
Identification of Disalicyloyl Curcumin as a Potential DNA Polymerase Inhibitor for Marek’s Disease Herpesvirus: A Computational Study Using Virtual Screening and Molecular Dynamics Simulations


 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
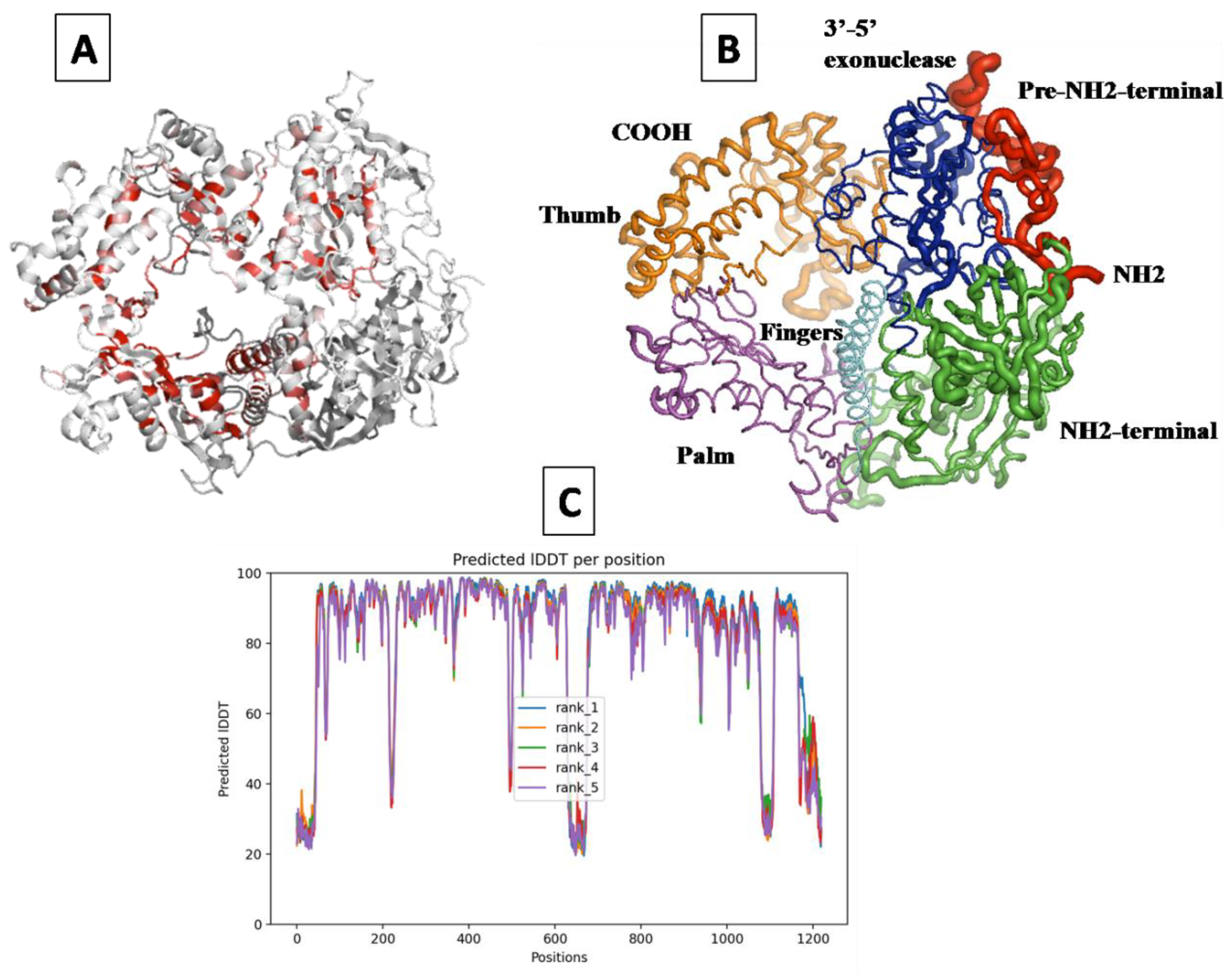
2.1. Prediction of the 3D Structure of MDV DNA Polymerase
2.2. Virtual Screening and Docking
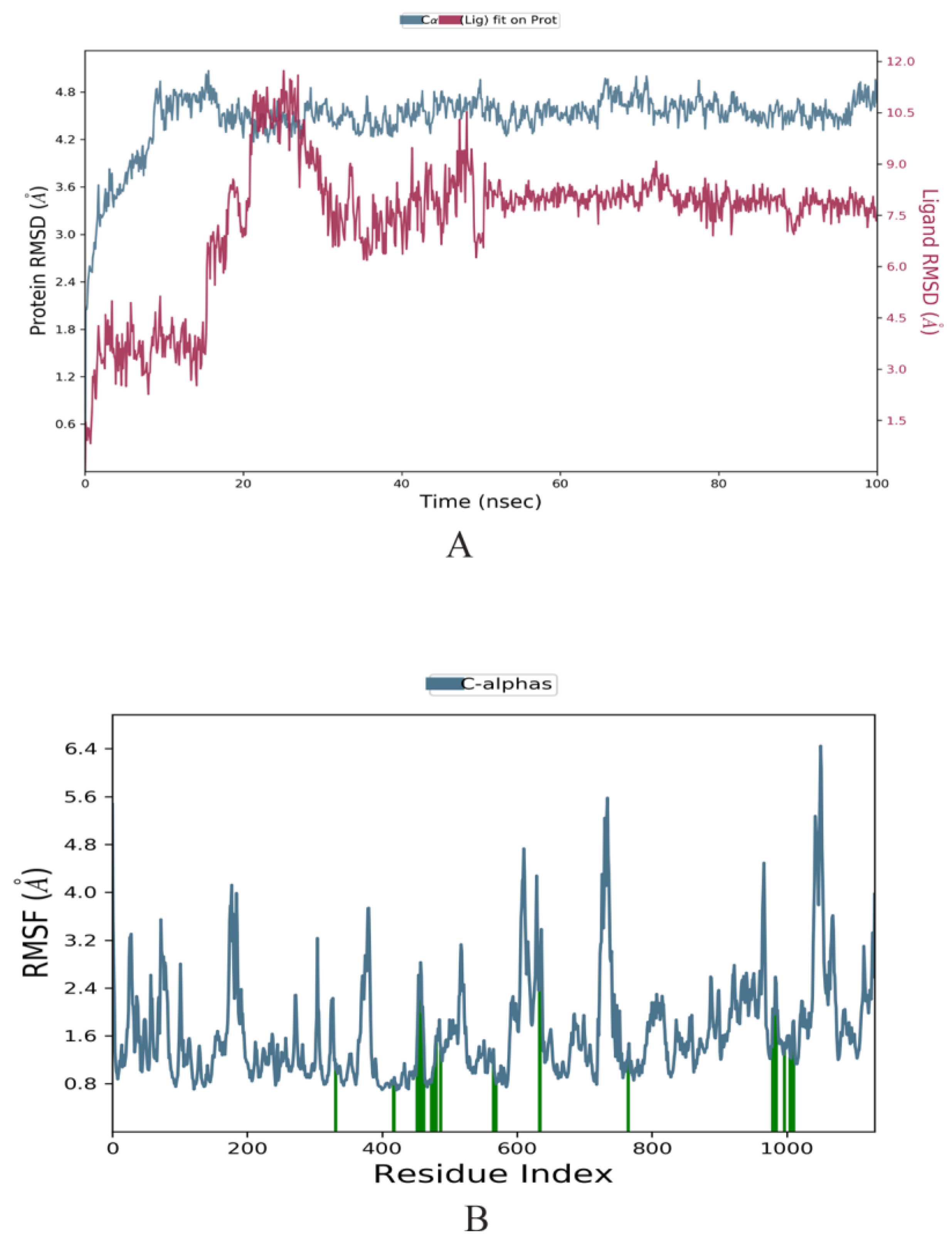
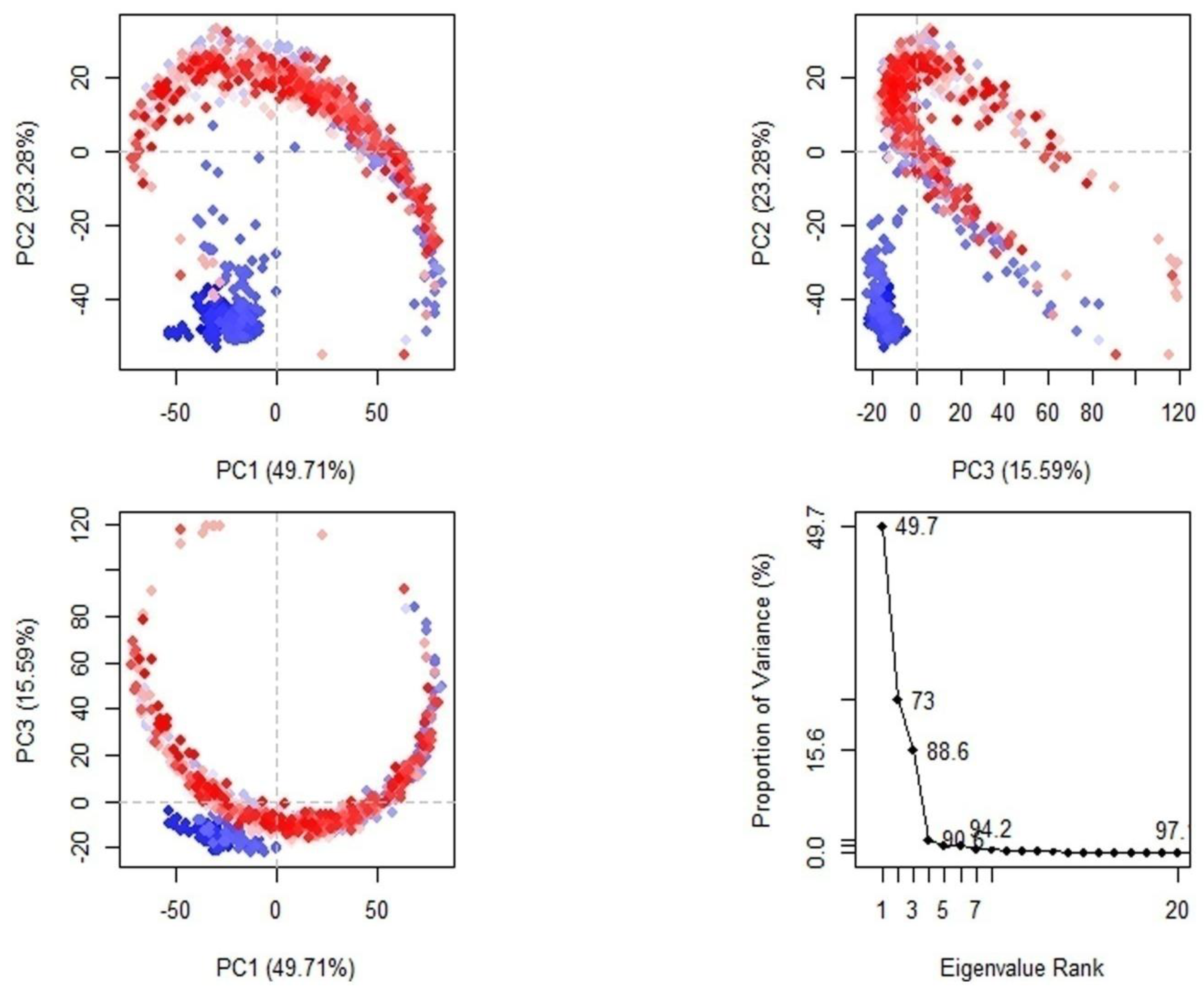
2.3. Molecular Dynamics Simulation
2.4. ADMET Prediction of Compounds
3. Discussion
4. Materials and Methods
4.1. Three-Dimensional Structural Prediction
4.2. Target Preparation and Active Site Analysis
4.3. Ligand Preparation
4.4. In Silico Screening of the Potential MDV DNA Polymerase Inhibitors
4.5. Molecular Dynamics Simulation
4.6. Post-MD Analysis
4.7. Toxicity Predictions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bertzbach, L.D.; Conradie, A.M.; You, Y.; Kaufer, B.B. Latest Insights into Marek’s Disease Virus Pathogenesis and Tumorigenesis. Cancers 2020, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Rozins, C.; Day, T.; Greenhalgh, S. Managing Marek’s disease in the egg industry. Epidemics 2019, 27, 52–58. [Google Scholar] [CrossRef]
- Yilmaz, A.; Turan, N.; Tali, E.B.H.E.; Aydin, O.; Umar, S.; Cakan, B.; Sadeyen, J.R.; Baigent, S.; Iqbal, M.; Nair, V.; et al. Molecular characterisation and phylogenetic analysis of Marek’s disease virus in Turkish layer chickens British poultry. Science 2020, 61, 523–530. [Google Scholar] [CrossRef]
- Dambrine, G.; Labaille, J.; Boissel, É.; Dupuy, C.; Rasschaert, D. Marek’s disease herpesvirus: A model of vaccine-dependent adaptation. L’herpèsvirus de la maladie de Marek (MDV): Un modèle d’adaptation à la pression vaccinale. Virologie 2016, 20, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Yehia, N.; El-Sayed, H.S.; Omar, S.E.; Erfan, A.; Amer, F. Genetic evolution of Marek’s disease virus in vaccinated poultry farms. Vet. World 2021, 14, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Kumar, M.; Mandeep, S.; Singh, Y.; Shukla, P. Systems Biology Approaches for Therapeutics Development against COVID-19. Front. Cell. Infect. Microbiol. 2020, 10, 560240. [Google Scholar] [CrossRef]
- Taylor, M.; Gerriets, V. Acyclovir. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Berdis, A.J. DNA polymerases as therapeutic targets. Biochemistry 2008, 47, 8253–8260. [Google Scholar] [CrossRef]
- Schalkwijk, H.H.; Snoeck, R.; Andrei, G. Acyclovir resistance in herpes simplex viruses: Prevalence and therapeutic alternatives. Biochem. Pharmacol. 2022, 206, 115322. [Google Scholar] [CrossRef]
- Yarchoan, R.; Pluda, J.M.; Perno, C.F.; Mitsuya, H.; Thomas, R.V.; Wyvill, K.M.; Broder, S. Initial clinical experience with dideoxynucleosides as single agents and in combination therapy. Ann. N. Y. Acad. Sci. 1990, 616, 328–343. [Google Scholar] [CrossRef]
- Anderson, V.R.; Perry, C.M. Fludarabine: A review of its use in non-Hodgkin’s lymphoma. Drugs 2007, 67, 1633–1655. [Google Scholar] [CrossRef]
- Ali, S.I.; Sheikh, W.M.; Rather, M.A.; Venkatesalu, V.; Bashir, S.M.; Nabi, S.U. Medicinal plants: Treasure for antiviral drug discovery. Phytotherapyresearch 2021, 35, 3447–3483. [Google Scholar] [CrossRef]
- Dhama, K.; Karthik, K.; Khandia, R.; Munjal, A.; Tiwari, R.; Rana, R.; Khurana, S.K.; Sana, U.; Khan, R.U.; Alagawany, M.; et al. Medicinal and Therapeutic Potential of Herbs and Plant Metabolites/Extracts Countering Viral Pathogens—Current Knowledge and Future Prospects. Curr. Drug Metab. 2018, 19, 236–263. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, M.I.; Ashraf, H.; Aslam, A.; Parveen, S.; Kamran, Z.; Naz, N.; Arshad, M.; Khalid, S.; Mukhtar, M. REPORT-Some ethanobotanically important plants from Cholistan area for anti avian influenza virus (AIV) H9N2 screening. Pak. J. Pharm. Sci. 2019, 32, 2751–2756. [Google Scholar] [PubMed]
- Murcia, P.; Donachie, W.; Palmarini, M. Viral Pathogens of Domestic Animals and Their Impact on Biology, Medicine and Agriculture. In Encyclopedia of Microbiology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 805–819. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- Ghalyanchilangeroudi, A.; Hosseini, H.; Nazarpak, H.; Molouki, A.; Dezfoulian, O.; Morshed, R. Molecular Characterization and Phylogenetic Analysis of Marek’s Disease Virus in Iran. Avian Dis. 2022, 66, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J. The simple chicken major histocompatibility complex: Life and death in the face of pathogens and vaccines. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2000, 355, 1077–1084. [Google Scholar] [CrossRef]
- Ashraf, A.; Mahboob, S.; Andleeb, R.; Ijaz, M.U.; Shah, M.S. Status updates of Newcastle disease and amelioration effects of medicinal plants against Newcastle disease virus: A review. Acta Virol. 2018, 62, 3–15. [Google Scholar] [CrossRef]
- Leyva-Diaz, A.A.; Hernandez-Patlan, D.; Solis-Cruz, B.; Adhikari, B.; Kwon, Y.M.; Latorre, J.D.; Hernandez-Velasco, X.; Fuente-Martinez, B.; Hargis, B.M.; Lopez-Arellano, R.; et al. Evaluation of curcumin and copper acetate against Salmonella Typhimurium infection, intestinal permeability, and cecal microbiota composition in broiler chickens. J. Anim. Sci. Biotechnol. 2021, 12, 23. [Google Scholar] [CrossRef]
- Lopresti, A.L. The Problem of Curcumin and Its Bioavailability: Could Its Gastrointestinal Influence Contribute to Its Overall Health-Enhancing Effects? Adv. Nutr. 2018, 9, 41–50. [Google Scholar] [CrossRef]
- Tulman, E.R.; Afonso, C.L.; Lu, Z.; Zsak, L.; Rock, D.L.; Kutish, G.F. The genome of a very virulent Marek’s disease virus. J. Virol. 2000, 74, 7980–7988. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O.; Djinovic-Carugo, K. Half a century of Ramachandran plots. Acta Crystallogr. 2013, D69, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Binkowski, T.A.; Naghibzadeh, S.; Liang, J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003, 31, 3352–3355. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Shaw, D.E. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Grant, B.J.; Rodrigues, A.P.; El Sawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3D: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S No | Compound | Binding Energy (Kcal/mol) |
|---|---|---|
| 1 | Disalicyloyl curcumin | −12.66 |
| 2 | Ferrocenyl curcumin | −11.04 |
| 3 | Curcumin dimer 1 | −10.54 |
| 4 | Curcumin dimer 2 | −10.26 |
| 5 | N-(4 Methoxyphenylpyrazole)Curcumin | −10.09 |
| 6 | 4-(4-Hydroxybenzylidene) curcumin | −9.91 |
| 7 | Phenyl Diphenoxyphosphinecarboxylate Oxide | −9.66 |
| 8 | Curcumin dimer 3 | −8.94 |
| 9 | Turmeric | −8.90 |
| 10 | Curcumine-difluorée | −8.12 |
| 11 | 4-Benzylidene Curcumin | −8.11 |
| 12 | Di-(tert-Butyl-dimethylsilyl) Curcumin | −8.01 |
| 13 | Withaferin A | −7.74 |
| 14 | 1-Benzothiophene–2-Sulfonamide | −6.60 |
| 15 | Phosphinecarboxylic acid, dihydroxy-, 4-chlorophenyl ester, oxide | −6.53 |
| 16 | Ribasphere | −6.48 |
| 17 | Sodium Phenyl Phenoxycarbonylphosphonate | −6.29 |
| 18 | Methyl [(4-acetylphenoxy)-methoxyphosphoryl] | −6.28 |
| 19 | Methyl [(4-acetylphenoxy)-methoxyphosphoryl] | −6.22 |
| 20 | Adenosine | −6.18 |
| 21 | Tert-butyl 6-fluoro–1H-pyrazolo[3,4-b] pyridine−3-carboxylate | −5.51 |
| 22 | (4-chlorophenoxy)carbonyl ethoxyphosphinic acid | −4.30 |
| 23 | Elvucitabine | −4.21 |
| 24 | 4-[(E)-(4-Chlorophenyl)methylideneamino]–3-(2,4-dichloro–5-fluorophenyl)–1H–1,2,4-triazole–5-thione | −4.08 |
| 25 | 6-Amino–3-[(E)-[2-fluoro–2 (hydroxyméthyl)cyclopropylidène]méthyl]–1,6-dihydropyrimidine–2-one | −3.80 |
| 26 | 8-Oxo-Dgtp | −3.65 |
| 27 | Acyclovir Triphosphate | −3.22 |
| 28 | DisodiumMethox phosphinecarboxylate Oxide | −3.15 |
| 29 | Isopropyl Dimethoxyphosphinecarboxylate Oxide | −3.07 |
| 30 | CyclopentylmethylDimethoxyphosphinecarboxylate Oxide | −3.04 |
| Compound | Structure 2D | Binding Affinity (kcal/mol) | H Bond Residues | Other Interactions | Diagrammatic Sketch Illustrating the Interactions |
|---|---|---|---|---|---|
| Disalicyloyl curcumin | 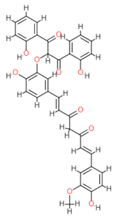 | −12.66 | Lys 501 Glu 521 | Gln 500 Lys 502 Ser 517 Tyr 1037 | 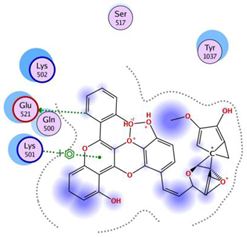 |
| Ferrocenyl curcumin | 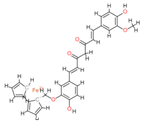 | −11.04 | Lys501 Lys 502 | Gln 500 Ser 517 Ile 518 Thr 520 Glu 521 Ile 603 | 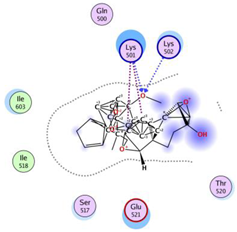 |
| Curcumin dimer 1 |  | −10.54 | Gln 500 Lys 501 Lys 502 Thr 520 Ser 517 | Phe 499 Ile 518 Gln 521 Gln 607 | 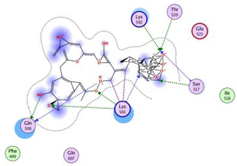 |
| Curcumin dimer 2 |  | −10.26 | Gln 500 | Phe 499 Lys 501 Lys 502 Gly 503 Ser 517 Ile 518 Glu 521 Ile 603 Phe 604 Gln 607 Tyr 799 Tyr 1037 | 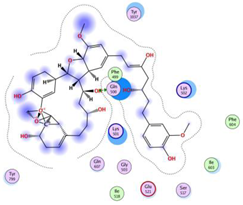 |
| N-(4 Methoxyphenylpyrazole) Curcumin |  | −10.09 | Lys 502 Ser 517 | Gln 500 Lys 501 Ile 518 Thr 520 Glu 521 Tyr 1037 |  |
| Property | Model Name | Disalicyloyl Curcumin | Ferrocenyl Curcumin | Curcumin Dimer 1 | Curcumin Dimer 2 | N-(4 Methoxyphenylpyrazole) Curcumin | Unit |
|---|---|---|---|---|---|---|---|
| Absorption | Water solubility | −3.182 | −4.172 | −2.984 | −2.967 | −4.139 | Numeric (log mol/L) |
| Caco2 permeability | 0.019 | −0.248 | −0.699 | −0.786 | 0.604 | Numeric (log Papp in 10−6 cm/s) | |
| Intestinal absorption (human) | 81.841 | 95.281 | 75.414 | 76.116 | 91.876 | Numeric (% Absorbed) | |
| Skin Permeability | −2.735 | −2.735 | −2.735 | −2.735 | −2.735 | Numeric (log Kp) | |
| P-glycoprotein substrate | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| P-glycoprotein I inhibitor | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| P-glycoprotein II inhibitor | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| Distribution | VDss (human) | −1.632 | −1.123 | −1.685 | −1.648 | −1.183 | Numeric (log L/kg) |
| Fraction unbound (human) | 0.168 | 0.01 | 0.149 | 0.141 | 0.052 | Numeric (Fu) | |
| BBB permeability | −1.532 | −0.668 | −1.929 | −2.042 | −0.765 | Numeric (log BB) | |
| CNS permeability | −3.504 | −2.685 | −3.123 | −3.079 | −2.647 | Numeric (log PS) | |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | Categorical (Yes/No) |
| CYP3A4 substrate | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| CYP1A2 inhibitior | No | No | No | No | No | Categorical (Yes/No) | |
| CYP2C19 inhibitior | No | Yes | No | No | Yes | Categorical (Yes/No) | |
| CYP2C9 inhibitior | Yes | Yes | No | No | Yes | Categorical (Yes/No) | |
| CYP2D6 inhibitior | No | No | No | No | No | Categorical (Yes/No) | |
| CYP3A4 inhibitior | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| Excretion | Total Clearance | −0.153 | 0.864 | −0.132 | −0.038 | 0.09 | Numeric (log ml/min/kg) |
| Renal OCT2 substrate | No | No | No | No | No | Categorical (Yes/No) | |
| Toxicity | AMES toxicity | No | No | No | No | No | Categorical (Yes/No) |
| Max. tolerated dose (human) | 0.37 | −0.034 | 0.391 | 0.377 | 0.087 | Numeric (log mg/kg/day) | |
| hERG I inhibitor | No | No | No | No | No | Categorical (Yes/No) | |
| hERG II inhibitor | Yes | Yes | Yes | Yes | Yes | Categorical (Yes/No) | |
| Oral Rat Acute Toxicity (LD50) | 2.638 | 2.016 | 2.529 | 2.53 | 2.522 | Numeric (mol/kg) | |
| Oral Rat Chronic Toxicity (LOAEL) | 2.508 | 2.08 | 3.069 | 3.331 | 2.226 | Numeric (log mg/kg_bw/day) | |
| Hepatotoxicity | No | Yes | No | No | Yes | Categorical (Yes/No) | |
| Skin Sensitisation | No | No | No | No | No | Categorical (Yes/No) | |
| T. pyriformis toxicity | 0.285 | 0.286 | 0.285 | 0.285 | 0.286 | Numeric (log ug/L) | |
| Minnow toxicity | −0.668 | −3.33 | −2.905 | −3.603 | −2.349 | Numeric (log mM) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherif, A.; Basharat, Z.; Yaseen, M.; Bhat, M.A.; Uddin, I.; Ziedan, N.I.; Mabood, F.; Sadfi-Zouaoui, N.; Messaoudi, A. Identification of Disalicyloyl Curcumin as a Potential DNA Polymerase Inhibitor for Marek’s Disease Herpesvirus: A Computational Study Using Virtual Screening and Molecular Dynamics Simulations. Molecules 2023, 28, 6576. https://doi.org/10.3390/molecules28186576
Cherif A, Basharat Z, Yaseen M, Bhat MA, Uddin I, Ziedan NI, Mabood F, Sadfi-Zouaoui N, Messaoudi A. Identification of Disalicyloyl Curcumin as a Potential DNA Polymerase Inhibitor for Marek’s Disease Herpesvirus: A Computational Study Using Virtual Screening and Molecular Dynamics Simulations. Molecules. 2023; 28(18):6576. https://doi.org/10.3390/molecules28186576
Chicago/Turabian StyleCherif, Aziza, Zarrin Basharat, Muhammad Yaseen, Mashooq Ahmad Bhat, Imad Uddin, Noha I. Ziedan, Fazal Mabood, Najla Sadfi-Zouaoui, and Abdelmonaem Messaoudi. 2023. "Identification of Disalicyloyl Curcumin as a Potential DNA Polymerase Inhibitor for Marek’s Disease Herpesvirus: A Computational Study Using Virtual Screening and Molecular Dynamics Simulations" Molecules 28, no. 18: 6576. https://doi.org/10.3390/molecules28186576
APA StyleCherif, A., Basharat, Z., Yaseen, M., Bhat, M. A., Uddin, I., Ziedan, N. I., Mabood, F., Sadfi-Zouaoui, N., & Messaoudi, A. (2023). Identification of Disalicyloyl Curcumin as a Potential DNA Polymerase Inhibitor for Marek’s Disease Herpesvirus: A Computational Study Using Virtual Screening and Molecular Dynamics Simulations. Molecules, 28(18), 6576. https://doi.org/10.3390/molecules28186576