
Kinetic Study on the Base-Catalyzed Imine-Enamine Tautomerism of a Chiral Biologically Active Isoxazoline Derivative by HPLC on Amylose Tris(3,5-dimethylphenylcarbamate) Chiral Stationary Phase

 ,
, 
 ,
, 

Abstract
:1. Introduction
2. Results
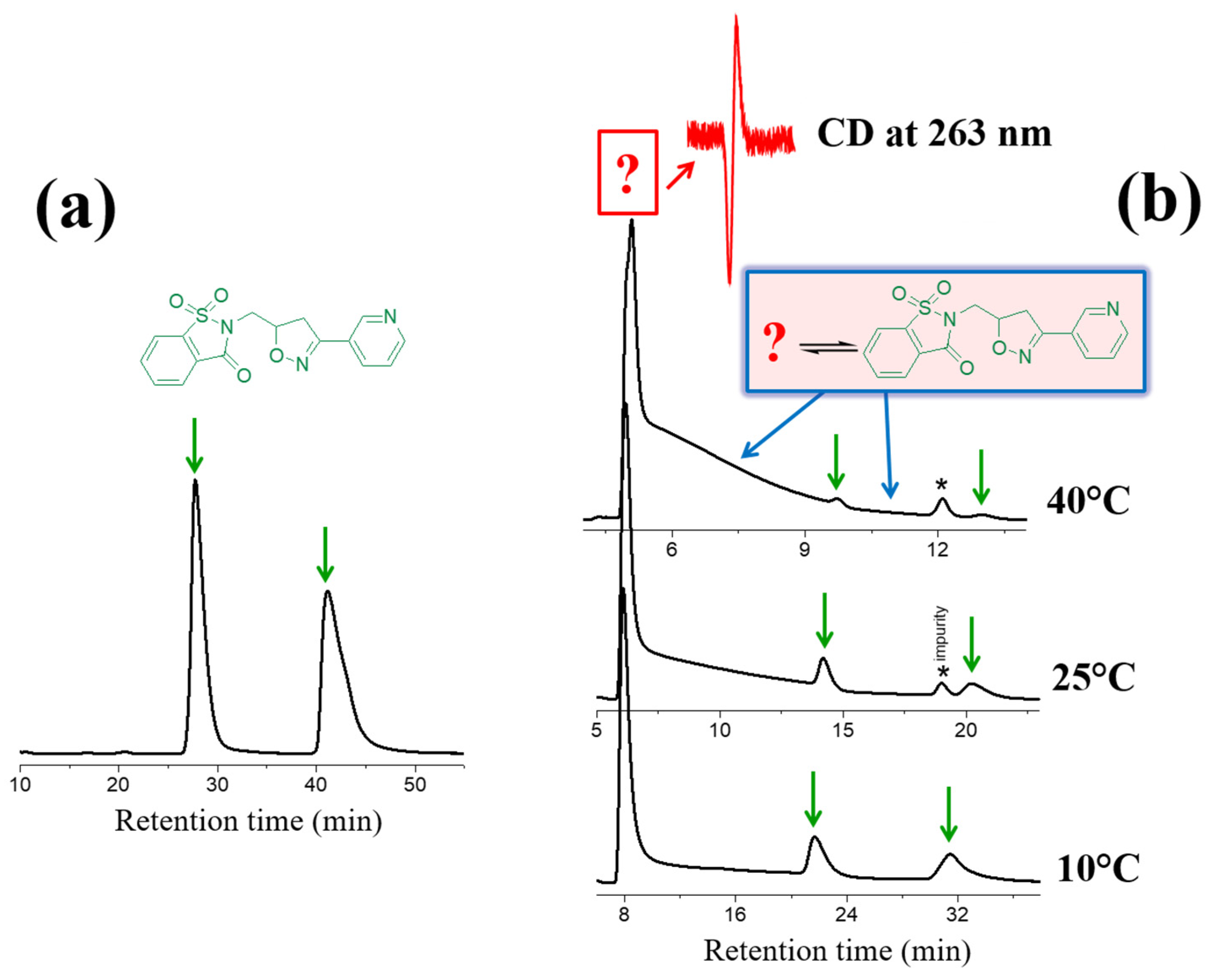
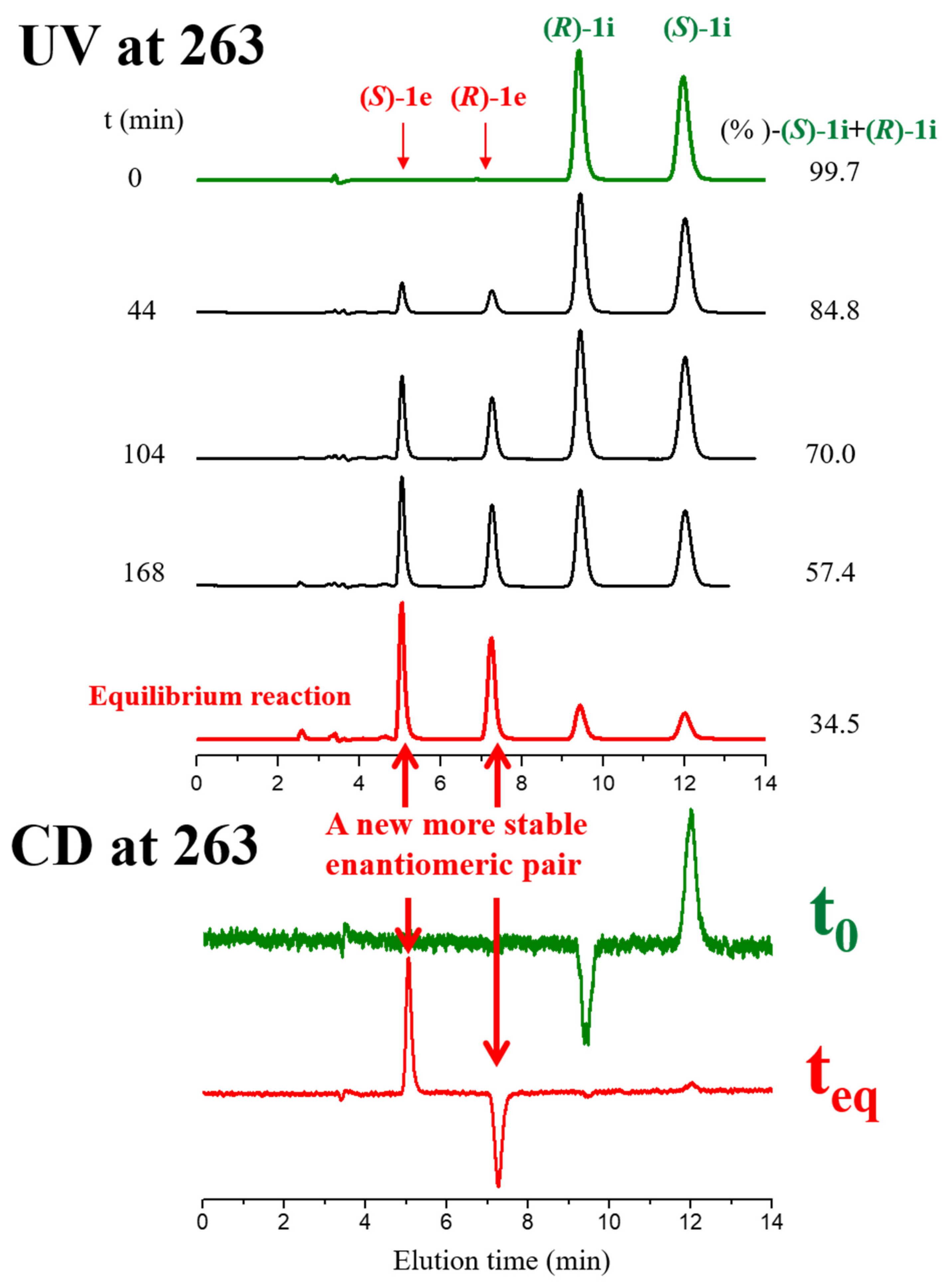
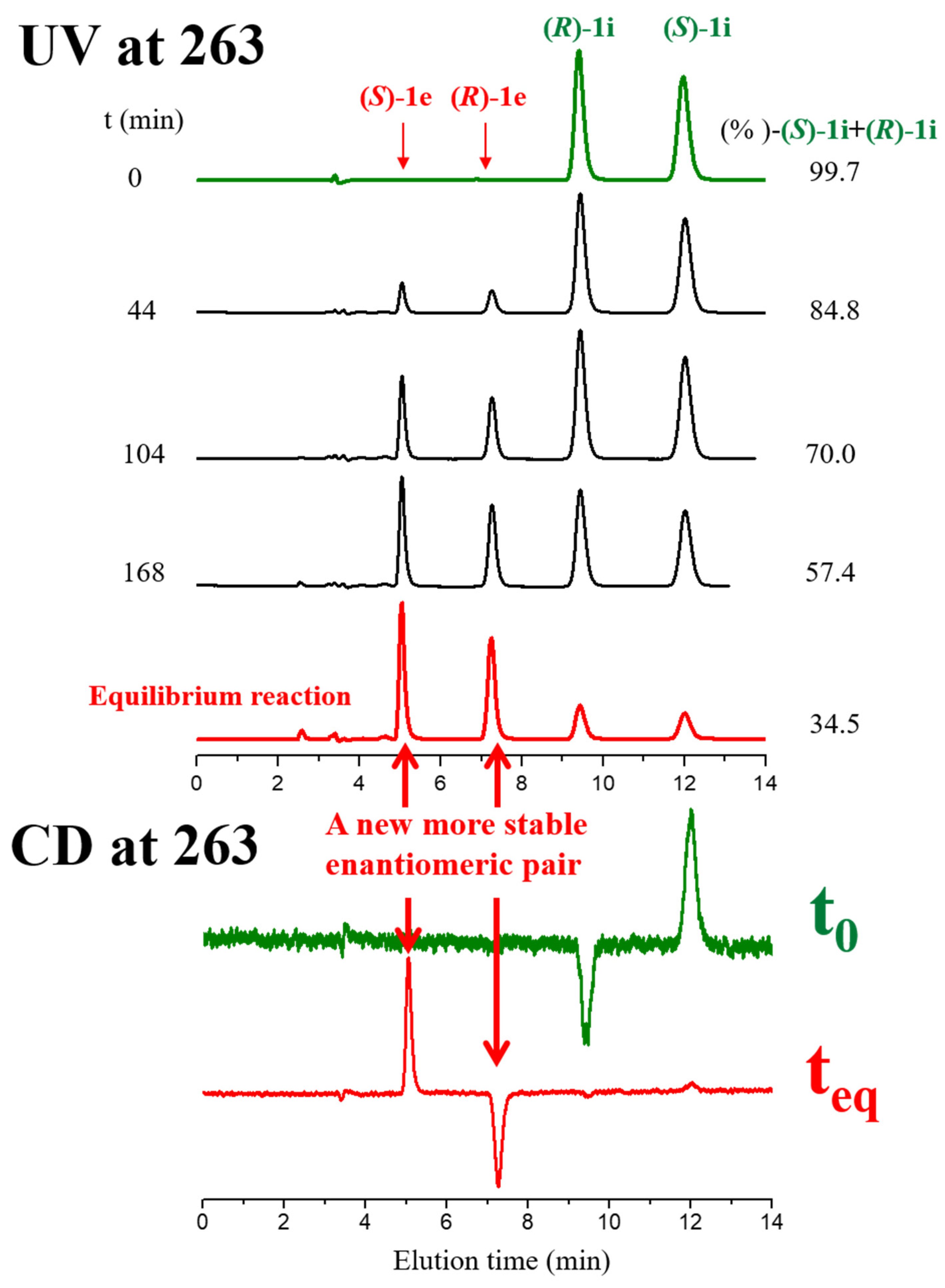
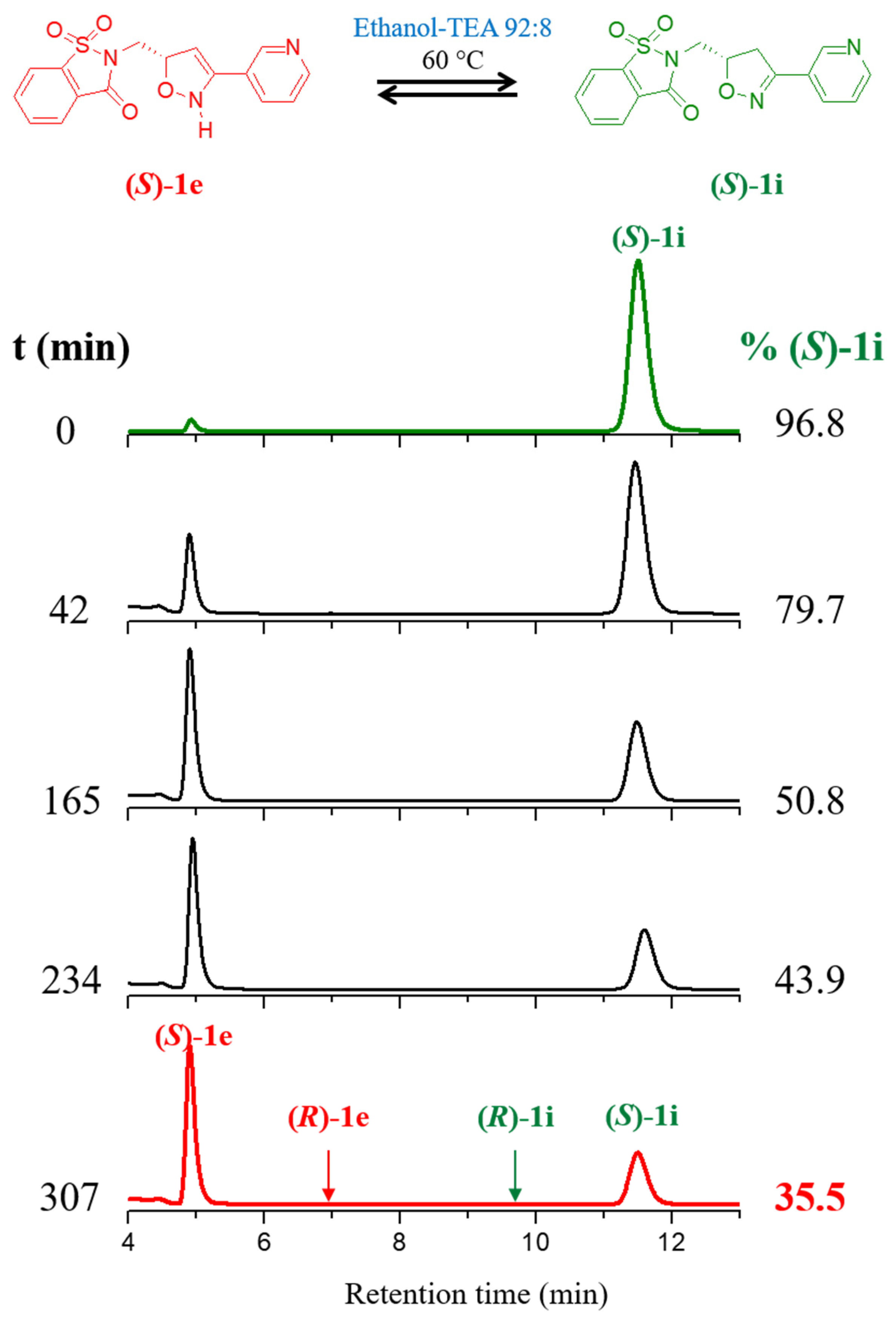
2.1. Base-Catalyzed Imino-Enamine Tautomerism Studied by HPLC on Chiral Stationary Phase
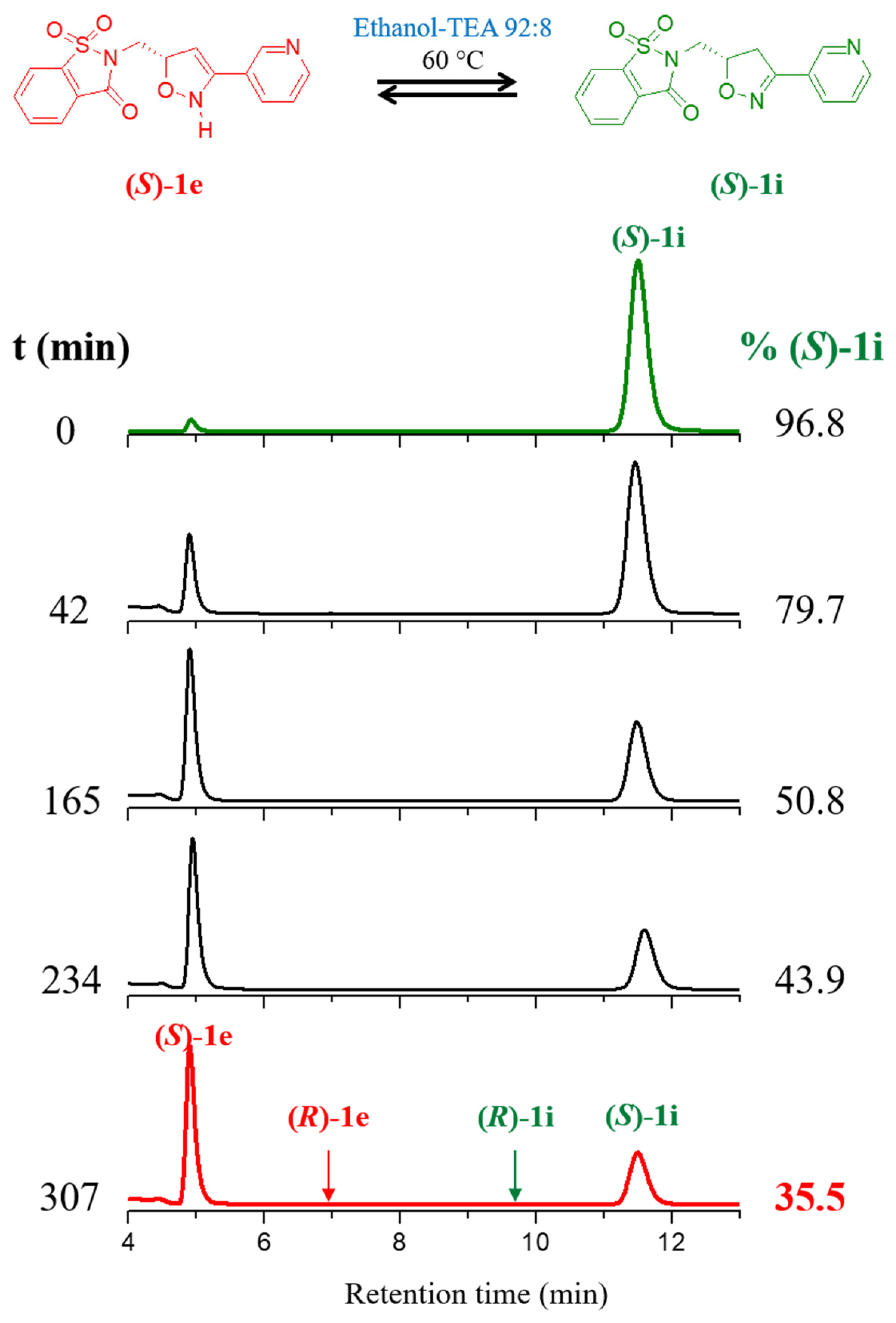
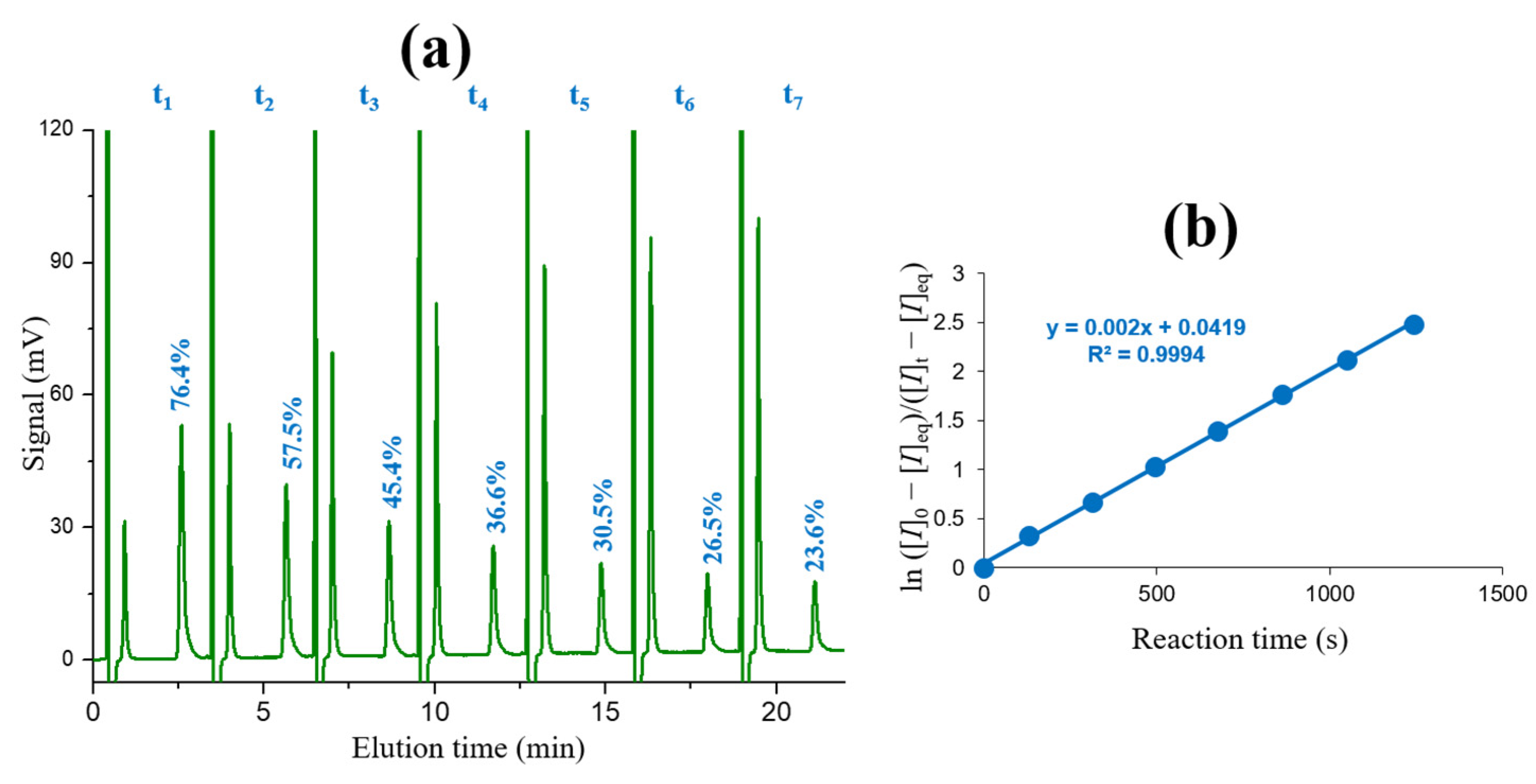
2.2. Kinetics of Base-Catalyzed Imino-Enamine Isomerization
3. Materials and Methods
3.1. Materials
3.2. Chromatographic Analysis
3.3. Chiroptical Analysis
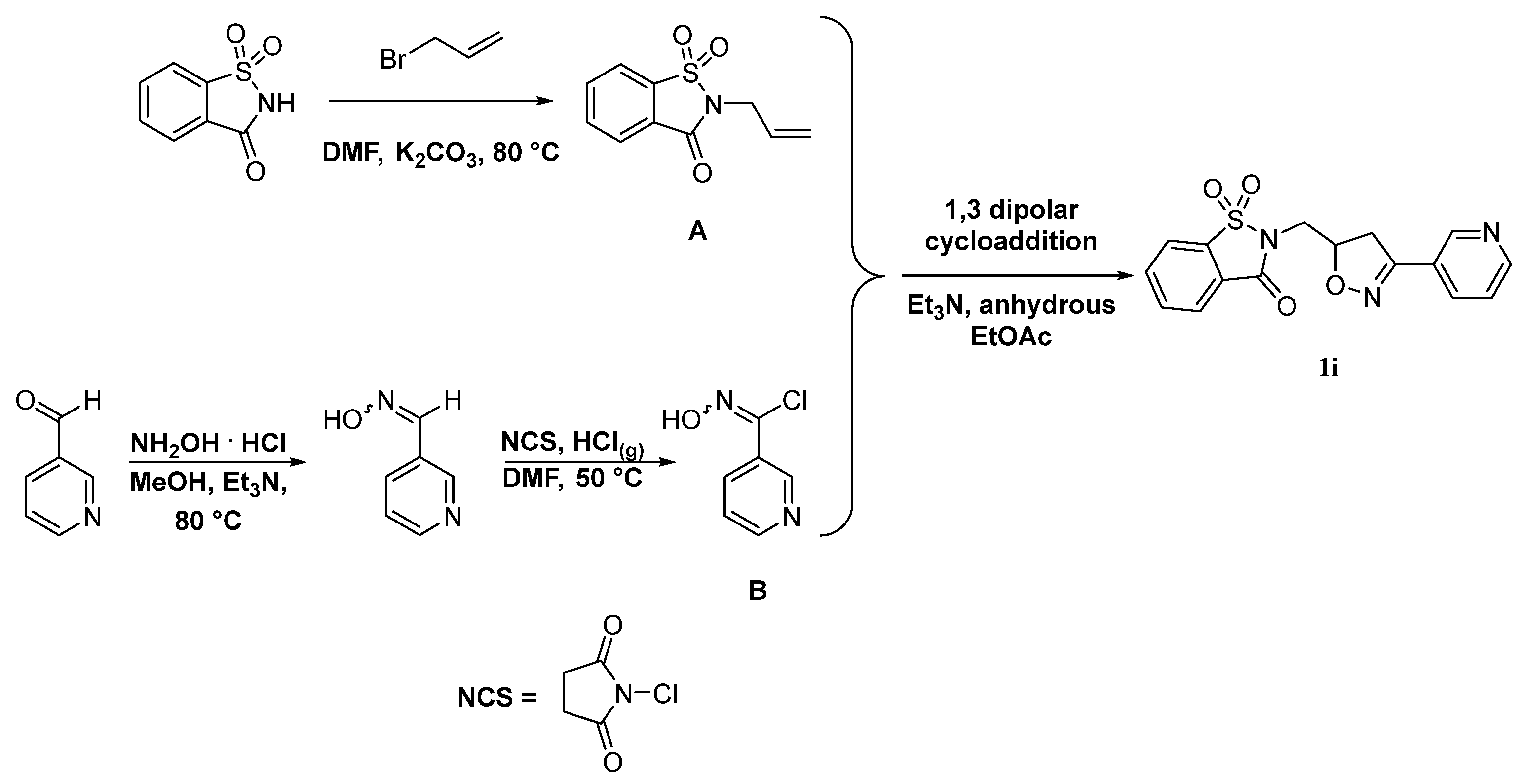
3.4. Synthesis of Compound 1i
3.5. Absolute Configuration Assignment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Cavazzini, A.; Pasti, L.; Massi, A.; Marchetti, N.; Dondi, F. Recent applications in chiral high performance liquid chromatography: A review. Anal. Chim. Acta 2011, 706, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Lämmerhofer, M. Chiral recognition by enantioselective liquid chromatography: Mechanisms and modern chiral stationary phases. J. Chromatogr. A 2010, 1217, 814–856. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C. Stereolabile chiral compounds: Analysis by dynamic chromatography and stopped-flow methods. Chem. Soc. Rev. 2005, 34, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Cirilli, R.; Ferretti, R.; La Torre, F.; Secci, D.; Bolasco, A.; Carradori, S.; Pierini, M. High-performance liquid chromatographic separation of enantiomers and diastereomers of 2-methylcyclohexanone thiosemicarbazone, and determination of absolute configuration and configurational stability. J. Chromatogr. A 2007, 1172, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, S.; Benincori, T.; Bonometti, V.; Cirilli, R.; Mussini, P.R.; Pierini, M.; Pilati, T.; Sannicolò, F. Steric and electronic effects on the configurational stability of residual chiral phosphorus-centered three-bladed propellers: Tris-aryl phosphanes. Chem. Eur. J. 2013, 19, 182–194. [Google Scholar] [CrossRef]
- Schurig, V. Peak coalescence phenomena in enantioselective chromatography. Chirality Pharmacol. Biol. Chem. Conseq. Mol. Asymmetry 1998, 10, 140–146. [Google Scholar]
- Trapp, O.; Schoetz, G.; Schurig, V. Determination of enantiomerization barriers by dynamic and stopped-flow chromatographic methods. Chirality 2001, 13, 403–414. [Google Scholar] [CrossRef]
- Trapp, O. Interconversion of stereochemically labile enantiomers (enantiomerization). Differ. Enantiomers II 2013, 341, 231–269. [Google Scholar]
- D’Acquarica, I.; Gasparrini, F.; Pierini, M.; Villani, C.; Zappia, G. Dynamic HPLC on chiral stationary phases: A powerful tool for the investigation of stereomutation processes. J. Sep. Sci. 2006, 29, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Storch, G.; Spallek, M.J.; Rominger, F.; Trapp, O. Tautomerization-Mediated Molecular Switching Between Six-and Seven-Membered Rings Stabilized by Hydrogen Bonding. Chem. Eur. J. 2015, 21, 8939–8945. [Google Scholar] [CrossRef]
- Cirilli, R.; Costi, R.; Di Santo, R.; La Torre, F.; Pierini, M.; Siani, G. Perturbing effects of chiral stationary phase on enantiomerization second-order rate constants determined by enantioselective dynamic high-performance liquid chromatography: A practical tool to quantify the accessible acid and basic catalytic sites bonded on chromatographic supports. Anal. Chem. 2009, 81, 3560–3570. [Google Scholar] [PubMed]
- Caccamese, S.; Principato, G. Resolution of the enantiomers of tetrahydrozoline by chiral HPLC. The racemization of the enantiomers via an imine–enamine tautomerism. Tetrahedron Asymmetry 1998, 9, 2939–2945. [Google Scholar] [CrossRef]
- De Camp, W.H. Chiral drugs: The FDA perspective on manufacturing and control. J. Pharm. Biomed. Anal. 1993, 11, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Shankar, R. 2-Isoxazolines: A synthetic and medicinal overview. ChemMedChem 2021, 16, 430–447. [Google Scholar] [CrossRef] [PubMed]
- D’Ascenzio, M.; Secci, D.; Carradori, S.; Zara, S.; Guglielmi, P.; Cirilli, R.; Pierini, M.; Poli, G.; Tuccinardi, T.; Angeli, A. 1, 3-Dipolar cycloaddition, HPLC enantioseparation, and docking studies of saccharin/isoxazole and saccharin/isoxazoline derivatives as selective carbonic anhydrase IX and XII inhibitors. J. Med. Chem. 2020, 63, 2470–2488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kientzy, C.; Franco, P.; Ohnishi, A.; Kagamihara, Y.; Kurosawa, H. Solvent versatility of immobilized 3, 5-dimethylphenylcarbamate of amylose in enantiomeric separations by HPLC. J. Chromatogr. A 2005, 1075, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Chankvetadze, B. Recent trends in preparation, investigation and application of polysaccharide-based chiral stationary phases for separation of enantiomers in high-performance liquid chromatography. Trends Analyt. Chem. 2020, 122, 115709. [Google Scholar] [CrossRef]
- Ikai, T.; Okamoto, Y. Structure control of polysaccharide derivatives for efficient separation of enantiomers by chromatography. Chem. Rev. 2009, 109, 6077–6101. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Solvent | T(°C) | k(I→E)(s−1) | k(E→I)(s−1) | t0.5(I→E)(min) | t0.5(E→I)(min) |
|---|---|---|---|---|---|
| Methanol/TEA (0.1%) | 20 | 1.64 × 10−4 | 2.89 × 10−5 | 70 | 400 |
| Methanol/TEA (0.1%) | 25 | 1.69 × 10−4 | 3.18 × 10−5 | 68 | 363 |
| Methanol/TEA (0.1%) | 30 | 3.36 × 10−4 | 7.07 × 10−5 | 34 | 163 |
| Methanol/TEA (0.1%) | 35 | 1.20 × 10−3 | 2.65 × 10−4 | 10 | 44 |
| Methanol/TEA (0.2%) | 20 | 2.66 × 10−4 | 4.70 × 10−5 | 43 | 246 |
| Methanol/TEA (0.2%) | 25 | 3.64 × 10−4 | 6.84 × 10−5 | 32 | 169 |
| Methanol/TEA (0.2%) | 30 | 8.06 × 10−4 | 1.65 × 10−4 | 14 | 70 |
| Methanol/TEA (0.2%) | 35 | 1.79 × 10−3 | 4.23 × 10−4 | 6 | 27 |
| Methanol/TEA (0.4%) | 20 | 4.39 × 10−4 | 7.75 × 10−5 | 26 | 149 |
| Methanol/TEA (0.4%) | 25 | 6.81 × 10−4 | 1.09 × 10−4 | 17 | 106 |
| Methanol/TEA (0.4%) | 30 | 1.65 × 10−3 | 3.37 × 10−4 | 7 | 34 |
| Methanol/TEA (0.4%) | 35 | 2.86 × 10−3 | 6.75 × 10−4 | 4 | 17 |
| Ethanol/TEA (10%) | 50 | 6.1 × 10−5 | 3.0 × 10−5 | 189 | 391 |
| Ethanol/TEA (10%) | 55 | 8.6 × 10−5 | 4.6 × 10−5 | 135 | 250 |
| Ethanol/TEA (10%) | 60 | 9.5 × 10−5 | 5.0 × 10−5 | 122 | 232 |
| Ethanol/TEA (10%) | 65 | 1.3 × 10−5 | 8.2 × 10−5 | 87 | 141 |
| Ethanol/TEA (4%) | 60 | 6.4 × 10−5 | 3.4 × 10−5 | 179 | 340 |
| Ethanol/TEA (6%) | 60 | 8.0 × 10−5 | 4.2 × 10−5 | 145 | 275 |
| Ethanol/TEA (8%) | 60 | 8.8 × 10−5 | 4.6 × 10−5 | 132 | 250 |
| 1-Propanol/TEA (10%) | 60 | The isomeric ratio remained unchanged within 24 h | |||
| Ethyl acetate/TEA (10%) | 60 | The isomeric ratio remained unchanged within 24 h | |||
| Acetonitrile/TEA (10%) | 60 | The isomeric ratio remained unchanged within 24 h | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadutto, D.; Guglielmi, P.; Carradori, S.; Secci, D.; Cirilli, R. Kinetic Study on the Base-Catalyzed Imine-Enamine Tautomerism of a Chiral Biologically Active Isoxazoline Derivative by HPLC on Amylose Tris(3,5-dimethylphenylcarbamate) Chiral Stationary Phase. Molecules 2023, 28, 6518. https://doi.org/10.3390/molecules28186518
Sadutto D, Guglielmi P, Carradori S, Secci D, Cirilli R. Kinetic Study on the Base-Catalyzed Imine-Enamine Tautomerism of a Chiral Biologically Active Isoxazoline Derivative by HPLC on Amylose Tris(3,5-dimethylphenylcarbamate) Chiral Stationary Phase. Molecules. 2023; 28(18):6518. https://doi.org/10.3390/molecules28186518
Chicago/Turabian StyleSadutto, Daniele, Paolo Guglielmi, Simone Carradori, Daniela Secci, and Roberto Cirilli. 2023. "Kinetic Study on the Base-Catalyzed Imine-Enamine Tautomerism of a Chiral Biologically Active Isoxazoline Derivative by HPLC on Amylose Tris(3,5-dimethylphenylcarbamate) Chiral Stationary Phase" Molecules 28, no. 18: 6518. https://doi.org/10.3390/molecules28186518
APA StyleSadutto, D., Guglielmi, P., Carradori, S., Secci, D., & Cirilli, R. (2023). Kinetic Study on the Base-Catalyzed Imine-Enamine Tautomerism of a Chiral Biologically Active Isoxazoline Derivative by HPLC on Amylose Tris(3,5-dimethylphenylcarbamate) Chiral Stationary Phase. Molecules, 28(18), 6518. https://doi.org/10.3390/molecules28186518