
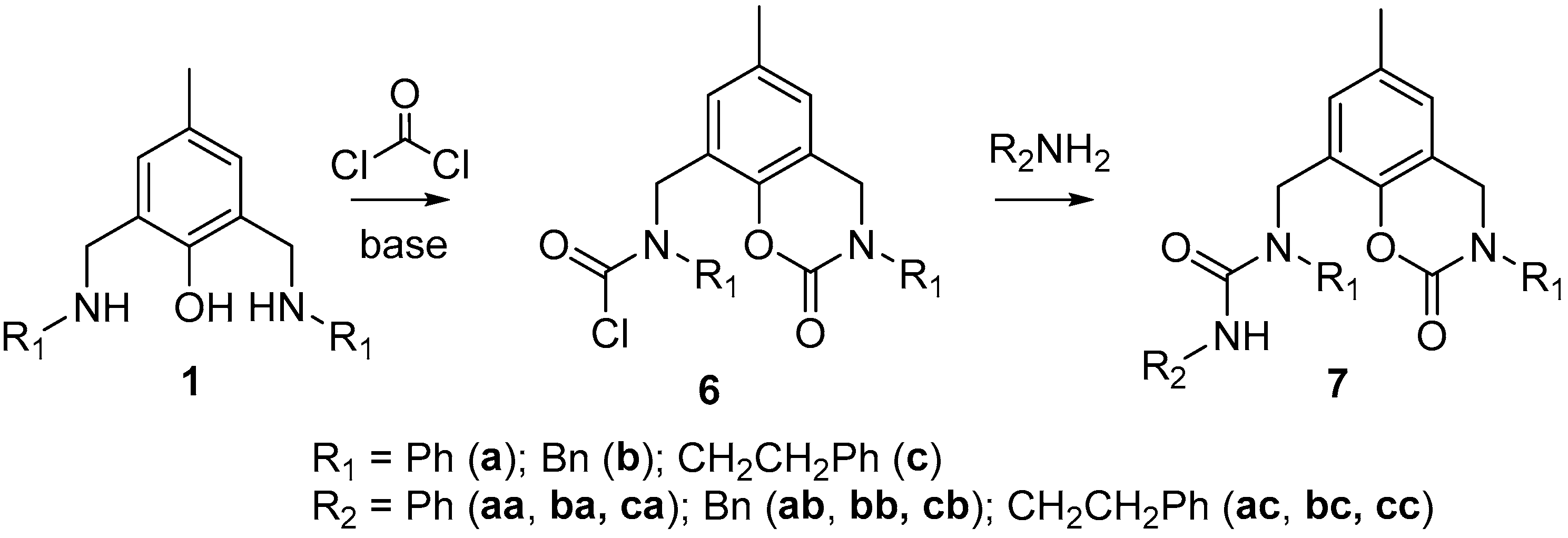
Polydentate N,O-Ligands Possessing Unsymmetrical Urea Fragments Attached to a p-Cresol Scaffold


Abstract
:1. Introduction
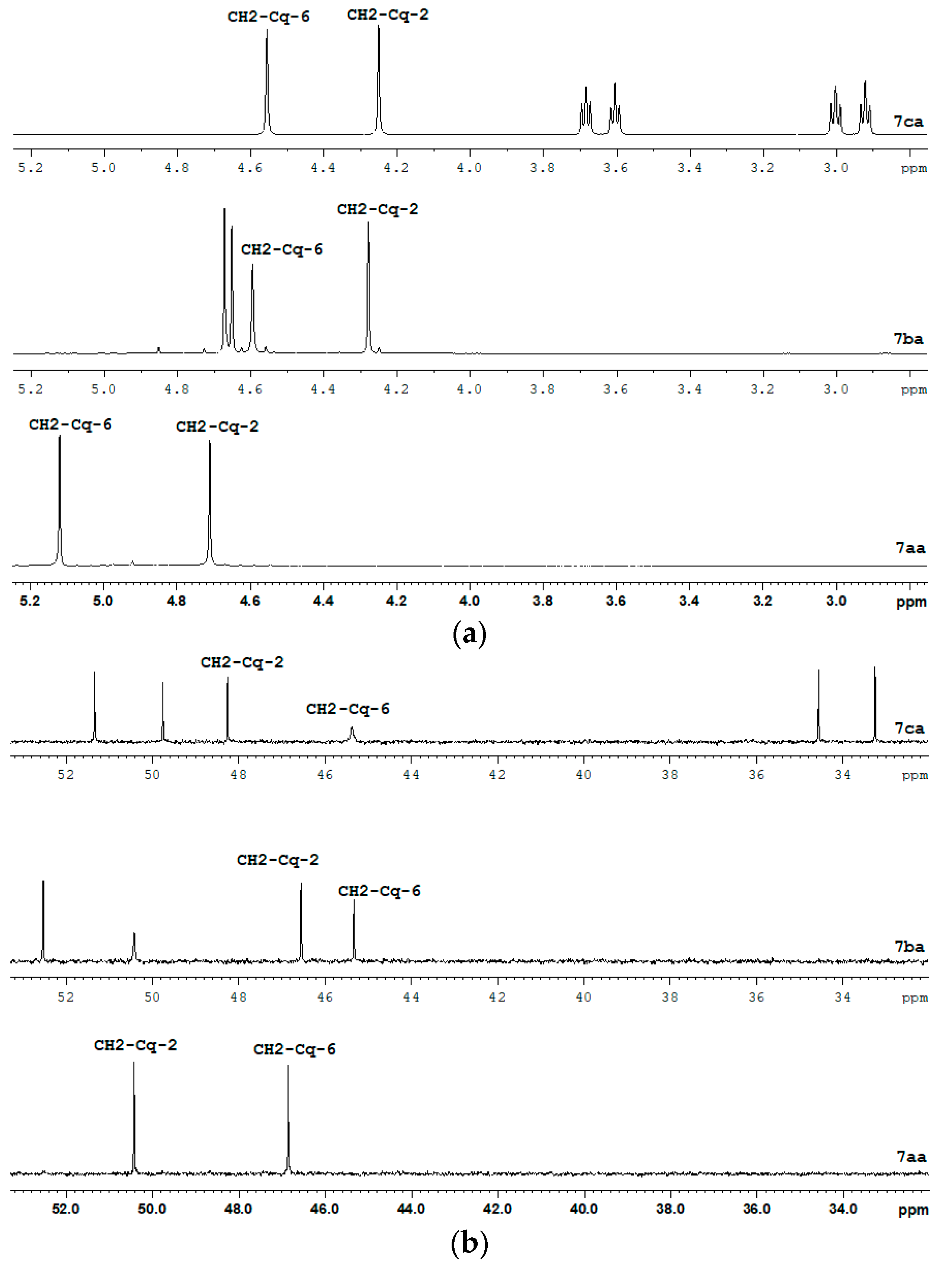
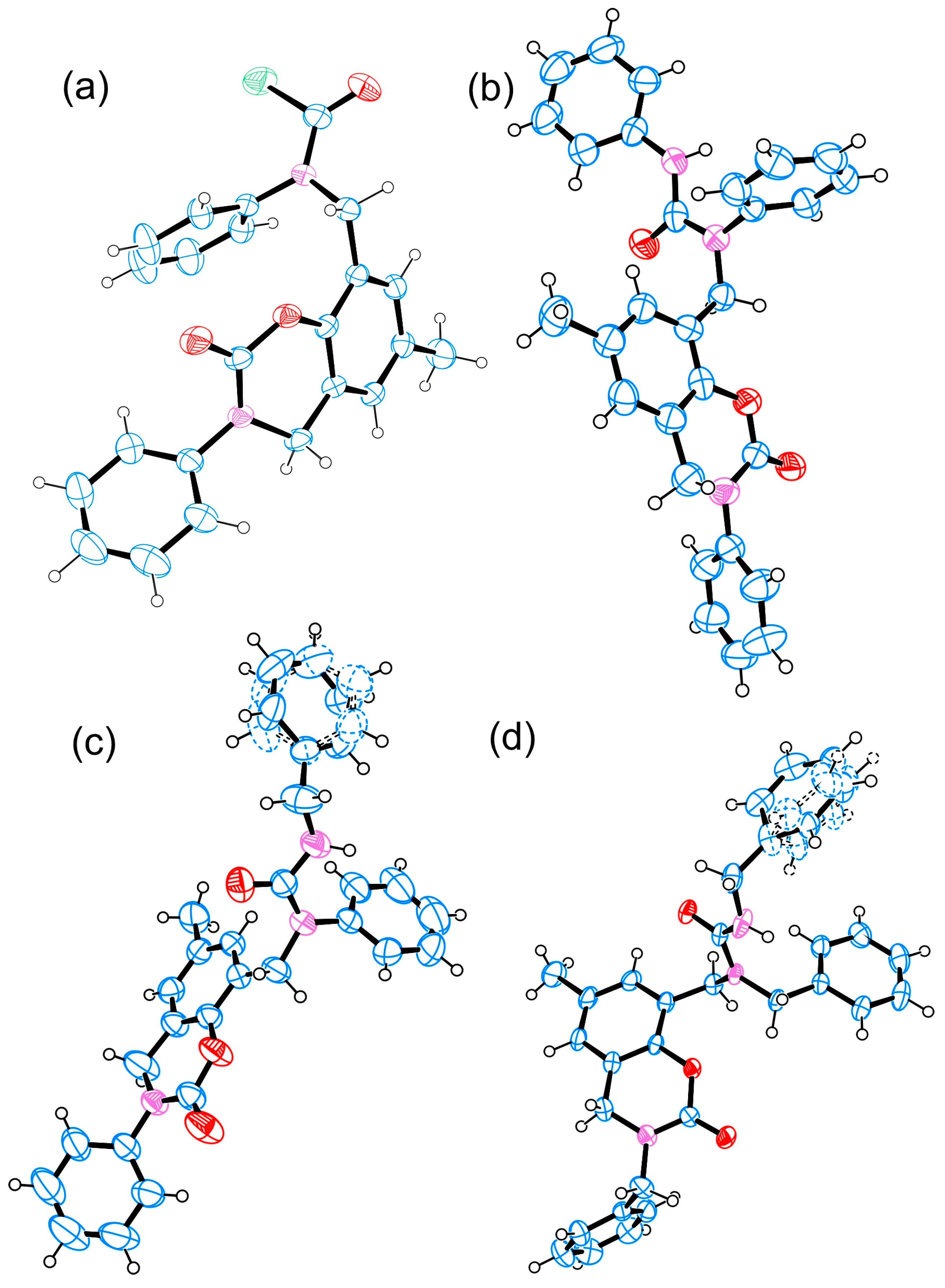
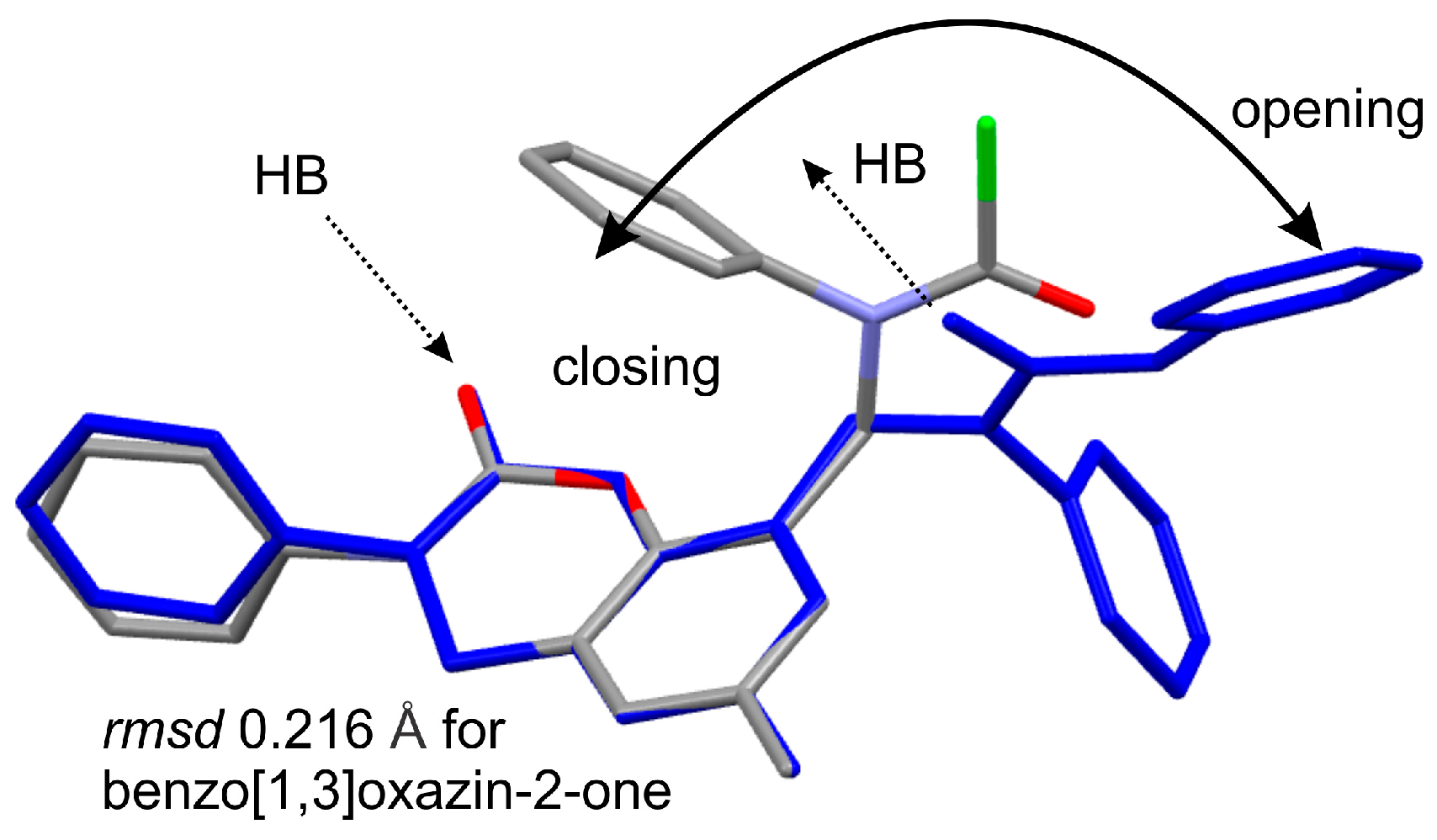
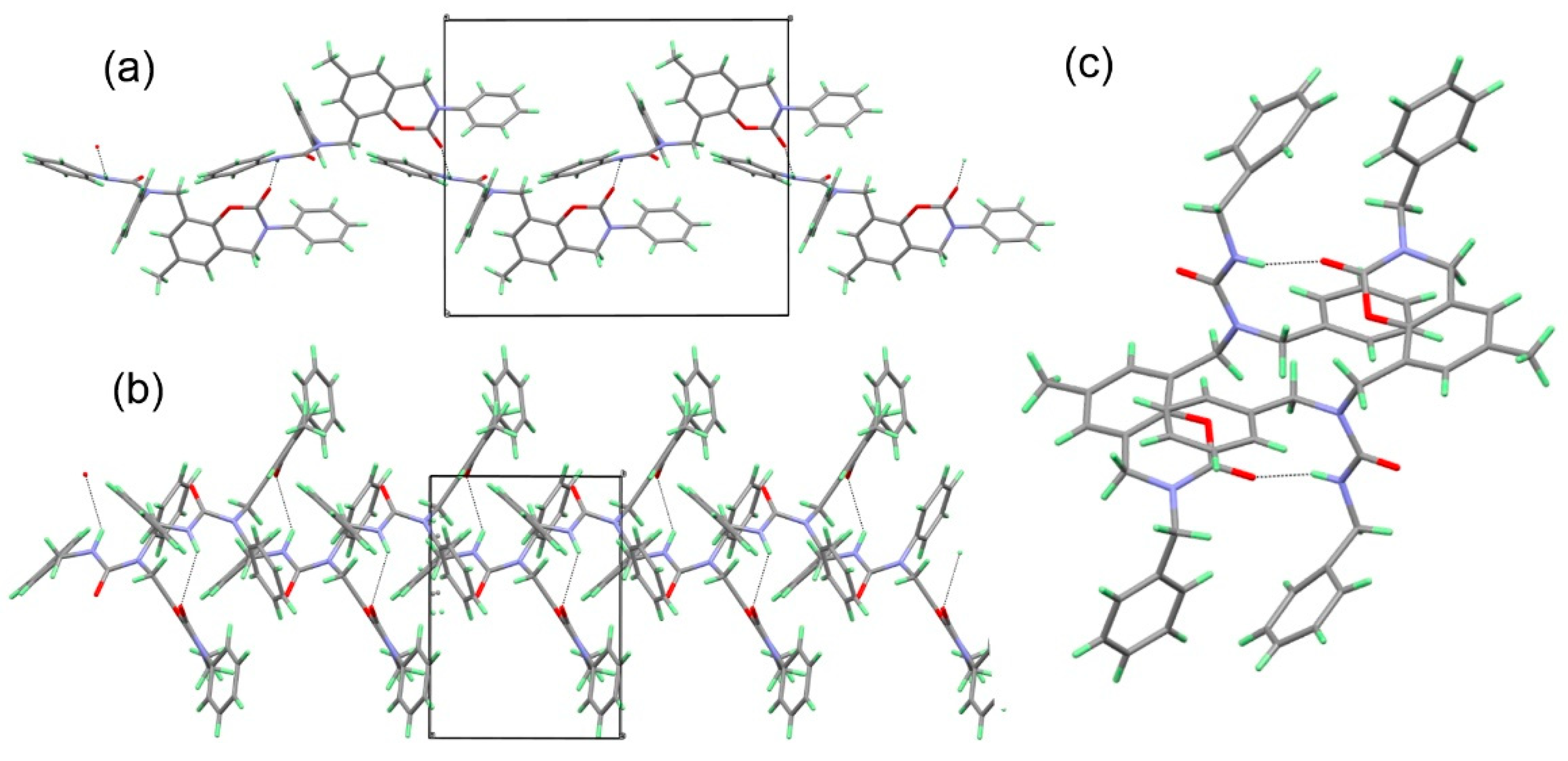
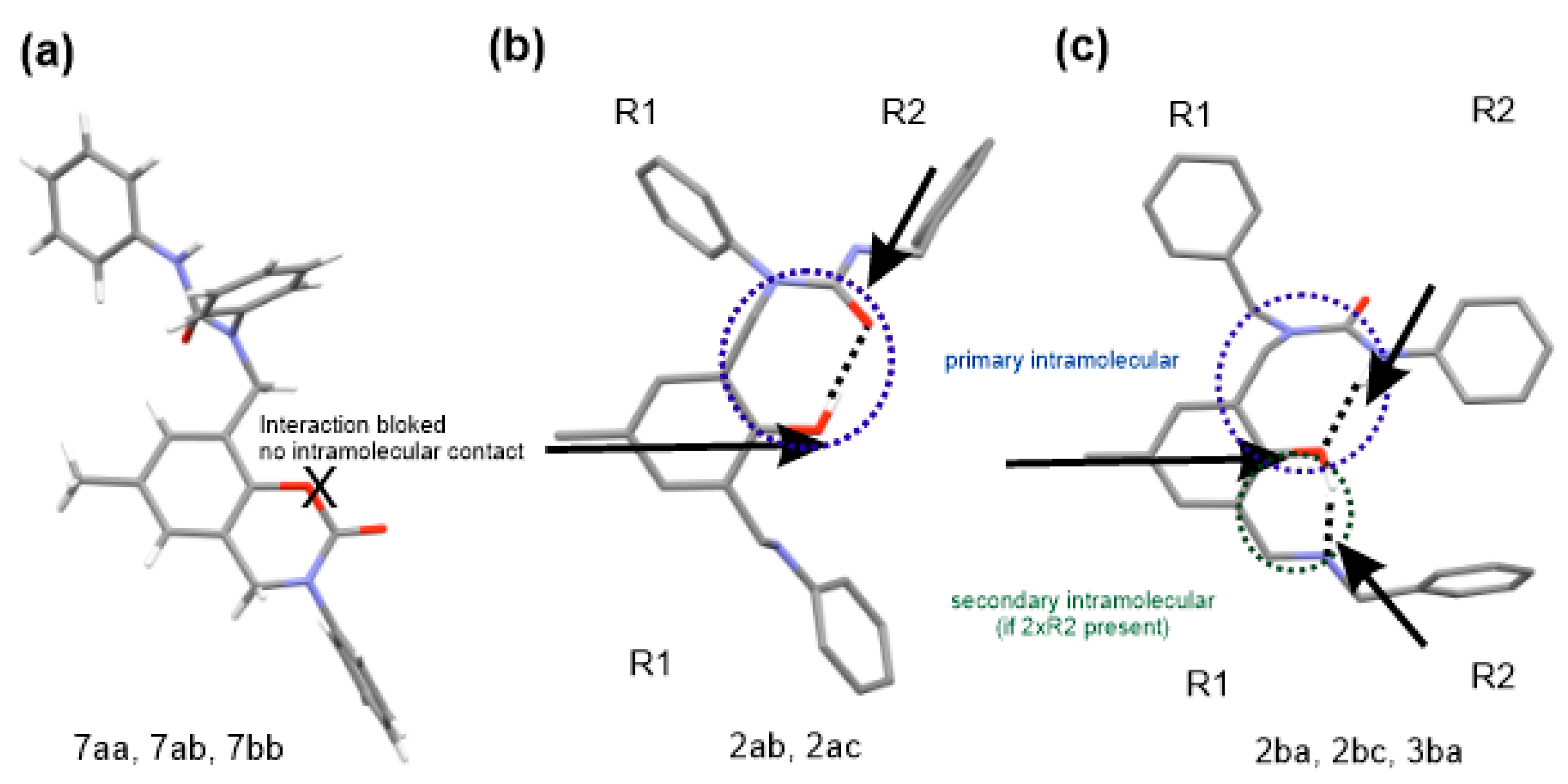
2. Results and Discussion
3. Materials and Methods
3.1. General
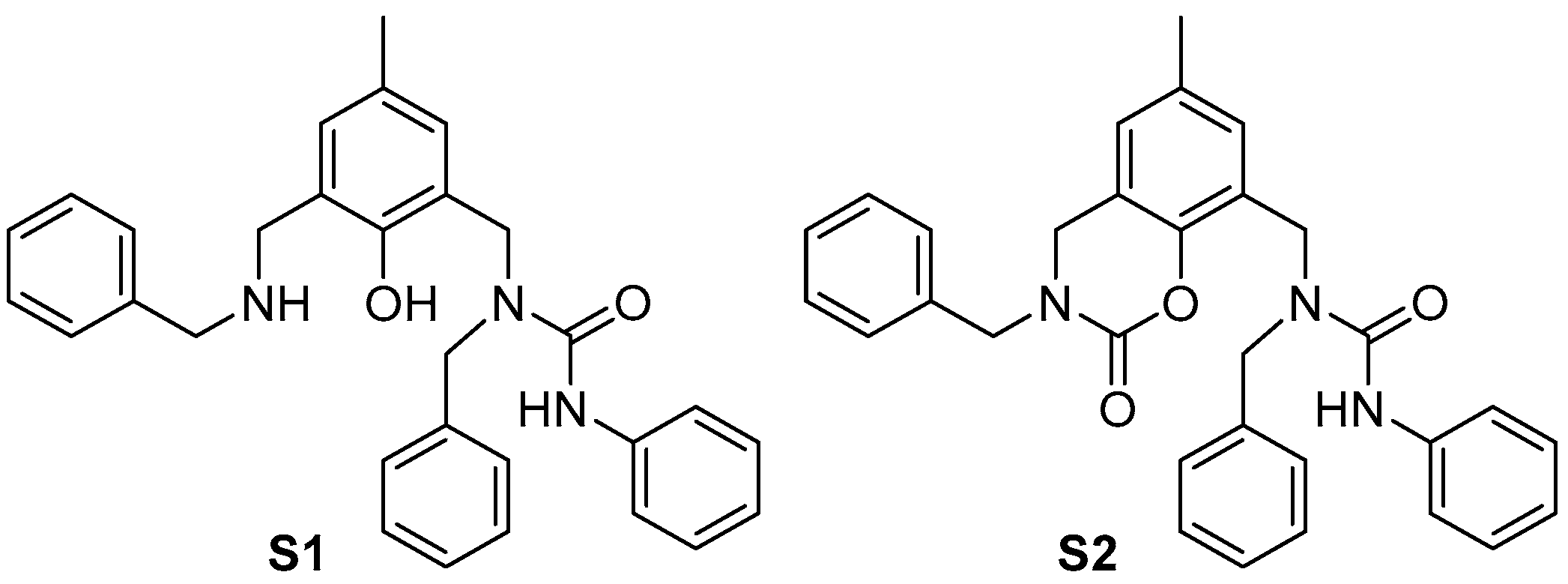
3.2. General Procedure for the Preparation of Bis-Amines 1
- 2,6-bis((benzylimino)methyl)-4-methylphenol: 99% yield; orange solid; m. p. 69.6–69.7 °C (lit. 68–72°C [46]).
- 2,6-bis((phenylethylimino)methyl)-4-methylphenol: 68% yield; orange oil; 1H NMR 2.28 (s, 3H, CH3), 3.01 (t, 4H, J 7.4, CH2-Ph), 3.85 (t, 4H, J 7.4, CH2-N), 7.19–7.24 (m, 6H, CH-2+6 and CH-4 Ph), 7.29 (t, 4H, J 7.5, CH-3+5 Ph), 7.45 (bs, 2H, CH-3 and CH-5), 8.438 (bs, 2H, CH=N), 13.92 (bs, 1H, OH).
- 1a: 99% yield; reddish oil; 1H NMR 2.32 (s, 3H, CH3), 4.39 (s, 4H, CH2), 6.82 (dd, 4H, J 7.6, 1.1, CH-2+H-6 Ph), 6.88 (tt, 2H, J 7.3, 1.1, CH-4 Ph), 7.03 (s, 2H, CH-3 and CH-5), 7.27 (td, 4H, J 7.4, 1.1, CH-3+5 Ph); 13C NMR 20.6 (CH3), 46.4 (CH2), 114.7 (CH-2+6 Ph), 119.3 (CH-4 Ph), 124.4 (Cq-2 and Cq-6), 128.8 (CH-3 and CH-5), 128.9 (Cq-4), 129.3 (CH-3+5 Ph), 147.9 (Cq-1 Ph), 152.8 (Cq-1).
- 1b: 98% yield; yellowish oil; NMR identical with the literature data [47].
- 1c: 88% yield; colourless oil; 1H NMR 2.20 (s, 3H, CH3), 2.86 (t, 4H, J 6.9, CH2-Ph), 2.92 (td, 4H, J 6.9, 1.3, CH2-N), 3.87 (s, 4H, CH2-bridge), 4.89 (bs, 2H, NH), 6.80 (s, 2H, CH-3 and CH-5), 7.19-7.22 (m, 6H, CH-2+6 and CH-4 Ph), 7.29 (td, 4H, J 7.4, 1.7, CH-3+5 Ph); 13C NMR 20.40 (CH3), 35.8 (CH2-Ph), 49.9 (CH2-N), 50.6 (CH2-bridge), 123.5 (Cq-2 and Cq-6), 126.3 (CH-4 Ph), 127.7 (Cq-4), 128.6 (CH-2+6 Ph), 128.7 (CH-3+5 Ph), 128.8 (CH-3 and CH-5), 139.4 (Cq-1 Ph), 154.1 (Cq-1).
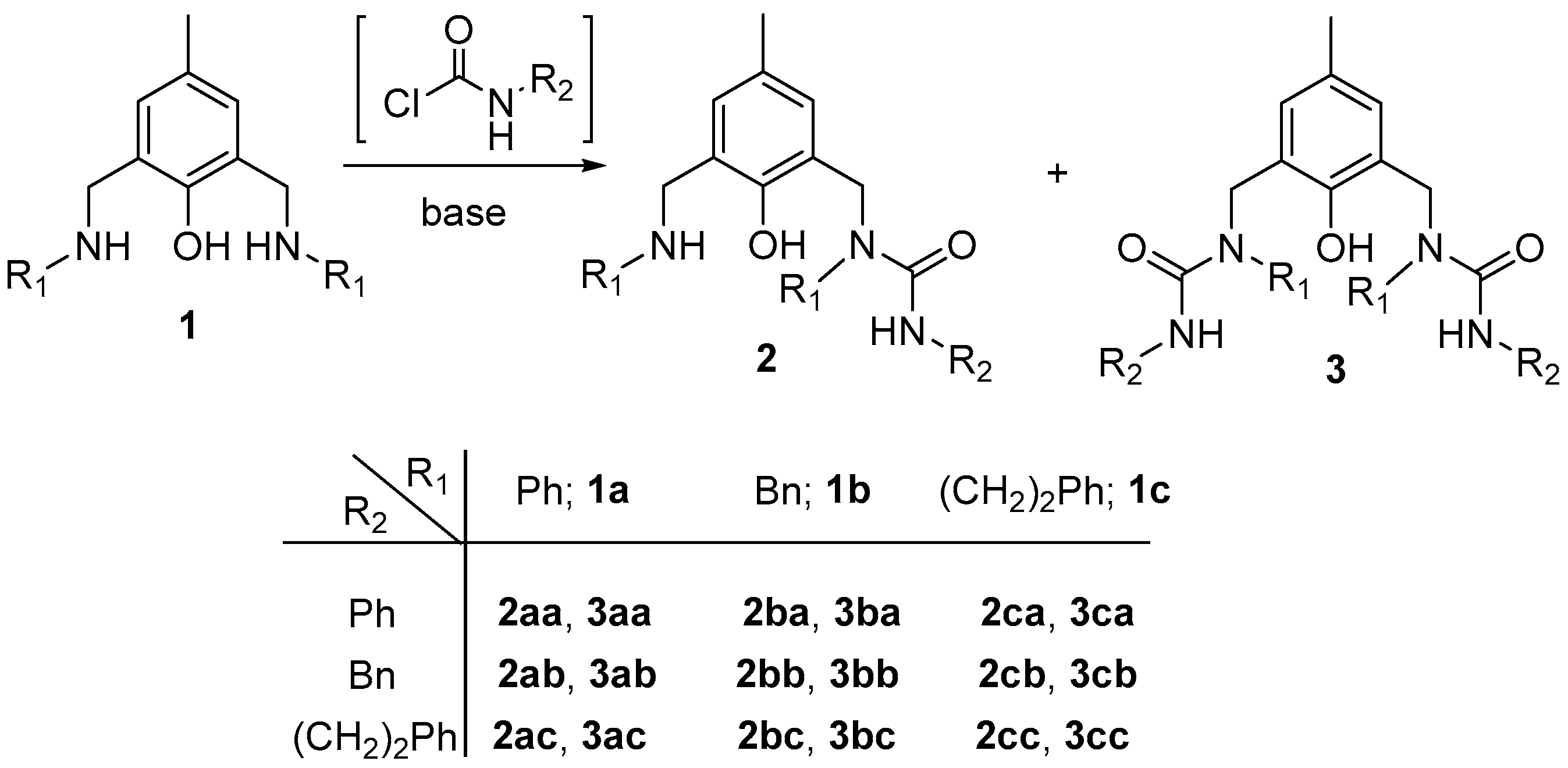
3.3. General Procedure for the Preparation of Ligands 2 and 3

- Ligand 2aa: Rf 0.65 (0.5% MeOH/DCM); colourless solid; m. p. 111.6–112.8 °C; 1H NMR 2.09 (s, 3H, CH3), 4.36 (s, 2H, CH2-Cq-6), 4.77 (s, 2H, CH2-Cq-2), 6.13 (s, 1H, NH-CO), 6.37 (d, 1H, J 2.0, CH-3), 6.70 (tt, 1H, J 7.3, 0.9, CH-4 Ph-N(6) of R1), 6.72 (dd, 2H, J 8.6, 0.9, CH-2+6 Ph-N(6) of R1), 7.03 (m, 1H, CH-4 Ph of R2), 7.04 (d, 1H, J 2.1, CH-5), 7.16 (ddt, 2H, J 8.5, 7.3, 1.9, CH-3+5 Ph-N(6) of R1), 7.19 (dd, 2H, J 8.5, 1.5, CH-2+6 Ph-N(2) of R1), 7.25 (m, 4H, CH-2+6 and CH-3+5 Ph of R2), 7.46 (tt, 1H, J 7.1, 1.3, CH-4 Ph-N(2) of R1), 7.48 (ddt, 2H, J 8.6, 7.0, 1.3, CH-3+5 Ph-N(2) of R1), 9.94 (bs, 1H, OH); 13C NMR 20.36 (CH3), 44.5 (CH2-Cq-6), 50.5 (CH2-Cq-2), 113.6 (CH-2+6 Ph-N(6) of R1), 117.6 (CH-4 Ph-N(6) of R1), 119.8 (CH-2+6 Ph of R2), 122.6 (Cq-2), 123.7 (CH-4 Ph of R2), 126.8 (Cq-6), 127.9 (Cq-4), 128.9 (CH-3+5 Ph of R2), 129.0 (CH-4 Ph-N(2) of R1), 129.1 (CH-2+6 Ph-N(2) of R1), 129.2 (CH-3+5 Ph-N(6) of R1), 130.1 (CH-5), 130.5 (CH-3+5 Ph-N(2) of R1), 130.9 (CH-3), 137.8 (Cq-1 Ph of R2), 140.2 (Cq-1 Ph-N(2) of R1), 148.3 (Cq-1 Ph-N(6) of R1), 152.0 (Cq-1), 156.1 (C=O); ESI MS m/z 226 [M-NPhCONHPh]+ (100), 371 [M − Cl]+ (36), 407 [M+1]+ (58), 429 [M + Na]+ (18).
- Ligand 3aa: Rf 0.35 (0.5% MeOH/DCM); pale brown solid; m. p. 91.1–91.3 °C; 1H NMR 2.07 (s, 3H, CH3), 4.88 (s, 4H, CH2-Cq-2 and CH2-Cq-6), 6.59 (s, 2H, NH-CO), 6.677 (s, 2H, CH-3 and CH-5), 7.00 (tt, 2H, J 7.4, 1.0, CH-4 Ph of R2), 7.22 (m, 8H, CH-2+6 Ph of R1 and CH-3+5 Ph of R2), 7.31 (dd, 4H, J 8.6, 1.0, CH-2+6 Ph of R2), 7.33 (tt, 2H, J 7.4, 1.1, CH-4 Ph of R1), 7.42 (t, 4H, J 7.6, CH-3+5 Ph of R1), 10.06 (bs, 1H, OH); 13C NMR ; 20.4 (CH3), 49.4 (CH2-Cq-2 and CH2-Cq-6), 119.6 (CH-2+6 Ph of R2), 123.2 (CH-4 Ph of R2), 124.0 (Cq-2 and Cq-6), 128.16 (Cq-4), 128.24 (CH-4 Ph of R1), 128.6 (CH-2+6 Ph of R1), 128.8 (CH-3+5 Ph of R2), 130.1 (CH-3+5 Ph of R1), 131.0 (CH-3 and CH-5), 138.5 (Cq-1 Ph of R2), 141.1 (Cq-1 Ph of R1), 151.2 (Cq-1), 155.5 (C=O); ESI MS m/z 557 [M + 1]+ (16), 579 [M + Na]+ (54), 595 [M + K]+ (7), 1135 [2M + Na]+ (100).
- Ligand 2ab: Rf 0.70 (1% MeOH/DCM); colourless solid; m. p. 137.2–137.3 °C; 1H NMR 2.09 (s, 3H, CH3), 4.37 (s, 2H, CH2-Cq-6), 4.40 (d, 2H, J 5.8, CH2 of R2), 4.58 (t, 1H, J 5.8, NH-CO), 4.72 (s, 2H, CH2-Cq-2), 6.34 (d, 1H, J 2.0, CH-3), 6.69 (tt, 1H, J 7.3, 1.0, CH-4 Ph-N(6) of R1), 6.72 (dd, 2H, J 8.4, 0.7, CH-2+6 Ph-N(6) of R1), 7.04 (d, 1H, J 1.8, CH-5), 7.11 (dd, 2H, J 8.6, 1.4, CH-2+6 Ph-N(2) of R1), 7.17 (m, 4H, CH-3+5 Ph-N(6) of R1 and CH-2+6 Ph of R2), 7.23 (tt, 1H, J 7.4, 1.9, CH-4 Ph of R2), 7.29 (td, 2H, J 7.6, 1.6, CH-3+5 Ph of R2), 7.36 (tt, 1H, J 7.3, 1.2, CH-4 Ph-N(2) of R1), 7.41 (ddt, 2H, J 8.5, 7.1, 1.1, CH-3+5 Ph-N(2) of R1), 10.24 (bs, 1H, OH); 13C NMR 20.38 (CH3), 44.4 (CH2-Cq-6), 44.9 (CH2 of R2), 50.8 (CH2-Cq-2), 113.5 (CH-2+6 Ph-N(6) of R1), 117.4 (CH-4 Ph-N(6) of R1), 123.0 (Cq-2), 126.9 (Cq-6), 127.3 (CH-2+6 Ph of R2), 127.4 (CH-4 Ph of R2), 127.7 (Cq-4), 128.63 (CH-4 Ph-N(2) of R1), 128.65 (CH-3+5 Ph of R2), 128.9 (CH-2+6 Ph-N(2) of R1), 129.1 (CH-3+5 Ph-N(6) of R1), 130.1 (CH-5), 130.4 (CH-3+5 Ph-N(2) of R1), 130.9 (CH-3), 138.8 (Cq-1 Ph of R2), 140.6 (Cq-1 Ph-N(2) of R1), 148.6 (Cq-1 Ph-N(6) of R1), 152.2 (Cq-1), 158.7 (C=O); ESI MS m/z 359 [M-PhCH2 + 1]+ (100), 452 [M + 1]+ (47), 474 [M + Na]+ (16), 490 [M + K]+ (2), 926 [2M + Na]+ (20).
- Ligand 3ab: Rf 0.27 (1% MeOH/DCM); colourless solid; m. p. 142.7–142.9 °C; 1H NMR 2.09 (s, 3H, CH3), 4.40 (d, 4H, J 5.8, CH2 of R2), 4.49 (t, 2H, J 5.8, NH-CO), 4.83 (s, 4H, CH2-Cq-2 and CH2-Cq-6), 6.68 (s, 2H, CH-3 and CH-5), 7.16 (dd, 4H, J 8.1, 1.4, CH-2+6 Ph of R1), 7.17 (m, 6H, CH-2+6 and CH-4 Ph of R2), 7.28 (m, 6H, CH-4 Ph of R1 and CH-3+5 Ph of R2), 7.35 (ddt, 4H, J 7.9, 7.4, 1.7, CH-3+5 Ph of R1), 10.02 (bs, 1H, OH); 13C NMR 20.5 (CH3), 44.4 (CH2-Cq-6), 44.8 (CH2 of R2), 49.6 (CH2-Cq-2 and CH2-Cq-6), 124.4 (Cq-2 and Cq-6), 127.2 (CH-4 Ph of R2), 127.3 (CH-2+6 Ph of R2), 127.67 (Cq-6), 127.69 (Cq-4), 128.5 (CH-2+6 Ph of R1), 128.6 (CH-3+5 Ph of R2), 129.9 (CH-3+5 Ph of R1), 130.2 (CH-3 and CH-5), 139.3 (Cq-1 Ph of R2), 141.6 (Cq-1 Ph of R1), 151.5 (Cq-1), 158.0 (C=O); ESI MS m/z 585 [M + 1]+ (43), 607 [M + Na]+ (89), 623 [M + K]+ (15), 1191 [2M + Na]+ (100).
- Ligand 2ac: Rf 0.55 (1% MeOH/DCM); colourless solid; m. p. 134.5–134.6 °C; 1H NMR 2.08 (s, 3H, CH3), 2.73 (t, 2H, J 6.8, CH2-Ph of R2), 3.42 (td, 2H, J 6.8, 5.9, CH2-N of R2), 4.22 (t, 1H, J 5.7, NH-CO), 4.37 (s, 2H, CH2-Cq-6), 4.67 (s, 2H, CH2-Cq-2), 6.33 (d, 1H, J 2.0, CH-3), 6.69 (tt, 1H, J 7.3, 1.0, CH-4 Ph-N(6) of R1), 6.72 (dd, 2H, J 8.6, 1.0, CH-2+6 Ph-N(6) of R1), 6.96 (m, 2H, CH-2+6 Ph-N(2) of R1), 7.02 (dd, 2H, J 8.4, 1.2, CH-2+6 Ph of R2), 7.04 (d, 1H, J 1.8, CH-5), 7.15-7.20 (m, 5H, CH-3+5 Ph-N(6) of R1 and CH-3+5 and CH-4 Ph of R2), 7.34 (m, 3H, CH-3+5 and CH-4 Ph-N(2) of R1), 10.28 (bs, 1H, OH); 13C NMR 20.3 (CH3), 36.0 (CH2-Ph of R2), 42.1 (CH2-N of R2), 44.4 (CH2-Cq-6), 50.5 (CH2-Cq-2), 113.4 (CH-2+6 Ph-N(6) of R1), 117.3 (CH-4 Ph-N(6) of R1), 123.0 (Cq-2), 126.4 (CH-4 Ph of R2), 126.9 (Cq-6), 127.6 (Cq-4), 128.4 (CH-4 Ph-N(2) of R1), 128.5 (CH-2+6 Ph of R2), 128.7 (CH-2+6 Ph-N(2) of R1), 128.8 (CH-3+5 Ph of R2), 129.1 (CH-3+5 Ph-N(6) of R1), 130.0 (CH-5), 130.1 (CH-3+5 Ph-N(2) of R1), 130.8 (CH-3), 138.7 (Cq-1 Ph of R2), 140.5 (Cq-1 Ph-N(2) of R1), 148.6 (Cq-1 Ph-N(6) of R1), 152.1 (Cq-1), 158.6 (C=O); ESI MS m/z 373 [M-PhCH2 + 1]+ (100), 466 [M + 1]+ (55), 489 [M + Na]+ (5), 504 [M + K]+ (1), 954 [2M + Na]+ (15).
- Ligand 3ac: Rf 0.21 (1% MeOH/DCM); colourless solid; m. p. 132.8–132.9 °C; 1H NMR 2.09 (s, 3H, CH3), 2.74 (t, 4H, J 6.9, CH2-Ph of R2), 3.42 (td, 4H, J 6.9, 6.0, CH2-N of R2), 4.42 (t, 2H, J 5.7, NH-CO), 4.37 (s, 4H, CH2-Cq-2 and CH2-Cq-6), 6.65 (d, 2H, J 2.0, CH-3 and CH-5), 7.02 (dd, 4H, J 8.8, 1.4, CH-2+6 Ph of R1), 7.06 (dd, 4H, J 8.5, 1.4, CH-2+6 Ph of R2), 7.16 (tt, 2H, J 7.3, 1.2, CH-4 Ph of R2), 7.20 (ddt 4H, J 8.3, 7.0, 1.2, CH-3+5 Ph of R2), 7.25 (tt, 2H, J 7.3, 1.3, CH-4 Ph of R1), 7.29 (ddt, 4H, J 8.4, 7.1, 1.3, CH-3+5 Ph of R1), 10.02 (bs, 1H, OH); 13C NMR 20.5 (CH3), 36.1 (CH2-Ph of R2), 42.1 (CH2-N of R2), 49.3 (CH2-Cq-2 and CH2-Cq-6), 124.4 (Cq-2 and Cq-6), 126.2 (CH-4 Ph of R2), 127.5 (Cq-4), 127.6 (CH-4 Ph of R1), 128.46 (CH-3+5 Ph of R2), 128.48 (CH-2+6 Ph of R1), 128.7 (CH-2+6 Ph of R2), 129.7 (CH-3+5 Ph of R1), 130.1 (CH-3 and CH-5), 139.1 (Cq-1 Ph of R2), 141.5 (Cq-1 Ph of R1), 151.4 (Cq-1), 157.9 (C=O); ESI MS m/z 613 [M + 1]+ (34), 635 [M + Na]+ (100), 652 [M + K]+ (10).
- Ligand 3ba: Rf 0.47 (1% MeOH/DCM); colourless solid; m. p. 181.5–181.6 °C; 1H NMR 2.24 (s, 3H, CH3), 4.50 (bs, 4H, CH2-Cq-2 and CH2-Cq-6), 4.62 (s, 4H, CH2 of R1), 6.90 (s, 2H, CH-3 and CH-5), 6.99 (bt, 2H, J 7.3, CH-4 Ph of R2), 7.19 (bt 4H, J 7.9, CH-3+5 Ph of R2), 7.30-7.37 (bm, 10H, CH Ph), 7.41 (bt 4H, J 7.7, CH-3+5 Ph of R1), 11.23 (bs, 1H, OH); 13C NMR 20.4 (CH3), 49.7 (CH2-Cq-2 and CH2-Cq-6), 49.8 (CH2- of R1), 119.9 (CH-2+6 Ph of R2), 123.1 (Cq-2 and Cq-6), 123.7 (CH-4 Ph of R2), 127.2 (CH-4 Ph of R1), 128.7 (Cq-4), 128.8 (CH-3+5 Ph of R2), 128.9 (CH-2+6 Ph of R1), 129.2 (CH-3+5 Ph of R1), 132.2 (CH-3 and CH-5), 138.6 (Cq-1 Ph of R1), 138.9 (Cq-1 Ph of R2), 151.6 (Cq-1), 157.2 (C=O); ESI MS m/z 226 [PhNHCONHCH2Ph]+ (28), 360 [M-PhCH2NHCONHPh+1]+ (95), 585 [M + 1]+ (100), 1170 [2M + 1]+ (59).
- Ligand 2bb: Rf 0.26 (3% MeOH/DCM); yellow solid; m. p. 121.0–121.2 °C; 1H NMR 2.21 (s, 3H, CH3), 3.72 (s, 2H, CH2-N(6) of R1), 3.87 (s, 2H, CH2-Cq-6), 4.34 (s, 2H, CH2-Cq-2), 4.45 (d, 2H, J 5.6, CH2-NH of R2), 4.66 (s, 2H, CH2-N(2) of R1), 6.01 (bs, 1H, NH-CO), 6.74 (d, 1H, J 1.3, CH-5), 6.82 (d, 1H, J 1.3, CH-3), 7.17 (dd, 2H, J 7.7, 1.1, CH-2+6 Ph of R2), 7.19-7.24 (m, 5H, CH Ph), 7.28 (m, 2H, 2 CH-4 Ph), 7.33 (m, 6H, CH Ph), 11.34 (bs, 1H, OH); 13C NMR 20.5 (CH3), 44.8 (CH2-Cq-2), 44.9 (CH2 of R2), 50.2 (CH2-N(2) of R1), 51.5 (CH2-Cq-6), 52.6 (CH2-N(6) of R1), 122.4 (Cq-6), 123.7 (Cq-2), 126.8 (CH Ph), 127.2 (CH-2+6 Ph of R2), 127.4 (2 CH Ph), 127.4 (Cq-4), 127.6 (CH Ph), 127.8 (CH Ph), 128.29 (2 CH Ph), 128.33 (2 CH Ph), 128.6 (2 CH Ph), 128.69 (2 CH Ph), 128.72 (CH-5), 129.5 (CH-3), 138.2 (Cq-1 Ph of N(6)-R1), 138.6 (Cq-1 Ph of N(2)-R1), 139.9 (Cq-1 Ph of R2), 153.2 (Cq-1), 158.8 (C=O); ESI MS m/z 480 [M + 1]+ (100), 502 [M + Na]+ (7), 982 [2M + Na]+ (20).
- Ligand 3bb: Rf 0.36 (1% MeOH/DCM); colourless solid; m. p. 152.6–152.7 °C; 1H NMR 2.20 (s, 3H, CH3), 4.39 (d, 4H, J 5.5, CH2-NH of R2), 4.42 (s, 4H, CH2-Cq-2 and CH2-Cq-6), 4.52 (d, 4H, J 5.5, CH2 of R1), 5.35 (bs, 1H, NH-CO), 6.83 (s, 2H, CH-3 and CH-5), 7.14 (d 4H, J 7.4, CH-2+6 Ph of R1), 7.20 (t, 2H, J 7.4, CH-4 Ph of R1), 7.26 (m, 8H, CH-3+5 Ph of R1 and CH-2+6 Ph of R2). 7.29 (t, 2H, J 7.3, CH-4 Ph of R2), 7.35 (dd 4H, J 7.6, 7.3, CH-3+5 Ph of R2), 10.71 (bs, 1H, OH); 13C NMR 20.4 (CH3), 45.0 (CH2 of R2), 46.9 (CH2-Cq-2 and CH2-Cq-6), 50.2 (CH2 of R1), 123.9 (Cq-2 and Cq-6), 127.0 (CH-2+6 Ph of R2), 127.1 (CH-4 Ph of R1), 127.3 (CH-2+6 Ph of R1), 127.6 (CH-4 Ph of R2), 128.2 (Cq-4), 128.5 (CH-3+5 Ph of R1), 128.9 (CH-3+5 Ph of R2), 130.9 (CH-3 and CH-5), 137.1 (Cq-1 Ph of R1), 139.2 (Cq-1 Ph of R2), 151.9 (Cq-1), 159.3 (C=O); ESI MS m/z 613 [M + 1]+ (26), 635 [M + Na]+ (100), 651 [M + K]+ (15).
- Ligand 2bc: Rf 0.22 (3% MeOH/DCM); yellow solid; m. p. 140.9–141.1 °C; 1H NMR 2.21 (s, 3H, CH3), 2.78 (t, 2H, J 7.0, CH2-Ph of R2), 3.49 (td, 2H, J 7.0, 5.7, CH2-N of R2), 3.78 (s, 2H, CH2-N(6) of R1), 3.92 (s, 2H, CH2-Cq-6), 4.30 (s, 2H, CH2-Cq-2), 4.55 (s, 2H, CH2-N(2) of R1), 5.45 (bs, 1H, NH-CO), 6.77 (bs, 1H, CH-5), 6.80 (bs, 1H, CH-3), 7.09 (dd, 2H, J 7.5, 1.1, CH-2+6 Ph of R2), 7.15 (tt, 1H, J 7.3, 1.0, CH-4 Ph of R2), 7.20 (ddt, 2H, J 7.5, 7.2, 1.0, CH-3+5 Ph of R2), 7.27 (m, 4H, CH Ph), 7.29 (dd, 2H, J 7.6, 1.2, CH-2+6 Ph), 7.33 (m, 4H, CH Ph), 11.13 (bs, 1H, OH); 13C NMR 20.6 (CH3), 36.4 (CH2-Ph of R2), 42.3 (CH2-N of R2), 45.2 (CH2-Cq-2), 50.0 (CH2-N(2) of R1), 51.4 (CH2-Cq-6), 52.6 (CH2-N(6) of R1), 122.5 (Cq-6), 123.6 (Cq-2), 126.1 (CH-4 Ph of R2), 127.2 (CH Ph), 127.55 (CH-2+6 Ph), 127.65 (CH Ph), 128.0 (Cq-4), 128.2 (CH Ph), 128.36 (2 CH Ph), 128.39 (CH-3+5 Ph of R2), 128.6 (2 CH Ph), 128.7 (2 CH Ph), 128.8 (CH-2+6 Ph of R2), 128.9 (CH-5), 129.4 (CH-3), 138.2 (Cq-1 Ph of N(2) R2), 138.3 (Cq-1 Ph of N(6) R2), 139.6 (Cq-1 Ph of R2), 153.2 (Cq-1), 159.0 (C=O); ESI MS m/z 387 [M-PhCH2 + 1]+ (8), 494 [M + 1]+ (100), 516 [M + Na]+ (4), 987 [2M + 1]+ (14), 1010 [2M + Na + 1]+ (8).
- Ligand 3bc: Rf 0.34 (1% MeOH/DCM); colourless solid; m. p. 131.6–131.7 °C; 1H NMR 2.24 (s, 3H, CH3), 2.88 (t, 4H, J 7.1, CH2-Ph of R2), 3.49 (bt, 4H, J 7.1, CH2-N of R2), 4.24 (d, 4H, J 4.9, CH2 of R1), 4.34 (s, 4H, CH2-Cq-2 and CH2-Cq-6), 4.89 (bs, 2H, NH-CO), 6.88 (s, 2H, CH-3 and CH-5), 7.12 (d, 4H, J 7.3, CH-2+6 Ph of R1), 7.17 (m, 6H, CH Ph), 7.22-7.28 (m, 10H, CH Ph), 10.79 (bs, 1H, OH); 13C NMR 20.5 (CH3), 36.4 (CH2-Ph of R2), 45.0 (CH2 of R1), 46.6 (CH2-Cq-2 and CH2-Cq-6), 49.2 (CH2-N of R2), 123.8 (Cq-2 and Cq-6), 126.7 (CH Ph), 127.1 (CH Ph), 127.5 (CH-2+6 Ph of R1), 128.1 (Cq-4), 128.5 (2 CH Ph), 128.8 (4 CH Ph), 130.6 (CH-3 and CH-5), 139.09 (Cq-1 Ph), 139.11 (Cq-1 Ph), 151.8 (Cq-1), 159.1 (C=O); ESI MS m/z 641 [M + 1]+ (39), 663 [M + Na]+ (100), 679 [M + K]+ (15).
- Ligand 2ca: Rf 0.24 (1% MeOH/DCM); colourless oil; 1H NMR 2.23 (s, 3H, CH3), 2.82 (t, 2H, J 6.9, CH2-Ph of N(6) R1), 2.93 (m, 4H, CH2-Ph of N(2) R1 and CH2-N of N(6) R1), 3.63 (t, 2H, J 7.5, CH2-N of N(2) R1), 3.95 (s, 2H, CH2-Cq-6), 4.35 (s, 2H, CH2-Cq-2), 6.77 (d, 1H, J 1.6, CH-5), 6.94 (d, 1H, J 1.7, CH-3), 6.97 (tt, 1H, J 7.3, 1.1, CH-4 Ph of R1), 7.14 (dd, 2H, J 8.2, 1.2, CH-2+6 Ph of R2), 7.21 (m, 2H, CH-4 Ph of R1 and CH-4 Ph of R2), 7.24–7.32 (m, 8H, CH-3+5 Ph and CH-2+6 Ph of R1), 7.36 (bd, 2H, J 7.6, CH-2+6 Ph of R1); 13C NMR 20.5 (CH3), 34.4 (CH2-Ph of N(2) R1), 35.4 (CH2-Ph of N(6) R1), 46.4 (CH2-Cq-2), 49.3 (CH2-N of N(6) R1), 49.6 (CH2-N of N(2) R1), 52.0 (CH2-Cq-6), 119.3 (CH-2+6 Ph of R1), 122.1 (CH-4 Ph of R1), 122.3 (Cq-6), 123.8 (Cq-2), 126.6 (CH-4 Ph of R2), 128.5 (Cq-4), 128.5 (CH-4 Ph of R1), 128.58 (2 CH Ph), 128.62 (2 CH Ph), 128.68 (2 CH Ph), 128.73 (2 CH Ph), 129.0 (2 CH Ph), 129.1 (CH-5), 130.1 (CH-3), 138.6 (Cq-1 Ph of N(6) R1), 139. 7 (Cq-1 Ph of N(2) R1), 140.1 (Cq-1 Ph of R2), 153.2 (Cq-1), 156.0 (C=O); ESI MS m/z 494 [M + 1]+ (100), 496 [M + Na]+ (8), 532 [M + K]+ (1), 987 [2M + 1]+ (17), 1010 [2M + Na + 1]+ (17).
- Ligand 3ca: Rf 0.35 (1% MeOH/DCM); yellow solid; m. p. 118.5–118.8°C; 1H NMR 2.30 (s, 3H, CH3), 2.97 (t, 4H, J 6.9, CH2-Ph of R1), 3.60 (t, 4H, J 6.9, CH2-N of R1), 4.45 (bs, 4H, CH2-Cq-2 and CH2-Cq-6), 6.95 (m, 3H, CH Ph), 6.99 (s, 2H, CH-3 and CH-5), 7.15 (m, 6H, CH Ph), 7.28 (m, 7H, CH Ph), 7.36 (m, 4H, CH Ph), 11.35 (bs, 1H, OH); 13C NMR 20.5 (CH3), 34.1 (CH2-Ph of R1), 46.7 (CH2-Cq-2 and CH2-Cq-6), 49.2 (CH2-N of R1), 119.8 (Cq-2 and Cq-6), 123.1 (2 CH-4 Ph), 128.6 (Cq-4), 128.8 (8 CH Ph), 128.96 (2 CH-4 Ph), 129.03 (8 CH Ph), 131.9 (CH-3 and CH-5), 138.8 (Cq-1 Ph of R2), 139.3 (Cq-1 Ph of R1), 153.6 (Cq-1), 158.2 (C=O); ESI MS m/z 494 [M-PhNHCO + 1]+ (23), 613[M + 1]+ (32), 635 [M + Na]+ (100), 651 [M + K]+ (13).
- Ligand 2cb: Rf 0.14 (2% MeOH/DCM); colourless solid; m. p. 90.3–90.5 °C; 1H NMR 2.20 (s, 3H, CH3), 2.77 (t, 2H, J 6.9, CH2-Ph of N(6) R1), 2.86 (t, 2H, J 6.9, CH2-N of N(6) R1), 2.91 (dd, 2H, J 7.8, 7.4, CH2-Ph of N(2) R1), 3.61 (dd, 2H, J 7.7, 7.5, CH2-N of N(2) R1), 3.86 (s, 2H, CH2-Cq-6), 4.30 (s, 2H, CH2-Cq-2), 4.38 (d, 2H, J 5.5, CH2 of R2), 5.66 (bs, 1H, NH-CO), 6.73 (d, 1H, J 1.7, CH-5), 6.89 (d, 1H, J 1.7, CH-3), 7.14 (dd, 2H, J 8.1, 1.2, CH-2+6 Ph), 7.17-7.30 (m, 13H, CH Ph); 13C NMR 20.5 (CH3), 34.8 (CH2-Ph of N(2) R1), 35.5 (CH2-Ph of N(6) R1), 44.8 (CH2 of R2), 45.880 (CH2-Cq-2), 49.5 (CH2-N of N(6) R1), 49.9 (CH2-N of N(2) R1), 52.0 (CH2-Cq-6), 122.2 (Cq-6), 124.0 (Cq-2), 126.3 (CH Ph), 126.6 (CH Ph), 126.9 (CH Ph), 127.4 (2 CH Ph), 128.3 (Cq-4), 128.4 (2 CH Ph), 128.6 (2 CH Ph), 128.69 (2 CH Ph), 128.71 (CH-5), 128.73 (2 CH Ph), 129.0 (2 CH Ph), 129.1 (CH-3), 138.9 (Cq-1 Ph of N(6) R1), 139.6 (Cq-1 Ph of N(2) R1), 139.9 (Cq-1 Ph of R2), 153.2 (Cq-1), 158.5 (C=O); ESI MS m/z 508 [M + 1]+ (100), 530 [M + Na]+ (9), 1037 [2M + Na]+ (100).
- Ligand 3cb: Rf 0.41 (2% MeOH/DCM); colourless solid; m. p. 151.6–152.0 °C; 1H NMR 2.24 (s, 3H, CH3), 2.88 (t, 4H, J 7.1, CH2-Ph of R1), 3.49 (bt, 4H, J 7.0, CH2-N of R1), 4.24 (d, 4H, J 4.4, CH2 of R2), 4.34 (s, 4H, CH2-Cq-2 and CH2-Cq-6), 4.89 (bs, 2H, NH-CO), 6.88 (s, 2H, CH-3 and CH-5), 7.12 (d, 4H, J 7.3, CH-2+6 Ph), 7.17 (m, 6H, CH Ph), 7.25 (m, 10H, CH Ph), 10.79 (bs, 1H, OH); 13C NMR 20.5 (CH3), 34.4 (CH2-Ph of R1), 45.0 (CH2 of R2), 46.6 (CH2-Cq-2 and CH2-Cq-6), 49.2 (CH2-N of R1), 123.8 (Cq-2 and Cq-6), 126.7 (2 CH-4 Ph), 127.1 (2 CH-4 Ph), 127.5 (4 CH Ph), 128.1 (Cq-4), 128.5 (4 CH Ph), 128.8 (8 CH Ph), 130.6 (CH-3 and CH-5), 138.7 (Cq-1 Ph of R1), 139.1 (Cq-1 Ph of R2), 151.8 (Cq-1), 159.1 (C=O); ESI MS m/z 641 [M + 1]+ (18), 663 [M + Na]+ (100), 679 [M + K]+ (14).
- Ligand 2cc: Rf 0.11 (2% MeOH/DCM); colourless oil; 1H NMR 2.204 (s, 3H, CH3), 2.73 (t, 2H, J 7.1, CH2-Ph of R2), 2.84 (t, 2H, J 7.4, CH2-Ph of N(2) R1), 2.87 (dd, 2H, J 7.0, 6.7, CH2-Ph of N(6) R1), 2.96 (dd, 2H, J 7.0, 6.6, CH2-N of N(6) R1), 2.73 (td, 2H, J 7.0, 5.8, CH2-N of R2), 3.61 (bt, 2H, J 7.4, CH2-N of N(2) R1), 3.92 (s, 2H, CH2-Cq-6), 4.24 (s, 2H, CH2-Cq-2), 4.95 (bs, 1H, NH-CO), 6.77 (bs, 1H, CH-5), 6.84 (d, 1H, J 1.6, CH-3), 7.14-7.19 (m, 7H, CH Ph), 7.21-7.24 (m, 4H, CH Ph), 7.2707.31 (m, 4H, CH Ph); 13C NMR 20.5 (CH3), 34.5 (CH2-Ph of N(2) R1), 35.2 (CH2-Ph of N(6) R1), 36.4 (CH2-Ph of R2), 42.1 (CH2-N of R2), 45.9 (CH2-Cq-2), 49.35 (CH2-N of N(6) R1), 49.44 (CH2-N of N(2) R1), 51.6 (CH2-Cq-6), 122.0 (Cq-6), 123.8 (Cq-2), 126.2 (CH-4 Ph), 126.4 (CH-4 Ph), 126.6 (CH-4 Ph), 128.2 (Cq-4), 128.4 (2 CH Ph), 128.6 (2 CH Ph), 128.68 (2 CH Ph), 128.73 (2 CH Ph), 128.80 (2 CH Ph), 128.85 (2 CH Ph), 129.0 (CH-5), 129.4 (CH-3), 138.6 (Cq-1 Ph), 139.4 (Cq-1 Ph), 139.5 (Cq-1 Ph), 153.1 (Cq-1), 158.6 (C=O); ESI MS m/z 522 [M + 1]+ (100), 544 [M + Na]+ (4), 1043 [2M + 1]+ (8), 1066 [2M + Na + 1]+ (2).
- Ligand 3cc: Rf 0.29 (1% MeOH/DCM); colourless solid; m. p. 107.2–107.3 °C; 1H NMR 2.22 (s, 3H, CH3), 2.73 (t, 4H, J 7.1, CH2-Ph of R2), 2.80 (t, 4H, J 7.2, CH2-Ph of R1), 3.35 (td, 4H, J 6.8, 6.1, CH2-N of R2), 3.40 (bd, 4H, J 7.1, CH2-N of R1), 4.28 (s, 4H, CH2-Cq-2 and CH2-Cq-6), 4.67 (bs, 1H, NH-CO), 6.84 (s, 2H, CH-3 and CH-5), 7.12 (m, 8H, CH-2+6 Ph), 7.18 (m, 2H, CH-4 Ph), 7.21-7.28 (m, 10H, CH Ph), 10.77 (bs, 1H, OH); 13C NMR 20.5 (CH3), 34.3 (CH2-Ph of R1), 36.2 (CH2-Ph of R2), 42.1 (CH2-N of R2), 46.6 (CH2-Cq-2 and CH2-Cq-6), 49.1 (CH2-N of R1), 124.0 (Cq-2 and Cq-6), 126.3 (CH Ph), 126.6 (CH Ph), 128.0 (Cq-4), 128.3 (CH Ph), big common signals for CH Ph at 128.5, 128.7, 128.7 and 128.8, 130.5 (CH-3 and CH-5), 139.0 (Cq-1 Ph), 139.2 (Cq-1 Ph), 151.7 (Cq-1), 158.9 (C=O); ESI MS m/z 669 [M + 1]+ (58), 691 [M + Na]+ (100), 707 [M + K]+ (11).
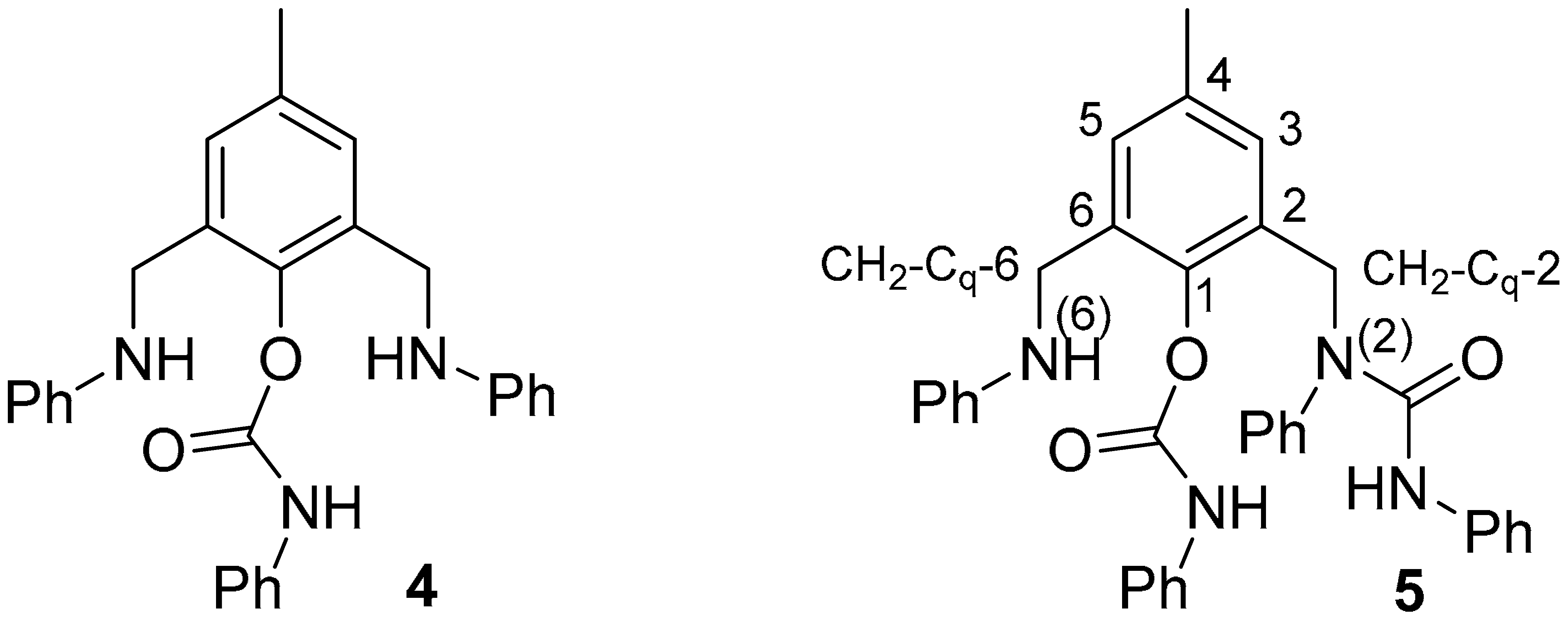
- Ligand 4: Rf 0.30 (DCM); pale red solid; m. p. 149.8–150.0 °C; 1H NMR 2.28 (s, 3H, CH3), 3.80 (bs, 2H, NH), 4.27 (s, 4H, CH2), 6.63 (d, 4H, J 7.9, CH-2+6 Ph of R1), 6.71 (t, 2H, J 7.3, CH-4 Ph of R1), 7.08 (bt, 1H, J 6.7, CH-4 Ph of R2), 7.14 (dd, 4H, J 8.5, 7.4, CH-3+5 Ph of R1), 7.17 (s, 2H, CH-3 and CH-5), 7.28 (m, 4H, CH-2+6 and CH-3+5 Ph of R2); 13C NMR 20.2 (CH3), 43.8 (CH2), 113.1 (CH-2+6 Ph of R1), 117.9 (CH-4 Ph of R1), 118.9 (CH-2+6 Ph of R2), 124.1 (CH-4 Ph of R2), 129.0 (CH-3+5 Ph of R2), 129.2 (CH-3+5 Ph of R1), 129.3 (CH-3 and CH-5), 131.9 (Cq-2 and Cq-6), 136.5 (Cq-4), 137.0 (Cq-1 Ph of R2), 144.6 (Cq-1), 147.7 (Cq-1 Ph of R1), 151.6 (C=O); ESI MS m/z 226 [M-CONHPh-NPh]+ (100), 319 [M-CONPh + 1]+ (14), 438 [M + 1]+ (41), 876 [2M + 1]+ (3).
- Ligand 5: Rf 0.38 (0.5% MeOH/DCM); colourless solid; m. p. 112.6–112.7 °C; 1H NMR 2.16 (s, 3H, CH3), 4.17 (bs, 1H, NH), 4.27 (s, 2H, CH2-Cq-6), 4.96 (s, 2H, CH2-Cq-2), 6.14 (s, 1H, NH-CON), 6.61 (dd, 2H, J 8.4, 0.9, CH-2+6 Ph-N(6) of R1), 6.68 (tt, 1H, J 7.3, 1.0, CH-4 Ph-N(6) of R1), 6.74 (bs, 1H, CH-3), 6.95 (tt, 1H, J 6.8, 1.8, CH-4 Ph), 6.99 (tt, 1H, J 7.4, 1.0, CH-4 Ph of O R2), 7.15, m, 11H, CH-5 and 10 CH Ph), 7.28 (dd, 2H, J 8.4, 0.9, CH-2+6 Ph of O R2), 7.32 (tt, 1H, J 7.4, 1.3, CH-4 Ph), 7.36 (ddd, 2H, J 8.4, 7.0, 1.4, CH-3+5 Ph), 7.64 (bs, 1H, NH-COO); 13C NMR 20.9 (CH3), 43.4 (CH2-Cq-6), 48.5 (CH2-Cq-2), 112.9 (CH-2+6 Ph-N(6) of R1), 117.5 (CH-4 Ph-N(6) of R1), 118.9 (CH-2+6 Ph of O R2), 119.6 (CH-2+6 Ph of N R2), 123.0 (CH-4 Ph), 123.5 (CH-4 Ph of O R2), 128.4 (CH-4 Ph), 128.7 (2 CH Ph), 128.8 (2 CH Ph), 129.0 (2 CH Ph), 129.2 (2 CH Ph), 129.4 (CH-5), 130.1 (CH-3+5 Ph), 130.3 (Cq-2), 130.7 (CH-3), 132.7 (Cq-6), 135.7 (Cq-4), 137.5 (Cq-1 Ph), 138.5 (Cq-1 Ph of N R2), 140.5 (Cq-1 Ph), 145.1 (Cq-1), 148.2 (Cq-1 Ph of N R2), 151.7 (C=O-O), 154.2 (C=O-N); ESI MS m/z 557 [M + 1]+ (98), 579 [M + Na]+ (100), 595 [M + K]+ (14), 1135 [2M + Na]+ (47).
3.4. General Procedure for the Preparation of Ligands 7
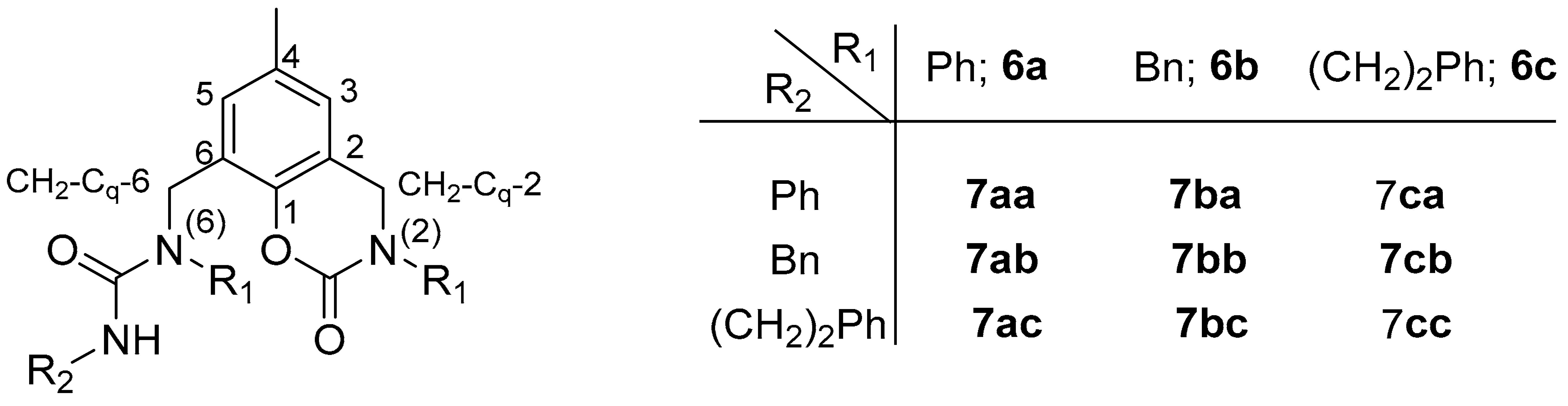
- Ligand 6a: 49% yield; Rf 0.65 (1% MeOH/DCM); red solid; m. p. 178.3–178.6 °C; 1H NMR 2.37 (s, 3H, CH3), 4.71 (s, 2H, CH2-Cq-2), 5.08 (s, 2H, CH2-Cq-6), 6.86 (bs, 1H, CH-3), 7.17 (bdd, 2H, J 7.3, 1.0, CH-2+6 Ph-N(2)), 7.28 (bs, 1H, CH-5), 7.30 (bm, 3H, CH-2+6 and CH-4 Ph-N(6)), 7.37 (bm, 3H, CH-3+5 and CH-4 Ph-N(2)), 7.42 (bdd, 2H, J 8.0, 7.5, (bm, 3H, CH-3+5 Ph-N(6)); 13C NMR 20.9 (CH3), 50.0 (CH2-Cq-6), 50.3 (CH2-Cq-2), 118.0 (Cq-2), 122.9 (Cq-6), 125.0 (CH-2+6 Ph-N(6)), 125.5 (CH-3), 127.2 (CH-4 Ph-N(6)), 128.3 (CH-2+6 Ph-N(2)), 128.7 (CH-4 Ph-N(2)), 129.38 (CH-3+5 Ph-N(2)), 129.41 (CH-3+5 Ph-N(6)), 129.6 (CH-5), 134.3 (Cq-4), 141.5 (Cq-1 Ph-N(2)), 144.6 (Cq-1 Ph-N(6)), 145.6 (Cq-1), 149.8 (O=C-O), 150.0 (O=C-Cl); ESI MS m/z 252 [M-NPhCOCl]+ (73), 345 [M-NHPh]+ (100), 438 [M + 1]+ (49), 876 [2M + 1]+ (14).
- Ligand 6c: 39% yield; Rf 0.51 (1% MeOH/DCM); colourless oil; Some NMR signals are duplicated due to hindered rotation; depicted as “A” and “B”; A:B = 1.3:1; 1H NMR 2.26 (s, 3H, CH3, A), 2.28 (s, 3H, CH3, B), 2.96 (m, 4H, CH2-Ph of N(6) R1, A+B), 3.00 (t, 4H, J 7.4, CH2-Ph of N(2) R1, A+B), 3.60 (t, 2H, J 7.8, CH2-N of N(6) R1, B), 3.69 (t, 4H, J 7.6, CH2-N of N(2) R1, A+B), 3.72 (t, 2H, J 7.8, CH2-N of N(6) R1, A), 4.25 (s, 2H, CH2-Cq-2, A), 4.26 (s, 2H, CH2-Cq-2, B), 4.57 (s, 2H, CH2-Cq-6, A), 4.71 (s, 2H, CH2-Cq-6, B), 6.73 (s, 2H, CH-3, A+B), 6.93 (s, 1H, CH-5, B), 7.05 (s, 1H, CH-5, A), 7.20-7.24 (m, 12H, CH Ph, A+B), 7.27-7.33 (m, 8H, CH Ph, A+B); 13C NMR 20.8 (CH3, A), 20.9 (CH3, B), 33.3 (CH2-Ph of N(2) R1, A+B), 33.5 (CH2-Ph of N(6) R1, B), 34.8 (CH2-Ph of N(6) R1, A), 46.6 (CH2-Cq-6, A), 48.26 (CH2-Cq-2, B), 48.30 (CH2-Cq-2, A), 48.7 (CH2-Cq-6, B), 51.4 (CH2-N of N(2) R1, A+B), 51.7 (CH2-N of N(6) R1, B), 52.8 (CH2-N of N(6) R1, A), 117.3 (Cq-2, A), 117.4 (Cq-2, B), 122.9 (Cq-6, B), 123.0 (Cq-6, A), 125.2 (CH-3, B), 125.5 (CH-3, A), 126.7 (CH Ph), 126.8 (CH Ph), 127.5 (CH-5, B), 128.68 (CH Ph), 128.73 (CH Ph), 128.8 (CH Ph), 128.9 (CH Ph), 129.0 (CH Ph), 129.9 (CH-5, A), 134.0 (Cq-4, A+B), 137.6 (Cq-1 Ph-N(6), A), 137.8 (Cq-1 Ph-N(6), B), 138.4 (Cq-1 Ph-N(2), A+B), 145.2 (Cq-1, A), 145.7 (Cq-1, B), 150.1 (O=C-Cl, A+B), 150.1 (O=C-O, A+B); ESI MS m/z 401 [M-COCl + 1]+ (100), 463 [M + 1]+ (1.5), 925 [2M + 1]+ (2).
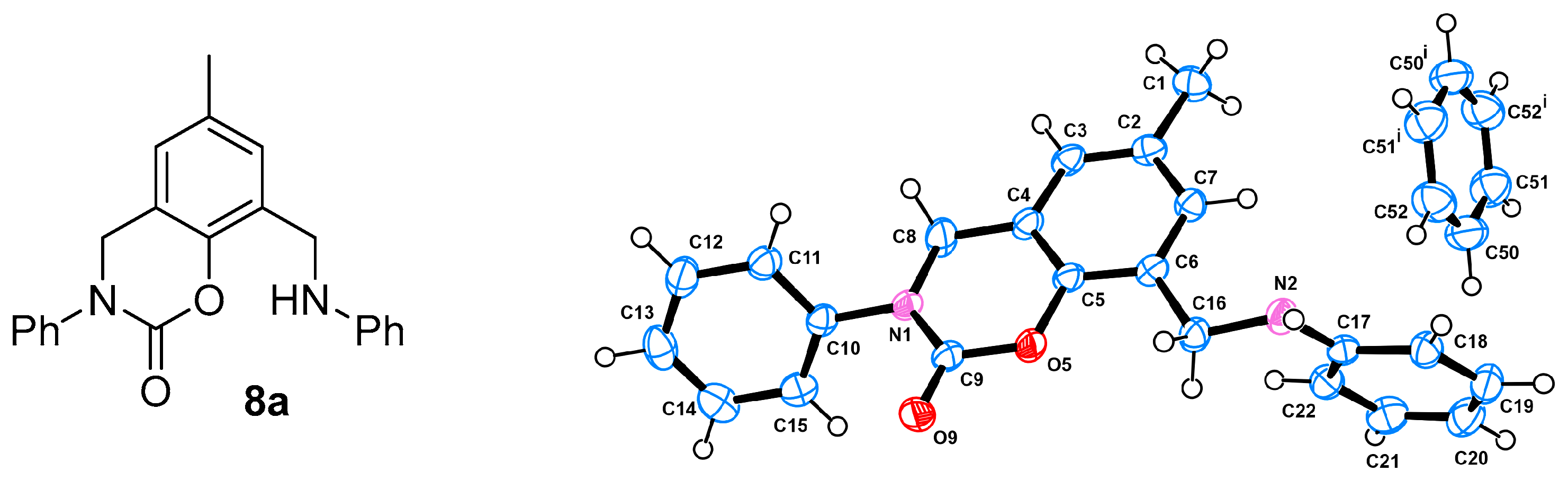
- Ligand 8a: 2% yield; Rf 0.54 (1% MeOH/DCM); deep red solid; m. p. 135.8–135.9 °C; 1H NMR 2.27 (s, 3H, CH3), 4.46 (s, 2H, CH2-Cq-6), 4.76 (s, 2H, CH2-Cq-2), 6.67 (dd, 2H, J 8.6, 1.0, CH-2+6 Ph-N(6)), 6.71 (tt, 1H, J 7.3, 1.0, CH-4 Ph-N(6)), 6.80 (bs, 1H, CH-3), 7.16 (m, 3H, CH-5 and CH-3+5 Ph-N(6)), 7.31 (tt, 1H, J 7.3, 1.2, CH-4 Ph-N(2)), 7.38 (dd, 2H, J 8.4, 1.2, CH-2+6 Ph-N(2)), 7.44 (dd, 2H, J 8.4, 7.4, CH-3+5 Ph-N(2)); 13C NMR 20.8 (CH3), 42.4 (CH2-Cq-6), 50.6 (CH2-Cq-2), 113.2 (CH-2+6 Ph-N(6)), 117.66 (Cq-2), 117.70 (CH-4 Ph-N(6)), 124.4 (CH-3), 125.3 (CH-2+6 Ph-N(2)), 126.7 (Cq-6), 127.3 (CH-4 Ph-N(2)), 128.9 (CH-5), 129.2 (CH-3+5 Ph-N(6)), 129.4 (CH-3+5 Ph-N(2)), 133.9 (Cq-4), 141.7 (Cq-1 Ph-N(2)), 145.6 (Cq-1), 147.6 (Cq-1 Ph-N(6)), 150.4 (C=O).
- Ligand 7aa: Rf 0.62 (3% MeOH/DCM); colourless solid; m. p. 235.2–235.3 °C; 1H NMR 2.34 (s, 3H, CH3), 4.71 (s, 2H, CH2-Cq-2), 5.12 (s, 2H, CH2-Cq-6), 6.36 (s, 1H, NH), 6.80 (bs, 1H, CH-3), 7.01 (tt, 1H, J 7.4, 1.1, CH-4 Ph of R2), 7.26 (m, 3H, CH Ph), 7.29–7.37 (m, 9H, 8 CH Ph and CH-5 at 7.326), 7.43 (m, 4H, CH Ph); 13C NMR 21.0 (CH3), 46.9 (CH2-Cq-6), 50.4 (CH2-Cq-2), 117.6 (Cq-2), 119.3 (CH-2+6 Ph of R2), 123.0 (CH-4 Ph of R2), 124.6 (CH-3), 125.1 (2 CH Ph), 125.6 (Cq-6), 127.1 (CH-4 Ph), 128.2 (CH-4 Ph), 128.3 (2 CH Ph), 128.8 (2 CH Ph), 129.3 (CH-5), 129.3 (2 CH Ph), 130.2 (2 CH Ph), 134.1 (Cq-4), 138.7 (Cq-1 Ph of R2), 141.3 (Cq-1 Ph of R1), 141.7 (Cq-1 Ph of R1), 145.4 (Cq-1), 150.2 (O=C-O), 154.5 (O=C-N); ESI MS m/z 226 [PhNHCONHPhCH3]+ (52), 345 [M-CONHPh + 1]+ (73), 464 [M + 1]+ (100), 486 [M + Na]+ (5), 928 [2M + 1]+ (3).
- Ligand 7ab: Rf 0.42 (3% MeOH/DCM); colourless solid; m. p. 130.9–131.0 °C; 1H NMR 2.34 (s, 3H, CH3), 4.46 (d, 2H, J 4.7, CH2 of R2), 4.72 (s, 2H, CH2-Cq-2), 4.78 (bs, 1H, NH), 5.08 (s, 2H, CH2-Cq-6), 6.79 (bs, 1H, CH-3), 7.25 (m, 5H, CH Ph), 7.27-7.33 (m, 7H, 6 CH Ph and CH-5 at 7.298), 7.36 (bt, 2H, J 7.6, CH-3+5 Ph), 7.47 (bt, 2H, J 7.7, CH-3+5 Ph); 13C NMR 21.0 (CH3), 44.8 (CH2 of R2), 47.0 (CH2-Cq-6), 50.5 (CH2-Cq-2), 117.6 (Cq-2), 124.4 (CH-3), 125.1 (2 CH Ph), 126.0 (Cq-6), 127.1 (CH-4 Ph), 127.2 (2 CH Ph), 127.3 (2 CH Ph), 127.8 (CH-4 Ph), 128.2 (CH-4 Ph), 128.6 (2 CH Ph), 129.2 (CH-5), 129.3 (CH-3+5 Ph), 130.0 (CH-3+5 Ph), 134.0 (Cq-4), 139.5 (Cq-1 Ph of R2), 141.69 (Cq-1 Ph of R1), 141.73 (Cq-1 Ph of R1), 145.4 (Cq-1), 150.3 (O=C-O), 157.3 (O=C-N); ESI MS m/z 226 [PhNHCONHCH2Ph]+ (90), 345 [M-CONHCH2Ph+1]+ (71), 359 [M-CONPh]+ (68), 478 [M + 1]+ (100), 500 [M + Na]+ (7), 956 [2M + 1]+ (8).
- Ligand 7ac: Rf 0.41 (3% MeOH/DCM); colourless oil; 1H NMR 2.35 (s, 3H, CH3), 2.79 (t, 2H, J 6.8, CH2-Ph of R2), 3.48 (td, 2H, J 6.8, 5.8, CH2-N of R2), 4.40 (t, 1H, J 5.5, NH), 4.70 (s, 2H, CH2-Cq-2), 5.04 (s, 2H, CH2-Cq-6), 6.78 (bs, 1H, CH-3), 7.10 (m, 4H, 2 CH-2+6 Ph), 7.19 (tt, 1H, J 7.4, 1.2, CH-4 Ph), 7.24 (tt, 2H, J 7.4, 1.2, CH-3+5 Ph of R2), 7.26(d, 1H, J 1.1, CH-5), 7.27-7.32 (m, 6H, CH Ph), 7.41 (ddt, 2H, J 8.4, 7.4, 1.9, CH-3+5 Ph); 13C NMR 21.0 (CH3), 36.1 (CH2-Ph of R2), 42.0 (CH2-N of R2), 46.7 (CH2-Cq-6), 50.4 (CH2-Cq-2), 117.6 (Cq-2), 124.3 (CH-3), 125.1 (2 CH Ph), 126.1 (Cq-6), 126.3 (CH-4 Ph), 127.1 (CH-4 Ph), 127.6 (CH-4 Ph), 128.2 (2 CH Ph), 128.5 (2 CH Ph), 128.8 (2 CH Ph), 129.1 (CH-5), 129.3 (CH-3+5 Ph of R1), 129.8 (2 CH Ph), 133.9 (Cq-4), 139.2 (Cq-1 Ph of R2), 141.6 (Cq-1 Ph of R1), 141.7 (Cq-1 Ph of R1), 145.3 (Cq-1), 150.3 (O=C-O), 157.2 (O=C-N); ESI MS m/z 492 [M + 1]+ (10), 514 [M + Na]+ (35), 1006 [2M + Na]+ (47).
- Ligand 7bb: Rf 0.43 (2% MeOH/DCM); colourless solid; m. p. 109.8–110.4 °C; 1H NMR 2.24 (s, 3H, CH3), 4.24 (s, 2H, CH2-Cq-2), 4.45 (s, 2H, CH2 of R2), 4.54 (s, 2H, CH2-Cq-6), 4.62 (s, 2H, CH2-N(6) of R1), 4.64 (s, 2H, CH2-N(2) of R1), 4.97 (bs, 1H, NH), 6.71 (bs, 1H, CH-3), 6.98 (bs, 1H, CH-5), 7.17 (dd, 2H, J 7.8, 1.2, CH-2+6 Ph), 7.21 (tt, 1H, J 7.4, 1.2, CH-4 Ph), 7.25 (tt, 2H, J 7.5, 1.2, CH-3+5 Ph of R2), 7.27 (m, 3H, CH Ph), 7.31–7.36 (m, 7H, CH Ph); 13C NMR 20.8 (CH3), 44.88 (CH2 of R2), 44.92 (CH2-Cq-6), 46.5 (CH2-Cq-2), 50.9 (s, 2H, CH2-N(6) of R1), 52.5 (s, 2H, CH2-N(2) of R1), 116.9 (Cq-2), 124.4 (Cq-6), 124.9 (CH-3), 127.0 (CH-4 Ph), 127.4 (2 CH Ph), 127.4 (CH-4 Ph), 127.4 (CH-4 Ph), 128.1 (CH-5), 128.2 (2 CH Ph), 128.3 (2 CH Ph), 128.4 (2 CH Ph), 128.7 (2 CH Ph), 128.9 (2 CH Ph), 134.0 (Cq-4), 135.2 (Cq-1 Ph of N(2) R1), 137.7 (Cq-1 Ph of N(6) R1), 139.5 (Cq-1 Ph of R2), 145.2 (Cq-1), 150.5 (O=C-O), 158.3 (O=C-N); ESI MS m/z 373 [M-CONCH2Ph]+ (93), 506 [M + 1]+ (100), 528 [M + Na]+ (9), 1012 [2M + 1]+ (38).
- Ligand 7bc: Rf 0.39 (2% MeOH/DCM); colourless oil; 1H NMR 2.247 (s, 3H, CH3), 2.77 (t, 2H, J 6.9, CH2-Ph of R2), 3.50 (bt, 2H, J 6.8, CH2-N of R2), 4.26 (s, 2H, CH2-Cq-2), 4.46 (s, 2H, CH2-Cq-6), 4.54 (s, 2H, CH2-N(6) of R1), 4.63 (bs, 1H, NH), 4.66 (s, 2H, CH2-N(2) of R1), 6.71 (bs, 1H, CH-3), 6.94 (bs, 1H, CH-5), 7.06 (dd, 2H, J 7.7, 1.2, CH-2+6 Ph), 7.12 (tt, 1H, J 7.3, 1.3, CH-4 Ph), 7.19 (m, 4H, CH-3+5 Ph of R2 and 2 CH Ph), 7.25 (tt, 1H, J 7.3, 1.1, CH-4 Ph), 7.29–7.37 (m, 7H, CH Ph); 13C NMR 20.8 (CH3), 36.3 (CH2-Ph of R2), 42.2 (CH2-N of R2), 44.9 (CH2-Cq-6), 46.6 (CH2-Cq-2), 50.7 (CH2-N(6) of R1), 52.5 (s, 2H, CH2-N(2) of R1), 116.8 (Cq-2), 124.5 (Cq-6), 124.8 (CH-3), 126.1 (CH-4 Ph), 127.3 (2 CH Ph), 127.4 (CH-4 Ph), 128.1 (CH-5), 128.2 (CH-4 Ph), 128.3 (2 CH Ph), 128.49 (2 CH Ph), 128.70 (2 CH Ph), 128.71 (2 CH Ph), 128.9 (2 CH Ph), 133.9 (Cq-4), 135.3 (Cq-1 Ph of N(2) R1), 137.6 (Cq-1 Ph of N(6) R1), 139.3 (Cq-1 Ph of R2), 145.2 (Cq-1), 150.6 (O=C-O), 158.3 (O=C-N); ESI MS m/z 520 [M + 1]+ (14), 542 [M + Na]+ (48), 558 [M + K]+ (2), 1062 [2M + Na]+ (100).
- Ligand 7ca: Rf 0.60 (3% MeOH/DCM); colourless solid; m. p. 133.1-133.2 °C; 1H NMR 2.27 (s, 3H, CH3), 2.92 (t, 2H, J 7.2, CH2-Ph of N(6) R1), 3.00 (t, 2H, J 7.4, CH2-Ph of N(2) R1), 3.61 (t, 2H, J 7.2, CH2-N of N(6) R1), 3.68 (t, 2H, J 7.4, CH2-N of N(2) R1), 4.25 (s, 2H, CH2-Cq-2), 4.56 (s, 2H, CH2-Cq-6), 6.53 (bs, 1H, NH), 6.72 (bs, 1H, CH-3), 6.98 (bt, 1H, J 7.1, CH-4 Ph), 7.02 (bs, 1H, CH-5), 7.24 (m, 8H, CH Ph), 7.30 (m, 6H, CH Ph); 13C NMR 20.82 (CH3), 33.2 (CH2-Ph of N(2) R1), 34.6 (CH2-Ph of N(6) R1), 45.4 (CH2-Cq-6), 48.3 (CH2-Cq-2), 49.8 (CH2-N of N(6) R1), 51.3 (CH2-N of N(2) R1), 117.2 (Cq-2), 128.4 (CH-2+6 Ph of R2), 122.6 (CH-4 Ph of R2), 124.4 (Cq-6), 125.1 (CH-3), 126.6 (CH-4 Ph), 126.7 (CH-4 Ph), 128.70 (2 CH Ph), 128.71 (2 CH Ph), 128.78 (2 CH Ph), 128.79 (2 CH Ph), 128.97 (2 CH Ph), 129.03 (CH-5), 134.0 (Cq-4), 138.3 (Cq-1 Ph of N(2) R1), 139.3 (Cq-1 Ph of N(6) R1 and Cq-1 Ph of R2), 145.5 (Cq-1), 150.1 (O=C-O), 155.8 (O=C-N); ESI MS m/z 520 [M + 1]+ (11), 542 [M + Na]+ (54), 558 [M + K]+ (4), 1062 [2M + Na]+ (100).
- Ligand 7cb: Rf 0.39 (3% MeOH/DCM); colourless oil; 1H NMR 2.236 (s, 3H, CH3), 2.90 (dd, 2H, J 7.6, 7.4, CH2-Ph of N(6) R1), 2.98 (dd, 2H, J 7.6, 7.4, CH2-Ph of N(2) R1), 3.57 (dd, 2H, J 7.6, 7.4, CH2-N of N(6) R1), 3.66 (dd, 2H, J 7.6, 7.4, CH2-N of N(2) R1), 4.23 (s, 2H, CH2-Cq-2), 4.36 (s, 2H, CH2 of R2), 4.49 (s, 2H, CH2-Cq-6), 4.71 (bs, 1H, NH), 6.68 (bs, 1H, CH-3), 6.95 (bs, 1H, CH-5), 7.19 (m, 5H, CH Ph), 7.24 (m, 5H, CH Ph), 7.28 (m, 5H, CH Ph); 13C NMR 20.8 (CH3), 33.2 (CH2-Ph of N(2) R1), 34.8 (CH2-Ph of N(6) R1), 44.8 (CH2 of R2), 46.0 (CH2-Cq-6), 48.2 (CH2-Cq-2), 49.9 (CH2-N of N(6) R1), 51.3 (CH2-N of N(2) R1), 117.1 (Cq-2), 124.6 (Cq-6), 124.7 (CH-3), 126.4 (CH-4 Ph), 126.7 (CH-4 Ph), 127.0 (CH-4 Ph), 127.5 (2 CH Ph), 128.1 (CH-5), 128.4 (2 CH Ph), 128.6 (2 CH Ph), 128.7 (2 CH Ph), 128.8 (2 CH Ph), 128.9 (2 CH Ph), 133.9 (Cq-4), 138.4 (Cq-1 Ph of R2), 139.1 (Cq-1 Ph of N(2) R1), 139.6 (Cq-1 Ph of N(6) R1), 145.3 (Cq-1), 150.1 (O=C-O), 158.0 (O=C-N); ESI MS m/z 534 [M+1]+ (14), 556 [M + Na]+ (83), 572 [M + K]+ (5), 1090 [2M + Na]+ (100).
- Ligand 7cc: Rf 0.40 (3% MeOH/DCM); colourless oil; 1H NMR 2.237 (s, 3H, CH3), 2.74 (t, 2H, J 7.0, CH2-Ph of R2), 2.82 (dd, 2H, J 7.7, 7.4, CH2-Ph of N(6) R1), 3.00 (dd, 2H, J 7.7, 7.3, CH2-Ph of N(2) R1), 3.44 (dd, 2H, J 7.0, 6.8, CH2-N of R2), 3.46 (dd, 2H, J 7.8, 7.5, CH2-N of N(6) R1), 3.68 (dd, 2H, J 7.7, 7.2, CH2-N of N(2) R1), 4.24 (s, 2H, CH2-Cq-2), 4.40 (bs, 1H, NH), 4.42 (s, 2H, CH2-Cq-6), 6.68 (bs, 1H, CH-3), 6.91 (bs, 1H, CH-5), 7.14 (m, 4H, CH Ph), 7.19 (m, 2H, 2 CH-4 Ph), 7.24 (m, 5H, CH Ph), 7.29 (m, 4H, CH Ph); 13C NMR 20.8 (CH3), 33.3 (CH2-Ph of N(2) R1), 34.7 (CH2-Ph of N(6) R1), 36.2 (CH2-Ph of R2), 42.0 (CH2-N of R2), 44.8 (CH2-Cq-6), 48.3 (CH2-Cq-2), 49.7 (CH2-N of N(6) R1), 51.3 (CH2-N of N(2) R1), 117.0 (Cq-2), 124.6 (CH-3), 124.8 (Cq-6), 126.2 (CH-4 Ph), 126.4 (CH-4 Ph), 126.7 (CH-4 Ph), 128.2 (CH-5), 128.5 (2 CH Ph), 128.6 (2 CH Ph), 128.7 (2 CH Ph), 128.77 (2 CH Ph), 128.80 (2 CH Ph), 128.85 (2 CH Ph), 133.8 (Cq-4), 138.4 (Cq-1 Ph of N(2) R1), 139.0 (Cq-1 Ph of N(6) R1), 139.4 (Cq-1 Ph of R2), 145.3 (Cq-1), 150.16 (O=C-O), 158.0 (O=C-N); ESI MS m/z 548 [M + 1]+ (18), 570 [M + Na]+ (100), 586 [M + K]+ (7), 1118 [2M + Na]+ (99).
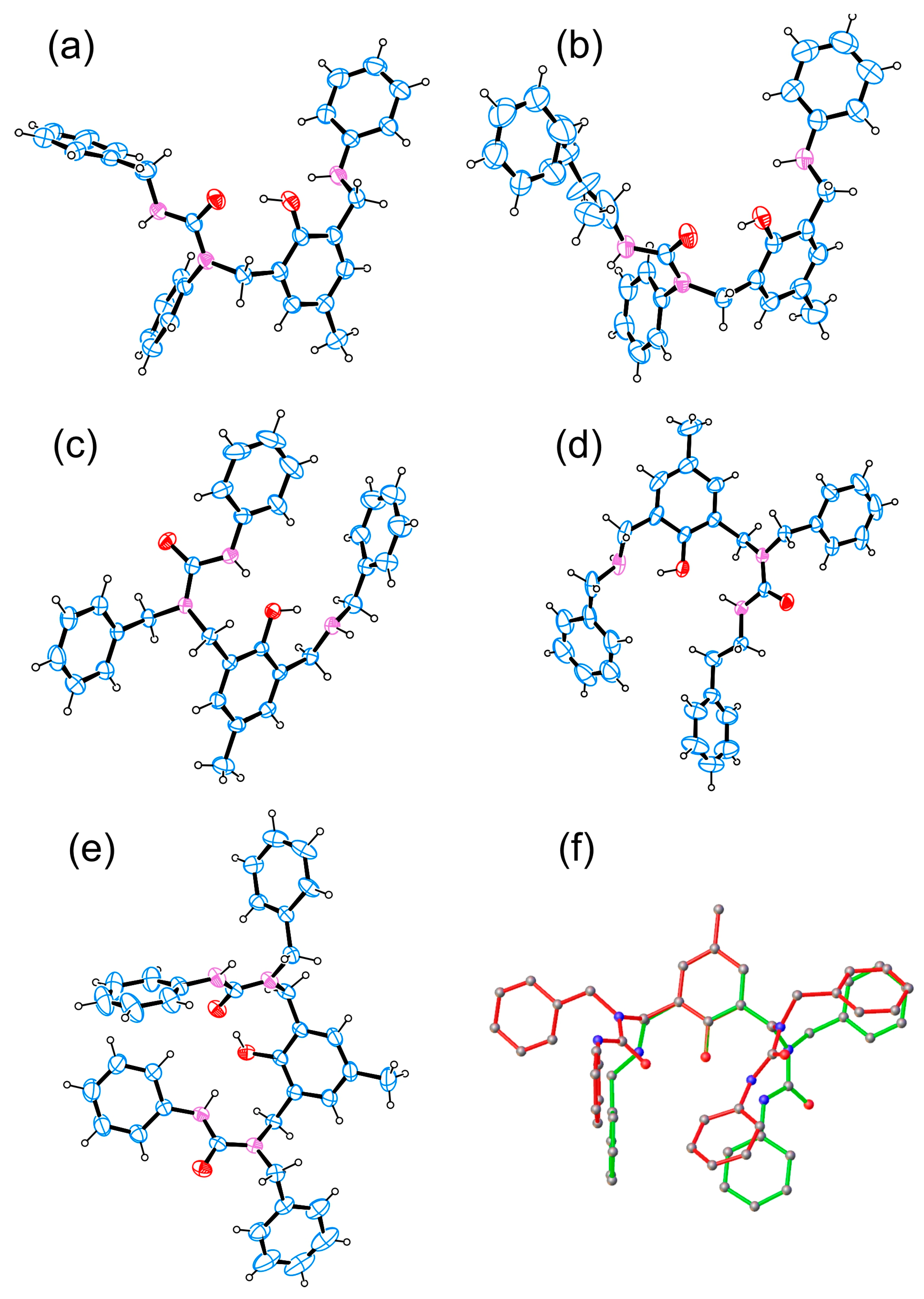
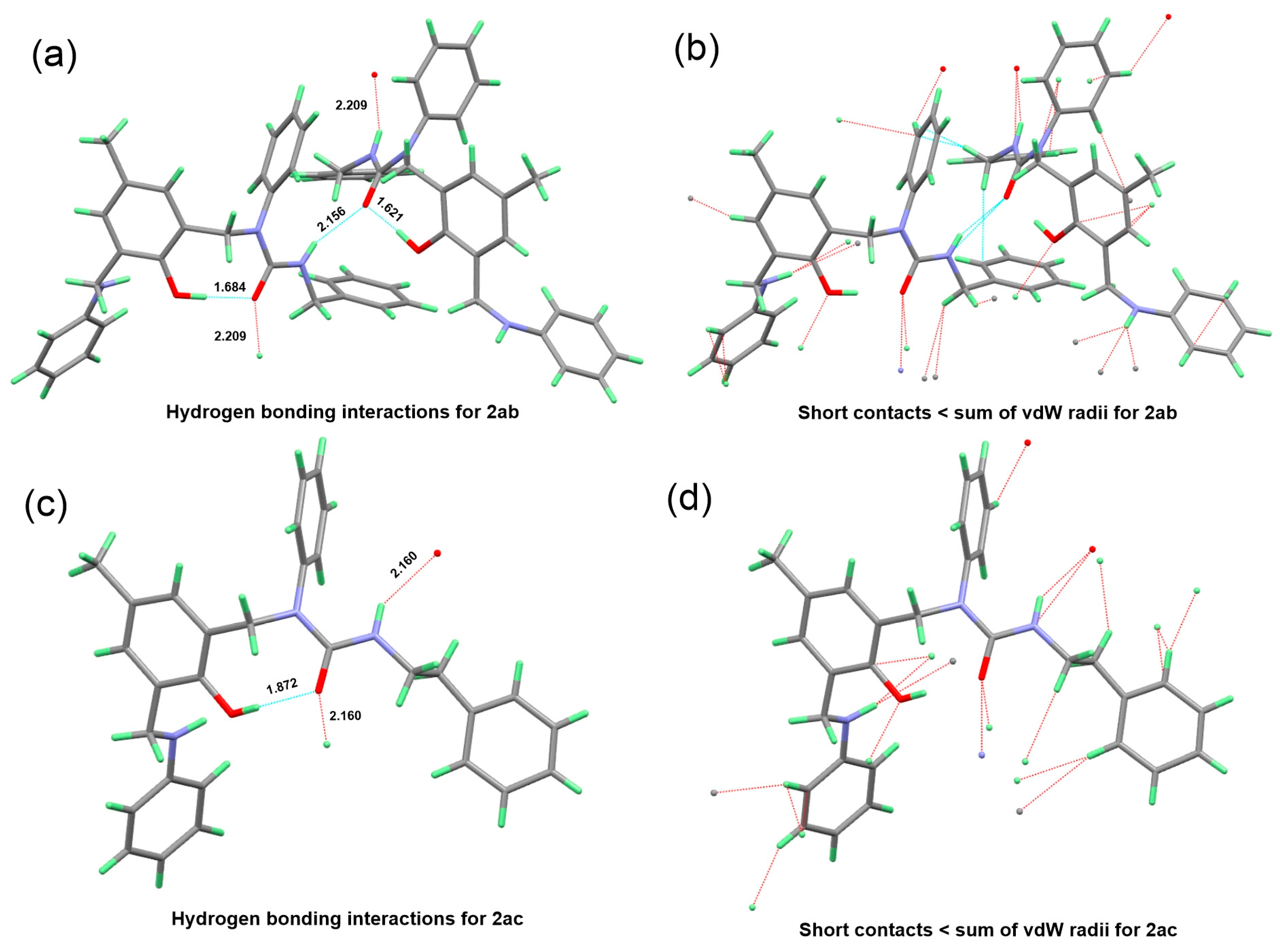
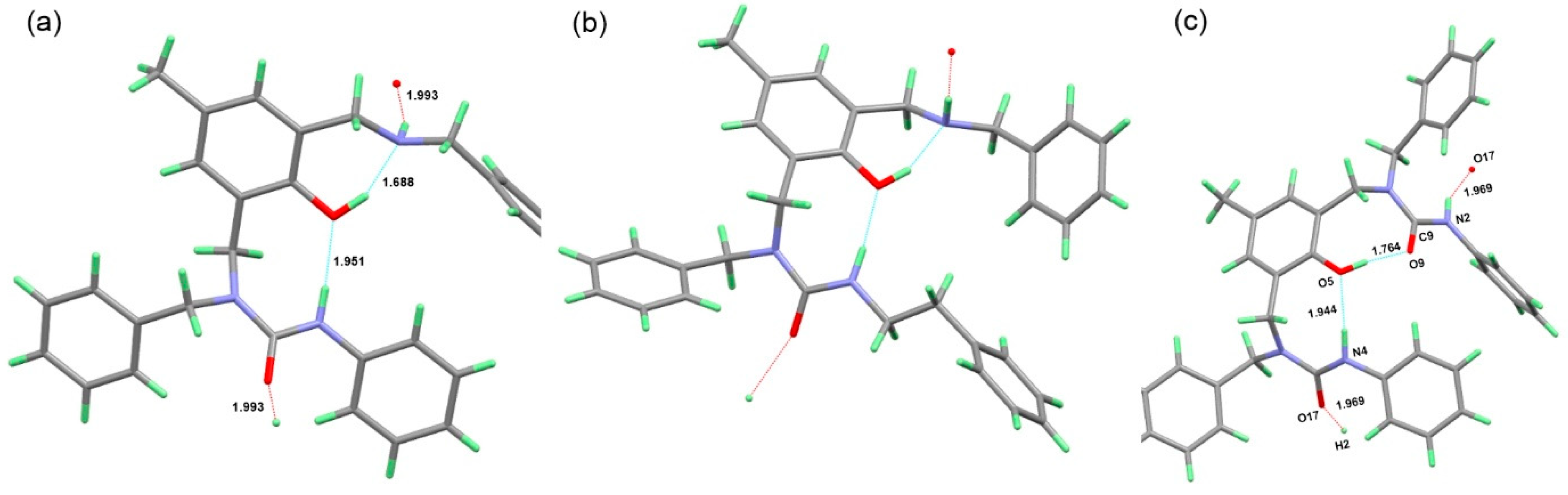
3.5. Crystallography
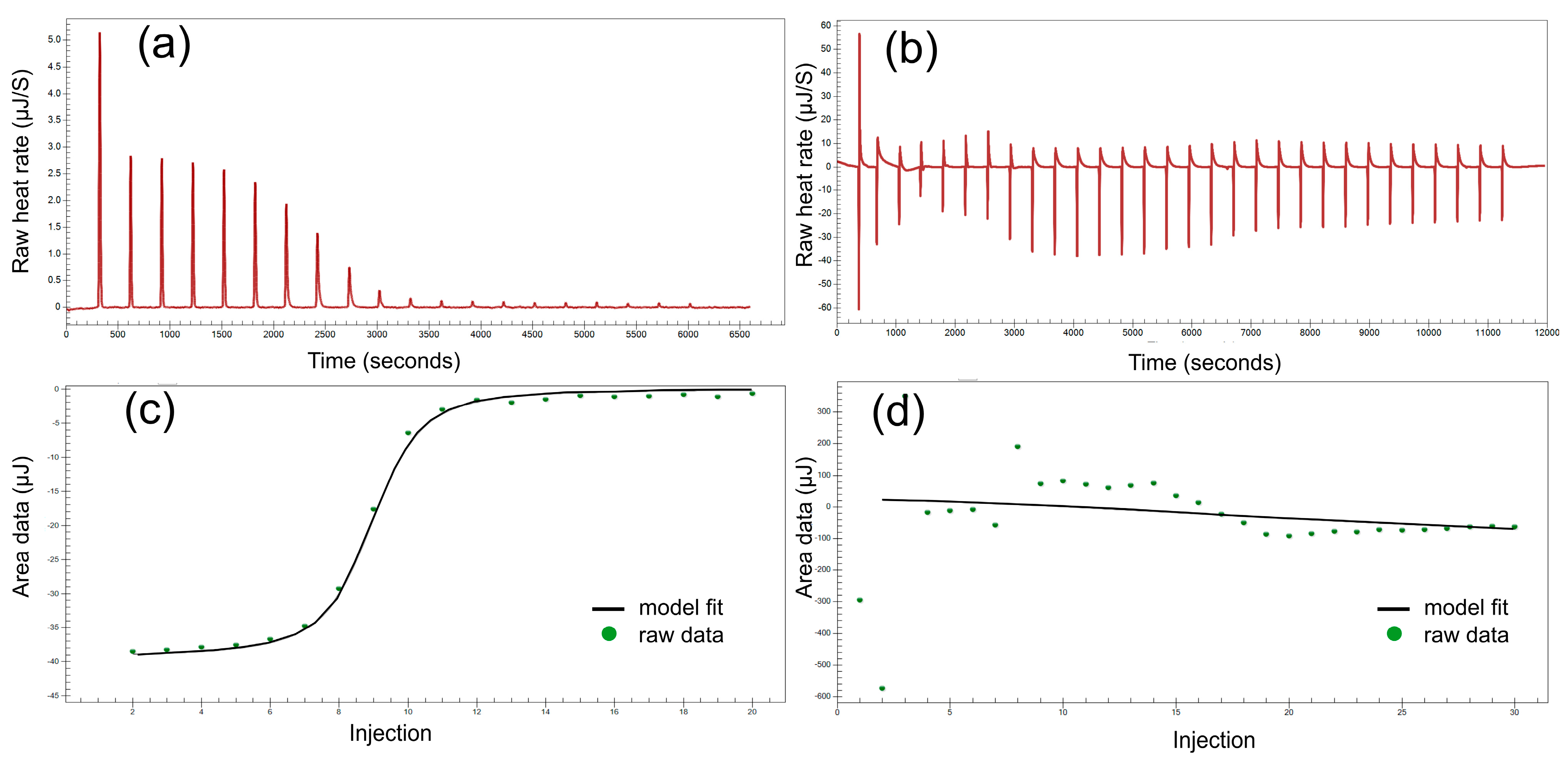
3.6. Isothermal Titration Calorimetry (ITC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cox, M. Solvent Extraction Principles and Practice, 2nd ed.; Rydberg, J., Cox, M., Musikas, C., Choppin, G.R., Eds.; Marcel Dekker Inc.: New York, NY, USA, 2004; Chapter 11; pp. 455–505. [Google Scholar]
- Kislik, V.S. Solvent Extraction. Classical and Novel Approaches; Elsevier B.V.: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Regel-Rosocka, M.; Alguacil, F.J. Recent trends in metals extraction. Rev. Metal. 2013, 49, 292–316. [Google Scholar] [CrossRef]
- Wilson, A.M.; Bailey, P.J.; Tasker, P.A.; Turkington, J.R.; Grant, R.A.; Love, J.B. Solvent extraction: The coordination chemistry behind extractive metallurgy. Chem. Soc. Rev. 2014, 43, 123–134. [Google Scholar] [CrossRef] [PubMed]
- El-Nadi, Y.A. Solvent extraction and its applications on ore processing and recovery of metals: Classical approach. Sep. Pur. Rev. 2017, 46, 195–215. [Google Scholar] [CrossRef]
- Moyer, B.A. (Ed.) Ion Exchange and Solvent Extraction: Vol. 23, Changing the Landscape in Solvent Extraction; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2020. [Google Scholar]
- Narayanan, R.P.; Palantavida, S. Selective extraction and solid state complexation of iron(III) with bis(β-diketone) ligand. Mater. Today Proc. 2021, 41, 638–643. [Google Scholar] [CrossRef]
- Zhang, J.; Wenzel, M.; Steup, J.; Schaper, G.; Hennersdorf, F.; Du, H.; Zheng, S.; Lindoy, L.F.; Weigand, J.J. 4-Phosphoryl pyrazolones for highly selective lithium separation from alkali metal ions. Chem. Eur. J. 2022, 28, e202103640. [Google Scholar] [CrossRef]
- Fu, F.; Wang, Q. Removal of heavy metal ions from wastewaters: A review. J. Environ. Manag. 2011, 92, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-w.; Yang, T.; Chen, Q.-d.; Shen, X.-h. Extraction behaviors of ionic liquid systems and application perspectives in reprocessing of spent nuclear fuel. J. Nucl. Radiochem. 2015, 37, 286–309. [Google Scholar]
- Alyapyshev, M.Y.; Babain, V.A.; Ustynyuk, Y.A. Recovery of minor actinides from high-level wastes: Modern trends. Russ. Chem. Rev. 2016, 85, 943–961. [Google Scholar] [CrossRef]
- Mohapatra, P.K. Actinide ion extraction using room temperature ionic liquids: Opportunities and challenges for nuclear fuel cycle applications. Dalton Trans. 2017, 46, 1730–1747. [Google Scholar] [CrossRef]
- Leoncini, A.; Huskens, J.; Verboom, W. Ligands for f-element extraction used in the nuclear fuel cycle. Chem. Soc. Rev. 2017, 46, 7229–7273. [Google Scholar] [CrossRef]
- Fan, Y.; Li, Y.; Shu, X.; Wu, R.; Chen, S.; Jin, Y.; Xu, C.; Chen, J.; Huang, C.; Xia, C. Complexation and separation of trivalent actinides and lanthanides by a novel DGA derived from macrocyclic crown ether: Synthesis, extraction, and spectroscopic and density functional theory studies. ACS Omega 2021, 6, 2156–2166. [Google Scholar] [CrossRef]
- Nyamato, G.S. Perspectives and prospects of chelation extraction of heavy metals from wastewater: A review. Water Sci. Technol. 2023, 88, 47–61. [Google Scholar] [CrossRef]
- Lehn, J.M. Supramolecular Chemistry: Concepts and Perspectives; Wiley: Hoboken, NJ, USA, 1995. [Google Scholar]
- Atmani, C.; El Hajj, F.; Benmansour, S.; Marchivie, M.; Triki, S.; Conan, F.; Patinec, V.; Handel, H.; Dupouy, G.; Gómez-García, C.J. Guidelines to design new spin crossover materials. Coord. Chem. Rev. 2010, 254, 1559–1569. [Google Scholar] [CrossRef]
- Easmon, J. Copper and iron complexes with antitumour activity. Expert Opin. Therap. Patents 2002, 12, 789–818. [Google Scholar] [CrossRef]
- Kundu, S.; Egboluche, T.K.; Hossain, M.A. Urea- and thiourea-based receptors for anion binding. Acc. Chem. Res. 2023, 56, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.H.; Dietz, M.L.; Chiarizia, R. Incorporating size selectivity into synergistic solvent extraction: A review of crown ether-containing systems. Ind. Eng. Chem. Res. 2000, 39, 3442–3464. [Google Scholar] [CrossRef]
- Wainwright, K.P. Applications for polyaza macrocycles with nitrogen-attached pendant arms. Adv. Inorg. Chem. 2001, 52, 293–334. [Google Scholar]
- Chaudhary, A.; Singh, R.V. Review: Metal complexes of polyaza and polyoxaaza macrocyclic ligands: A look into the past and present work. Rev. Inorg. Chem. 2008, 28, 35–76. [Google Scholar] [CrossRef]
- Marchetti, F.; Pettinari, R.; Pettinari, C. Recent advances in acylpyrazolone metal complexes and their potential applications. Coord. Chem. Rev. 2015, 303, 1–31. [Google Scholar] [CrossRef]
- Doidge, E.D.; Roebuck, J.W.; Healy, M.R.; Tasker, P.A. Phenolic pyrazoles: Versatile polynucleating ligands. Coord. Chem. Rev. 2015, 288, 98–117. [Google Scholar] [CrossRef]
- Pestov, A.V.; Slepukhin, P.A.; Charushin, V.N. Copper and nickel chelate complexes with polydentate N,O-ligands: Structure and magnetic properties of polynuclear complexes. Russ. Chem. Rev. 2015, 84, 310–333. [Google Scholar] [CrossRef]
- Atanassova, M.; Kurteva, V. Synergism as a phenomenon in solvent extraction of 4f-elements with calixarenes. RSC Adv. 2016, 6, 11303–11324. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, X.; Zhao, S.; He, P.; Luo, T.; Jiang, J.; Liang, W.; Cai, J.; Xu, H. A new photoresponsive bis (crown ether) for extraction of metal ions. ChemistrySelect 2019, 4, 10316–10319. [Google Scholar] [CrossRef]
- Florés, O.; Velic, D.; Mabrouk, N.; Bettaïeb, A.; Tomasoni, C.; Robert, J.-M.; Paul, C.; Goze, C.; Roussakis, C.; Bodio, E. Rapid synthesis and antiproliferative properties of polyazamacrocycle-based bi- and tetra-gold(i) phosphine dithiocarbamate complexes. ChemBioChem 2019, 20, 2255–2261. [Google Scholar] [CrossRef] [PubMed]
- Götzke, L.; Schaper, G.; März, J.; Kaden, P.; Huittinen, N.; Stumpf, T.; Kammerlander, K.K.K.; Brunner, E.; Hahn, P.; Mehnert, A.; et al. Coordination chemistry of f-block metal ions with ligands bearing bio-relevant functional groups. Coord. Chem. Rev. 2019, 386, 267–309. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Baun, C.; Nielsen, K.M.; Thisgaard, H.; Jensen, A.I.; Zhuravlev, F. Design, synthesis, computational, and preclinical evaluation of natTi/45Ti-labeled urea-based glutamate PSMA ligand. Molecules 2020, 25, 1104. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, J.; Callegari, E.; Baranyai, Z.; Fraccarollo, A.; Cossi, M.; Tei, L. Semi-rigid (aminomethyl) piperidine-based pentadentate ligands for Mn(II) complexation. Molecules 2021, 26, 5993. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Mei, L.; Huang, Z.-W.; Chi, X.-W.; Geng, J.-S.; Hu, K.-Q.; Yu, J.-P.; Jiao, C.-S.; Zhang, M.; Chai, Z.-F.; et al. Coordination-Adaptive Polydentate Pseudorotaxane Ligand for Capturing Multiple Uranyl Species. Inorg. Chem. 2022, 61, 3058–3071. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Oliferenko, A.; Lomaka, A.; Karelson, M. Six-membered cyclic ureas as HIV-1 protease inhibitors: A QSAR study based on CODESSA PRO approach. Bioorg. Med. Chem. Lett. 2002, 12, 3453–3457. [Google Scholar] [CrossRef]
- Kane, J.L., Jr.; Hirth, B.H.; Liang, B.; Gourlie, B.B.; Nahill, S.; Barsomian, G. Ureas of 5-aminopyrazole and 2-aminothiazole inhibit growth of gram-positive bacteria. Bioorg. Med. Chem. Lett. 2003, 13, 4463–4466. [Google Scholar] [CrossRef]
- Brugel, T.A.; Maier, J.A.; Clark, M.P.; Sabat, M.; Golebiowski, A.; Bookland, R.G.; Laufersweiler, M.J.; Laughlin, S.K.; VanRens, J.C.; De, B.; et al. Development of N-2,4-pyrimidine-N-phenyl-N′-phenyl ureas as inhibitors of tumor necrosis factor alpha (TNF-α) synthesis. Part 1. Bioorg. Med. Chem. Lett. 2006, 16, 3510–3513. [Google Scholar] [CrossRef]
- Kuhl, O. N-phosphino carboxylic acid amides, lactams and ureas: Synthesis, properties and applications. Coord. Chem. Rev. 2006, 250, 2867–2915. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Zhang, H.-Z.; Cui, S.-F.; Lv, J.-S.; Yan, C.-Y.; Wan, K.; Zhang, Y.-Y.; Zhang, S.-L.; Cai, G.-X.; Geng, R.-X.; et al. Recent developments in organometallic supramolecular complexes as anticancer drugs. In Advances in Anticancer Agents in Medicinal Chemistry; Prudhomme, M., Ed.; Bentham Books: Potomac, MD, USA, 2013; Volume 2, Chapter 2; pp. 46–129. [Google Scholar]
- Elkamhawy, A.; Viswanath, A.N.I.; Pae, A.N.; Kim, H.Y.; Heo, J.C.; Park, W.K.; Lee, C.O.; Yang, H.; Kim, K.H.; Nam, D.H.; et al. Discovery of potent and selective cytotoxic activity of new quinazoline-ureas against TMZ-resistant glioblastoma multiforme (GBM). Eur. J. Med. Chem. 2015, 103, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Maurer, D.; Breit, B. Urea-substituted tetramethylcyclopentadienyl ligands for supramolecularly accelerated RhIII-catalyzed ortho-C−H olefination of benzoic acid derivatives. Chem. Eur. J. 2021, 27, 2643–2648. [Google Scholar] [CrossRef]
- AlNajjar, Y.T.; Gabr, M.; ElHady, A.K.; Salah, M.; Wilms, G.; Abadi, A.H.; Becker, W.; Abdel-Halim, M.; Engel, M. Discovery of novel 6-hydroxybenzothiazole urea derivatives as dual Dyrk1A/α-synuclein aggregation inhibitors with neuroprotective effects. Eur. J. Med. Chem. 2022, 227, 113911. [Google Scholar] [CrossRef]
- Atanassova, M.P.; Todorova, S.E.; Kurteva, V.B.; Todorova, N.I. Insights into the synergistic selectivity of 4f-ions implementing 4-acyl-5-pyrazolone and two new unsymmetrical NH-urea containing ring molecules in an ionic liquid. Sep. Purif. Technol. 2018, 204, 328–335. [Google Scholar] [CrossRef]
- Todorova, S.; Kurteva, V.; Shivachev, B.; Nikolova, R.P. Crystal structures of novel polydentate N,O-ligands. Acta Cryst. 2016, A72, s403. [Google Scholar] [CrossRef]
- Todorova, S.; Atanassova, M.; Kurteva, V. Data on the synthesis and characterization of two novel polydentate ligands possessing unsymmetrical NH-urea fragment. Data Brief 2018, 20, 933–939. [Google Scholar] [CrossRef]
- Ameerunisha, S.; Srinivas, B.; Zacharias, P.S. Specific transport of copper(II) ions across liquid membrane by Schiff base ligands. Bull. Chem. Soc. Jpn. 1994, 67, 263–266. [Google Scholar] [CrossRef]
- Rozwadowski, Z.; Dziembowska, T.; Schroeder, G.; Brzezinski, B. Studies of intramolecular hydrogen bonds in di-Schiff bases of 2-hydroxy-5-methyl isophthaldehyde. J. Mol. Struct. 1998, 444, 221–225. [Google Scholar] [CrossRef]
- Grzybowski, J.J.; Urbach, F.L. Binuclear metal complexes. 2. A unique mononuclear copper(I) complex with a potentially binucleating Schiff base ligand. Inorg. Chem. 1980, 19, 2604–2608. [Google Scholar]
- Beer, P.D.; Fox, O.D. Redox-Active Self Assembled Metallomacrocycles. British. UK Patent Application, GB 2364305 A, 23 January 2002. [Google Scholar]
- Rigaku, O.D. Crysalis Pro; Rigaku Oxford Diffraction Ltd.: Yarnton, UK, 2015. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conditions a | Products | ||||
|---|---|---|---|---|---|---|
| 2 | Yield, % | 3 | Yield, % | Total, % | ||
| 1 | DIPEA, 1a:CC 1:1.5, toluene, 24 h | 2aa | 15 | 3aa | - | 51 b |
| 2 | 1a:CC 1:1, benzene, 2 h | 19 | traces | 19 | ||
| 3 | 1a:CC 1:2, toluene, 24 h | 39 | 13 | 52 | ||
| 4 | 1a:CC 1:3, toluene, 24 h | 35 | 22 | 57 | ||
| 5 | 1a:CC 1:1, toluene, 2 h | 2ab | 30 | 3ab | traces | 30 |
| 6 | 1a:CC 1:2, toluene, 24 h | 32 | 23 | 55 | ||
| 7 | 1a:CC 1:1, benzene, 2 h | 2ac | traces | 3ac | - | - |
| 8 | 1a:CC 1:2, benzene, 2 h | 17 | traces | 17 | ||
| 9 | 1a:CC 1:2, toluene, 2 h | 39 | 8 | 47 | ||
| 10 | 1a:CC 1:2, DCE, 2 h | 30 | 32 | 62 | ||
| 11 | 1b:CC 1:2, toluene, 24 h | 2bac | 21 | 3ba | - | 21 |
| 12 | 1b:CC 1:2, benzene, 24 h | 37 | 7 | 44 | ||
| 13 | 1b:CC 1:4, toluene, 24 h | 19 | 17 | 36 | ||
| 14 | 1b:CC 1:1, toluene, 2 h | 2bb | 34 | 3bb | traces | 34 |
| 15 | 1b:CC 1:2, toluene, 24 h | - | 46 | 46 | ||
| 16 | 1b:CC 1:1, benzene, 2 h | 2bc | 26 | 3bc | - | 26 |
| 17 | 1b:CC 1:2, benzene, 2 h | 16 | - | 16 | ||
| 18 | 1b:CC 1:2, toluene, 24 h | 17 | 28 | 45 | ||
| 19 | 1b:CC 1:2, DCE, 2 h | 8 | 27 | 35 | ||
| 20 | 1c:CC 1:1.5, toluene, 24 h | 2ca | 44 | 3ca | 17 | 61 |
| 21 | 1c:CC 1:2, toluene, 24 h | 20 | 43 | 63 | ||
| 22 | 1c:CC 1:2, toluene, 24 h | 2cb | 15 | 3cb | 43 | 58 |
| 23 | 1c:CC 1:3, toluene, 4 h | - | 43 | 43 | ||
| 24 | 1c:CC 1:1, DCE, 2 h | 7 | traces | 7 | ||
| 25 | 1c:CC 1:2, DCE, 2 h | 18 | 16 | 34 | ||
| 26 | 1c:CC 1:1, benzene, 2 h | 2cc | 42 | 3cc | 21 | 63 |
| 27 | 1c:CC 1:2, benzene, 2 h | - | 19 | 19 | ||
| 28 | 1c:CC 1:2, toluene, 24 h | 20 | 39 | 59 | ||
| 29 | 1c:CC 1:2, DCE, 2 h | - | 38 | 38 | ||
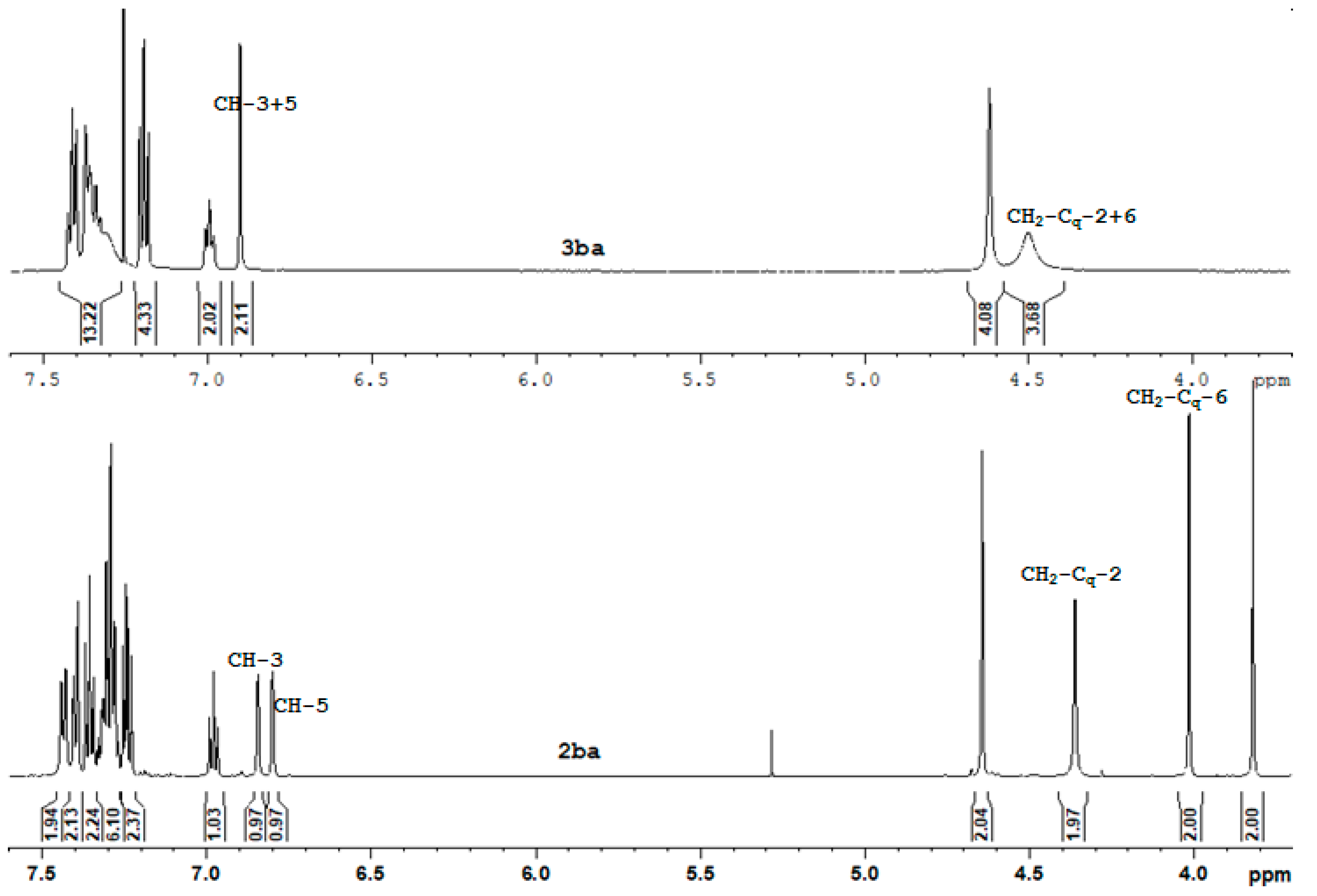
| Lig. | CH-3 CH-3 | CH-5 CH-5 | CH2-Cq-2 CH2-Cq-2 | CH2-Cq-6 CH2-Cq-6 |
|---|---|---|---|---|
| 2aa | 6.372 130.90 | 7.042 130.14 | 4.773 50.47 | 4.363 44.54 |
| 3aa | 6.667/131.03 | 4.876/49.40 | ||
| 2ab | 6.344 130.90 | 7.042 130.08 | 4.723 50.81 | 4.372 44.43 |
| 3ab | 6.678/130.23 | 4.828/49.63 | ||
| 2ac | 6.327 130.83 | 7.038 129.99 | 4.667 50.51 | 4.374 44.36 |
| 3ac | 6.647/130.07 | 4.778/49.29 | ||
| 2ba * | 6.843 130.79 | 6.800 129.16 | 4.362 45.21 | 4.015 51.62 |
| 3ba | 6.900/132.16 | 4.501 (br)/49.72 | ||
| 2bb | 6.822 129.46 | 6.741 128.72 | 4.336 44.76 | 3.869 51.46 |
| 3bb | 6.833/130.94 | 4.416/46.94 | ||
| 2bc | 6.802 129.43 | 6.770 128.93 | 4.295 45.21 | 3.922 51.44 |
| 3bc | 6.879/130.64 | 4.893/46.64 | ||
| 2ca | 6.939 130.11 | 6.771 129.08 | 4.349 46.39 | 3.952 52.00 |
| 3ca | 6.989/131.93 | 4.446/46.66 | ||
| 2cb | 6.886 129.06 | 6.726 128.71 | 4.297 45.80 | 3.861 52.00 |
| 3cb | 6.879/130.64 | 4.335/46.64 | ||
| 2cc | 6.840 129.36 | 6.769 129.04 | 4.244 45.87 | 3.925 51.62 |
| 3cc | 6.844/130.46 | 4.284/46.57 | ||
| Starting Chloride | Products | |||||
|---|---|---|---|---|---|---|
| Compd. | Yield, % | Compd. | Yield, % | Compd. | Yield, % | |
| 6a | 7aa | 73 | 7ab | 84 | 7ac | 84 |
| 6b | 7ba a | 87 | 7bb | 79 | 7bc | 71 |
| 6c | 7ca | 72 | 7cb | 71 | 7cc | 68 |
| Lig. | CH-3 CH-3 | CH-5 CH-5 | CH2-Cq-2 CH2-Cq-2 | CH2-Cq-6 CH2-Cq-6 |
|---|---|---|---|---|
| 7aa | 6.798 124.56 | 7.326 129.30 | 4.710 50.43 | 5.120 46.86 |
| 7ab | 6.791 124.35 | 7.298 129.15 | 4.715 50.49 | 5.084 47.05 |
| 7ac | 6.777 124.26 | 7.260 129.09 | 4.704 50.45 | 5.037 46.74 |
| 7ba * | 6.749 125.33 | 7.015 129.30 | 4.279 46.56 | 4.595 45.33 |
| 7bb | 6.709 124.87 | 6.978 128.11 | 4.240 46.54 | 4.537 44.92 |
| 7bc | 6.707 124.80 | 6.938 128.13 | 4.260 46.57 | 4.464 44.87 |
| 7ca | 6.720 125.10 | 7.020 129.03 | 4.251 48.26 | 4.556 45.38 |
| 7cb | 6.685 124.69 | 6.948 128.09 | 4.229 48.24 | 4.490 44.95 |
| 7cc | 6.681 124.62 | 6.907 128.16 | 4.244 48.26 | 4.425 44.80 |
| Cell Contents [mM] | Syringe Contents [mM] | ΔH [kJ·mol−1] | Ka.106 [M−1] | n |
|---|---|---|---|---|
| EDTA 0.15 | CaCl2 0.95 | −16.54 ± 0.36 | 1.30 ± 4.91 | 0.886 |
| 2ba 0.20 | PbCl2 1.20 | 62.95 ± 9.85 | 0.31 ± 0.25 | 0.869 |
| 2ba 0.20 | CaCl2 1.20 | −100.00 ± 7.72 | 0.024 ± .059 | 1.856 |
| 2bc 0.20 | PbCl2 1.10 | −16.62 ± 0.44 | 0.14 ± 0.33 | 0.545 |
| 2cb 0.30 | PbCl2 1.50 | −37.27 ± 20.16 | 0.17 ± 0.60 | 0.201 * |
| 3bb 0.25 | KCl 1.10 | −13.48 ± 0.45 | 1.00 ± 0.58 | 0.444 |
| 3cc 0.20 | KCl 1.20 | −16.06 ± 1.48 | 0.54 ± 0.71 | 0.424 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorova, S.E.; Rusew, R.I.; Shivachev, B.L.; Kurteva, V.B. Polydentate N,O-Ligands Possessing Unsymmetrical Urea Fragments Attached to a p-Cresol Scaffold. Molecules 2023, 28, 6540. https://doi.org/10.3390/molecules28186540
Todorova SE, Rusew RI, Shivachev BL, Kurteva VB. Polydentate N,O-Ligands Possessing Unsymmetrical Urea Fragments Attached to a p-Cresol Scaffold. Molecules. 2023; 28(18):6540. https://doi.org/10.3390/molecules28186540
Chicago/Turabian StyleTodorova, Stanislava E., Rusi I. Rusew, Boris L. Shivachev, and Vanya B. Kurteva. 2023. "Polydentate N,O-Ligands Possessing Unsymmetrical Urea Fragments Attached to a p-Cresol Scaffold" Molecules 28, no. 18: 6540. https://doi.org/10.3390/molecules28186540
APA StyleTodorova, S. E., Rusew, R. I., Shivachev, B. L., & Kurteva, V. B. (2023). Polydentate N,O-Ligands Possessing Unsymmetrical Urea Fragments Attached to a p-Cresol Scaffold. Molecules, 28(18), 6540. https://doi.org/10.3390/molecules28186540