
Development of Masitinib Derivatives with Enhanced Mpro Ligand Efficiency and Reduced Cytotoxicity

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
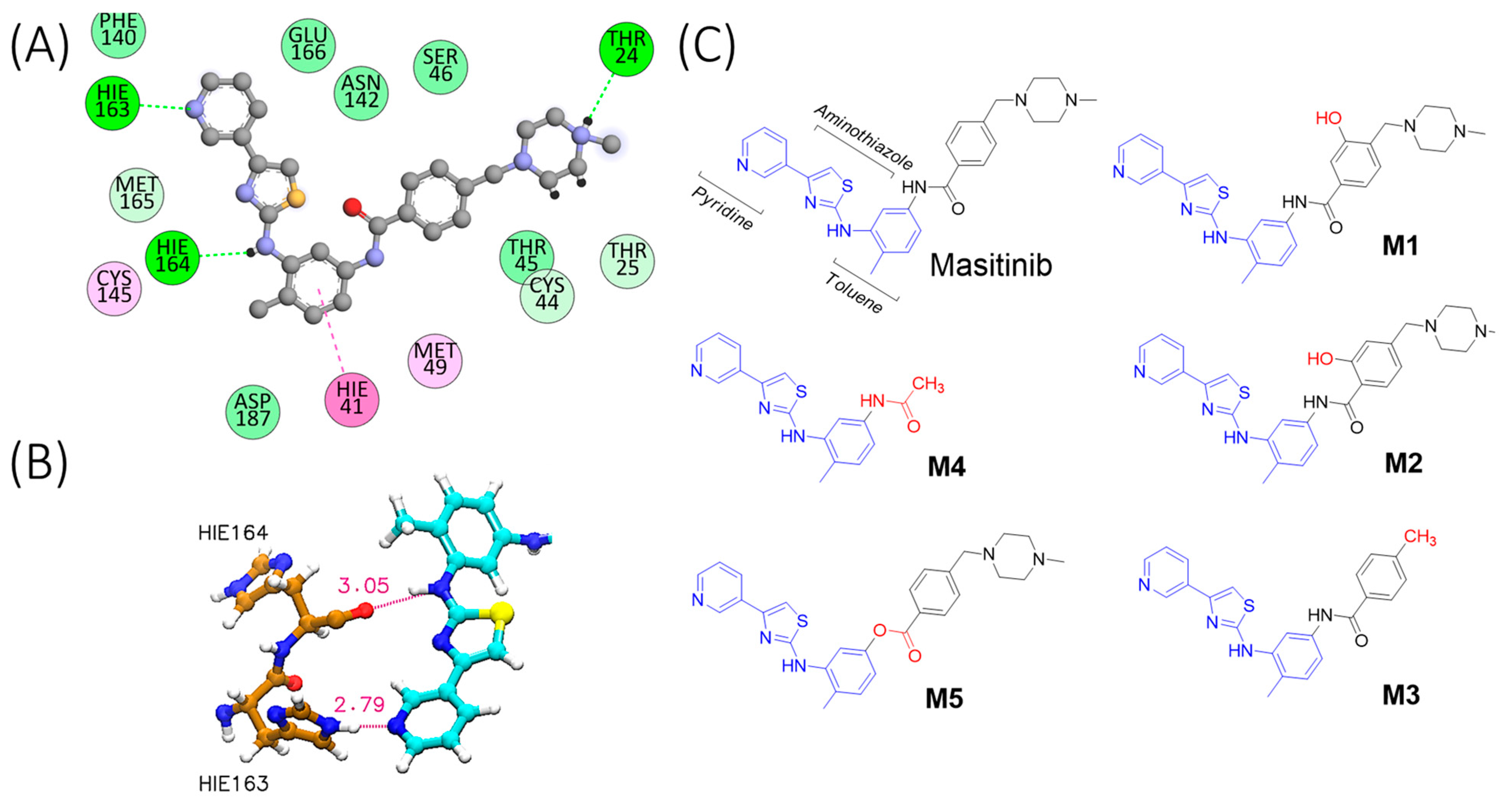
2.1. Molecular Simulation of Mpro-Masitinib Complex: Exploration of Ligand–Protein Key Interactions
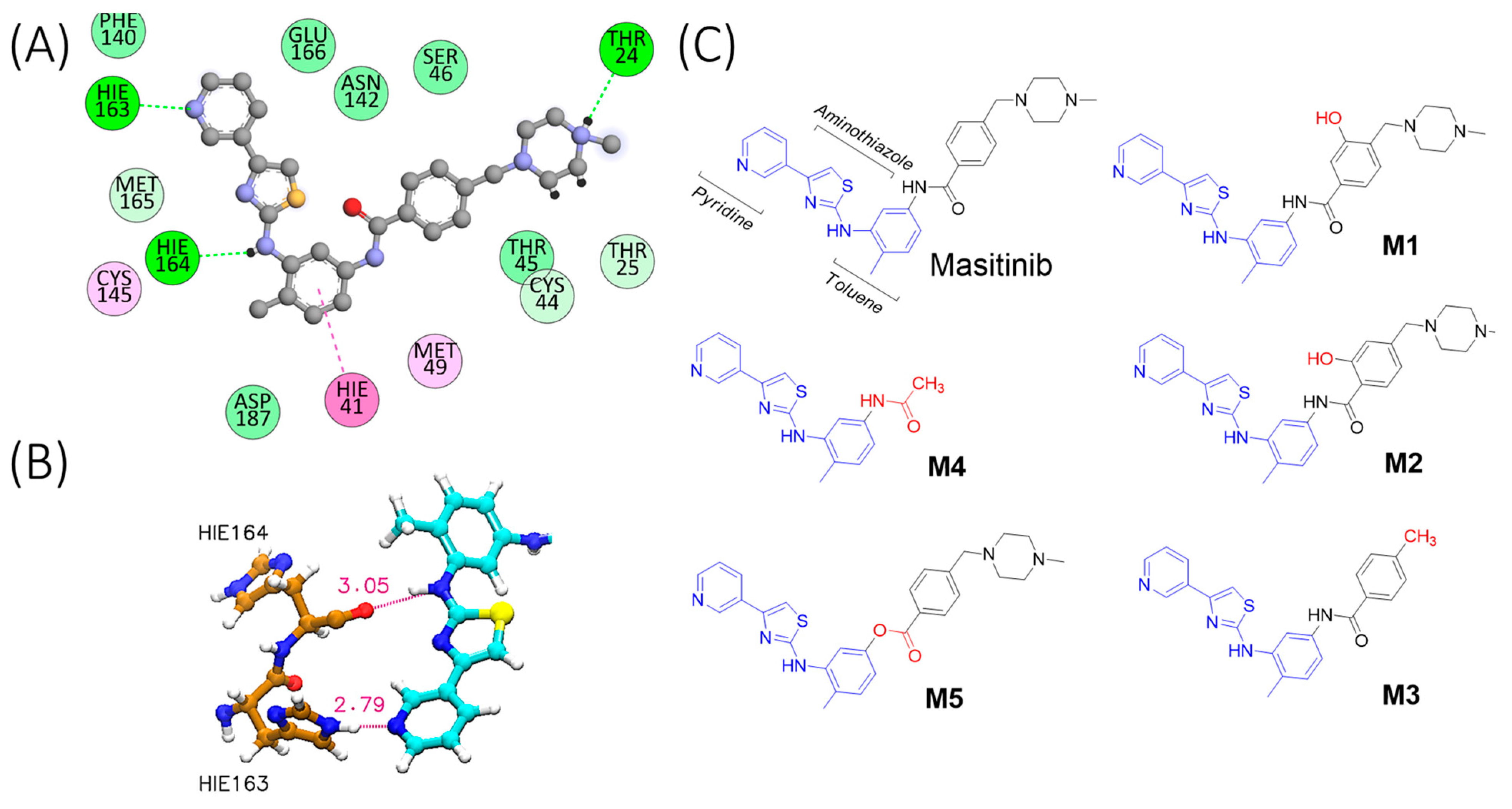
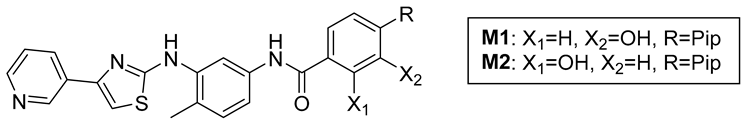
2.2. Compound Synthesis
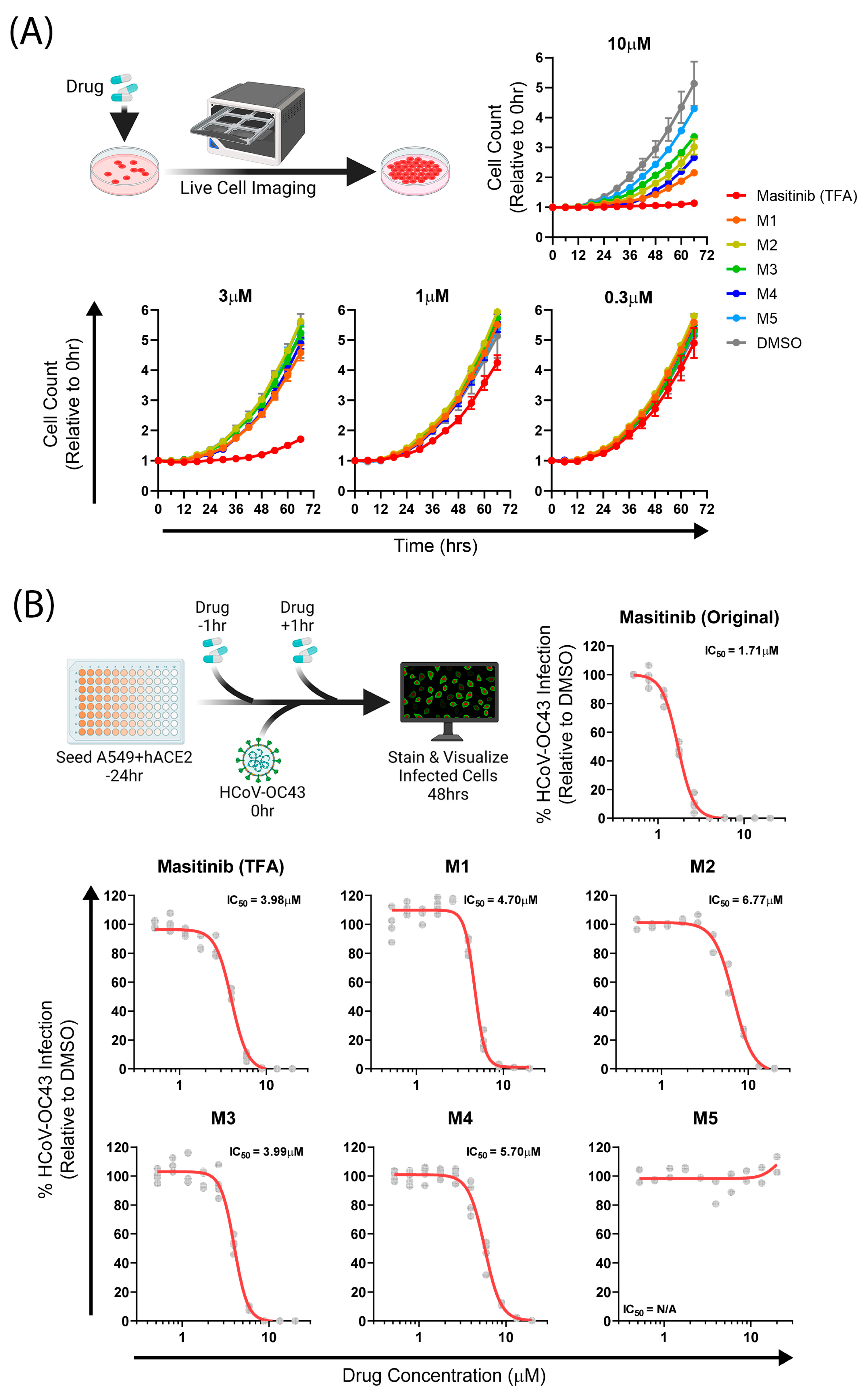
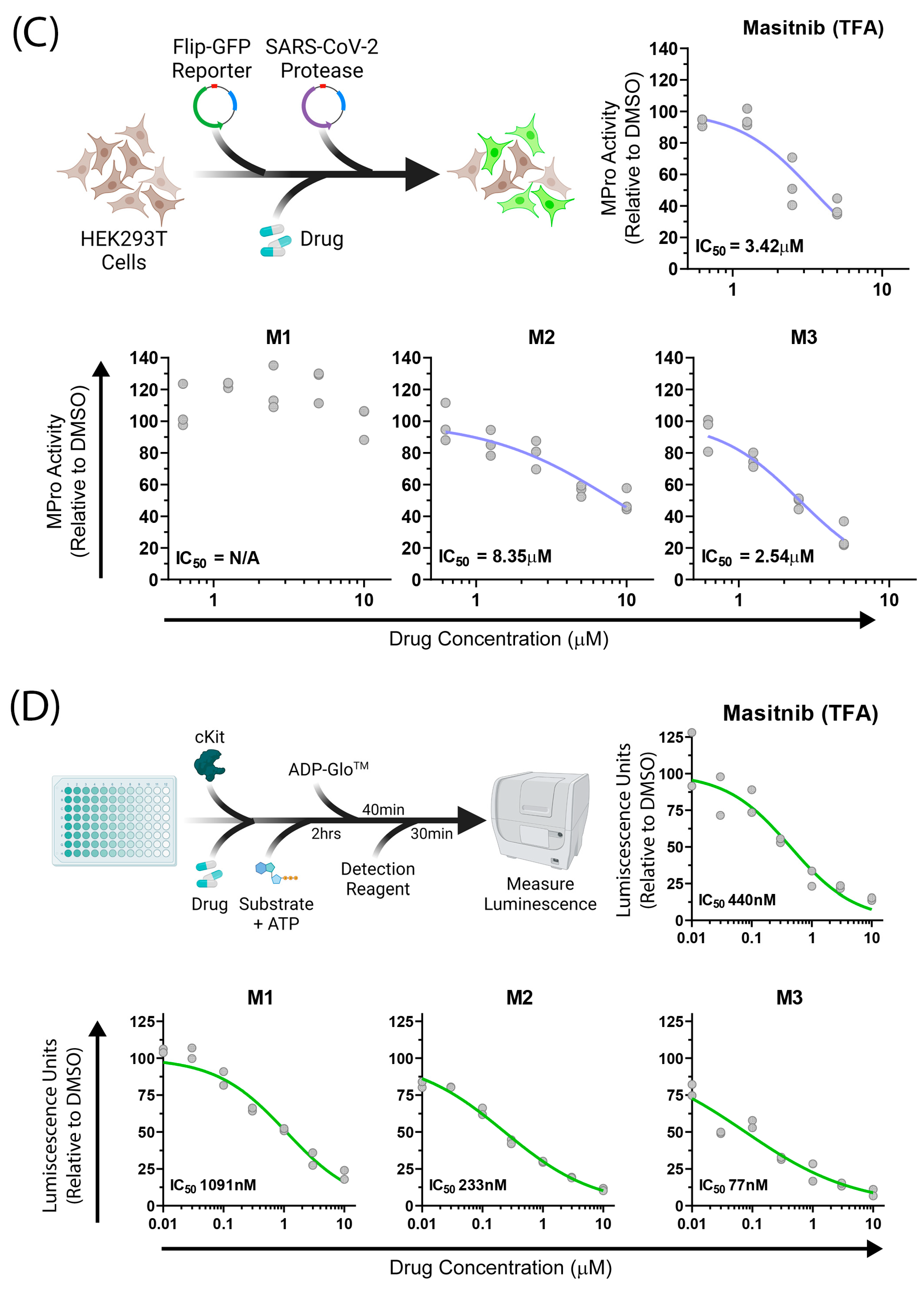
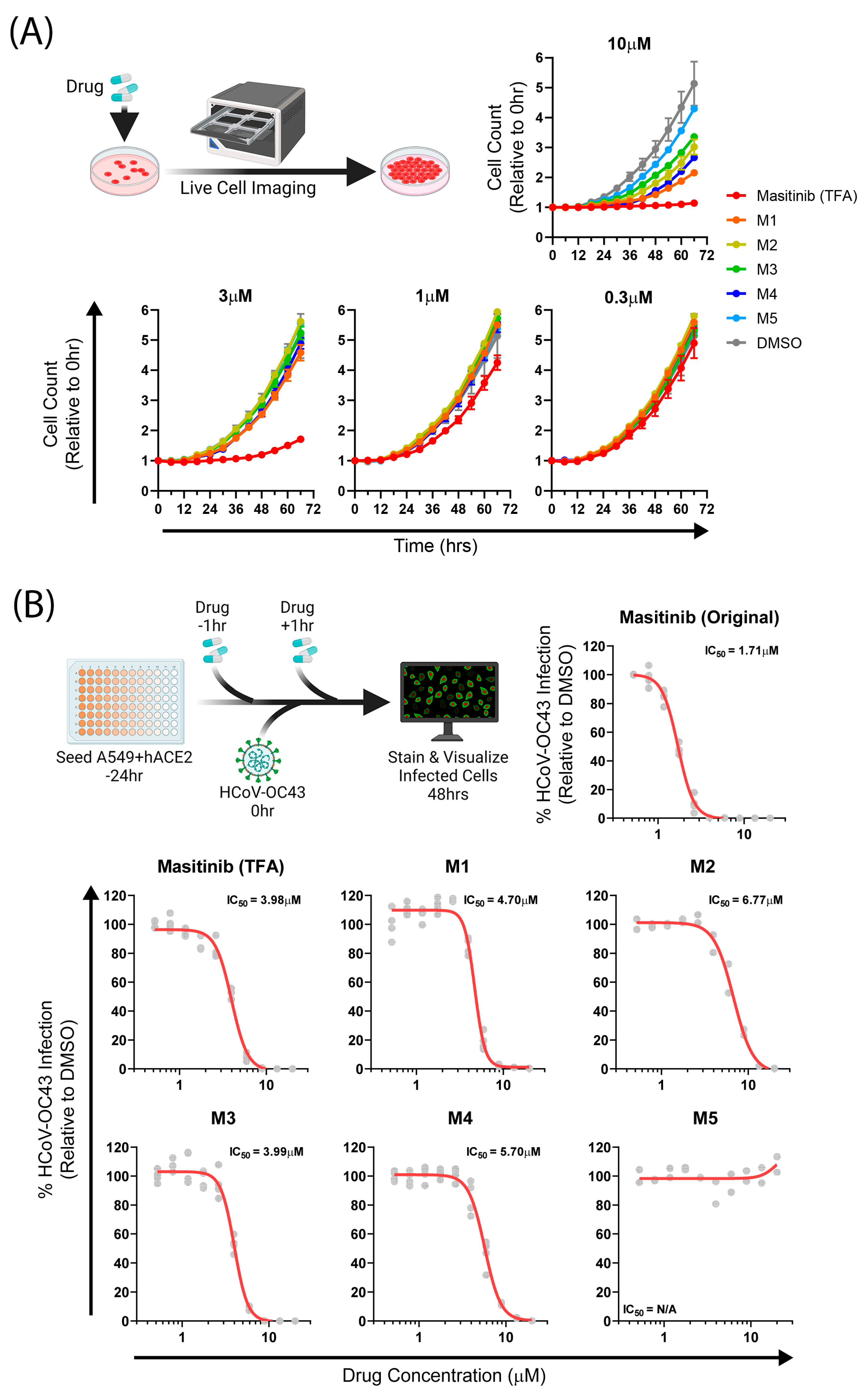
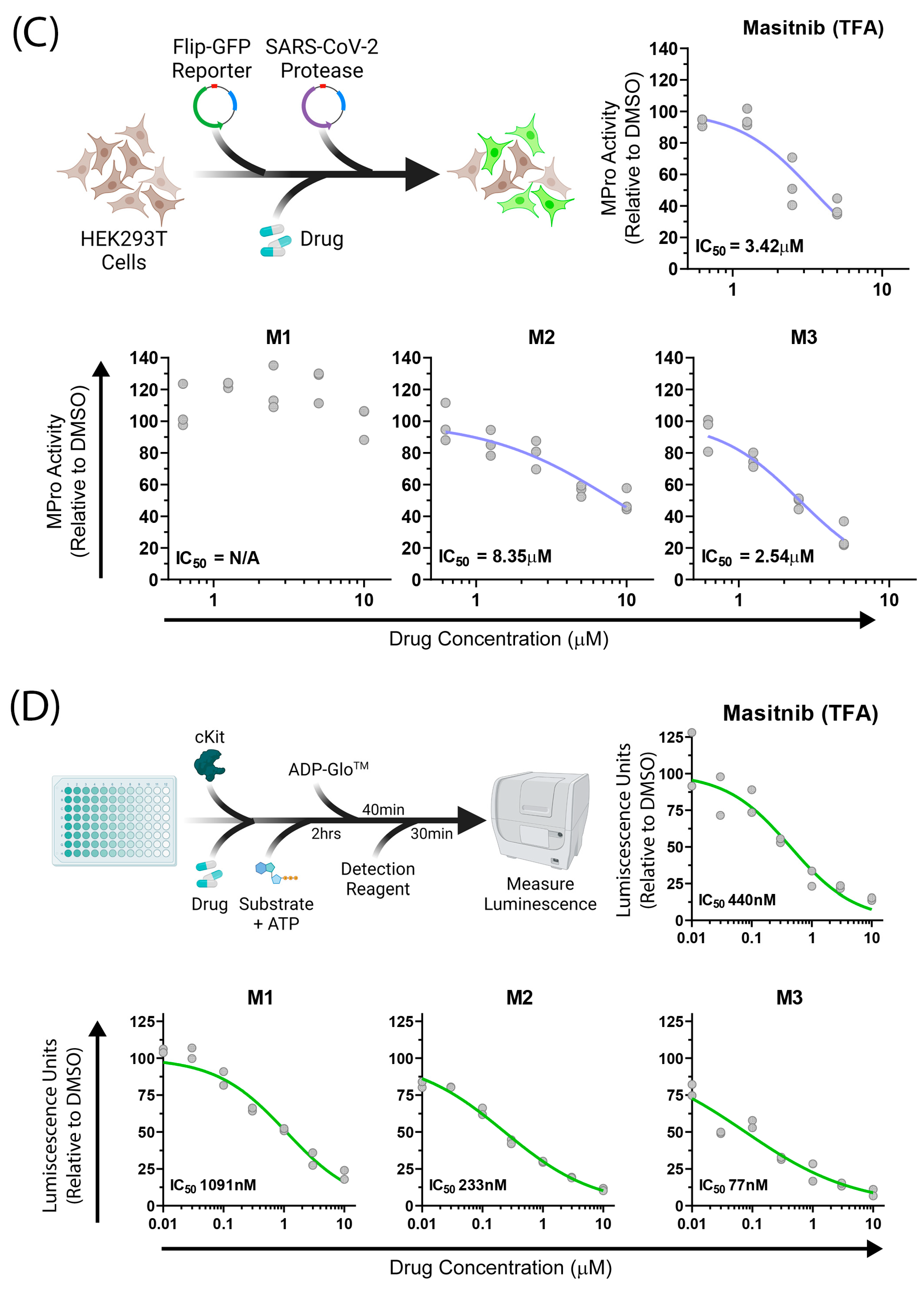
2.3. Biological Assays
3. Discussion and Conclusions
4. Material and Methods
4.1. Molecular Docking
4.2. Molecular Dynamics
4.3. TI Calculations
4.4. LIE-D Calculations
4.5. Hydrogen Bond Analysis
4.6. Cells
4.7. Viruses
4.8. HCoV-OC43 Infections
4.9. Compound Toxicity Assays
4.10. SARS-CoV-2 Mpro Activity
4.11. Materials
4.12. Instrumentation
4.13. Synthetic Procedures and Characterization

- Methyl 4-((4-methylpiperazin-1-yl)methyl)benzoate: 1H-NMR (400 MHz, CDCl3, 298 K): 7.98 (d, 2H), 7.40 (d, 2H), 3.91 (s, 3H), 3.55 (s, 2H), 2.75–2.33 (br s, 8H), 2.29 (s, 3H). 13C{1H} NMR (298 K, CDCl3, 101 MHz): 167.08, 143.84, 129.57, 128.97, 128.94, 62.62, 55.09, 53.13, 52.05, 46.00. ESI-MS: M + H+ = 249.1 expected, 249.0 observed.
- Methyl 2-methoxy-4-((4-methylpiperazin-1-yl)methyl)benzoate: 1H-NMR (400 MHz, DMSO-d6, 298 K): 7.61 (d, 1H), 7.07 (s, 1H), 6.96 (d, 1H), 3.82 (s, 3H), 3.76 (s, 3H), 3.50 (s, 2H), 2.48–2.22 (br s, 8H), 2.16 (s, 3H). 13C{1H} NMR (293 K, CDCl3, 101 MHz): 166.62, 159.33, 145.01, 131.63, 120.62, 118.56, 112.34, 62.75, 56.05, 55.13, 53.18, 51.94, 46.04. ESI-MS: M + H+ = 279.2 expected, 279.0 observed.
- Methyl 3-methoxy-4-((4-methylpiperazin-1-yl)methyl)benzoate: 1H-NMR (400 MHz, CDCl3, 298 K): 7.60 (d, 1H), 7.49 (s, 1H), 7.43 (d, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.57 (s, 2H), 2.60–2.38 (br s, 8H), 2.27 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3, 298 K): 167.10, 157.53, 131.99, 129.78, 129.73, 121.81, 111.01, 55.80, 55.57, 55.15, 53.16, 52.10, 46.01. ESI-MS: M + H+ = 279.2 expected, 279.1 observed.
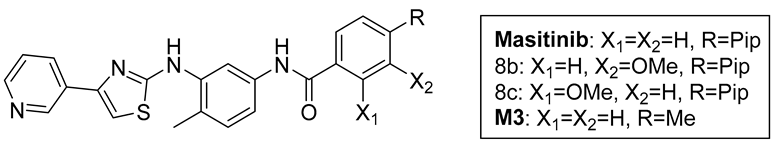
- Masitinib trifluoroacetate salt: Following the general procedure above, 6 (115 mg, 407 µmol), AlMe3 (2M in toluene, 0.82 mL, 1.63 mmol), and 7a (101 mg, 407 µmol) were reacted to prepare masitinib (85.1 mg) in a 42% yield. NMR spectra matched with literature precedent [19]. 1H NMR (500 MHz, DMSO-d6, 373 K): 9.76 (br s, 1H), 9.10 (d, J = 1.5 Hz, 1H), 8.48 (dd, J = 4.7, 1.6 Hz, 1H), 8.32 (d, J = 2.1 Hz, 1H), 8.25 (dt, J = 8.3, 2.1 Hz, 1H), 7.97 (d, J = 8.2 Hz, 2H), 7.47 (d, J = 8.3 Hz, 2H), 7.40 (m, 2H), 7.29 (s, 1H), 7.20 (d, J = 8.3 Hz, 1H), 3.75 (br s, 2H), 3.21 (br s, 4H), 2.79 (br s, 3H), 2.75 (br s, 4H), 2.30 (s, 3H). ESI-MS: M + H+ = 499.2 expected, 499.1 observed.
- M1 trifluoroacetate salt: Following the general procedure, 6 (180 mg, 637 µmol), AlMe3 (2M in toluene, 0.82 mL, 1.63 mmol), and 7b (177 mg, 637 µmol) were reacted to prepare crude 8b (crude yield 226 mg, 67%). A Schlenk flask was charged with a portion of crude 8b (70 mg, 132 µmol) and reacted with BBr3 (0.43 g, 0.16 mL, 1.70 mmol) according to the general procedure to obtain M1 as a yellow solid (81 mg, 24% over two steps). 1H NMR (400 MHz, DMSO-d6, 293 K): 9.75 (br s, 1H), 9.14 (br s, 1H), 9.24 (d, J = 1.7 Hz, 1H), 8.57 (m, 1H), 8.35 (d, J = 7.7 Hz, 1H), 8.20 (s, 1H), 7.49 (dd, J = 5.2, 2.6 Hz, 1H), 7.44 (s, 2H), 7.43 (s, 1H), 7.34 (d, J = 7.6 Hz, 1H), 7.20 (d, J = 8.2 Hz, 1H), 3.85 (br s, 2H) 3.24 (br s, 4H), 2.89 (br s, 4H), 2.77 (s, 3H), 2.29 (s, 3H). 13C {1H} NMR (DMSO-d6, 126 MHz, 298 K): 166.3, 165.3, 158.2 (q, J = 34 Hz), 156.6, 156.3, 145.9, 145.2, 144.1, 139.0, 137.6, 137.5, 136.6, 136.2, 131.5, 130.5, 125.0, 124.2, 124.1, 118.2, 116.2 (q, J = 294 Hz), 115.7, 115.0, 114.9, 113.2, 106.3, 51.9, 48.6, 45.1, 42.9, 17.6. ESI-MS: M+H+ = 515.2 expected, 515.1 observed.
- M2 trifluoroacetate salt: Following the general procedure, 6 (58 mg, 204 µmol), AlMe3 (2M in toluene, 0.82 mL, 1.63 mmol), and 7b (56.5 mg, 204 µmol) were reacted to prepare crude 8c (crude yield 90 mg, 83%). A Schlenk flask was charged with crude 8c (90 mg) and reacted with BBr3 (0.43 g, 0.16 mL, 1.70 mmol) according to the general procedure to obtain M2 as a yellow solid (57 mg, 54% over two steps). 1H NMR (400 MHz, DMSO-d6, 298 K): 12.11 (br s, 1H), 10.38 (br s, 1H), 9.54 (br s), 9.26 (d, J = 1.6 Hz, 1H), 8.77 (s, 1H), 8.64 (m, 2H), 8.03 (d, J = 8.1 Hz, 1H), 7.69 (dd, J = 8.1, 5.3 Hz, 1H), 7.65 (s, 1H), 7.22 (s, 1H), 6.99 (s, 1H), 6.96 (d, J = 8.1 Hz), 3.73 (br s, 2H), 3.42 (br s, 4H), 3.06 (br s, 4H), 2.81 (s, 3H), 2.29 (s, 3H). 13C {1H} NMR (DMSO-d6, 126 MHz, 298 K): 166.3, 165.6, 158.6 (q, J = 35 Hz), 145.1, 143.6, 142.4, 139.2, 137.9, 136.6, 132.2, 130.6, 129.3, 125.7, 124.2, 120.3, 118.4, 117.1, 116.1 (q, J = 295 Hz), 115.8, 113.3, 107.3, 59.4, 51.4, 48.7, 42.1, 17.8. ESI-MS: M + H+ = 515.2 expected, 515.1 observed.
- M3 trifluoroacetate salt: Following the general procedure, 6 (130 mg, 461 µmol), AlMe3 (2M in toluene, 0.92 mL, 1.84 mmol), and 7d (75 mg, 507 µmol) were reacted to prepare M3 (36 mg) in a 15% yield. 1H NMR (500 MHz, DMSO-d6, 298 K): 10.15 (s, 1H), 9.55 (br s, 1H), 9.33 (d, J = 1.7 Hz, 1H), 8.80 (s, 1H), 8.79 (d, J = 1.8 Hz), 8.69 (dd, J = 5.2, 1.2 Hz, 1H), 7.92 (d, J = 8.2 Hz, 2H), 7.83 (dd, J = 8.2, 5.2 Hz, 1H), 7.70 (s, 1H), 7.34 (d, J = 8.1 Hz, 3H), 7.19 (d, J = 8.3 Hz, 1H), 2.39 (s, 3H), 2.27 (s, 3H). 13C {1H} NMR (126 MHz, DMSO d6, 298 K): 165.9, 165.2, 158.3 (q, J = 35 Hz), 145.1, 143.6, 143.5, 142.4, 141.5, 139.0, 138.1, 137.9, 137.8, 132.2, 130.4, 128.9, 127.8, 125.7, 123.6, 115.9 (q, J = 291 Hz), 115.5, 113.1, 112.9, 107.2, 21.0, 17.6. ESI-MS: M + H+ = 401.1 expected, 401.0 observed.

Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Papain-like Protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Drayman, N.; DeMarco, J.K.; Jones, K.A.; Azizi, S.A.; Froggatt, H.M.; Tan, K.; Maltseva, N.I.; Chen, S.; Nicolaescu, V.; Dvorkin, S.; et al. Masitinib Is a Broad Coronavirus 3CL Inhibitor That Blocks Replication of SARS-CoV-2. Science 2021, 373, 931–936. [Google Scholar] [CrossRef]
- WHO Solidarity Consortium. Repurposed Antiviral Drugs for Covid 19- Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Mandal, K. Repurposing an Antiviral Drug against SARS-CoV-2 Main Protease. Angew. Chem. 2021, 60, 23492–23494. [Google Scholar] [CrossRef]
- Pinzi, L.; Tinivella, A.; Caporuscio, F.; Rastelli, G. Drug Repurposing and Polypharmacology to Fight SARS-CoV-2 Through Inhibition of the Main Protease. Front. Pharmacol. 2021, 12, 636989. [Google Scholar] [CrossRef]
- Marforio, T.D.; Mattioli, E.J.; Zerbetto, F.; Calvaresi, M. Fullerenes against COVID-19: Repurposing C60 and C70 to Clog the Active Site of SARS-CoV-2 Protease. Molecules 2022, 27, 1916. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Bai, Y.; Cui, J.; Zong, Z.; Gao, Y.; Zheng, Z. Computer-Aided Drug Design Boosts RAS Inhibitor Discovery. Molecules 2022, 27, 5710. [Google Scholar] [CrossRef] [PubMed]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current Trends in Computer Aided Drug Design and a Highlight of Drugs Discovered via Computational Techniques: A Review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Shultz, M.D. Improving the plausibility of success with inefficient metrics. ACS Med. Chem. Lett. 2014, 5, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.; Thumar, R.; Patel, B.; Athar, M.; Jha, P.C.; Patel, D. Structural Differences In 3C-like protease (Mpro) From SARS-CoV and SARS-CoV-2: Molecular Insights for Drug Repurposing Against COVID-19 Revealed by Molecular Dynamics Simulations. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dassault Systemes BIOVIA. Discovery Studio Visualizer, v20.1.0.19295; Dassault Systemes: San Diego, CA, USA, 2020. [Google Scholar]
- Miranda, W.E.; Noskov, S.Y.; Valiente, P.A. Improving the LIE Method for Binding Free Energy Calculations of Protein–Ligand Complexes. J. Chem. Inf. Model. 2015, 55, 1867–1877. [Google Scholar] [CrossRef]
- Ciufolini, M.; Wermuth, C.; Gielthen, B.; Moussy, A. 2-(3-Aminoaryl)amino-4-aryl-thiazoles and Their Use as c-Kit Inhibitors. W.O. Patent 2004/014903 A1, 19 February 2004. [Google Scholar]
- Arnon, R.; Miller, A. (Eds.) Chapter 21-Emerging Therapies for Multiple Sclerosis. In Translational Neuroimmunology in Multiple Sclerosis; Academic Press: Cambridge, MA, USA, 2016; pp. 285–304. ISBN 9780128019146. [Google Scholar] [CrossRef]
- Jason, G. Tyrosine Kinase Inhibitors in the Treatment of Eosinophilic Neoplasms and Systemic Mastocytosis. Hematol. Oncol. Clin. N. Am. 2017, 31, 643–661. [Google Scholar]
- Roe, M.K.; Junod, N.A.; Young, A.R.; Beachboard, D.C.; Stobart, C.C. Targeting novel structural and functional features of coronavirus protease nsp5 (3CL(pro), M(pro)) in the age of COVID-19. J. Gen. Virol. 2021, 102, 001558. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, C.; Chang, D.; Wang, Y.; Dong, X.; Jiao, T.; Zhao, Z.; Ren, L.; Dela Cruz, C.S.; Sharma, L.; et al. Identification of Potent and Safe Antiviral Therapeutic Candidates Against SARS-CoV-2. Front. Immunol. 2020, 11, 586572. [Google Scholar] [CrossRef]
- Jornada, D.H.; Dos Santos Fernandes, G.F.; Chiba, D.E.; De Melo, T.R.F.; Dos Santos, J.L.; Chung, M.C. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2016, 21, 42. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, M.; Puccini, P.; Gervasi, P.G.; Longo, V. Carboxylesterases and arylacetamide deacetylase comparison in human A549, H460, and H727 pulmonary cells. Life Sci. 2021, 277, 119486. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Fielding, B.; Gamieldien, J. Human Coronavirus OC43 3CL Protease and the Potential of ML188 as a Broad-Spectrum Lead Compound: Homology Modelling and Molecular Dynamic Studies. BMC Struct. Biol. 2015, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comput. Aided Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247260. [Google Scholar] [CrossRef]
- Byléhn, F.; Menéndez, C.A.; Perez-Lemus, G.R.; Alvarado, W.; de Pablo, J.J. Modeling the Binding Mechanism of Remdesivir, Favilavir, and Ribavirin to SARS-CoV-2 RNA-Dependent RNA Polymerase. ACS Cent. Sci. 2021, 7, 164–174. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Protein | Molecule | Acceptor | Donor | Fraction |
|---|---|---|---|---|
| Mpro (SARS-CoV-2) | M(wt)1 | His164 (O) | LIG (N1) | 0.82 |
| LIG (N3) | His163 (NE2) | 0.56 | ||
| Thr24 | LIG | 0.07 | ||
| M(wt)0 | His164 (O) | LIG (N1) | 0.94 | |
| LIG (N3) | His163 (NE2) | 0.53 | ||
| LIG | Thr24 | 0.12 | ||
| Mpro (OC43) | M(wt)1 | Gln164 (O) | LIG (N1) | 0.75 |
| LIG (N3) | His163 (NE2) | 0.44 | ||
| Asn24 | LIG | 0.35 | ||
| M(wt)0 | Gln164 (O) | LIG (N1) | 0.56 | |
| LIG (N3) | His163 (NE2) | 0.20 | ||
| LIG | Glu166 | 0.12 |
| Ligand | LIE-D (kcal/mol) | TI (kcal/mol) | S.D |
|---|---|---|---|
| Mwt | −12.52 | −8.80 | 0.80 |
| M1 | −12.22 | −9.28 | 1.28 |
| M2 | −13.41 | −10.06 | 1.47 |
| M3 | −11.42 | −8.35 | 1.13 |
| M4 | −10.95 | −6.66 | 1.78 |
| M5 | −11.57 |
| Doubling Time (h) | Dose | ||||
|---|---|---|---|---|---|
| 10 µM | 3 µM | 1 µM | 0.3 µM | ||
| COMPOUNDS | DMSO | 23.13 | ND | ND | ND |
| MWT | 347.8 | 74.57 | 24.94 | 22.72 | |
| M1 | 52.78 | 23.72 | 21.89 | 21.75 | |
| M2 | 33.63 | 21.57 | 21.44 | 21.52 | |
| M3 | 30.76 | 22.49 | 21.49 | 22.13 | |
| M4 | 37.36 | 22.28 | 21.47 | 22.01 | |
| M5 | 25.19 | 21.83 | 21.95 | 22.37 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menendez, C.A.; Mohamed, A.; Perez-Lemus, G.R.; Weiss, A.M.; Rawe, B.W.; Liu, G.; Crolais, A.E.; Kenna, E.; Byléhn, F.; Alvarado, W.; et al. Development of Masitinib Derivatives with Enhanced Mpro Ligand Efficiency and Reduced Cytotoxicity. Molecules 2023, 28, 6643. https://doi.org/10.3390/molecules28186643
Menendez CA, Mohamed A, Perez-Lemus GR, Weiss AM, Rawe BW, Liu G, Crolais AE, Kenna E, Byléhn F, Alvarado W, et al. Development of Masitinib Derivatives with Enhanced Mpro Ligand Efficiency and Reduced Cytotoxicity. Molecules. 2023; 28(18):6643. https://doi.org/10.3390/molecules28186643
Chicago/Turabian StyleMenendez, Cintia A., Adil Mohamed, Gustavo R. Perez-Lemus, Adam M. Weiss, Benjamin W. Rawe, Guancen Liu, Alex E. Crolais, Emma Kenna, Fabian Byléhn, Walter Alvarado, and et al. 2023. "Development of Masitinib Derivatives with Enhanced Mpro Ligand Efficiency and Reduced Cytotoxicity" Molecules 28, no. 18: 6643. https://doi.org/10.3390/molecules28186643
APA StyleMenendez, C. A., Mohamed, A., Perez-Lemus, G. R., Weiss, A. M., Rawe, B. W., Liu, G., Crolais, A. E., Kenna, E., Byléhn, F., Alvarado, W., Mendels, D., Rowan, S. J., Tay, S., & de Pablo, J. J. (2023). Development of Masitinib Derivatives with Enhanced Mpro Ligand Efficiency and Reduced Cytotoxicity. Molecules, 28(18), 6643. https://doi.org/10.3390/molecules28186643