
Design, Synthesis, and Biological Evaluation of Novel 3-Cyanopyridone/Pyrazoline Hybrids as Potential Apoptotic Antiproliferative Agents Targeting EGFR/BRAFV600E Inhibitory Pathways

 ,
,  , , , and
, , , and
Abstract
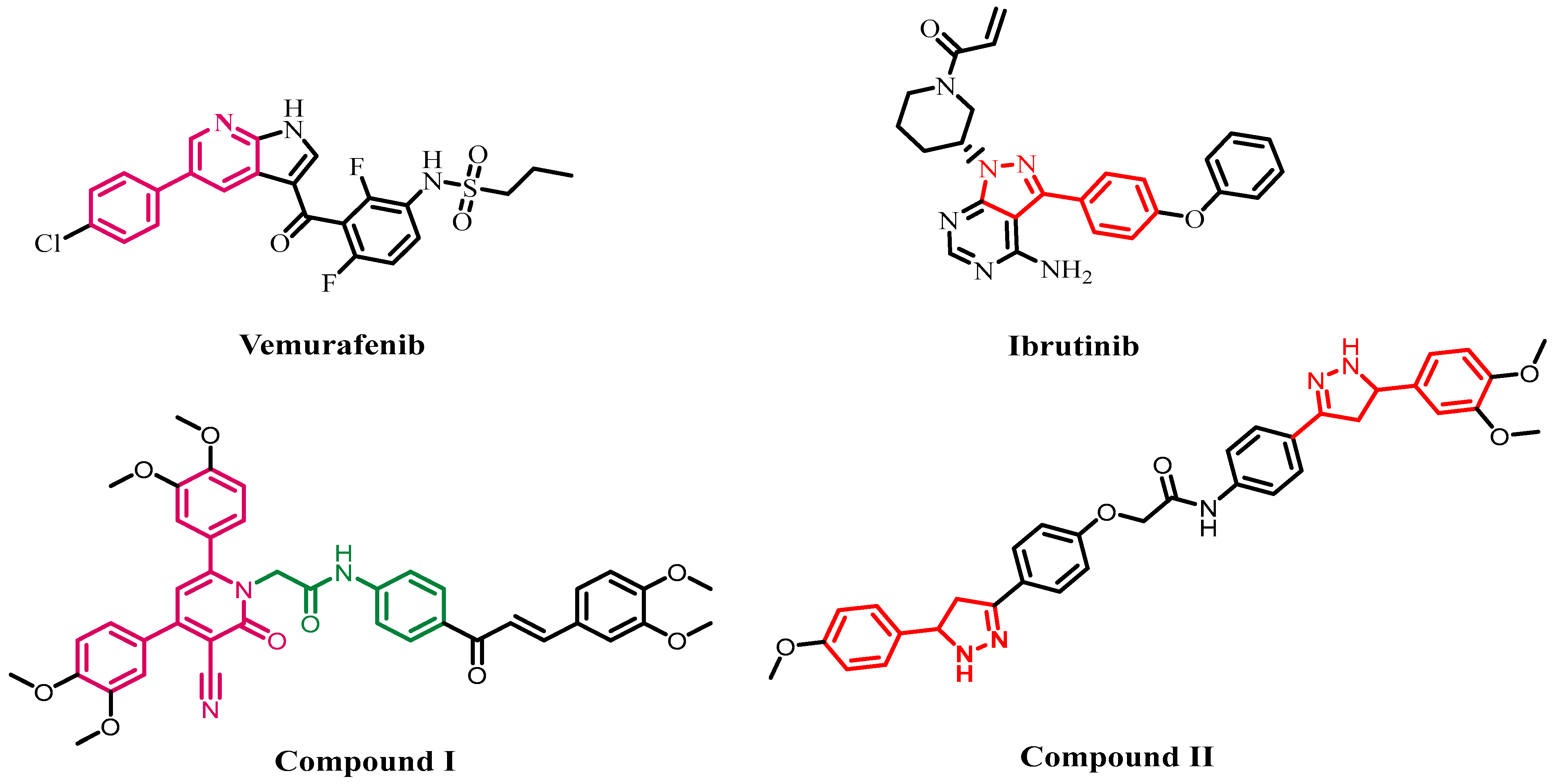
:1. Introduction
2. Results and Discussion
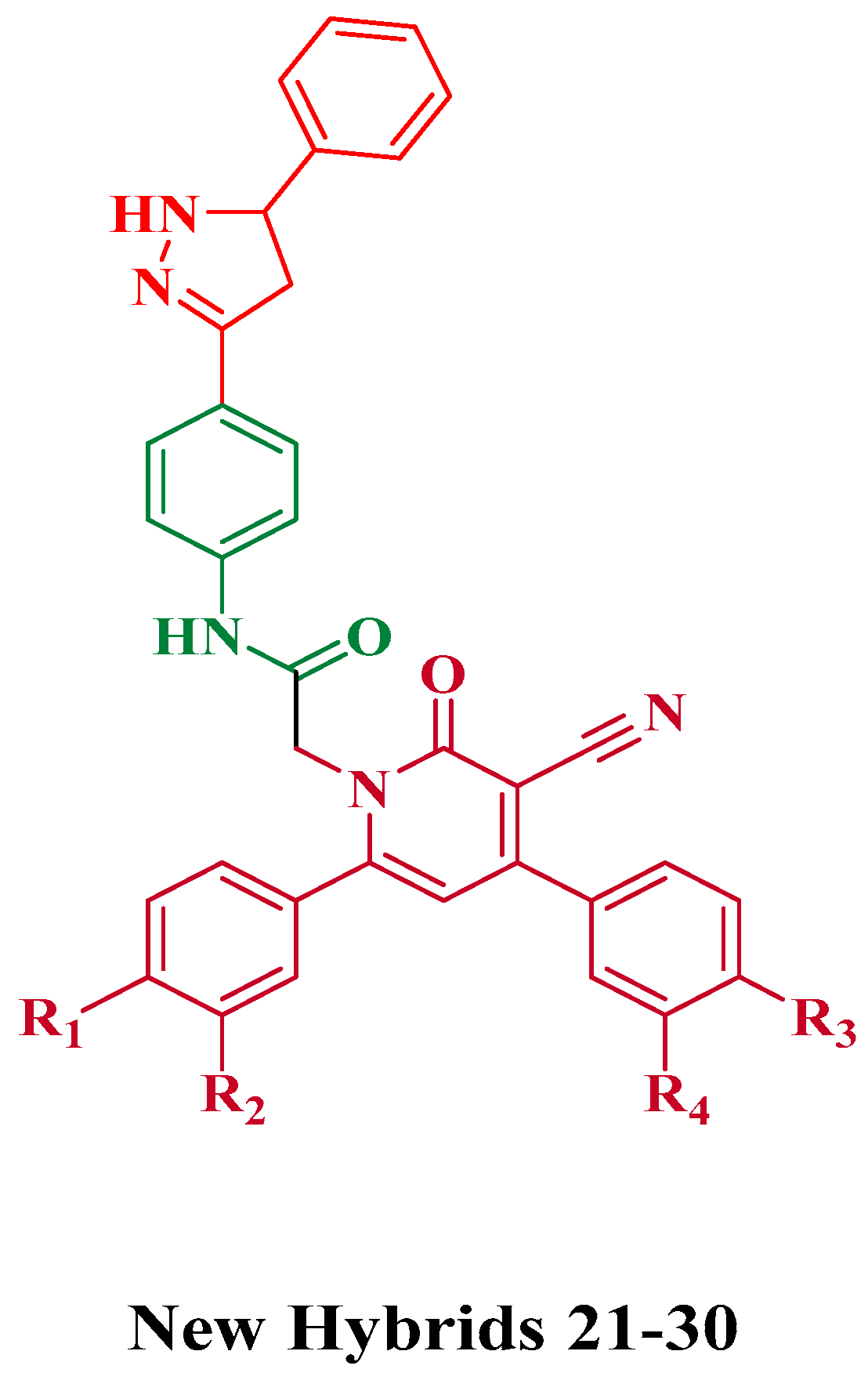
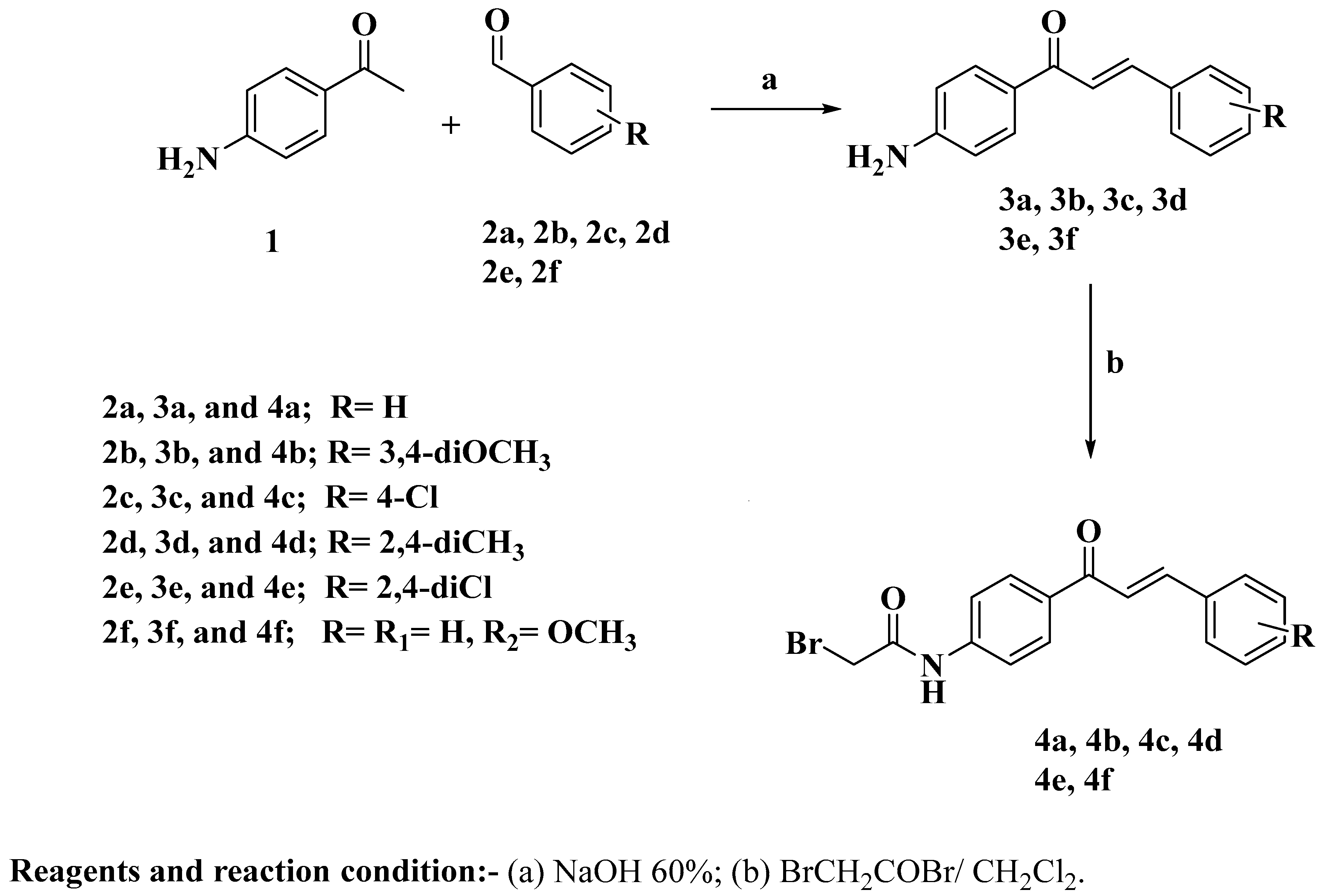
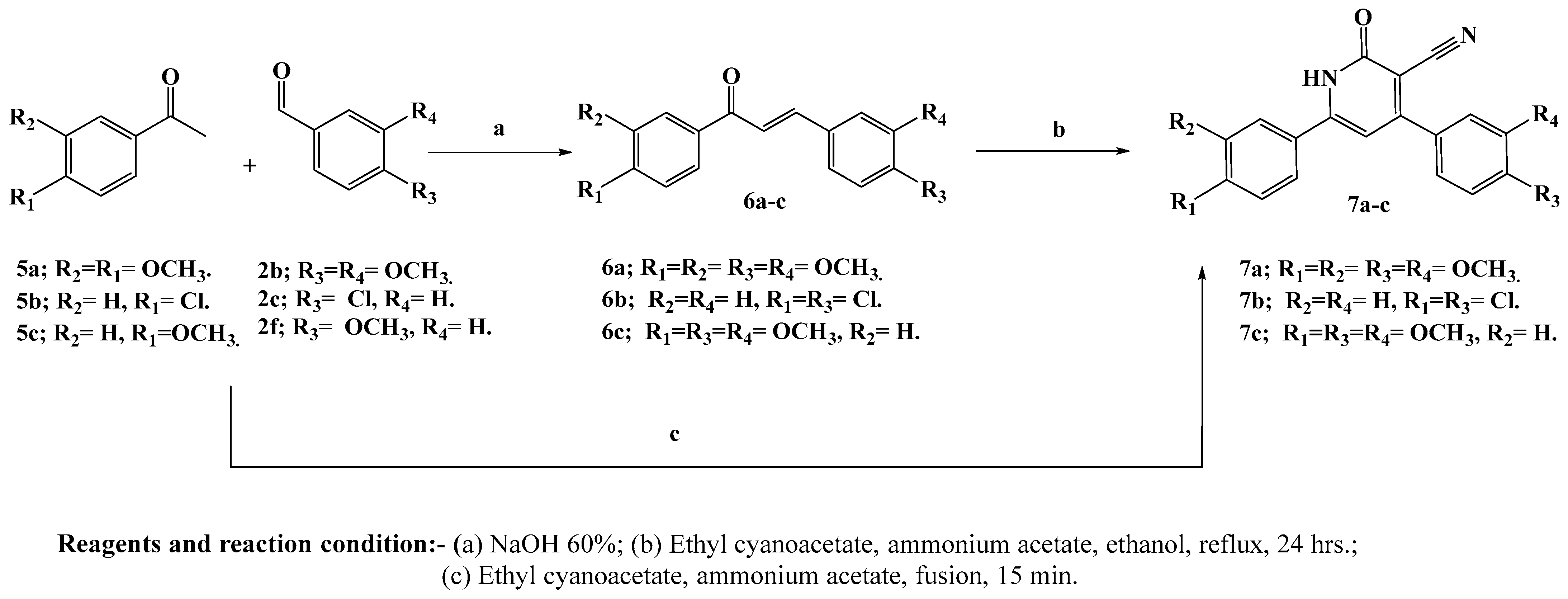
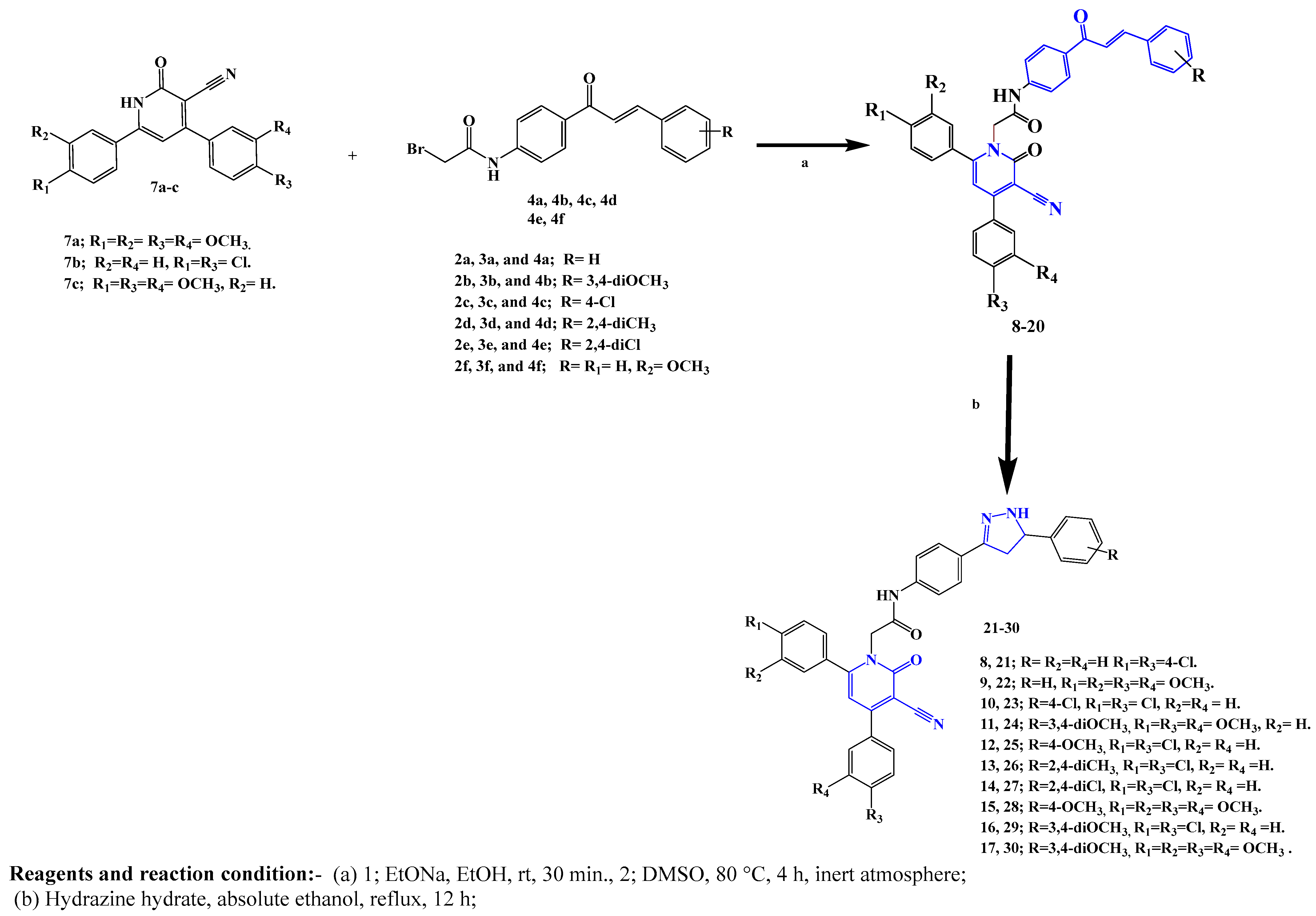
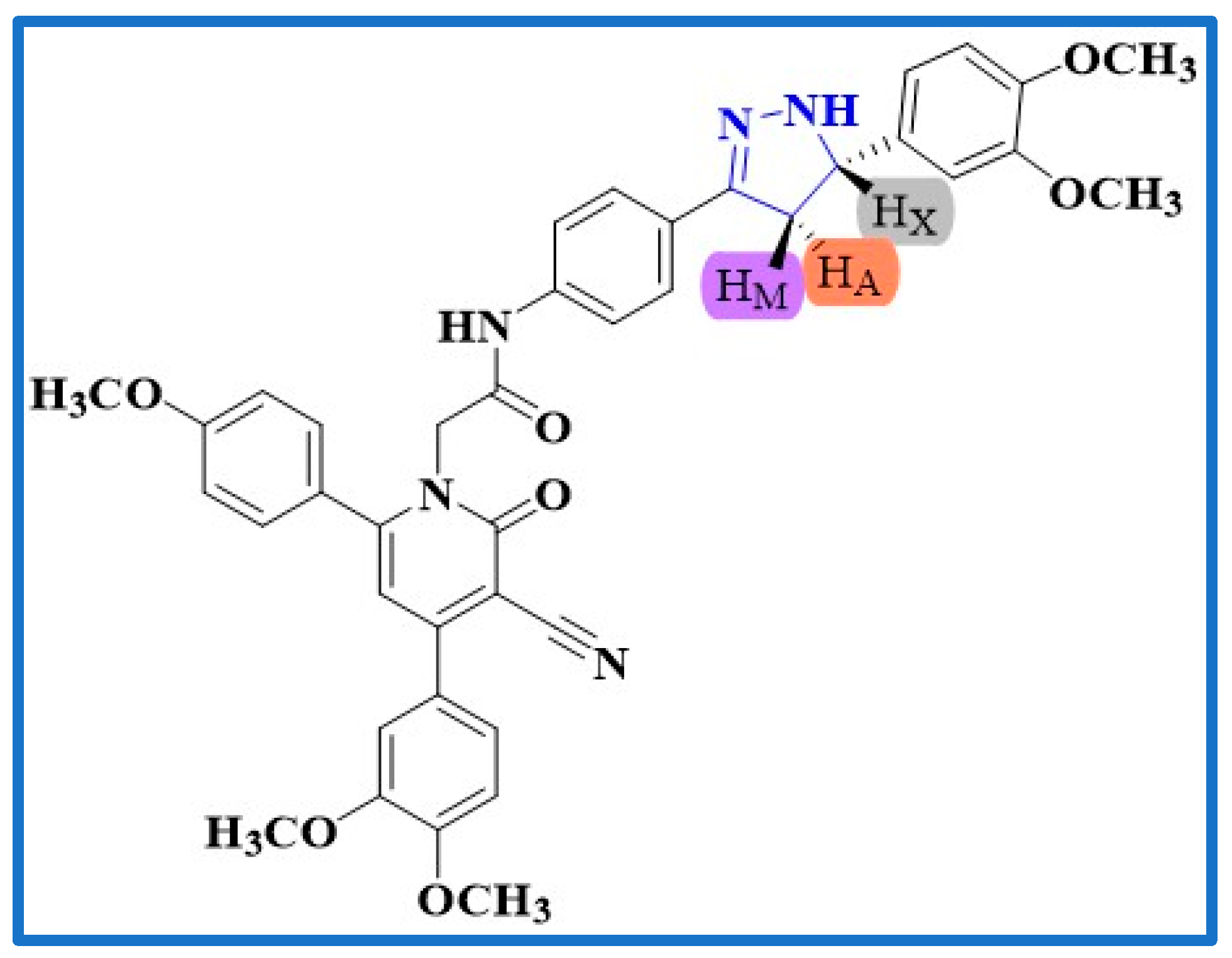
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Evaluation of Cell Viability
2.2.2. Evaluation of Antiproliferative Activity
2.2.3. Evaluation of EGFR Inhibitory Activity
2.2.4. Evaluation of BRAFV600E Inhibitory Activity
2.2.5. Valuation of Apoptotic Activity
2.2.6. Caspase 3 Activation Assay
2.2.7. Caspase-8, Bax, and Bcl-2 Levels Assay
2.3. In Silico Studies
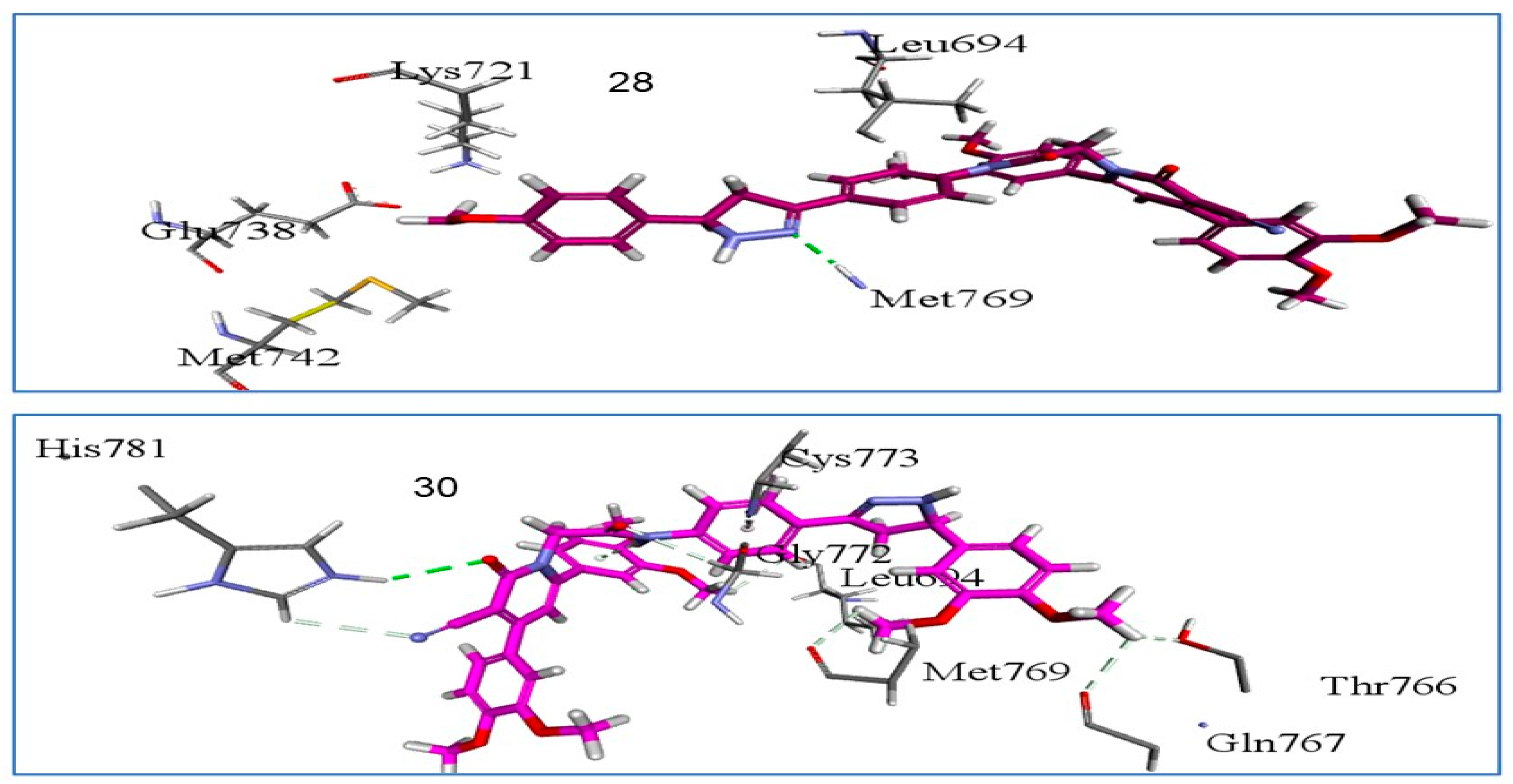
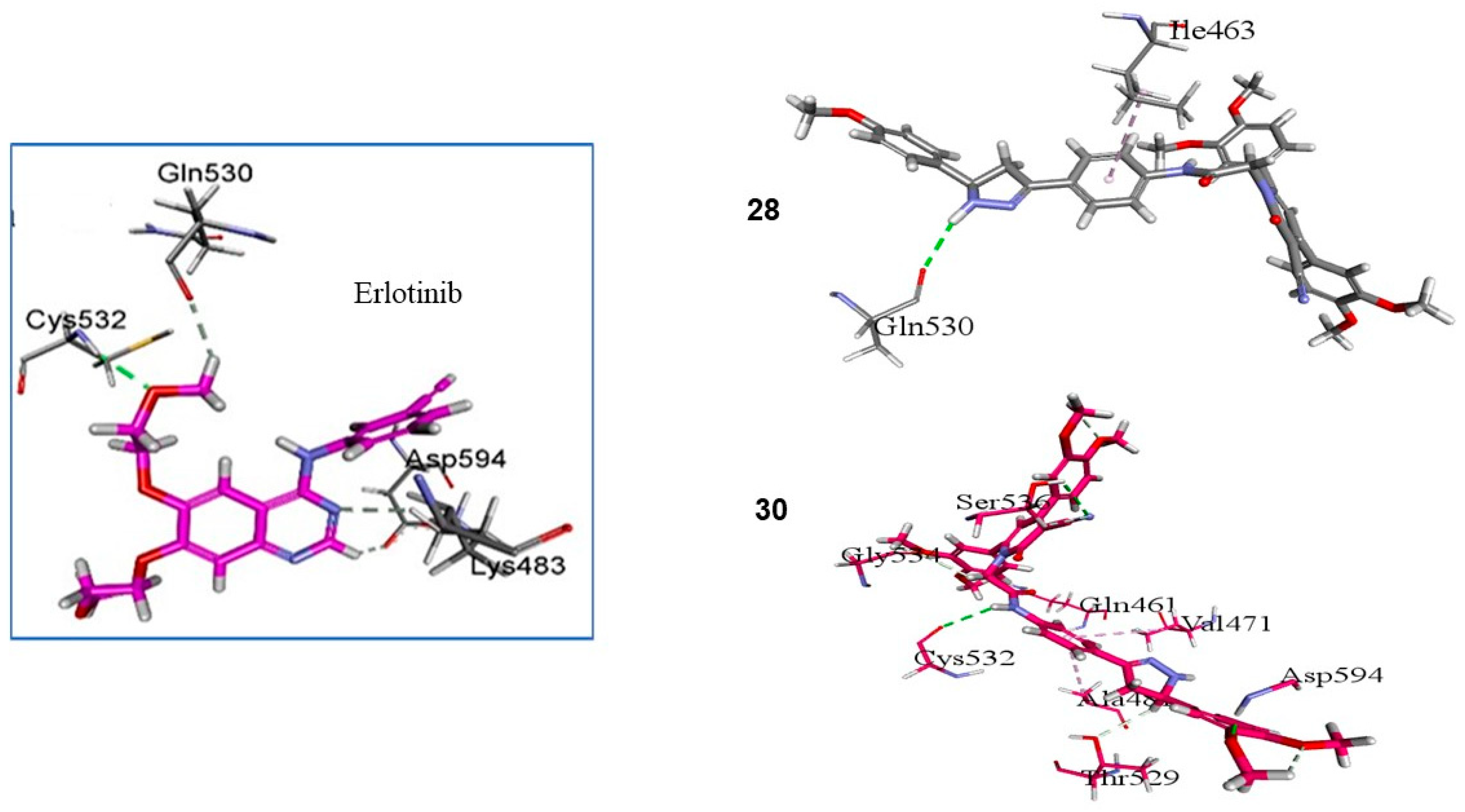
2.3.1. Docking Study
2.3.2. ADMET Studies
2.3.3. In Silico Toxicity Predictions
2.4. Structure–Activity Relationship (SAR)

3. Materials and Methods
3.1. Chemistry
General Procedure for the Synthesis of Compounds 21–30
- 2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (21). White crystals (0.71 g, 82% yield); m.p 273–274 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.30 (1H, s, O=C-NH), 8.30 (2H, d, J = 8.50 Hz, Ar-H), 8.21 (1H, s, pyrazoline NH), 7.77 (3H, m, Ar-H), 7.73–7.69 (5H, m, Ar-H), 7.64 (2H, d, J = 8.50 Hz, Ar-H), 7.60–7.55 (5H, m, Ar-H), 7.36 (1H, s, Ar-H), 5.42 (2H, s, NCH2), 4.85–4.83 (1H, m, pyrazoline H), 3.44 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.84 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 167.17, 164.82, 157.12, 156.35, 154.68, 153.64, 147.69, 138.06, 136.75, 136.21, 136.03, 135.61, 131.27, 131.00, 130.88, 130.53, 129.77, 129.52, 129.42, 129.25, 129.14, 128.87, 127.11, 112.77, 109.67, 92.86, 66.33, 64.10, 40.82; Anal. Calcd. For C35H25Cl2N5O2 (618.52): C, 67.97; H, 4.07; N, 11.32. Found: C, 67.66; H, 4.11; N, 11.45.
- 2-(3-Cyano-4,6-bis(3,4-dimethoxyphenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (22). White crystals (0.81 g, 86% yield); m.p 262–263 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.50 (1H, s, O=C-NH), 7.85–7.81 (2H, m, pyrazoline NH + Ar-H), 7.70 (1H, s, Ar-H), 7.62 (2H, d, J = 8.50 Hz, Ar-H), 7.57 (2H, d, J = 8.50 Hz, Ar-H), 7.49 (1H, s, Ar-H), 7.33–7.39 (6H, m, Ar-H), 7.26 (1H, s, Ar-H), 7.18 (1H, d, J = 9.00 Hz, Ar-H), 6.96 (1H, d, J = 9.00 Hz, Ar-H), 5.19 (2H, s, NCH2), 4.81 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.88 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.63 (3H, s, OCH3), 3.43 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.80 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.79, 163.77, 157.10, 156.56, 151.61, 150.87, 149.32, 149.18, 148.86, 143.54, 139.03, 129.43, 128.98, 128.86, 128.56, 127.58, 127.10, 126.53, 121.99, 121.38, 119.52, 116.30, 113.73, 112.75, 112.26, 111.98, 110.70, 91.30, 65.97, 64.10, 56.20, 56.18, 56.08, 55.82, 41.14. Anal. Calcd. For C39H35N5O6 (669.74): C, 69.94; H, 5.27; N, 10.46. Found: C, 70.03; H, 5.16; N, 10.39.
- 2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (23). White powder (0.81 g, 89% yield); m.p 286–287 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.56 (1H, s, O=C-NH), 8.24 (2H, d, J = 8.50 Hz, Ar-H), 7.91 (1H, s, pyrazoline NH), 7.81 (2H, d, J = 8.50 Hz, Ar-H), 7.70–7.60 (8H, m, Ar-H), 7.85–7.81 (5 H, m, Ar-H), 5.20 (2H, s, NCH2), 4.81–4.85 (1H, m, pyrazoline H), 3.45 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.79 (1H, dd, J = 11.23 and 3.03 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.57, 163.68, 156.20, 155.94, 148.99, 142.59, 138.93, 136.23, 135.73, 135.51, 134.85, 132.03, 131.09, 129.72, 129.44, 129.31, 129.01, 128.93, 128.80, 126.61, 119.71, 115.45, 114.80, 93.08, 66.26, 63.33, 41.10; Anal. Calcd. For C35H24Cl3N5O2 (652.96): C, 64.38; H, 3.70; N, 10.73. Found: C, 64.46; H, 3.82; N, 10.59.
- 2-(3-Cyano-4-(3,4-dimethoxyphenyl)-6-(4-methoxyphenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (24). White crystals (0.86 g, 88% yield); m.p 244–245 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.50 (1H, s, O=C-NH), 8.16 (2H, d, J = 8.5 Hz, Ar-H), 7.77 (1H, s, pyrazoline NH), 7.58–7.66 (4H, m, Ar-H), 7.34–7.41 (3H, m, Ar-H + pyridine-C5–H), 7.16 (1H, d, J = 8.5 Hz, Ar-H), 6.88–6.99 (5H, m, Ar-H), 5.16 (2H, s, NCH2), 4.78 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.87 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.72 (3H, s, OCH3), 3.39 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.81 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.78, 163.81, 161.85, 157.01, 156.62, 150.90, 149.20, 149.13, 148.41, 138.92, 135.77, 129.69, 129.28, 129.10, 128.51, 126.51, 121.99, 119.65, 119.09, 116.27, 114.61, 113.49, 112.68, 112.23, 112.16, 112.12, 110.84, 91.22, 66.05, 64.00, 56.18, 56.16, 56.03, 55.88, 55.83, 40.83. Anal. Calcd. For C40H37N5O7 (699.76): C, 68.66; H, 5.33; N, 10.01. Found: C, 68.58; H, 5.21; N, 10.12.
- 2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (25). White powder (0.83 g, 92% yield); m.p 256–257 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.53 (1H, s, O=C-NH), 8.22 (2H, d, J = 8.50 Hz, Ar-H), 7.89 (1H, s, pyrazoline NH), 7.80 (2H, d, J = 8.50 Hz, Ar-H), 7.58–7.69 (6H, m, Ar-H), 7.46–7.40 (3H, m, Ar-H), 7.28 (2H, d, J = 8.50 Hz, Ar-H), 6.89 (2H, d, J = 8.50 Hz, Ar-H), 5.20 (2H, s, NCH2), 4.73–4.78 (1H, m, pyrazoline H), 3.79-385 (1H, m, pyrazoline H), 3.72 (3H, s, OCH3), 2.77 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.74, 163.20, 155.79, 155.38, 135.83, 135.36, 134.99, 134.32, 130.57, 129.19, 128.97, 128.82, 128.55, 127.78, 127.16, 126.49, 126.12, 125.70, 119.34, 115.00, 114.22, 113.78, 113.45, 92.59, 65.83, 63.26, 55.54, 39.85; Anal. Calcd. For C36H27Cl2N5O3 (648.54): C, 66.67; H, 4.20; N, 10.80. Found: C, 66.75; H, 4.36; N, 10.73.
- 2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(2,4-dimethylphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (26). White crystals (0.78 g, 86% yield); m.p 269–270 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.52 (1H, s, O=C-NH), 8.21 (2H, d, J = 8.50 Hz, Ar-H), 7.90 (1H, s, pyrazoline NH), 7.81–7.79 (3H, m, Ar-H), 7.69–7.58 (6H, Ar-H), 7.45 (2H, d, J = 8.50 Hz, Ar-H), 7.29 (1H, d, J = 8.50 Hz, Ar-H), 6.96 (2H, d, J = 8.50 Hz, Ar-H), 5.20 (2H, s, NCH2), 4.92 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.44 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.65 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H), 2.28 (3H, s, CH3), 2.22 (3H, s, CH3); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.54, 163.70, 156.21, 155.94, 148.44, 138.78, 138.54, 136.23, 136.16, 135.73, 135.53, 135.26, 134.87, 131.37, 131.11, 129.74, 129.50, 129.31, 126.92, 126.51, 126.06, 119.69, 119.63, 115.46, 114.83, 93.09, 66.27, 60.74, 40.47, 21.01, 19.45; Anal. Calcd. For C37H29Cl2N5O2 (646.57): C, 68.73; H, 4.52; N, 10.83. Found: C, 68.88; H, 4.46; N, 10.74.
- 2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(2,4-dichlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (27). White powder (0.78 g, 91% yield); m.p 291–292 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.52 (1H, s, O=C-NH), 8.22 (2H, d, J = 8.50 Hz, Ar-H), 7.91 (1H, s, pyrazoline NH), 7.82 (2H, d, J = 8.50 Hz, Ar-H), 7.70–7.57 (9H, m, Ar-H), 7.47–7.43 (3H, m, Ar-H), 5.20 (2H, s, NCH2), 5.05 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.58 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.73 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.77, 163.21, 155.72, 155.44, 139.68, 138.59, 135.75, 135.28, 135.02, 134.35, 132.71, 132.31, 131.64, 130.62, 130.08, 129.24, 128.96, 128.82, 128.20, 127.75, 127.57, 126.24, 119.21, 114.93, 114.33, 92.61, 65.79, 59.88, 40.15; Anal. Calcd. For C35H23Cl4N5O2 (687.40): C, 61.16; H, 3.37; N, 10.19. Found: C, 61.23; H, 3.43; N, 10.25.
- 2-(3-Cyano-4,6-bis(3,4-dimethoxyphenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (28). White powder (0.81 g, 83% yield); m.p 256–257 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.53 (1H, s, O=C-NH), 8.17 (2H, d, J = 8.5 Hz, Ar-H), 7.77 (1H, s, pyrazoline NH), 7.65 (2H, d, J = 8.50 Hz, Ar-H), 7.60 (2H, d, J = 8.50 Hz, Ar-H), 7.41 (1H, s, Ar-H), 7.36 (2H, d, J = 8.5 Hz, Ar-H), 7.17 (1H, s, Ar-H), 6.99–6.88 (5H, m, Ar-H), 5.16 (2H, s, NCH2), 4.76 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.87 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.73 (3H, s, OCH3), 3.44 (1H, dd, J = 16.23 and 3.09 Hz, pyrazoline H), 2.81 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.99, 163.36, 161.47, 156.62, 156.27, 153.09, 150.53, 148.82, 143.48, 142.79, 129.47, 129.27, 128.86, 128.62, 128.10, 127.37, 121.86, 121.62, 118.93, 115.84, 114.22, 113.18, 112.30, 111.86, 111.36, 110.05, 92.02, 90.84, 65.67, 62.13, 55.86, 55.83, 55.81, 55.78, 55.44, 38.89. Anal. Calcd. For C40H37N5O7 (699.76): C, 68.66; H, 5.33; N, 10.01. Found: C, 68.58; H, 5.26; N, 10.09.
- 2-(3-Cyano-4,6-bis(4-chlorophenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (29). White crystals (0.79 g, 83% yield); m.p 243–245 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.53 (1H, s, O=C-NH), 8.23 (2H, d, J = 8.50 Hz, Ar-H), 7.91 (1H, s, pyrazoline NH), 7.82 (2H, d, J = 8.50 Hz, Ar-H), 7.70–7.58 (7H, m, Ar-H),), 7.47–7.40 (3H, m, Ar-H), 6.99 (1H, s, Ar-H), 6.89 (1H, d, J = 8.50 Hz, Ar-H), 5.20 (2H, s, NCH2), 4.76 (1H, dd, J = 11.02 and 3.06 Hz, pyrazoline H), 3.74 (3H, s, OCH3), 3.72 (3H, s, OCH3), 3.37 (1H, m, pyrazoline H), 2.80 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.09, 163.25, 155.74, 155.41, 148.73, 148.62, 147.98, 138.38, 135.76, 135.30, 135.02, 134.34, 130.60, 129.23, 128.95, 128.81, 128.55, 126.07, 119.27, 118.83, 118.64, 114.96, 114.29, 111.70, 110.41, 92.60, 65.81, 63.57, 55.56, 55.42, 40.64. Anal. Calcd. For C37H29Cl2N5O4 (678.57): C, 65.49; H, 4.31; N, 10.32. Found: C, 65.57; H, 4.44; N, 10.40.
- 2-(3-Cyano-4,6-bis(3,4-dimethoxyphenyl)-2-oxopyridin-1(2H)-yl)-N-(4-(5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide (30). White powder (0.89 g, 87% yield); m.p 233–234 °C; 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.48 (1H, s, O=C-NH), 7.81–7.57 (8H, m, pyrazoline NH + 7 Ar-H), 7.36 (2H, d, J = 8.50 Hz, Ar-H), 7.17 (1H, d, J = 8.50 Hz, Ar-H), 6.88–6.99 (4H, m, Ar-H), 5.19 (2H, s, NCH2), 4.76-4.73 (1H, m, pyrazoline H), 3.87 (3H, s, OCH3), 3.85 (3H, s, OCH3), 3.78 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.72 (3H, s, OCH3), 3.63 (3H, s, OCH3), 2.81 (1H, dd, J = 16.20 and 11.01 Hz, pyrazoline H); 13C NMR (125 MHz, DMSO-d6) δ (ppm): 166.42, 163.36, 156.67, 156.08, 151.18, 150.46, 148.91, 148.75, 148.02, 138.60, 135.33, 129.02, 128.67, 128.13, 126.09, 121.58, 120.91, 119.14, 118.96, 115.92, 113.23, 112.29, 111.97, 111.50, 110.44, 110.27, 90.86, 65.57, 63.63, 55.74, 55.71, 55.61, 55.58, 55.46, 55.39, 40.66..Anal. Calcd. For C41H39N5O8 (729.79): C, 67.48; H, 5.39; N, 9.60.
3.2. Biology
3.2.1. Cell Viability Assay
3.2.2. Antiproliferative Assay
3.2.3. EGFR Inhibitory Assay
3.2.4. BRAFV600E Inhibitory Assay
3.2.5. Apoptotic Markers Assays
3.2.6. Docking Study
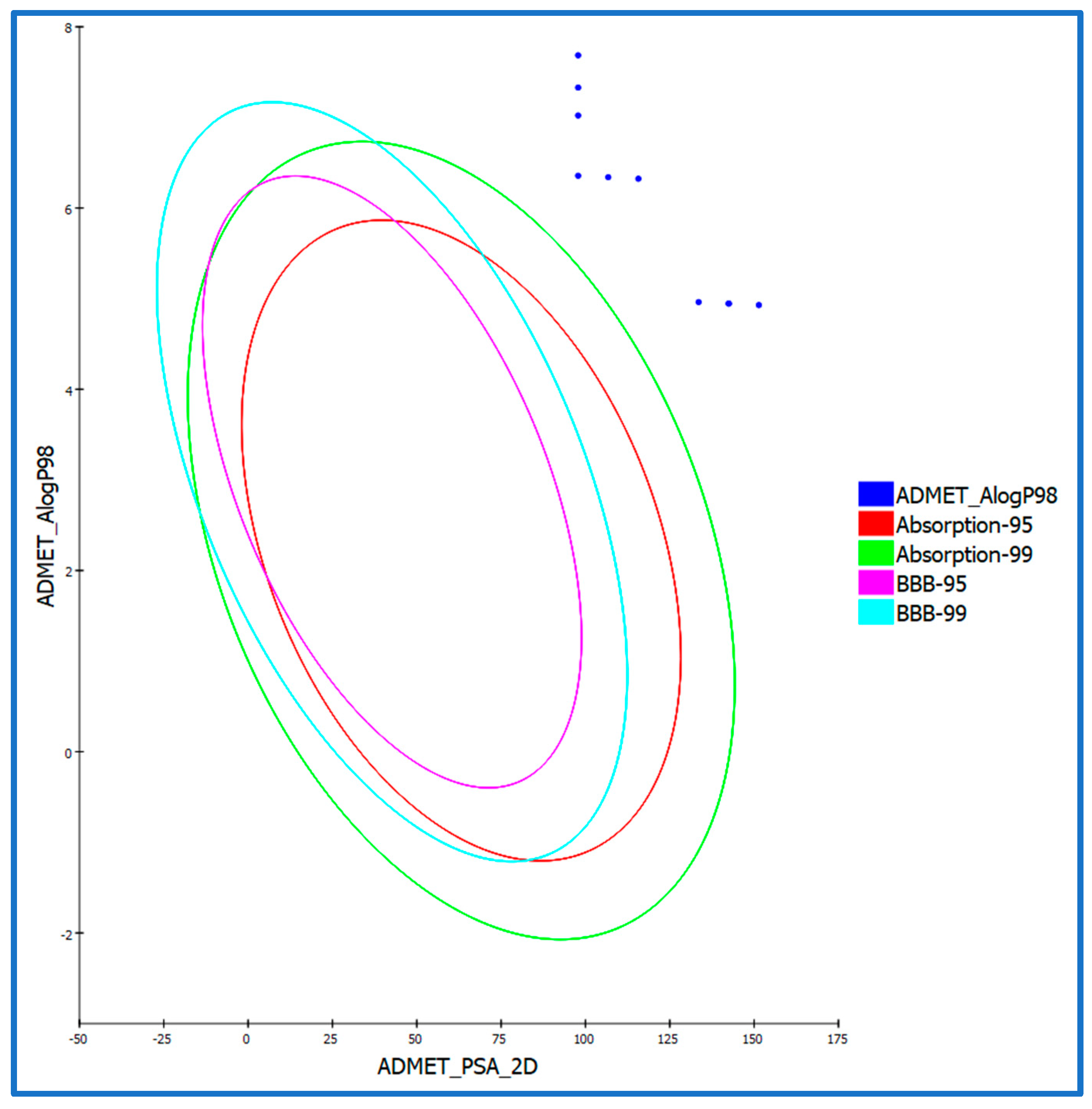
3.2.7. In Silico ADMET Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; Wolfe, C. The global burden of cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Abbott, M.; Ustoyev, Y. Cancer and the immune system: The history and background of immunotherapy. In Seminars in Oncology Nursing; Elsevier: Amsterdam, The Netherlands, 2019; p. 150923. [Google Scholar]
- Sheikh, A.; Md, S.; Kesharwani, P. Rgd engineered dendrimer nanotherapeutic as an emerging targeted approach in cancer therapy. J. Control. Release 2021, 340, 221–242. [Google Scholar] [CrossRef]
- Shah, S.C.; Kayamba, V.; Peek, R.M., Jr.; Heimburger, D. Cancer control in low-and middle-income countries: Is it time to consider screening? J. Glob. Oncol. 2019, 5, 1–8. [Google Scholar] [CrossRef]
- Balata, H.; Fong, K.M.; Hendriks, L.E.; Lam, S.; Ostroff, J.S.; Peled, N.; Wu, N.; Aggarwal, C. Prevention and early detection for nsclc: Advances in thoracic oncology 2018. J. Thorac. Oncol. 2019, 14, 1513–1527. [Google Scholar] [CrossRef]
- Amjad, M.T.; Chidharla, A.; Kasi, A. Cancer Chemotherapy. Available online: https://europepmc.org/article/nbk/nbk564367 (accessed on 5 September 2023).
- Mosaa, Z.A.; Al-Majidi, M.; Bader, A.T.; Boulhaoua, M.; Ahmad, I. Bioconjugates: A new class of therapeutics for cancer treatment. J. Univ. Babylon Pure Appl. Sci. 2023, 31, 161–175. [Google Scholar] [CrossRef]
- Gomaa, H.A.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.; Mohamed, F.A.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L. Optimization and sar investigation of novel 2, 3-dihydropyrazino [1, 2-a] indole-1, 4-dione derivatives as egfr and brafv600e dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef]
- Epstein, J.B.; Thariat, J.; Bensadoun, R.J.; Barasch, A.; Murphy, B.A.; Kolnick, L.; Popplewell, L.; Maghami, E. Oral complications of cancer and cancer therapy: From cancer treatment to survivorship. CA A Cancer J. Clin. 2012, 62, 400–422. [Google Scholar] [CrossRef]
- London, M.; Gallo, E. Epidermal growth factor receptor (egfr) involvement in epithelial-derived cancers and its current antibody-based immunotherapies. Cell Biol. Int. 2020, 44, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant iii (egfr v iii): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, T.; Zhang, J. Natural tyrosine kinase inhibitors acting on the epidermal growth factor receptor: Their relevance for cancer therapy. Pharmacol. Res. 2020, 161, 105164. [Google Scholar] [CrossRef]
- Shi, K.; Wang, G.; Pei, J.; Zhang, J.; Wang, J.; Ouyang, L.; Wang, Y.; Li, W. Emerging strategies to overcome resistance to third-generation egfr inhibitors. J. Hematol. Oncol. 2022, 15, 1–44. [Google Scholar] [CrossRef]
- Reda, M.; Ngamcherdtrakul, W.; Gu, S.; Bejan, D.S.; Siriwon, N.; Gray, J.W.; Yantasee, W. Plk1 and egfr targeted nanoparticle as a radiation sensitizer for non-small cell lung cancer. Cancer Lett. 2019, 467, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to egfr-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Gold, K.A.; Lee, H.Y.; Kim, E.S. Targeted therapies in squamous cell carcinoma of the head and neck. Cancer: Interdiscip. Int. J. Am. Cancer Soc. 2009, 115, 922–935. [Google Scholar] [CrossRef]
- Dong, R.-F.; Zhu, M.-L.; Liu, M.-M.; Xu, Y.-T.; Yuan, L.-L.; Bian, J.; Xia, Y.-Z.; Kong, L.-Y. Egfr mutation mediates resistance to egfr tyrosine kinase inhibitors in nsclc: From molecular mechanisms to clinical research. Pharmacol. Res. 2021, 167, 105583. [Google Scholar] [CrossRef]
- Crispo, F.; Notarangelo, T.; Pietrafesa, M.; Lettini, G.; Storto, G.; Sgambato, A.; Maddalena, F.; Landriscina, M. Braf inhibitors in thyroid cancer: Clinical impact, mechanisms of resistance and future perspectives. Cancers 2019, 11, 1388. [Google Scholar] [CrossRef]
- Youssif, B.G.; Gouda, A.M.; Moustafa, A.H.; Abdelhamid, A.A.; Gomaa, H.A.; Kamal, I.; Marzouk, A.A. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of brafv600e/p38α with potential antiproliferative activity. J. Mol. Struct. 2022, 1253, 132218. [Google Scholar] [CrossRef]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Gouda, A.M.; Youssif, B.G.; Tateishi, H.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M. Design and synthesis of novel quinoline/chalcone/1, 2, 4-triazole hybrids as potent antiproliferative agent targeting egfr and brafv600e kinases. Bioorg. Chem. 2021, 106, 104510. [Google Scholar] [CrossRef]
- Notarangelo, T.; Sisinni, L.; Condelli, V.; Landriscina, M. Dual egfr and braf blockade overcomes resistance to vemurafenib in braf mutated thyroid carcinoma cells. Cancer Cell Int. 2017, 17, 1–9. [Google Scholar] [CrossRef]
- Van Emburgh, B.O.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Siena, S.; Bardelli, A. Acquired resistance to egfr-targeted therapies in colorectal cancer. Mol. Oncol. 2014, 8, 1084–1094. [Google Scholar] [CrossRef]
- Tandon, R.; Kapoor, S.; Vali, S.; Senthil, V.; Nithya, D.; Venkataramanan, R.; Sharma, A.; Talwadkar, A.; Ray, A.; Bhatnagar, P.K. Dual epidermal growth factor receptor (egfr)/insulin-like growth factor-1 receptor (igf-1r) inhibitor: A novel approach for overcoming resistance in anticancer treatment. Eur. J. Pharmacol. 2011, 667, 56–65. [Google Scholar] [CrossRef]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R., Jr.; Dayyani, F.; Gopal, Y.V.; Jiang, Z.-Q.; Wistuba, I.I.; Tang, X.M. Resistance to braf inhibition in braf-mutant colon cancer can be overcome with pi3k inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef]
- Tan, L.; Zhang, J.; Wang, Y.; Wang, X.; Wang, Y.; Zhang, Z.; Shuai, W.; Wang, G.; Chen, J.; Wang, C. Development of dual inhibitors targeting epidermal growth factor receptor in cancer therapy. J. Med. Chem. 2022, 65, 5149–5183. [Google Scholar] [CrossRef]
- Del Curatolo, A.; Conciatori, F.; Cesta Incani, U.; Bazzichetto, C.; Falcone, I.; Corbo, V.; D’Agosto, S.; Eramo, A.; Sette, G.; Sperduti, I. Therapeutic potential of combined braf/mek blockade in braf-wild type preclinical tumor models. J. Exp. Clin. Cancer Res. 2018, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Begunov, R.; Sokolov, A. Biological activity of condensed pyridine derivatives with a bridgehead nitrogen atom. Pharm. Chem. J. 2023, 56, 1553–1567. [Google Scholar] [CrossRef]
- Teague, S.J. Synthesis of heavily substituted 2-aminopyridines by displacement of a 6-methylsulfinyl group. J. Org. Chem. 2008, 73, 9765–9766. [Google Scholar] [CrossRef] [PubMed]
- Mamedov, I.; Naghiyev, F.; Maharramov, A.; Uwangue, O.; Farewell, A.; Sunnerhagen, P.; Erdelyi, M. Antibacterial activity of 2-amino-3-cyanopyridine derivatives. Mendeleev Commun. 2020, 30, 498–499. [Google Scholar] [CrossRef]
- Al-Omar, M.A.; Amr, A.E.G.E.; Al-Salahi, R.A. Anti-inflammatory, analgesic, anticonvulsant and antiparkinsonian activities of some pyridine derivatives using 2,6-disubstituted isonicotinic acid hydrazides. Arch. Pharm. 2010, 343, 648–656. [Google Scholar] [CrossRef]
- Ismail, M.M.; Farrag, A.M.; Harras, M.F.; Ibrahim, M.H.; Mehany, A.B. Apoptosis: A target for anticancer therapy with novel cyanopyridines. Bioorg. Chem. 2020, 94, 103481. [Google Scholar] [CrossRef] [PubMed]
- Kotb, E.R.; El-Hashash, M.; Salama, M.A.; Kalf, H.S.; Abdel Wahed, N.A. Synthesis and reactions of some novel nicotinonitrile derivatives for anticancer and antimicrobial evaluation. Acta Chim. Slov. 2009, 56, 908–919. [Google Scholar]
- Ryad, N.; My, A.-S.; Ismail, M.M.; El Meligie, S. Design, synthesis and screening of 4,6-diaryl pyridine and pyrimidine derivatives as potential cytotoxic molecules. Chem. Pharm. Bull. 2018, 66, 939–952. [Google Scholar]
- Bass, A.K.; Nageeb, E.-S.M.; El-Zoghbi, M.S.; Mohamed, M.F.; Badr, M.; Abuo-Rahma, G.E.-D.A. Utilization of cyanopyridine in design and synthesis of first-in-class anticancer dual acting pim-1 kinase/hdac inhibitors. Bioorg. Chem. 2022, 119, 105564. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, M.E.; El-Miligy, M.M.; Fahmy, S.M.; Mahran, M.A.; Hazzaa, A.A. Design, synthesis and docking study of pyridine and thieno [2, 3-b] pyridine derivatives as anticancer pim-1 kinase inhibitors. Bioorg. Chem. 2018, 80, 674–692. [Google Scholar] [CrossRef]
- Abouzid, K.A.; Al-Ansary, G.H.; El-Naggar, A.M. Eco-friendly synthesis of novel cyanopyridine derivatives and their anticancer and pim-1 kinase inhibitory activities. Eur. J. Med. Chem. 2017, 134, 357–365. [Google Scholar] [CrossRef]
- Tarazi, H.; El-Gamal, M.I.; Oh, C.-H. Discovery of highly potent v600e-b-raf kinase inhibitors: Molecular modeling study. Bioorg. Med. Chem. 2019, 27, 655–663. [Google Scholar] [CrossRef]
- Ayala-Aguilera, C.C.; Valero, T.; Lorente-Macías, Á.; Baillache, D.J.; Croke, S.; Unciti-Broceta, A. Small molecule kinase inhibitor drugs (1995–2021): Medical indication, pharmacology, and synthesis. J. Med. Chem. 2021, 65, 1047–1131. [Google Scholar] [CrossRef]
- Motati, D.R.; Amaradhi, R.; Ganesh, T. Azaindole therapeutic agents. Bioorg. Med. Chem. 2020, 28, 115830. [Google Scholar] [CrossRef]
- Abou-Zied, H.A.; Beshr, E.A.; Gomaa, H.A.; Mostafa, Y.A.; Youssif, B.G.; Hayallah, A.M.; Abdel-Aziz, M. Discovery of new cyanopyridine/chalcone hybrids as dual inhibitors of egfr/brafv600e with promising antiproliferative properties. Arch. Der Pharm. 2023, 356, 2200464. [Google Scholar] [CrossRef] [PubMed]
- Farooq, S.; Ngaini, Z. One-pot and two-pot synthesis of chalcone based mono and bis-pyrazolines. Tetrahedron Lett. 2020, 61, 151416. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, H.I.; Kazaz, C.; Levent, S.; Gulcin, I. Synthesis, structure elucidation, and in vitro pharmacological evaluation of novel polyfluoro substituted pyrazoline type sulfonamides as multi-target agents for inhibition of acetylcholinesterase and carbonic anhydrase i and ii enzymes. Bioorg. Chem. 2020, 96, 103627. [Google Scholar] [CrossRef] [PubMed]
- Matiadis, D.; Sagnou, M. Pyrazoline hybrids as promising anticancer agents: An up-to-date overview. Int. J. Mol. Sci. 2020, 21, 5507. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Eid, N.M.; George, R.F. Facile synthesis of some pyrazoline-based compounds with promising anti-inflammatory activity. Future Med. Chem. 2018, 10, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.M.; El-Gazzar, M.G.; Aly, H.M.; Othman, R.A. Novel anticancer fused pyrazole derivatives as egfr and vegfr-2 dual tk inhibitors. Front. Chem. 2020, 7, 917. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Eltrombopag: A review of its use in treatment-refractory chronic primary immune thrombocytopenia. Drugs 2011, 71, 1333–1353. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef]
- Ibraheem, F.; Ahmad, M.; Ashfaq, U.A.; Aslam, S.; Ali Khan, Z.; Sultan, S. Synthesis, molecular docking and anti-diabetic studies of novel benzimidazole-pyrazoline hybrid molecules. Pak. J. Pharm. Sci. 2020, 33, 847–855. [Google Scholar]
- Beyhan, N.; Kocyigit-Kaymakcioglu, B.; Gümrü, S.; Aricioglu, F. Synthesis and anticonvulsant activity of some 2-pyrazolines derived from chalcones. Arab. J. Chem. 2017, 10, S2073–S2081. [Google Scholar] [CrossRef]
- Revanasiddappa, B.; Jisha, M.; Kumar, M.V.; Kumar, H. Synthesis, antibacterial and antifungal evlaution of novel pyrazoline derivatives. Dhaka Univ. J. Pharm. Sci. 2018, 17, 221–226. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Abou-Zied, H.A.; Beshr, E.A.M.; Youssif, B.G.M.; Hayallah, A.M.; Abdel-Aziz, M. Design, synthesis, antiproliferative actions, and dft studies of new bis–pyrazoline derivatives as dual egfr/brafv600e inhibitors. Int. J. Mol. Sci. 2023, 24, 9104. [Google Scholar]
- Abou-Zied, H.A.; Youssif, B.G.; Mohamed, M.F.; Hayallah, A.M.; Abdel-Aziz, M. Egfr inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Mahmoud, M.A.; Mostafa, Y.A.; Raslan, A.E.; Youssif, B.G. Novel piperine-carboximidamide hybrids: Design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzym. Inhib. Med. Chem. 2023, 38, 376–386. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Mostafa, Y.A.; Abdelrahman, M.H.; El-Bahrawy, A.H.; Trembleau, L.; Youssif, B.G. Synthesis and biological evaluation of indole-2-carboxamides with potent apoptotic antiproliferative activity as egfr/cdk2 dual inhibitors. Pharmaceuticals 2022, 15, 1006. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Mohammed, A.F.; Abdel Rahman, F.E.-Z.S.; Abdelrahman, M.H.; Gu, X.; Trembleau, L.; Youssif, B.G. Design, synthesis, apoptotic, and antiproliferative effects of 5-chloro-3-(2-methoxyvinyl)-indole-2-carboxamides and pyrido [3, 4-b] indol-1-ones as potent egfrwt/egfrt790m inhibitors. J. Enzym. Inhib. Med. Chem. 2023, 38, 2218602. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, T.F. Chapter XXV—Proliferative disorders. In Significant Pharmaceuticals Reported in Us Patents; DeRosa, T.F., Ed.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2007; pp. 497–608. [Google Scholar]
- Maghraby, M.T.E.; Salem, O.I.; Youssif, B.G.; Sheha, M.M. Design, synthesis, and modelling study of new 1, 2, 3-triazole/chalcone hybrids with antiproliferative action as epidermal growth factor receptor inhibitors. Chem. Biol. Drug Des. 2023, 101, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Mohammed, A.F.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G. Design, synthesis, and antiproliferative activity of new 5-chloro-indole-2-carboxylate and pyrrolo [3, 4-b] indol-3-one derivatives as potent inhibitors of egfrt790m/brafv600e pathways. Molecules 2023, 28, 1269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Villa-Pulgarin, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela-M, R.E.; Cuesta-Marban, A.; Müller, I.; Modolell, M.; Revuelta, J.L. Mitochondria and lipid raft-located fof1-atp synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef]
- Bao, H.; Zhang, Q.; Zhu, Z.; Xu, H.; Ding, F.; Wang, M.; Du, S.; Du, Y.; Yan, Z. Bhx, a novel pyrazoline derivative, inhibits breast cancer cell invasion by reversing the epithelial-mesenchymal transition and down-regulating wnt/β-catenin signalling. Sci. Rep. 2017, 7, 9153. [Google Scholar] [CrossRef] [PubMed]
- Martin, S. Caspases: Executioners of apoptosis. Pathobiol. Hum. Dis. 2014, 2014, 145–152. [Google Scholar]
- Wall, D.M.; McCormick, B.A. Bacterial secreted effectors and caspase-3 interactions. Cell. Microbiol. 2014, 16, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Youssif, B.G.; Mohamed, A.M.; Osman, E.E.A.; Abou-Ghadir, O.F.; Elnaggar, D.H.; Abdelrahman, M.H.; Treamblu, L.; Gomaa, H.A. 5-chlorobenzofuran-2-carboxamides: From allosteric cb1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hafliger, E.; Boccaccino, A.; Lapeyre-Prost, A.; Perret, A.; Gallois, C.; Antista, M.; Pilla, L.; Lecomte, T.; Scartozzi, M.; Soularue, E. Encorafenib plus cetuximab treatment in braf v600e-mutated metastatic colorectal cancer patients pre-treated with an anti-egfr: An ageo-gono case series. Eur. J. Cancer 2022, 168, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.A.; Tüzün, B.; Alsaif, N.A.; Khan, A.A.; Naglah, A.M. Synthesis, characterization, molecular modeling against egfr target and adme/t analysis of novel purine derivatives of sulfonamides. J. Mol. Struct. 2022, 1257, 132600. [Google Scholar] [CrossRef]
- Umar, A.B.; Uzairu, A.; Shallangwa, G.A.; Uba, S. Qsar modelling and molecular docking studies for anti-cancer compounds against melanoma cell line sk-mel-2. Heliyon 2020, 6, e03640. [Google Scholar] [CrossRef]
- Wu, Z.; Lei, T.; Shen, C.; Wang, Z.; Cao, D.; Hou, T. Admet evaluation in drug discovery. 19. Reliable prediction of human cytochrome p450 inhibition using artificial intelligence approaches. J. Chem. Inf. Model. 2019, 59, 4587–4601. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Sattar, N.E.; Badawy, E.H.; AbdEl-Hady, W.H.; Abo-Alkasem, M.I.; Mandour, A.A.; Ismail, N.S. Design and synthesis of new cdk2 inhibitors containing thiazolone and thiazolthione scafold with apoptotic activity. Chem. Pharm. Bull. 2021, 69, 106–117. [Google Scholar] [CrossRef]
- Rai, H.; Barik, A.; Singh, Y.P.; Suresh, A.; Singh, L.; Singh, G.; Nayak, U.Y.; Dubey, V.K.; Modi, G. Molecular docking, binding mode analysis, molecular dynamics, and prediction of admet/toxicity properties of selective potential antiviral agents against sars-cov-2 main protease: An effort toward drug repurposing to combat covid-19. Mol. Divers. 2021, 25, 1905–1927. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.P.; Roy, K. Qsar studies of cyp2d6 inhibitor aryloxypropanolamines using 2d and 3d descriptors. Chem. Biol. Drug Des. 2009, 73, 442–455. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of kinase inhibitors using a bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef]
- Goodrnan, G.; Wilson, R. Comparison of the dependence of the td50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal. 1992, 12, 525–533. [Google Scholar] [CrossRef]
- Gonella Diaza, R.; Manganelli, S.; Esposito, A.; Roncaglioni, A.; Manganaro, A.; Benfenati, E. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ. Res. 2015, 26, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, K.R. The draize eye test. Surv. Ophthalmol. 2001, 45, 493–515. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.S.; Bokhtia, R.M.; Al-Mahmoudy, A.M.; Taher, E.S.; AlAwadh, M.A.; Elagawany, M.; Abdel-Aal, E.H.; Panda, S.; Gouda, A.M.; Asfour, H.Z. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2, 4-dione as hiv-1 fusion inhibitors. Bioorg. Chem. 2020, 99, 103782. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, H.A.; El-Sherief, H.A.; Hussein, S.; Gouda, A.M.; Salem, O.I.; Alharbi, K.S.; Hayallah, A.M.; Youssif, B.G. Novel 1, 2, 4-triazole derivatives as apoptotic inducers targeting p53: Synthesis and antiproliferative activity. Bioorg. Chem. 2020, 105, 104369. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (nM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average IC50 (GI50) | ||
| 21 | 90 | 26 ± 2 | 30 ± 3 | 28 ± 2 | 28 ± 2 | 28 |
| 22 | 89 | 32 ± 3 | 35 ± 3 | 34 ± 3 | 32 ± 3 | 33 |
| 23 | 91 | 29 ± 2 | 34 ± 3 | 32± 3 | 32 ± 3 | 32 |
| 24 | 90 | 27 ± 2 | 31 ± 3 | 29± 2 | 28 ± 2 | 29 |
| 25 | 88 | 40 ± 3 | 45 ± 4 | 42 ± 4 | 42 ± 4 | 42 |
| 26 | 92 | 35 ± 3 | 40 ± 4 | 36 ± 3 | 36 ± 3 | 37 |
| 27 | 88 | 36 ± 3 | 42 ± 4 | 38 ± 3 | 38 ± 3 | 38 |
| 28 | 89 | 25 ± 2 | 30 ± 3 | 26 ± 2 | 26 ± 2 | 27 |
| 29 | 91 | 27 ± 2 | 31 ± 3 | 30 ± 3 | 30 ± 3 | 30 |
| 30 | 90 | 23 ± 2 | 28 ± 2 | 24 ± 2 | 24 ± 2 | 25 |
| Erlotinib | ND | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
| Compd. | EGFR Inhibition IC50 ± SEM (nM) | BRAFV600E Inhibition IC50 ± SEM (nM) |
|---|---|---|
| 21 | 72 ± 5 | 73 ± 6 |
| 24 | 73 ± 5 | 77 ± 6 |
| 28 | 70 ± 5 | 69 ± 6 |
| 29 | 75 ± 5 | 80 ± 7 |
| 30 | 68 ± 4 | 65 ± 5 |
| Erlotinib | 80 ± 5 | ± 5 |
| Compd. No. | Caspase-3 | Caspase-8 | Bax | Bcl-2 | ||||
|---|---|---|---|---|---|---|---|---|
| Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Reduction | |
| 21 | 530 ± 5 | 8 | ND | ND | ND | ND | ND | ND |
| 28 | 595 ± 5 | 9 | 2.25 | 25 | 325 | 36 | 0.80 | 6 |
| 30 | 710 ± 6 | 11 | 2.50 | 28 | 345 | 38 | 0.65 | 8 |
| Staurosporine | 465 ± 4 | 7 | 1.85 | 21 | 288 | 32 | 1.00 | 5 |
| Control | 65 | 1 | 0.09 | 1 | 9 | 1 | 5.00 | 1 |
| Comp. ID | PSA | PPB a | Absorption Level b | CYP2D6 Prediction c | BBB Level d | Solubility Level e | AlogP98 |
|---|---|---|---|---|---|---|---|
| 21 | 97.832 | Yes | 2 | No | 4 | 1 | 6.356 |
| 22 | 133.553 | No | 2 | No | 4 | 2 | 4.962 |
| 23 | 97.832 | Yes | 3 | No | 4 | 1 | 7.021 |
| 24 | 142.483 | No | 2 | No | 4 | 3 | 4.945 |
| 25 | 106.763 | Yes | 2 | No | 4 | 1 | 6.34 |
| 26 | 97.832 | Yes | 3 | No | 4 | 1 | 7.329 |
| 27 | 97.832 | Yes | 3 | No | 4 | 1 | 7.685 |
| 28 | 142.483 | No | 2 | No | 4 | 2 | 4.945 |
| 29 | 115.693 | Yes | 2 | No | 4 | 1 | 6.323 |
| 30 | 151.413 | Yes | 3 | No | 4 | 3 | 4.929 |
| Erlotinib | 71.052 | Yes | 0 | No | 1 | 2 | 4.309 |
| Comp. ID | FDA Rodent Carcinogenicity (Mouse, Female) | Rat Maximum Tolerated Dose (Feed) a | Rat Oral LD50 a | Ocular Irritancy | Skin Irritancy |
|---|---|---|---|---|---|
| 21 | Non-Carcinogen | 0.0760 | 1.27741 | Mild | Non-Irritant |
| 22 | Non-Carcinogen | 0.0312 | 8.48863 | Mild | Non-Irritant |
| 23 | Non-Carcinogen | 0.0597 | 1.67537 | Moderate-Severe | Non-Irritant |
| 24 | Non-Carcinogen | 0.0293 | 14.0883 | Mild | Non-Irritant |
| 25 | Non-Carcinogen | 0.0351 | 0.932102 | Mild | Non-Irritant |
| 26 | Non-Carcinogen | 0.0582 | 4.49508 | Mild | Non-Irritant |
| 27 | Non-Carcinogen | 0.0468 | 2.05463 | Mild | Non-Irritant |
| 28 | Non-Carcinogen | 0.0293 | 14.0883 | Mild | Non-Irritant |
| 29 | Non-Carcinogen | 0.0329 | 5.06156 | Mild | Non-Irritant |
| 30 | Non-Carcinogen | 0.0274 | 12.7428 | Mild | Non-Irritant |
| Erlotinib | Non-Carcinogen | 0.0828 | 0.662169 | Mild | Non-Irritant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wahaibi, L.H.; Abou-Zied, H.A.; Hisham, M.; Beshr, E.A.M.; Youssif, B.G.M.; Bräse, S.; Hayallah, A.M.; Abdel-Aziz, M. Design, Synthesis, and Biological Evaluation of Novel 3-Cyanopyridone/Pyrazoline Hybrids as Potential Apoptotic Antiproliferative Agents Targeting EGFR/BRAFV600E Inhibitory Pathways. Molecules 2023, 28, 6586. https://doi.org/10.3390/molecules28186586
Al-Wahaibi LH, Abou-Zied HA, Hisham M, Beshr EAM, Youssif BGM, Bräse S, Hayallah AM, Abdel-Aziz M. Design, Synthesis, and Biological Evaluation of Novel 3-Cyanopyridone/Pyrazoline Hybrids as Potential Apoptotic Antiproliferative Agents Targeting EGFR/BRAFV600E Inhibitory Pathways. Molecules. 2023; 28(18):6586. https://doi.org/10.3390/molecules28186586
Chicago/Turabian StyleAl-Wahaibi, Lamya H., Hesham A. Abou-Zied, Mohamed Hisham, Eman A. M. Beshr, Bahaa G. M. Youssif, Stefan Bräse, Alaa M. Hayallah, and Mohamed Abdel-Aziz. 2023. "Design, Synthesis, and Biological Evaluation of Novel 3-Cyanopyridone/Pyrazoline Hybrids as Potential Apoptotic Antiproliferative Agents Targeting EGFR/BRAFV600E Inhibitory Pathways" Molecules 28, no. 18: 6586. https://doi.org/10.3390/molecules28186586
APA StyleAl-Wahaibi, L. H., Abou-Zied, H. A., Hisham, M., Beshr, E. A. M., Youssif, B. G. M., Bräse, S., Hayallah, A. M., & Abdel-Aziz, M. (2023). Design, Synthesis, and Biological Evaluation of Novel 3-Cyanopyridone/Pyrazoline Hybrids as Potential Apoptotic Antiproliferative Agents Targeting EGFR/BRAFV600E Inhibitory Pathways. Molecules, 28(18), 6586. https://doi.org/10.3390/molecules28186586