R. Modeling of the burst release from PLGA micro- and nanoparticles as function of physicochemical parameters and formulation characteristics. Int. J. Pharm. 2017, 532, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.D. Analysis of initial burst in PLGA microparticles. Expert Opin. Drug Deliv. 2008, 5, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, P.; Borhan, A.; Abidian, M.R. Microencapsulation of Chemotherapeutics into Monodisperse and Tunable Biodegradable Polymers via Electrified Liquid Jets: Control of Size, Shape, and Drug Release. Adv. Mater. 2013, 25, 4555–4560. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.-Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Palazzo, A.; Hennink, W.E.; Kok, R.J. Effect of Particle Size on Drug Loading and Release Kinetics of Gefitinib-Loaded PLGA Microspheres. Mol. Pharm. 2017, 14, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.J.; Kim, J.Y.; Choo, J.; Lee, E.K.; Han, C.S.; Beebe, D.J.; Seong, G.H.; Lee, S.H. Continuous Fabrication of Biocatalyst Immobilized Microparticles Using Photopolymerization and Immiscible Liquids in Microfluidic Systems. Langmuir 2005, 21, 3738–3741. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Im, J.S.; Jin, X.; Kim, T.M.; Choi, J.W. In Vitro and In Vivo Evaluation of Drug-Encapsulated Lignin Nanoparticles for Release Control. ACS Sustain. Chem. Eng. 2022, 10, 5792–5802. [Google Scholar] [CrossRef]
- Ogunsipe, A. Solvent Effects on the Spectral Properties of Rhodamine 6G: Estimation of Ground and Excited State Dipole Moments. J. Solut. Chem. 2018, 47, 203–219. [Google Scholar] [CrossRef]
- Su, Y.; Liu, J.; Tan, S.; Liu, W.; Wang, R.; Chen, C. PLGA sustained-release microspheres loaded with an insoluble small-molecule drug: Microfluidic-based preparation, optimization, characterization, and evaluation in vitro and in vivo. Drug Deliv. 2022, 29, 1437–1446. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
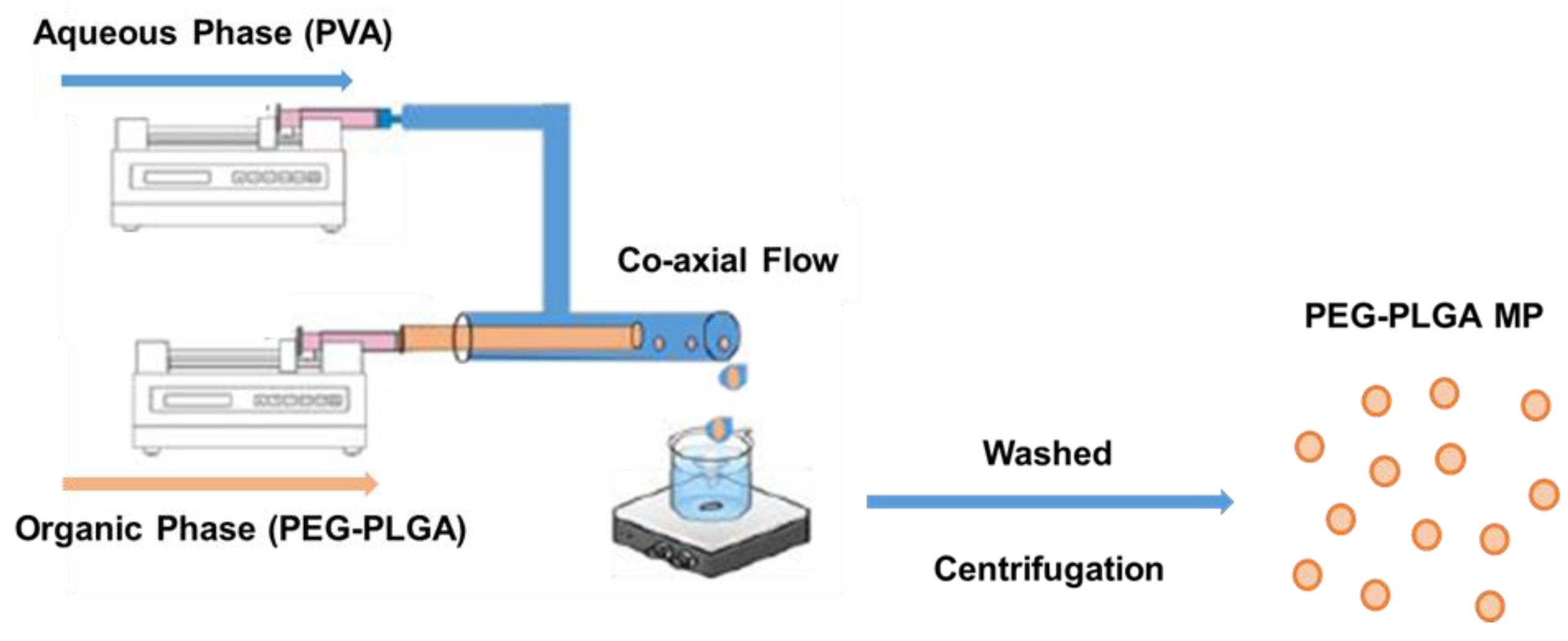{kind=link}
| Parameter | Formulation | Mean Size (µm ± sd) | %CV |
|---|---|---|---|
| Reference | Reference | 6.8 ± 0.1 | 1.5 |
| Stir Rate | 300 rpm | 23 ± 1.9 | 8.3 |
| 600 rpm | 11.6 ± 0.6 | 5.2 | |
| 1500 rpm | 4.9 ± 0.5 | 10.2 | |
| Polymer (PEG-PLGA) concentration | 1% w/v | 12.5 ± 0.6 | 4.8 |
| 5% w/v | 17.2 ± 1 | 5.8 | |
| Surfactant (PVA) concentration | 0.5% w/v | 16.7 ± 0.7 | 4.2 |
| 2.5% w/v | 7.5 ± 0.3 | 4 | |
| Organic/aqueous phase volume Ratio | 0.01% v/v | 8.8 ± 0.4 | 4.5 |
| 0.02% v/v | 10.6 ± 0.6 | 5.7 | |
| Organic/aqueous phase flow rate Ratio | 0.01 | 7.6 ± 0.4 | 5.3 |
| 0.025 | 6.6 ± 0.3 | 4.5 | |
| Organic solvent | 25% w/v ACN/DCM | 4.7 ± 0.8 | 17 |
| Formulation | Formulation Parameters | Mean Size (µm ± sd) | CV |
|---|---|---|---|
| REF-FM | 0.005 PVR, 5% PVA, 1000 rpm | 6.7 ± 3.1 | 0.45 |
| 0.01 PVR-FM | 0.01 PVR, 5% PVA, 1500 rpm | 5.7 ± 1.5 | 0.27 |
| 0.02 PVR-FM | 0.02 PVR, 7% PVA, 1500 rpm | 6.1 ± 2.2 | 0.36 |
| Formulation | Formulation Parameters | Size (μm) | CV |
|---|---|---|---|
| REF-FM | 0.1% w/v PC, 5% PVA,1000 rpm | 6.7 ± 3.1 | 0.45 |
| 1% PC-FM | 1% w/v PC, 5% PVA, 1500 rpm | 7.1 ± 2.7 | 0.38 |
| 5% PC-FM | 5% w/v PC, 7% PVA, 1500 rpm | 7.2 ± 4.1 | 0.57 |
| Formulation | Encapsulation Efficiency (%) | Drug Loading (%) |
|---|---|---|
| REF-FM with Rho | 16.73 ± 0.83 | 0.43 ± 0.08 |
| REF-FM with Coum | 36.33 ± 2.34 | 0.74 ± 0.09 |
| 1% PC-FM with Rho | 39.53 ± 5.13 | 0.21 ± 0.03 |
| 5% PC-FM with Rho | 58.2 ± 3.99 | 0.04 ± 0.004 |
| 0.01 PVR-FM | 20.55 ± 1.07 | 0.32 ± 0.04 |
| 0.02 PVR-FM | 23.3 ± 1.14 | 0.23 ± 0.02 |
| Formulation | Encapsulation Efficiency (%) | Drug Loading (%) |
|---|---|---|
| REF-FM with Rho | 8.97 ± 0.42 | 1.06 ± 0.08 |
| REF-FM with Coum | 24.05 ± 0.54 | 2.23 ± 0.33 |
| 1% PC-OPT FM with Rho | 34.6 ± 1.32 | 0.84 ± 0.02 |
| Parameter. | Condition |
|---|---|
| Stir rate/speed (rpm) | 300, 600, 1000, 1500 |
| Polymer concentration (% w/v) | 0.1, 1, 0.5, 2.5, 5 |
| Surfactant concentration (% w/v) | 0.5, 2.5, 5 |
| Organic/aqueous volume ratio (% v/v) | 0.005, 0.01, 0.02 |
| Organic/aqueous flow rate ratio (mL/h) | 0.005, 0.01, 0.025 |
| Organic solvent choice (% v/v) | 25% acetonitrile in DCM, DCM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagoe, P.N.K.; Velázquez, E.J.M.; Espiritusanto, Y.M.; Gilbert, A.; Orado, T.; Wang, Q.; Jain, E. Fabrication of PEG-PLGA Microparticles with Tunable Sizes for Controlled Drug Release Application. Molecules 2023, 28, 6679. https://doi.org/10.3390/molecules28186679
Sagoe PNK, Velázquez EJM, Espiritusanto YM, Gilbert A, Orado T, Wang Q, Jain E. Fabrication of PEG-PLGA Microparticles with Tunable Sizes for Controlled Drug Release Application. Molecules. 2023; 28(18):6679. https://doi.org/10.3390/molecules28186679
Chicago/Turabian StyleSagoe, Paul Nana Kwame, Eduardo José Machado Velázquez, Yohely Maria Espiritusanto, Amelia Gilbert, Thalma Orado, Qiu Wang, and Era Jain. 2023. "Fabrication of PEG-PLGA Microparticles with Tunable Sizes for Controlled Drug Release Application" Molecules 28, no. 18: 6679. https://doi.org/10.3390/molecules28186679
APA StyleSagoe, P. N. K., Velázquez, E. J. M., Espiritusanto, Y. M., Gilbert, A., Orado, T., Wang, Q., & Jain, E. (2023). Fabrication of PEG-PLGA Microparticles with Tunable Sizes for Controlled Drug Release Application. Molecules, 28(18), 6679. https://doi.org/10.3390/molecules28186679