

Artificial Small Molecules as Cofactors and Biomacromolecular Building Blocks in Synthetic Biology: Design, Synthesis, Applications, and Challenges

 , , , , and
, , , , and 
Abstract
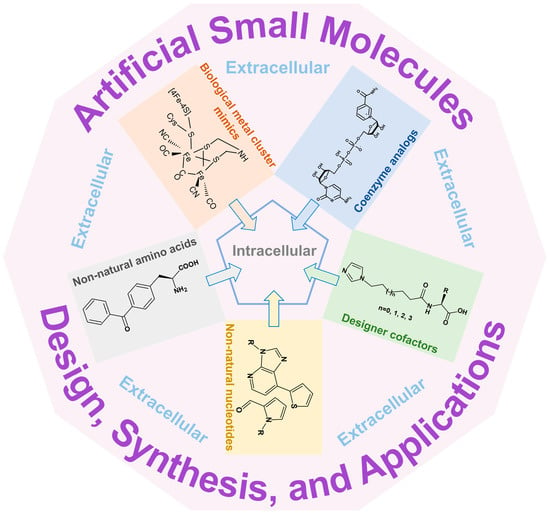
1. Introduction
2. Biological Metal Cluster Mimics
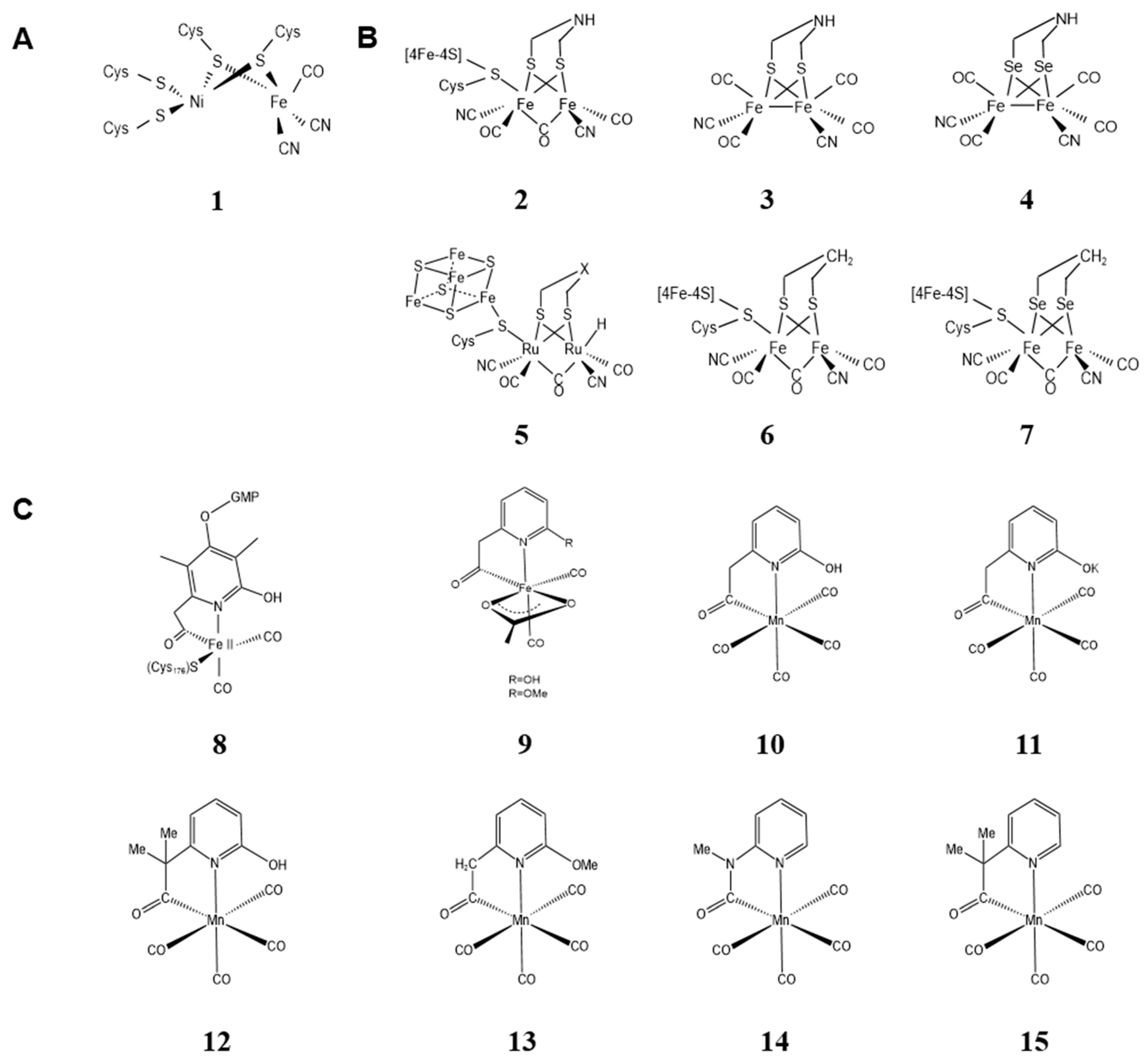
2.1. Metal Clusters of Artificial Hydrogenases
2.1.1. Metal Clusters of Artificial [Ni-Fe] Hydrogenases
2.1.2. Metal Clusters of Artificial [Fe-Fe] Hydrogenases
2.1.3. Metal Clusters of Artificial [Fe] Hydrogenases

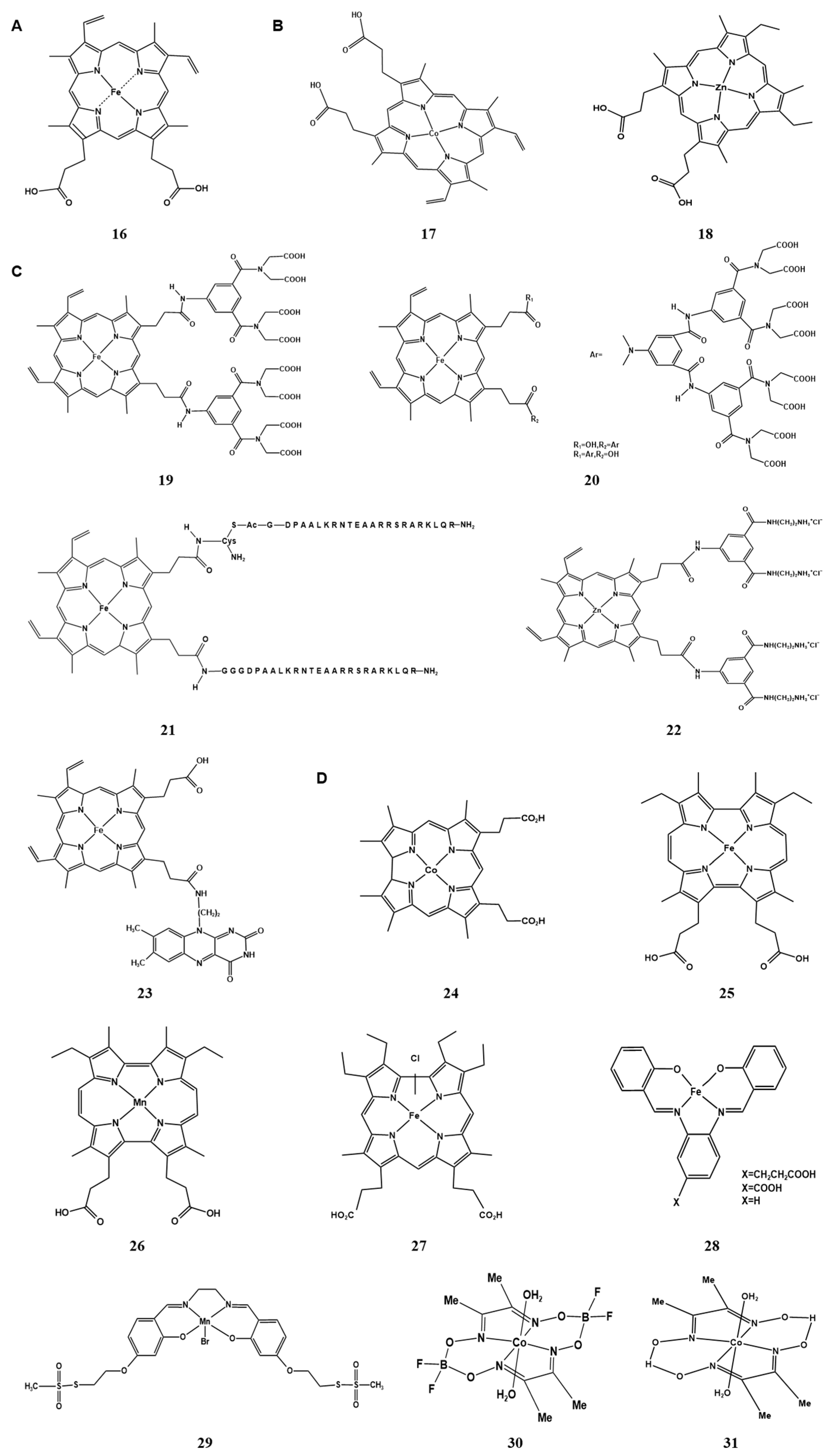
2.2. Metal Clusters of Artificial Hemoprotein
2.2.1. Metal Substitution of Heme in the Cofactor
2.2.2. Modification of Peripheral Functional Groups of Porphyrin Ligands
2.2.3. Providing Non-Natural Porphyrin/Non-Porphyrin Cofactor Scaffolds

2.3. Metal Clusters of the Artificial Photosynthesis System
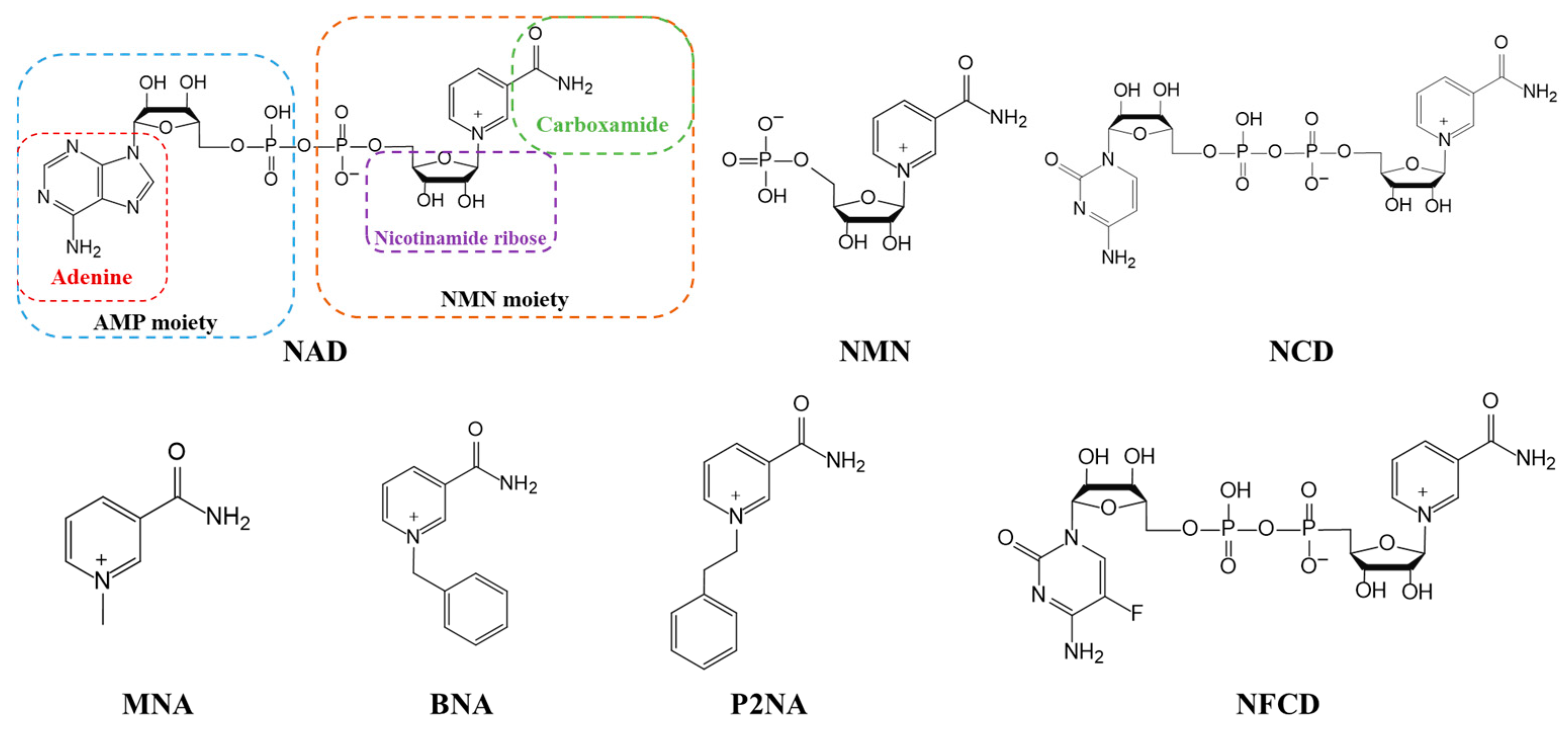
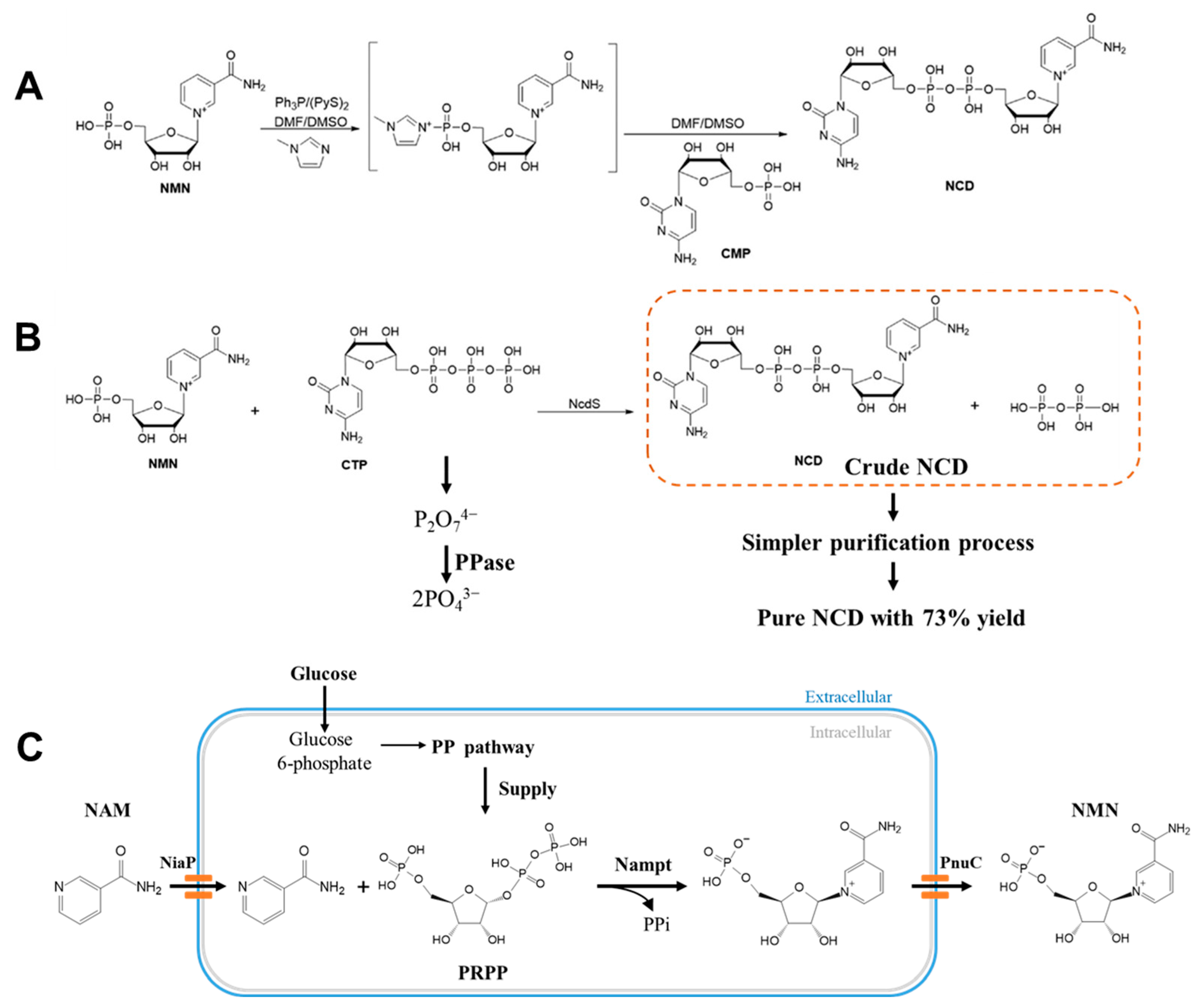
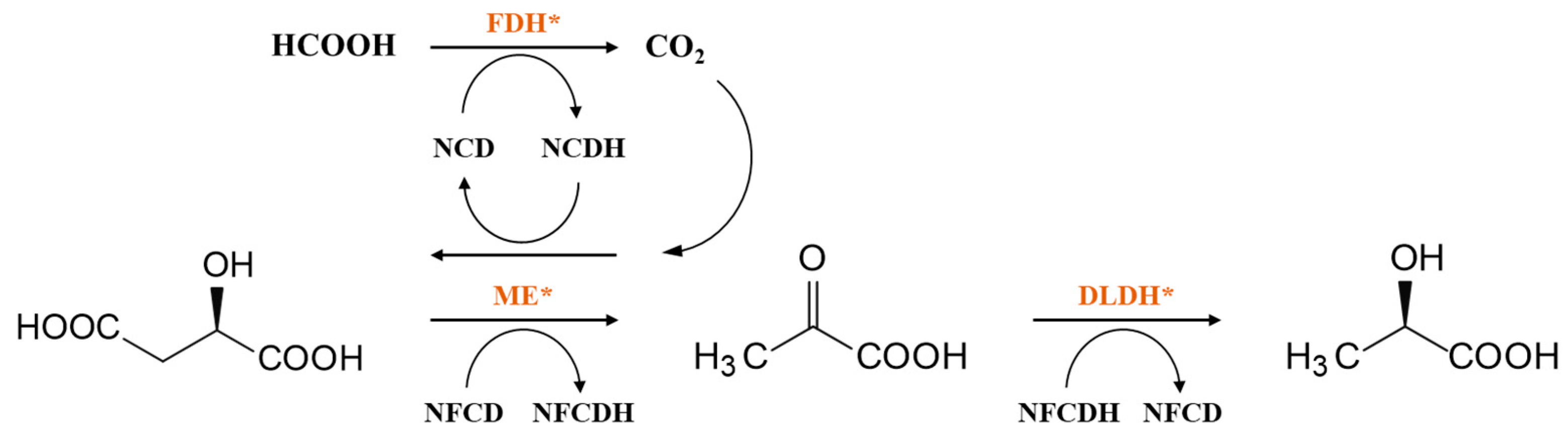
3. Coenzyme Analogs
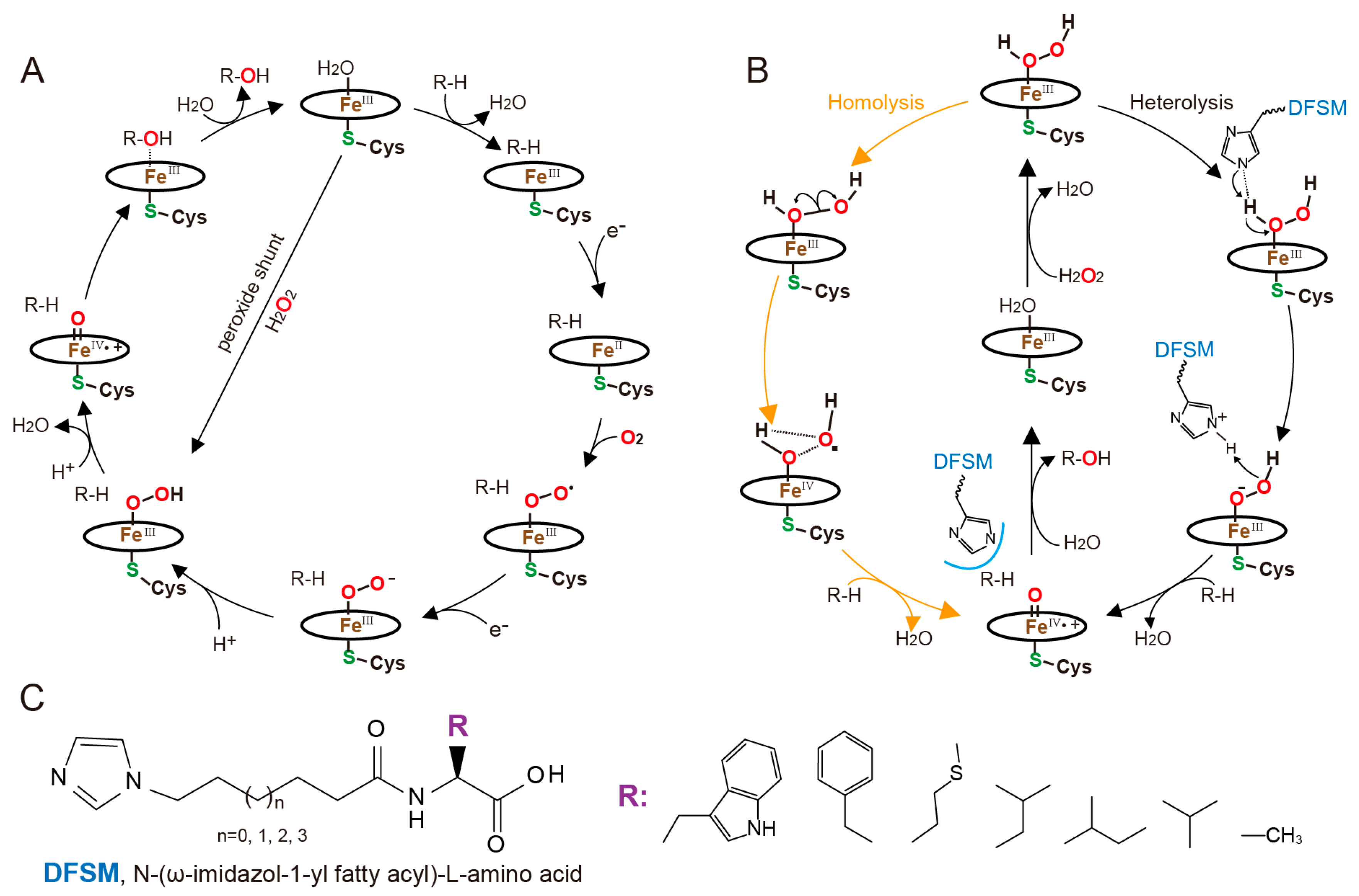
4. Designer Cofactors
5. XNAs and nnAAs
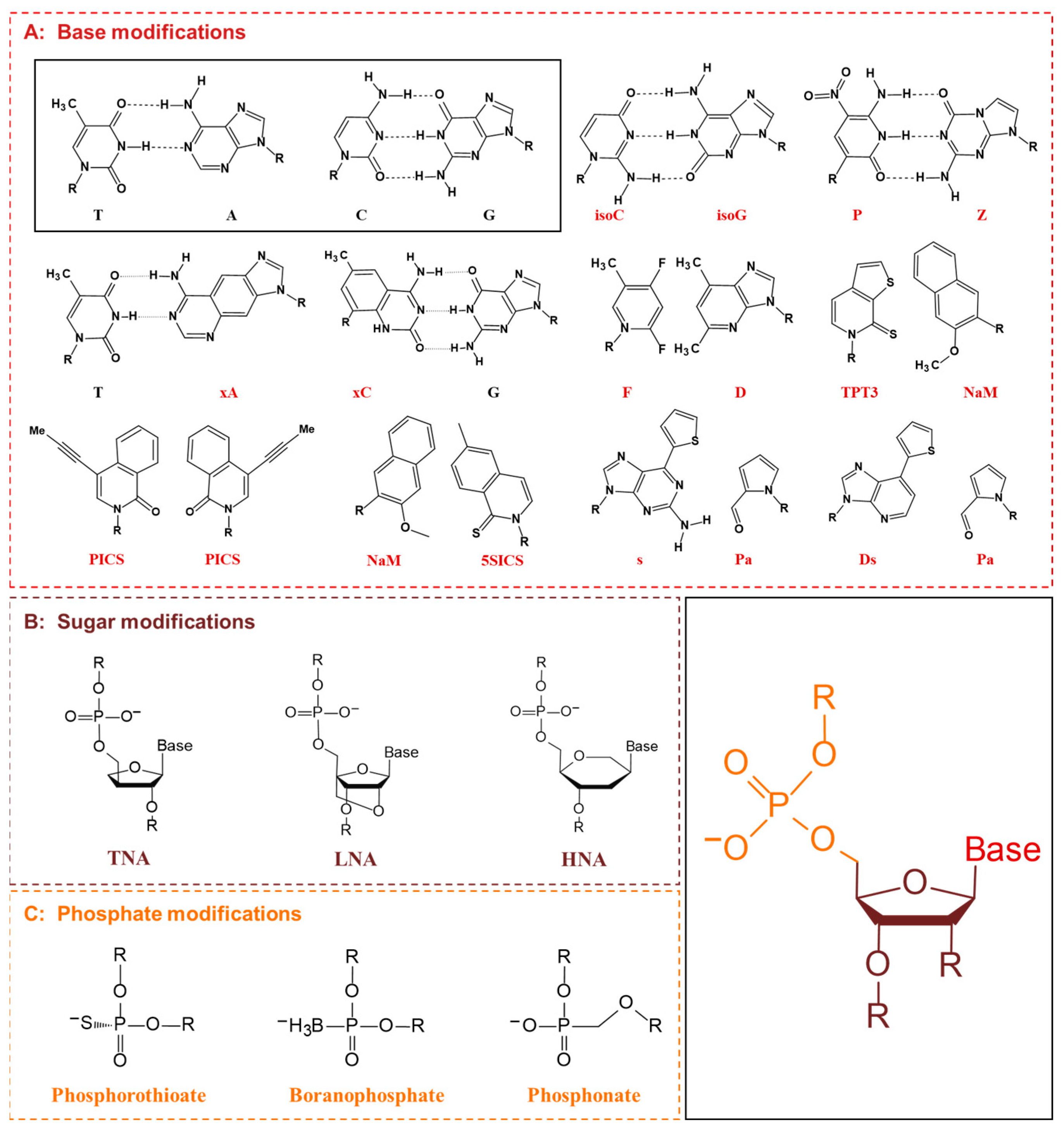
5.1. XNA Engineering
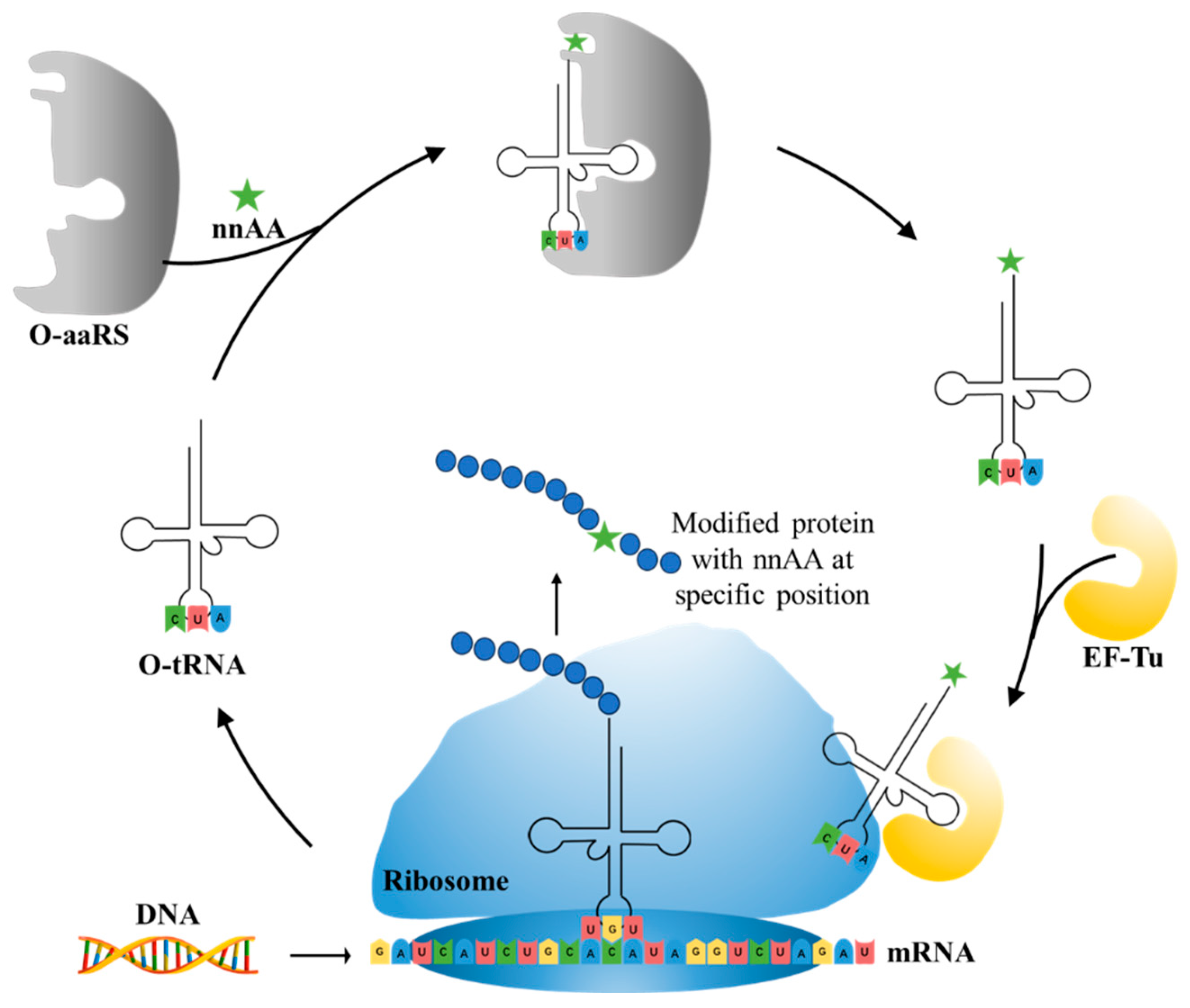
5.2. nnAAs Incorporation
5.3. Applications in Synthetic Biology
5.3.1. Enzymes Engineering
Enzyme Activity
Enzyme Stability
Stereoselectivity and Regioselectivity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviations | nnAA | Structural Formula | Target Protein | Reference |
|---|---|---|---|---|
| pBzF | p-benzoylphenylalanine |  | TAm | [204] |
| metA | [222] | |||
| pCNF | p-cyanophenylalanine |  | TK | [206] |
| pNF | p-nitrophenylalanine |  | NTR | [207] |
| AcrF | p-acrylamido-phenylalanine |  | TEM-1 β-lactamase | [208] |
| pFF | 4-fluorophenylalanine |  | PTE | [220] |
| pPa | p-propargyloxy-phenylalanine |  | GFP | [210] |
| mFF | m-fluorophenylalanine |  | PvuII | [209] |
| BiF | 4-phenyl-l-phenylalanine |  | diketoreductase | [225] |
| BuOF | O-tert-butyl-l-tyrosine |  | ||
| SetY | O-(2-mercaptoethyl)-l-tyrosine |  n = 1, SetY n = 2, SprY n = 3, SbuY | β-lactamase | [221] |
| SprY | O-(3-mercaptoethyl)-l-tyrosine | |||
| SbuY | O-(4-mercaptoethyl)-l-tyrosine | |||
| mFY | 3-fluorotyrosine |  | TAm | [205] |
| — | 3-chloro-l-tyrosine |  | glutathione S-transferase | [224] |
| — | 3-bromo-l-tyrosine |  | ||
| ImiTyr | 2-amino-3-(4-hydroxy-3-(1H-imidazol-1-yl) phenyl) propanoic acid |  | HCO | [216] |
| MtTyr | 2-amino-3-(4-hydroxy-3-(methylthio) phenyl)-propanoic acid |  | myoglobin | [217] |
| OMeTyr | O-methyltyrosine |  | squalene-hopene cyclase | [215] |
| pAcF | p-acetyl-phenylalanine |  | cytochrome P450 CYP102A1-139-3 | [227] |
| NapA | 3-(2-naphthyl)-alanine |  | ||
| BpyAla | (2,2′-bipyridin-5-yl) alanine |  | noncatalytic catabolite activator protein | [218] |
| LmrR | [226] | |||
| Hco | l-(7-hydroxycoumarin-4-yl) ethylglycine |  | arPTE | [214] |
| Nle | norleucine |  | cytochrome P450 | [211] |
| lipase | [212] | |||
| Dpc | 2,3-dihydroxypropyl cysteine |  | NAL | [213] |
| NMH | Nδ-methyl histidine |  | BH32 scaffold protein | [219] |
5.3.2. Cellular Process Controlling
5.3.3. Chassis Strain Engineering and Tracking
6. Summary, Challenge, and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- King, E.; Maxel, S.; Li, H. Engineering natural and noncanonical nicotinamide cofactor-dependent enzymes: Design principles and technology development. Curr. Opin. Biotechnol. 2020, 66, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Vignais, P.M.; Billoud, B. Occurrence, classification, and biological function of hydrogenases: An overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef] [PubMed]
- Canaguier, S.; Artero, V.; Fontecave, M. Modelling NiFe hydrogenases: Nickel-based electrocatalysts for hydrogen production. Dalton Trans. 2008, 3, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Song, L.C.; Li, J.P.; Xie, Z.J.; Song, H.B. Synthesis, Structural Characterization, and Electrochemical Properties of Dinuclear Ni/Mn Model Complexes for the Active Site of [NiFe]-Hydrogenases. Inorg. Chem. 2013, 52, 11618–11626. [Google Scholar] [CrossRef]
- Gu, Y.; Bloomer, B.J.; Liu, Z.; Chen, R.; Clark, D.S.; Hartwig, J.F. Directed evolution of artificial metalloenzymes in whole cells. Angew. Chem. Int. Ed. 2022, 61, e202110519. [Google Scholar] [CrossRef]
- Wan, L.; Wang, X.; Hu, Y.; Li, Q.; Zhao, Z.K. Gram-scale biocatalytic preparation of the non-natural cofactor nicotinamide cytosine dinucleotide. Tetrahedron Lett. 2022, 88, 153568. [Google Scholar] [CrossRef]
- Liu, Y.; Yasawong, M.; Yu, B. Metabolic engineering of Escherichia coli for biosynthesis of beta-nicotinamide mononucleotide from nicotinamide. Microb. Biotechnol. 2021, 14, 2581–2591. [Google Scholar] [CrossRef]
- Shoji, S.; Yamaji, T.; Makino, H.; Ishii, J.; Kondo, A. Metabolic design for selective production of nicotinamide mononucleotide from glucose and nicotinamide. Metab. Eng. 2021, 65, 167–177. [Google Scholar] [CrossRef]
- Black, W.B.; Zhang, L.; Mak, W.S.; Maxel, S.; Cui, Y.; King, E.; Fong, B.; Sanchez Martinez, A.; Siegel, J.B.; Li, H. Engineering a nicotinamide mononucleotide redox cofactor system for biocatalysis. Nat. Chem. Biol. 2020, 16, 87–94. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Upp, D.M.; Lewis, J.C.; Job Zhang, Y.P. A High-Throughput Method for Directed Evolution of NAD(P)(+)-Dependent Dehydrogenases for the Reduction of Biomimetic Nicotinamide Analogues. ACS Catal. 2019, 9, 11709–11719. [Google Scholar] [CrossRef]
- Wang, J.; Guo, X.; Wan, L.; Liu, Y.; Xue, H.; Zhao, Z.K. Engineering Formaldehyde Dehydrogenase from Pseudomonas putida to Favor Nicotinamide Cytosine Dinucleotide. Chembiochem 2022, 23, e202100697. [Google Scholar] [CrossRef]
- Xu, J.; Wang, C.; Cong, Z. Strategies for Substrate-Regulated P450 Catalysis: From Substrate Engineering to Co-catalysis. Chemistry 2019, 25, 6853–6863. [Google Scholar] [CrossRef]
- Zhang, Y.; Ptacin, J.L.; Fischer, E.C.; Aerni, H.R.; Caffaro, C.E.; San Jose, K.; Feldman, A.W.; Turner, C.R.; Romesberg, F.E. A semi-synthetic organism that stores and retrieves increased genetic information. Nature 2017, 551, 644–647. [Google Scholar] [CrossRef]
- Vargas-Rodriguez, O.; Sevostyanova, A.; Soll, D.; Crnkovic, A. Upgrading aminoacyl-tRNA synthetases for genetic code expansion. Curr. Opin. Chem. Biol. 2018, 46, 115–122. [Google Scholar] [CrossRef]
- Arnold, F.H. Innovation by Evolution: Bringing New Chemistry to Life (Nobel Lecture). Angew. Chem. Int. Ed. 2019, 58, 14420–14426. [Google Scholar] [CrossRef]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.F.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Kohler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef]
- Kleinhaus, J.T.; Wittkamp, F.; Yadav, S.; Siegmund, D.; Apfel, U.P. [FeFe]-Hydrogenases: Maturation and reactivity of enzymatic systems and overview of biomimetic models. Chem. Soc. Rev. 2021, 50, 1668–1784. [Google Scholar] [CrossRef]
- Ogata, H.; Nishikawa, K.; Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature 2015, 520, 571–574. [Google Scholar] [CrossRef]
- Xuan, J.S.; He, L.L.; Wen, W.; Feng, Y.A. Hydrogenase and Nitrogenase: Key Catalysts in Biohydrogen Production. Molecules 2023, 28, 1392. [Google Scholar] [CrossRef]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef]
- Ohki, Y.; Yasumura, K.; Ando, M.; Shimokata, S.; Tatsumi, K. A model for the CO-inhibited form of [NiFe] hydrogenase: Synthesis of CO3Fe(micro-StBu)3NiSC6H3-2,6-(mesityl)2 and reversible CO addition at the Ni site. Proc. Natl. Acad. Sci. USA 2010, 107, 3994–3997. [Google Scholar] [CrossRef] [PubMed]
- Tard, C.; Pickett, C.J. Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases. Chem. Rev. 2009, 109, 2245–2274. [Google Scholar] [CrossRef] [PubMed]
- Schilter, D.; Rauchfuss, T.B.; Stein, M. Connecting [NiFe]- and [FeFe]-hydrogenases: Mixed-valence nickel-iron dithiolates with rotated structures. Inorg. Chem. 2012, 51, 8931–8941. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.E.; Whaley, C.M.; Rauchfuss, T.B.; Gray, D.L. Nickel−Iron Dithiolato Hydrides Relevant to the [NiFe]-Hydrogenase Active Site. J. Am. Chem. Soc. 2009, 131, 6942–6943. [Google Scholar] [CrossRef]
- Canaguier, S.; Fourmond, V.; Perotto, C.U.; Fize, J.; Pecaut, J.; Fontecave, M.; Field, M.J.; Artero, V. Catalytic hydrogen production by a Ni-Ru mimic of NiFe hydrogenases involves a proton-coupled electron transfer step. Chem. Commun. 2013, 49, 5004–5006. [Google Scholar] [CrossRef]
- Oudart, Y.; Artero, V.; Norel, L.; Train, C.; Pécaut, J.; Fontecave, M. Synthesis, crystal structure, magnetic properties and reactivity of a Ni–Ru model of NiFe hydrogenases with a pentacoordinated triplet (S = 1) NiII center. J. Organomet. Chem. 2009, 694, 2866–2869. [Google Scholar] [CrossRef]
- Razavet, M.; Artero, V.; Cavazza, C.; Oudart, Y.; Lebrun, C.; Fontecilla-Camps, J.C.; Fontecave, M. Tricarbonylmanganese(I)-lysozyme complex: A structurally characterized organometallic protein. Chem. Commun. 2007, 27, 2805–2807. [Google Scholar] [CrossRef]
- Slater, J.W.; Shafaat, H.S. Nickel-Substituted Rubredoxin as a Minimal Enzyme Model for Hydrogenase. J. Phys. Chem. Lett. 2015, 6, 3731–3736. [Google Scholar] [CrossRef]
- Slater, J.W.; Marguet, S.C.; Monaco, H.A.; Shafaat, H.S. Going beyond Structure: Nickel-Substituted Rubredoxin as a Mechanistic Model for the [NiFe] Hydrogenases. J. Am. Chem. Soc. 2018, 140, 10250–10262. [Google Scholar] [CrossRef]
- Daraosheh, A.Q.; Abul-Futouh, H.; Murakami, N.; Ziems, K.M.; Gorls, H.; Kupfer, S.; Grafe, S.; Ishii, A.; Celeda, M.; Mloston, G.; et al. Novel [FeFe]-Hydrogenase Mimics: Unexpected Course of the Reaction of Ferrocenyl alpha-Thienyl Thioketone with Fe-3(CO)(12). Materials 2022, 15, 2867. [Google Scholar] [CrossRef]
- Land, H.; Senger, M.; Berggren, G.; Stripp, S.T. Current State of [FeFe]-Hydrogenase Research: Biodiversity and Spectroscopic Investigations. ACS Catal. 2020, 10, 7069–7086. [Google Scholar] [CrossRef]
- Birrell, J.A.; Wrede, K.; Pawlak, K.; Rodriguez-Maciá, P.; Rüdiger, O.; Reijerse, E.J.; Lubitz, W. Artificial Maturation of the Highly Active Heterodimeric [FeFe] Hydrogenase fromDesulfovibrio desulfuricansATCC 7757. Isr. J. Chem. 2016, 56, 852–863. [Google Scholar] [CrossRef]
- Caserta, G.; Adamska-Venkatesh, A.; Pecqueur, L.; Atta, M.; Artero, V.; Roy, S.; Reijerse, E.; Lubitz, W.; Fontecave, M. Chemical assembly of multiple metal cofactors: The heterologously expressed multidomain [FeFe]-hydrogenase from Megasphaera elsdenii. Biochim. Biophys. Acta 2016, 1857, 1734–1740. [Google Scholar] [CrossRef]
- Siebel, J.F.; Adamska-Venkatesh, A.; Weber, K.; Rumpel, S.; Reijerse, E.; Lubitz, W. Hybrid [FeFe]-hydrogenases with modified active sites show remarkable residual enzymatic activity. Biochemistry 2015, 54, 1474–1483. [Google Scholar] [CrossRef]
- Esselborn, J.; Lambertz, C.; Adamska-Venkatesh, A.; Simmons, T.; Berggren, G.; Nothl, J.; Siebel, J.; Hemschemeier, A.; Artero, V.; Reijerse, E.; et al. Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic. Nat. Chem. Biol. 2013, 9, 607–609. [Google Scholar] [CrossRef]
- Kertess, L.; Wittkamp, F.; Sommer, C.; Esselborn, J.; Rudiger, O.; Reijerse, E.J.; Hofmann, E.; Lubitz, W.; Winkler, M.; Happe, T.; et al. Chalcogenide substitution in the [2Fe] cluster of [FeFe]-hydrogenases conserves high enzymatic activity. Dalton Trans. 2017, 46, 16947–16958. [Google Scholar] [CrossRef]
- Sommer, C.; Richers, C.P.; Lubitz, W.; Rauchfuss, T.B.; Reijerse, E.J. A [RuRu] Analogue of an [FeFe]-Hydrogenase Traps the Key Hydride Intermediate of the Catalytic Cycle. Angew. Chem. Int. Ed. 2018, 57, 5429–5432. [Google Scholar] [CrossRef]
- Adamska-Venkatesh, A.; Simmons, T.R.; Siebel, J.F.; Artero, V.; Fontecave, M.; Reijerse, E.; Lubitz, W. Artificially maturated [FeFe] hydrogenase from Chlamydomonas reinhardtii: A HYSCORE and ENDOR study of a non-natural H-cluster. Phys. Chem. Chem. Phys. 2015, 17, 5421–5430. [Google Scholar] [CrossRef]
- Sommer, C.; Rumpel, S.; Roy, S.; Fares, C.; Artero, V.; Fontecave, M.; Reijerse, E.; Lubitz, W. Spectroscopic investigations of a semi-synthetic [FeFe] hydrogenase with propane di-selenol as bridging ligand in the binuclear subsite: Comparison to the wild type and propane di-thiol variants. J. Biol. Inorg. Chem. 2018, 23, 481–491. [Google Scholar] [CrossRef]
- DiPrimio, D.J.; Holland, P.L. Repurposing metalloproteins as mimics of natural metalloenzymes for small-molecule activation. J. Inorg. Biochem. 2021, 219, 111430. [Google Scholar] [CrossRef]
- He, C.J.; Wang, M.; Zhang, X.F.; Wang, Z.; Chen, C.N.; Liu, J.H.; Akermark, B.; Sun, L.C. An unusual cyclization in a bis(cysteinyl-S) diiron complex related to the active site of Fe-only hydrogenases. Angew. Chem. Int. Ed. 2004, 43, 3571–3574. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.K.; Lichtenstein, B.R.; Dutta, A.; Gordon, G.; Dutton, P.L. Synthetic hydrogenases: Incorporation of an iron carbonyl thiolate into a designed peptide. J. Am. Chem. Soc. 2007, 129, 14844–14845. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Onoda, A.; Hayashi, T. A hydrogenase model system based on the sequence of cytochrome c: Photochemical hydrogen evolution in aqueous media. Chem. Commun. 2011, 47, 8229–8231. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Chen, D.; Hu, X. Synthesis and reactivity of mononuclear iron models of [Fe]-hydrogenase that contain an acylmethylpyridinol ligand. Chemistry 2014, 20, 1677–1682. [Google Scholar] [CrossRef]
- Bai, L.; Fujishiro, T.; Huang, G.; Koch, J.; Takabayashi, A.; Yokono, M.; Tanaka, A.; Xu, T.; Hu, X.; Ermler, U.; et al. Towards artificial methanogenesis: Biosynthesis of the [Fe]-hydrogenase cofactor and characterization of the semi-synthetic hydrogenase. Faraday Discuss 2017, 198, 37–58. [Google Scholar] [CrossRef]
- Hu, B.W.; Chen, D.F.; Hu, X.L. Reversible Dimerization of Mononuclear Models of [Fe]-Hydrogenase. Chem. Eur. J. 2013, 19, 6221–6224. [Google Scholar] [CrossRef][Green Version]
- Shima, S.; Chen, D.F.; Xu, T.; Wodrich, M.D.; Fujishiro, T.; Schultz, K.M.; Kahnt, J.; Ataka, K.; Hu, X.L. Reconstitution of [Fe]-hydrogenase using model complexes. Nat. Chem. 2015, 7, 995–1002. [Google Scholar] [CrossRef]
- Pan, H.J.; Huang, G.F.; Wodrich, M.D.; Tirani, F.F.; Ataka, K.; Shima, S.; Hu, X.L. A catalytically active [Mn]-hydrogenase incorporating a non-native metal cofactor. Nat. Chem. 2019, 11, 669–675. [Google Scholar] [CrossRef]
- Pan, H.J.; Huang, G.; Wodrich, M.D.; Tirani, F.F.; Ataka, K.; Shima, S.; Hu, X. Diversifying Metal-Ligand Cooperative Catalysis in Semi-Synthetic [Mn]-Hydrogenases. Angew. Chem. Int. Ed. Engl. 2021, 60, 13350–13357. [Google Scholar] [CrossRef]
- Goralski, S.T.; Rose, M.J. Emerging artificial metalloenzymes for asymmetric hydrogenation reactions. Curr. Opin. Chem. Biol. 2022, 66, 102096. [Google Scholar] [CrossRef]
- Matsuo, T.; Hayashi, T. Electron transfer and oxidase activities in reconstituted hemoproteins with chemically modified cofactors. J. Porphyr. Phthalocyanines 2009, 13, 1082–1089. [Google Scholar] [CrossRef]
- Oohora, K.; Hayashi, T. Reconstitution of Heme Enzymes with Artificial Metalloporphyrinoids. Methods Enzymol. 2016, 580, 439–454. [Google Scholar]
- Oohora, K.; Onoda, A.; Hayashi, T. Hemoproteins Reconstituted with Artificial Metal Complexes as Biohybrid Catalysts. Acc. Chem. Res. 2019, 52, 945–954. [Google Scholar] [CrossRef]
- Oohora, K.; Hayashi, T. Myoglobins engineered with artificial cofactors serve as artificial metalloenzymes and models of natural enzymes. Dalton Trans. 2021, 50, 1940–1949. [Google Scholar] [CrossRef]
- Key, H.M.; Dydio, P.; Clark, D.S.; Hartwig, J.F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. [Google Scholar] [CrossRef]
- Hyster, T.K.; Arnold, F.H. P450BM3-Axial Mutations: A Gateway to Non-Natural Reactivity. Isr. J. Chem. 2015, 55, 14–20. [Google Scholar] [CrossRef]
- Tyagi, V.; Bonn, R.B.; Fasan, R. Intermolecular carbene S-H insertion catalysed by engineered myoglobin-based catalystsdagger. Chem. Sci. 2015, 6, 2488–2494. [Google Scholar] [CrossRef]
- Dydio, P.; Key, H.M.; Nazarenko, A.; Rha, J.Y.-E.; Seyedkazemi, V.; Clark, D.S.; Hartwig, J.F. An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102–106. [Google Scholar] [CrossRef]
- Wolf, M.W.; Vargas, D.A.; Lehnert, N. Engineering of RuMb: Toward a Green Catalyst for Carbene Insertion Reactions. Inorg. Chem. 2017, 56, 5623–5635. [Google Scholar] [CrossRef]
- Perkins, L.J.; Weaver, B.R.; Buller, A.R.; Burstyn, J.N. De novo biosynthesis of a nonnatural cobalt porphyrin cofactor in E. coli and incorporation into hemoproteins. Proc. Natl. Acad. Sci. USA 2021, 118, e2017625118. [Google Scholar] [CrossRef]
- Sommer, D.J.; Vaughn, M.D.; Ghirlanda, G. Protein secondary-shell interactions enhance the photoinduced hydrogen production of cobalt protoporphyrin IX. Chem. Commun. 2014, 50, 15852–15855. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Dwaraknath, S.; Ouyang, W.O.; Matsumoto, C.J.; Ouchida, S.; Lu, Y. Engineering an Oxygen-Binding Protein for Photocatalytic CO(2) Reductions in Water. Angew. Chem. Int. Ed. Engl. 2023, 62, e202215719. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-H.; Du, K.-J.; He, B.; Gao, S.-Q.; Wen, G.-B.; Lin, Y.-W. Photo-induced DNA cleavage by zinc-substituted myoglobin with a redesigned active center. Inorg. Chem. Front. 2017, 4, 2033–2036. [Google Scholar] [CrossRef]
- Matsuo, T.; Fukumoto, K.; Watanabe, T.; Hayashi, T. Precise design of artificial cofactors for enhancing peroxidase activity of myoglobin: Myoglobin mutant H64D reconstituted with a “single-winged cofactor” is equivalent to native horseradish peroxidase in oxidation activity. Chem. Asian J. 2011, 6, 2491–2499. [Google Scholar] [CrossRef]
- Sato, H.; Hayashi, T.; Ando, T.; Hisaeda, Y.; Ueno, T.; Watanabe, Y. Hybridization of Modified-Heme Reconstitution and Distal Histidine Mutation to Functionalize Sperm Whale Myoglobin. J. Am. Chem. Soc. 2004, 126, 436–437. [Google Scholar] [CrossRef]
- Sakamoto, S.; Kudo, K. Design and Synthesis of Semi-Artificial Myoglobin Possessing DNA-Binding Peptides on Heme Propionates. Bull. Chem. Soc. Jpn. 2005, 78, 1749–1756. [Google Scholar] [CrossRef]
- Hayashi, T.; Ando, T.; Matsuda, T.; Yonemura, H.; Yamada, S.; Hisaeda, Y. Introduction of a specific binding domain on myoglobin surface by new chemical modification. J. Inorg. Biochem. 2000, 82, 133–139. [Google Scholar] [CrossRef]
- Matsuo, T.; Hayashi, T.; Hisaeda, Y. Reductive Activation of Dioxygen by a Myoglobin Reconstituted with a Flavohemin. J. Am. Chem. Soc. 2002, 124, 11234–11235. [Google Scholar] [CrossRef]
- Hayashi, T.; Morita, Y.; Mizohata, E.; Oohora, K.; Ohbayashi, J.; Inoue, T.; Hisaeda, Y. Co(II)/Co(I) reduction-induced axial histidine-flipping in myoglobin reconstituted with a cobalt tetradehydrocorrin as a methionine synthase model. Chem. Commun. 2014, 50, 12560–12563. [Google Scholar] [CrossRef]
- Anguera, G.; Sánchez-García, D. Porphycenes and Related Isomers: Synthetic Aspects. Chem. Rev. 2017, 117, 2481–2516. [Google Scholar] [CrossRef]
- Hayashi, T.; Dejima, H.; Matsuo, T.; Sato, H.; Murata, D.; Hisaeda, Y. Blue Myoglobin Reconstituted with an Iron Porphycene Shows Extremely High Oxygen Affinity. J. Am. Chem. Soc. 2002, 124, 11226–11227. [Google Scholar] [CrossRef]
- Matsuo, T.; Dejima, H.; Hirota, S.; Murata, D.; Sato, H.; Ikegami, T.; Hori, H.; Hisaeda, Y.; Hayashi, T. Ligand Binding Properties of Myoglobin Reconstituted with Iron Porphycene: Unusual O2 Binding Selectivity against CO Binding1. J. Am. Chem. Soc. 2004, 126, 16007–16017. [Google Scholar] [CrossRef]
- Hayashi, T.; Murata, D.; Makino, M.; Sugimoto, H.; Matsuo, T.; Sato, H.; Shiro, Y.; Hisaeda, Y. Crystal Structure and Peroxidase Activity of Myoglobin Reconstituted with Iron Porphycene. Inorg. Chem. 2006, 45, 10530–10536. [Google Scholar] [CrossRef]
- Matsuo, T.; Murata, D.; Hisaeda, Y.; Hori, H.; Hayashi, T. Porphyrinoid Chemistry in Hemoprotein Matrix: Detection and Reactivities of Iron(IV)-Oxo Species of Porphycene Incorporated into Horseradish Peroxidase. J. Am. Chem. Soc. 2007, 129, 12906–12907. [Google Scholar] [CrossRef]
- Oohora, K.; Kihira, Y.; Mizohata, E.; Inoue, T.; Hayashi, T. C(sp3)–H Bond Hydroxylation Catalyzed by Myoglobin Reconstituted with Manganese Porphycene. J. Am. Chem. Soc. 2013, 135, 17282–17285. [Google Scholar] [CrossRef]
- Oohora, K.; Meichin, H.; Kihira, Y.; Sugimoto, H.; Shiro, Y.; Hayashi, T. Manganese(V) Porphycene Complex Responsible for Inert C–H Bond Hydroxylation in a Myoglobin Matrix. J. Am. Chem. Soc. 2017, 139, 18460–18463. [Google Scholar] [CrossRef]
- Matsuo, T.; Hayashi, A.; Abe, M.; Matsuda, T.; Hisaeda, Y.; Hayashi, T. Meso-Unsubstituted Iron Corrole in Hemoproteins: Remarkable Differences in Effects on Peroxidase Activities between Myoglobin and Horseradish Peroxidase. J. Am. Chem. Soc. 2009, 131, 15124–15125. [Google Scholar] [CrossRef]
- Ueno, T.; Yokoi, N.; Unno, M.; Matsui, T.; Tokita, Y.; Yamada, M.; Ikeda-Saito, M.; Nakajima, H.; Watanabe, Y. Design of metal cofactors activated by a protein–protein electron transfer system. Proc. Natl. Acad. Sci. USA 2006, 103, 9416–9421. [Google Scholar] [CrossRef]
- Carey, J.R.; Ma, S.K.; Pfister, T.D.; Garner, D.K.; Kim, H.K.; Abramite, J.A.; Wang, Z.; Guo, Z.; Lu, Y. A Site-Selective Dual Anchoring Strategy for Artificial Metalloprotein Design. J. Am. Chem. Soc. 2004, 126, 10812–10813. [Google Scholar] [CrossRef]
- Bacchi, M.; Berggren, G.; Niklas, J.; Veinberg, E.; Mara, M.W.; Shelby, M.L.; Poluektov, O.G.; Chen, L.X.; Tiede, D.M.; Cavazza, C.; et al. Cobaloxime-Based Artificial Hydrogenases. Inorg. Chem. 2014, 53, 8071–8082. [Google Scholar] [CrossRef]
- Chen, C.H.; Li, Y.X.; Zhao, G.Q.; Yao, R.Q.; Zhang, C.X. Natural and Artificial Mn4Ca Cluster for the Water Splitting Reaction. Chemsuschem 2017, 10, 4403–4408. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.J.M. Manganese-based Materials Inspired by Photosynthesis for Water-Splitting. Materials 2011, 4, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Faiella, M.; Roy, A.; Sommer, D.; Ghirlanda, G. De novo design of functional proteins: Toward artificial hydrogenases. Biopolymers 2013, 100, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Tanaka, K. Artificial Molecular Photosynthetic Systems: Towards Efficient Photoelectrochemical Water Oxidation. Chempluschem 2016, 81, 1028–1044. [Google Scholar] [CrossRef]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef]
- Zong, R.; Thummel, R.P. A new family of Ru complexes for water oxidation. J. Am. Chem. Soc. 2005, 127, 12802–12803. [Google Scholar] [CrossRef]
- Rotzinger, F.P.; Munavalli, S.; Comte, P.; Hurst, J.K.; Gratzel, M.; Pern, F.J.; Frank, A.J. A molecular water-oxidation catalyst derived from ruthenium diaqua bis(2,2′-bipyridyl-5,5′-dicarboxylic acid). J. Am. Chem. Soc. 1987, 109, 6619–6626. [Google Scholar] [CrossRef]
- McDaniel, N.D.; Coughlin, F.J.; Tinker, L.L.; Bernhard, S. Cyclometalated Iridium(III) Aquo Complexes: Efficient and Tunable Catalysts for the Homogeneous Oxidation of Water. J. Am. Chem. Soc. 2008, 130, 210–217. [Google Scholar] [CrossRef]
- Hull, J.F.; Balcells, D.; Blakemore, J.D.; Incarvito, C.D.; Eisenstein, O.; Brudvig, G.W.; Crabtree, R.H. Highly Active and Robust Cp* Iridium Complexes for Catalytic Water Oxidation. J. Am. Chem. Soc. 2009, 131, 8730–8731. [Google Scholar] [CrossRef]
- Lalrempuia, R.; McDaniel, N.D.; Muller-Bunz, H.; Bernhard, S.; Albrecht, M. Water Oxidation Catalyzed by Strong Carbene-Type Donor-Ligand Complexes of Iridium. Angew. Chem. Int. Ed. 2010, 49, 9765–9768. [Google Scholar] [CrossRef]
- Ellis, W.C.; McDaniel, N.D.; Bernhard, S.; Collins, T.J. Fast Water Oxidation Using Iron. J. Am. Chem. Soc. 2010, 132, 10990–10991. [Google Scholar] [CrossRef]
- Fillol, J.L.; Codola, Z.; Garcia-Bosch, I.; Gomez, L.; Pla, J.J.; Costas, M. Efficient water oxidation catalysts based on readily available iron coordination complexes. Nat. Chem. 2011, 3, 807–813. [Google Scholar] [CrossRef]
- Yin, Q.S.; Tan, J.M.; Besson, C.; Geletii, Y.V.; Musaev, D.G.; Kuznetsov, A.E.; Luo, Z.; Hardcastle, K.I.; Hill, C.L. A Fast Soluble Carbon-Free Molecular Water Oxidation Catalyst Based on Abundant Metals. Science 2010, 328, 342–345. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Borau-Garcia, J.; Berlinguette, C.P. Electrochemical evidence for catalytic water oxidation mediated by a high-valent cobalt complex. Chem. Commun. 2011, 47, 4249–4251. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, M.T.; Hou, C.; Ke, Z.F.; Lu, T.B. Homogeneous electrocatalytic water oxidation at neutral pH by a robust macrocyclic nickel(II) complex. Angew. Chem. Int. Ed. Engl. 2014, 53, 13042–13048. [Google Scholar] [CrossRef]
- Luo, G.Y.; Huang, H.H.; Wang, J.W.; Lu, T.B. Further Investigation of a Nickel-Based Homogeneous Water Oxidation Catalyst with Two cis Labile Sites. ChemSusChem 2016, 9, 485–491. [Google Scholar] [CrossRef]
- Garrido-Barros, P.; Grau, S.; Drouet, S.; Benet-Buchholz, J.; Gimbert-Suriñach, C.; Llobet, A. Can Ni Complexes Behave as Molecular Water Oxidation Catalysts? ACS Catal. 2019, 9, 3936–3945. [Google Scholar] [CrossRef]
- Koepke, S.J.; Light, K.M.; VanNatta, P.E.; Wiley, K.M.; Kieber-Emmons, M.T. Electrocatalytic Water Oxidation by a Homogeneous Copper Catalyst Disfavors Single-Site Mechanisms. J. Am. Chem. Soc. 2017, 139, 8586–8600. [Google Scholar] [CrossRef]
- Hu, Q.-Q.; Su, X.-J.; Zhang, M.-T. Electrocatalytic Water Oxidation by an Unsymmetrical Di-Copper Complex. Inorg. Chem. 2018, 57, 10481–10484. [Google Scholar] [CrossRef]
- Liu, Y.; Han, Y.; Zhang, Z.; Zhang, W.; Lai, W.; Wang, Y.; Cao, R. Low overpotential water oxidation at neutral pH catalyzed by a copper(ii) porphyrin. Chem. Sci. 2019, 10, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Conlan, B.; Cox, N.; Su, J.H.; Hillier, W.; Messinger, J.; Lubitz, W.; Dutton, P.L.; Wydrzynski, T. Photo-catalytic oxidation of a di-nuclear manganese centre in an engineered bacterioferritin ‘reaction centre’. Biochim. Biophys. Acta 2009, 1787, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Ennist, N.M.; Zhao, Z.; Stayrook, S.E.; Discher, B.M.; Dutton, P.L.; Moser, C.C. De novo protein design of photochemical reaction centers. Nat. Commun. 2022, 13, 4937. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.L.; Espiritu, E.; Edwardraja, S.; Simmons, C.R.; Williams, J.C.; Ghirlanda, G.; Allen, J.P. Design of dinuclear manganese cofactors for bacterial reaction centers. Biochim. Biophys. Acta 2016, 1857, 539–547. [Google Scholar] [CrossRef]
- Wu, H.; Tian, C.; Song, X.; Liu, C.; Yang, D.; Jiang, Z. Methods for the regeneration of nicotinamide coenzymes. Green Chem. 2013, 15, 1773–1789. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Estrella, M.A.; Du, J.; Chen, L.; Rath, S.; Prangley, E.; Chitrakar, A.; Aoki, T.; Schedl, P.; Rabinowitz, J.; Korennykh, A. The metabolites NADP(+) and NADPH are the targets of the circadian protein Nocturnin (Curled). Nat. Commun. 2019, 10, 2367. [Google Scholar] [CrossRef]
- Holm, A.K.; Blank, L.M.; Oldiges, M.; Schmid, A.; Solem, C.; Jensen, P.R.; Vemuri, G.N. Metabolic and transcriptional response to cofactor perturbations in Escherichia coli. J. Biol. Chem. 2010, 285, 17498–17506. [Google Scholar] [CrossRef]
- Nowak, C.; Pick, A.; Csepei, L.I.; Sieber, V. Characterization of Biomimetic Cofactors According to Stability, Redox Potentials, and Enzymatic Conversion by NADH Oxidase from Lactobacillus pentosus. Chembiochem 2017, 18, 1944–1949. [Google Scholar] [CrossRef]
- Löw, S.A.; Löw, I.M.; Weissenborn, M.J.; Hauer, B. Enhanced Ene-Reductase Activity through Alteration of Artificial Nicotinamide Cofactor Substituents. ChemCatChem 2016, 8, 911–915. [Google Scholar] [CrossRef]
- Halle, F.; Fin, A.; Rovira, A.R.; Tor, Y. Emissive Synthetic Cofactors: Enzymatic Interconversions of (tz) A Analogues of ATP, NAD(+), NADH, NADP(+), and NADPH. Angew. Chem. Int. Ed. Engl. 2018, 57, 1087–1090. [Google Scholar] [CrossRef]
- Anderson, B.M.; Kaplan, N.O. Enzymatic Studies with Analogues of Diphosphopyridine Nucleotide. J. Biol. Chem. 1959, 234, 1226–1232. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Q.; Wang, L.; Guo, X.; Wang, J.; Wang, Q.; Zhao, Z.K. Engineering D-Lactate Dehydrogenase to Favor an Non-natural Cofactor Nicotinamide Cytosine Dinucleotide. ChemBioChem 2020, 21, 1972–1975. [Google Scholar] [CrossRef]
- Guo, X.; Liu, Y.; Wang, Q.; Wang, X.; Li, Q.; Liu, W.; Zhao, Z.K. Non-natural Cofactor and Formate-Driven Reductive Carboxylation of Pyruvate. Angew. Chem. Int. Ed. Engl. 2020, 59, 3143–3146. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, Y.; Wang, L.; Guo, X.; Liu, W.; Li, Q.; Wang, X.; Xue, S.; Zhao, Z.K. Structural Insights into Phosphite Dehydrogenase Variants Favoring a Non-natural Redox Cofactor. ACS Catal. 2019, 9, 1883–1887. [Google Scholar] [CrossRef]
- Nowak, C.; Pick, A.; Lommes, P.; Sieber, V. Enzymatic Reduction of Nicotinamide Biomimetic Cofactors Using an Engineered Glucose Dehydrogenase: Providing a Regeneration System for Artificial Cofactors. ACS Catal. 2017, 7, 5202–5208. [Google Scholar] [CrossRef]
- You, C.; Huang, R.; Wei, X.; Zhu, Z.; Zhang, Y.P. Protein engineering of oxidoreductases utilizing nicotinamide-based coenzymes, with applications in synthetic biology. Synth. Syst. Biotechnol. 2017, 2, 208–218. [Google Scholar] [CrossRef]
- Zachos, I.; Nowak, C.; Sieber, V. Biomimetic cofactors and methods for their recycling. Curr. Opin. Chem. Biol. 2019, 49, 59–66. [Google Scholar] [CrossRef]
- Ji, D.; Wang, L.; Hou, S.; Liu, W.; Wang, J.; Wang, Q.; Zhao, Z.K. Creation of bioorthogonal redox systems depending on nicotinamide flucytosine dinucleotide. J. Am. Chem. Soc. 2011, 133, 20857–20862. [Google Scholar] [CrossRef]
- Dai, Z.; Zhang, X.N.; Nasertorabi, F.; Cheng, Q.; Pei, H.; Louie, S.G.; Stevens, R.C.; Zhang, Y. Facile chemoenzymatic synthesis of a novel stable mimic of NAD+. Chem. Sci. 2018, 9, 8337–8342. [Google Scholar] [CrossRef]
- Paul, C.E.; Gargiulo, S.; Opperman, D.J.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; Taglieber, A.; Arends, I.W.C.E.; Hollmann, F. Mimicking Nature: Synthetic Nicotinamide Cofactors for C=C Bioreduction Using Enoate Reductases. Org. Lett. 2013, 15, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, Y.J.; Wang, L.; Liu, W.; Liu, Y.; Peng, C.; Zhao, Z.K.; Drake, H.L. Engineering Escherichia coli Nicotinic Acid Mononucleotide Adenylyltransferase for Fully Active Amidated NAD Biosynthesis. Appl. Environ. Microbiol. 2017, 83, e00692-17. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, Y.; Guo, X.; Wang, Q.; Ning, S.; Li, Q.; Wang, J.; Wang, L.; Zhao, Z.K. Creating enzymes and self-sufficient cells for biosynthesis of the non-natural cofactor nicotinamide cytosine dinucleotide. Nat. Commun. 2021, 12, 2116. [Google Scholar] [CrossRef] [PubMed]
- Knaus, T.; Paul, C.E.; Levy, C.W.; de Vries, S.; Mutti, F.G.; Hollmann, F.; Scrutton, N.S. Better than Nature: Nicotinamide Biomimetics That Outperform Natural Coenzymes. J. Am. Chem. Soc. 2016, 138, 1033–1039. [Google Scholar] [CrossRef]
- Guarneri, A.; Westphal, A.H.; Leertouwer, J.; Lunsonga, J.; Franssen, M.C.R.; Opperman, D.J.; Hollmann, F.; Berkel, W.J.H.; Paul, C.E. Flavoenzyme-mediated Regioselective Aromatic Hydroxylation with Coenzyme Biomimetics. ChemCatChem 2020, 12, 1368–1375. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, X.; Liu, W.; Wang, J.; Kent Zhao, Z. Structural Insights into Malic Enzyme Variants Favoring an Unnatural Redox Cofactor. Chembiochem 2021, 22, 1765–1768. [Google Scholar] [CrossRef]
- Wang, L.; Ji, D.; Liu, Y.; Wang, Q.; Wang, X.; Zhou, Y.J.; Zhang, Y.; Liu, W.; Zhao, Z.K. Synthetic Cofactor-Linked Metabolic Circuits for Selective Energy Transfer. ACS Catal. 2017, 7, 1977–1983. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.; Liu, Y.; Li, Q.; Wang, J.; Liu, W.; Zhao, Z.K. Structure-Guided Design of Formate Dehydrogenase for Regeneration of a Non-Natural Redox Cofactor. Chemistry 2020, 26, 16611–16615. [Google Scholar] [CrossRef]
- Wang, J.; Guo, X.; Li, Q.; Wan, L.; Zhao, Z. Creation of non-natural cofactor-dependent methanol dehydrogenase. Synth. Biol. J. 2021, 2, 651–661. [Google Scholar]
- Rao, S.T.; Rossmann, M.G. Comparison of super-secondary structures in proteins. J. Mol. Biol. 1973, 76, 241–256. [Google Scholar] [CrossRef]
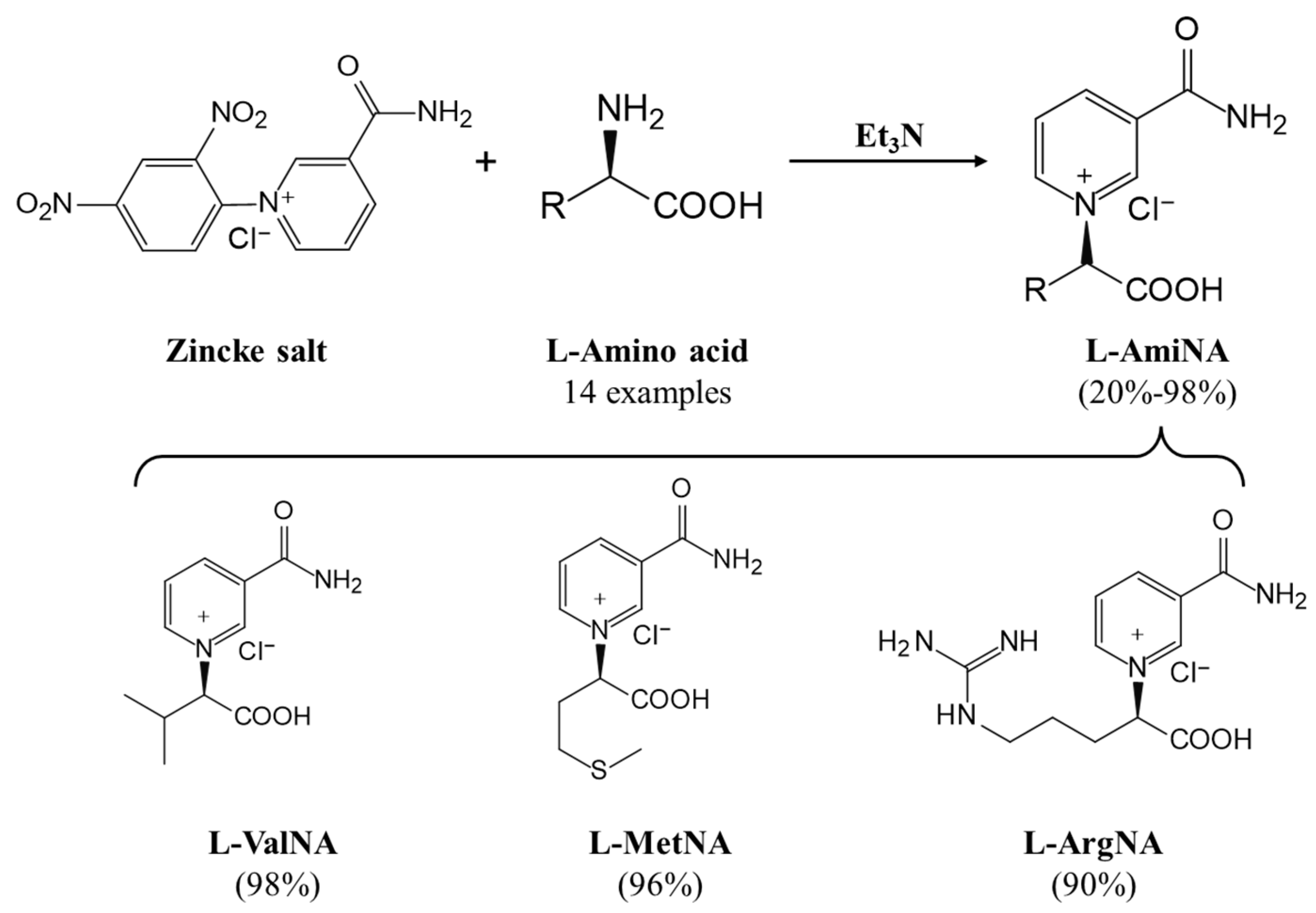
- Li, Q.; Liu, W.; Zhao, Z.K. Synthesis of proteogenic amino acid-based NAD analogs. Tetrahedron Lett. 2021, 72, 153073. [Google Scholar] [CrossRef]
- Ryan, J.D.; Fish, R.H.; Clark, D.S. Engineering cytochrome P450 enzymes for improved activity towards biomimetic 1,4-NADH cofactors. Chembiochem 2008, 9, 2579–2582. [Google Scholar] [CrossRef]
- Werck-Reichhart, D.; Feyereisen, R. Cytochromes P450: A success story. Genome Biol. 2000, 1, REVIEWS3003. [Google Scholar] [CrossRef]
- Coon, M.J. Cytochrome P450: Nature’s most versatile biological catalyst. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 1–25. [Google Scholar] [CrossRef]
- Zhang, X.W.; Li, S.Y. Expansion of chemical space for natural products by uncommon P450 reactions. Nat. Prod. Rep. 2017, 34, 1061–1089. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 enzymes in the generation of commercial products. Nat. Rev. Drug Discov. 2002, 1, 359–366. [Google Scholar] [CrossRef]
- Braunegg, G.; de Raadt, A.; Feichtenhofer, S.; Griengl, H.; Kopper, I.; Lehmann, A.; Weber, H.J. The concept of docking/protecting groups in biohydroxylation. Angew. Chem. Int. Ed. 1999, 38, 2763–2766. [Google Scholar] [CrossRef]
- Shoji, O.; Yanagisawa, S.; Stanfield, J.K.; Suzuki, K.; Cong, Z.Q.; Sugimoto, H.; Shiro, Y.; Watanabe, Y. Direct Hydroxylation of Benzene to Phenol by Cytochrome P450BM3 Triggered by Amino Acid Derivatives. Angew. Chem. Int. Ed. 2017, 56, 10324–10329. [Google Scholar] [CrossRef]
- Cong, Z.Q.; Shoji, O.; Kasai, C.; Kawakami, N.; Sugimoto, H.; Shiro, Y.; Watanabe, Y. Activation of Wild-Type Cytochrorne P450BM3 by the Next Generation of Decoy Molecules: Enhanced Hydroxylation of Gaseous Alkanes and Crystallographic Evidence. Acs Catal. 2015, 5, 150–156. [Google Scholar] [CrossRef]
- Di, S.; Fan, S.; Jiang, F.; Cong, Z. A Unique P450 Peroxygenase System Facilitated by a Dual-Functional Small Molecule: Concept, Application, and Perspective. Antioxidants 2022, 11, 529. [Google Scholar] [CrossRef]
- Fujishiro, T.; Shoji, O.; Nagano, S.; Sugimoto, H.; Shiro, Y.; Watanabe, Y. Crystal Structure of H2O2-dependent Cytochrome P450(SP alpha) with Its Bound Fatty Acid Substrate insight into the regioselective hydroxylation of fatty acids at the alpha position. J. Biol. Chem. 2011, 286, 29941–29950. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.N.; Chen, Z.F.; Chen, J.; Chen, J.F.; Wang, C.; Zhou, H.F.; Yao, L.S.; Shoji, O.; Watanabe, Y.; Cong, Z.Q. Dual-Functional Small Molecules for Generating an Efficient Cytochrome P450BM3 Peroxygenase. Angew. Chem. Int. Ed. 2018, 57, 7628–7633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, Y.; Chen, Q.; Dong, S.; Feng, Y.; Cong, Z.; Shaik, S.; Wang, B. H-Bonding Networks Dictate the Molecular Mechanism of H2O2 Activation by P450. ACS Catal. 2021, 11, 8774–8785. [Google Scholar] [CrossRef]
- Chen, Z.F.; Chen, J.; Ma, N.N.; Zho, H.F.; Cong, Z.Q. Selective hydroxylation of naphthalene using the H2O2-dependent engineered P450BM3 driven by dual-functional small molecules. J. Porphyr. Phthalocyanines 2018, 22, 831–836. [Google Scholar] [CrossRef]
- Peter, S.; Kinne, M.; Wang, X.; Ullrich, R.; Kayser, G.; Groves, J.T.; Hofrichter, M. Selective hydroxylation of alkanes by an extracellular fungal peroxygenase. FEBS J. 2011, 278, 3667–3675. [Google Scholar] [CrossRef]
- Chen, J.; Kong, F.; Ma, N.; Zhao, P.; Liu, C.; Wang, X.; Cong, Z. Peroxide-Driven Hydroxylation of Small Alkanes Catalyzed by an Artificial P450BM3 Peroxygenase System. ACS Catal. 2019, 9, 7350–7355. [Google Scholar] [CrossRef]
- Mahor, D.; Cong, Z.Q.; Weissenborn, M.J.; Hollmann, F.; Zhang, W.Y. Valorization of Small Alkanes by Biocatalytic Oxyfunctionalization. Chemsuschem 2022, 15, e202101116. [Google Scholar] [CrossRef]
- Wang, X.L.; Chen, J.; Ma, N.N.; Cong, Z.Q. Selective Hydroxylation of Alkanes Catalyzed by Cytochrome P450 Enzymes. Acta Chim. Sin. 2020, 78, 490–503. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Wang, C.L.; Ma, N.N.; Chen, J.; Liu, C.F.; Wang, F.; Xu, J.K.; Cong, Z.Q. Regioselective aromatic O-demethylation with an artificial P450BM3 peroxygenase system. Catal. Sci. Technol. 2020, 10, 1219–1223. [Google Scholar] [CrossRef]
- Zhao, P.X.; Chen, J.; Ma, N.N.; Chen, J.F.; Qin, X.Q.; Liu, C.F.; Yao, F.Q.; Yao, L.S.; Jin, L.Y.; Cong, Z.Q. Enabling highly (R)-enantioselective epoxidation of styrene by engineering unique non-natural P450 peroxygenases. Chem. Sci. 2021, 12, 6307–6314. [Google Scholar] [CrossRef]
- Chen, J.; Dong, S.; Fang, W.H.; Jiang, Y.P.; Chen, Z.F.; Qin, X.Q.; Wang, C.; Zhou, H.F.; Jin, L.Y.; Feng, Y.A.; et al. Regiodivergent and Enantioselective Hydroxylation of C-H bonds by Synergistic Use of Protein Engineering and Exogenous Dual-Functional Small Molecules. Angew. Chem. Int. Ed. 2022, 135, e202215088. [Google Scholar]
- Ma, N.; Fang, W.; Liu, C.; Qin, X.; Wang, X.; Jin, L.; Wang, B.; Cong, Z. Switching an Artificial P450 Peroxygenase into Peroxidase via Mechanism-Guided Protein Engineering. ACS Catal. 2021, 11, 8449–8455. [Google Scholar] [CrossRef]
- Wang, X.L.; Lin, X.D.; Jiang, Y.P.; Qin, X.Q.; Ma, N.N.; Yao, F.Q.; Dong, S.; Liu, C.F.; Feng, Y.G.; Jin, L.Y.; et al. Engineering Cytochrome P450BM3 Enzymes for Direct Nitration of Unsaturated Hydrocarbons. Angew. Chem. Int. Ed. 2023, 62, e202217678. [Google Scholar]
- Kong, F.H.; Chen, J.; Qin, X.Q.; Liu, C.F.; Jiang, Y.P.; Ma, L.; Xu, H.F.; Li, S.Y.; Cong, Z.Q. Evolving a P450BM3 Peroxygenase for the Production of Indigoid Dyes from Indoles. Chemcatchem 2022, 14, e202201151. [Google Scholar] [CrossRef]
- Zhao, P.; Kong, F.; Jiang, Y.; Qin, X.; Tian, X.; Cong, Z. Enabling Peroxygenase Activity in Cytochrome P450 Monooxygenases by Engineering Hydrogen Peroxide Tunnels. J. Am. Chem. Soc. 2023, 145, 5506–5511. [Google Scholar] [CrossRef]
- Kimoto, M.; Mitsui, T.; Yamashige, R.; Sato, A.; Yokoyama, S.; Hirao, I. A New Unnatural Base Pair System between Fluorophore and Quencher Base Analogues for Nucleic Acid-Based Imaging Technology. J. Am. Chem. Soc. 2010, 132, 15418–15426. [Google Scholar] [CrossRef]
- Kipper, K.; Lundius, E.G.; Curic, V.; Nikic, I.; Wiessler, M.; Lemke, E.A.; Elf, J. Application of Noncanonical Amino Acids for Protein Labeling in a Genomically Recoded Escherichia coli. ACS Synth. Biol. 2017, 6, 233–255. [Google Scholar] [CrossRef]
- Gerrits, M.; Budisa, N.; Merk, H. Site-Specific Chemoselective Pyrrolysine Analogues Incorporation Using the Cell-Free Protein Synthesis System. ACS Synth. Biol. 2019, 8, 381–390. [Google Scholar] [CrossRef]
- Saleh, A.M.; Wilding, K.M.; Calve, S.; Bundy, B.C.; Kinzer-Ursem, T.L. Non-canonical amino acid labeling in proteomics and biotechnology. J. Biol. Eng. 2019, 13, 43. [Google Scholar] [CrossRef]
- Xuan, W.; Yao, A.; Schultz, P.G. Genetically Encoded Fluorescent Probe for Detecting Sirtuins in Living Cells. J. Am. Chem. Soc. 2017, 139, 12350–12353. [Google Scholar] [CrossRef]
- Yang, B.; Tang, S.; Ma, C.; Li, S.T.; Shao, G.C.; Dang, B.; DeGrado, W.F.; Dong, M.Q.; Wang, P.G.; Ding, S.; et al. Spontaneous and specific chemical cross-linking in live cells to capture and identify protein interactions. Nat. Commun. 2017, 8, 2240. [Google Scholar] [CrossRef] [PubMed]
- Praveschotinunt, P.; Dorval Courchesne, N.M.; den Hartog, I.; Lu, C.; Kim, J.J.; Nguyen, P.Q.; Joshi, N.S. Tracking of Engineered Bacteria In Vivo Using Nonstandard Amino Acid Incorporation. ACS Synth. Biol. 2018, 7, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y. An engineered bacterium auxotrophic for an unnatural amino acid: A novel biological containment system. PeerJ 2015, 3, e1247. [Google Scholar] [CrossRef] [PubMed]
- Hauf, M.; Richter, F.; Schneider, T.; Faidt, T.; Martins, B.M.; Baumann, T.; Durkin, P.; Dobbek, H.; Jacobs, K.; Moglich, A.; et al. Photoactivatable Mussel-Based Underwater Adhesive Proteins by an Expanded Genetic Code. Chembiochem 2017, 18, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Karbalaei-Heidari, H.R.; Budisa, N. Combating Antimicrobial Resistance with New-To-Nature Lanthipeptides Created by Genetic Code Expansion. Front. Microbiol. 2020, 11, 590522. [Google Scholar] [CrossRef]
- Gohil, N.; Bhattacharjee, G.; Singh, V. Expansion of the Genetic Code. In Advances in Synthetic Biology; Singh, V., Ed.; Springer: Singapore, 2020; pp. 237–249. [Google Scholar]
- Piccirilli, J.A.; Benner, S.A.; Krauch, T.; Moroney, S.E.; Benner, S.A. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature 1990, 343, 33–37. [Google Scholar] [CrossRef]
- Yang, Z.; Hutter, D.; Sheng, P.; Sismour, A.M.; Benner, S.A. Artificially expanded genetic information system: A new base pair with an alternative hydrogen bonding pattern. Nucleic Acids Res. 2006, 34, 6095–6101. [Google Scholar] [CrossRef]
- Yang, Z.; Sismour, A.M.; Sheng, P.; Puskar, N.L.; Benner, S.A. Enzymatic incorporation of a third nucleobase pair. Nucleic Acids Res. 2007, 35, 4238–4249. [Google Scholar] [CrossRef]
- Delaney, J.C.; Gao, J.; Liu, H.; Shrivastav, N.; Essigmann, J.M.; Kool, E.T. Efficient replication bypass of size-expanded DNA base pairs in bacterial cells. Angew. Chem. Int. Ed. Engl. 2009, 48, 4524–4527. [Google Scholar] [CrossRef]
- Ogawa, A.K.; Wu, Y.; McMinn, D.L.; Liu, J.; Schultz, P.G.; Romesberg, F.E. Efforts toward the Expansion of the Genetic Alphabet: Information Storage and Replication with Unnatural Hydrophobic Base Pairs. J. Am. Chem. Soc. 2000, 122, 3274–3287. [Google Scholar] [CrossRef]
- Seo, Y.J.; Hwang, G.T.; Ordoukhanian, P.; Romesberg, F.E. Optimization of an Unnatural Base Pair toward Natural-Like Replication. J. Am. Chem. Soc. 2009, 131, 3246–3252. [Google Scholar] [CrossRef]
- Kimoto, M.; Mitsui, T.; Harada, Y.; Sato, A.; Yokoyama, S.; Hirao, I. Fluorescent probing for RNA molecules by an unnatural base-pair system. Nucleic Acids Res. 2007, 35, 5360–5369. [Google Scholar] [CrossRef]
- Eschenmoser, A. The TNA-Family of Nucleic Acid Systems: Properties and Prospects. Orig. Life Evol. Biosph. 2004, 34, 277–306. [Google Scholar] [CrossRef]
- Horhota, A.; Zou, K.; Ichida, J.K.; Yu, B.; McLaughlin, L.W.; Szostak, J.W.; Chaput, J.C. Kinetic Analysis of an Efficient DNA-Dependent TNA Polymerase. J. Am. Chem. Soc. 2005, 127, 7427–7434. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. Polymerase Chain Reaction and Transcription Using Locked Nucleic Acid Nucleotide Triphosphates. J. Am. Chem. Soc. 2008, 130, 8124–8125. [Google Scholar] [CrossRef]
- Vastmans, K.; Pochet, S.; Peys, A.; Kerremans, L.; Van Aerschot, A.; Hendrix, C.; Marlière, P.; Herdewijn, P. Enzymatic Incorporation in DNA of 1,5-Anhydrohexitol Nucleotides. Biochemistry 2000, 39, 12757–12765. [Google Scholar] [CrossRef]
- Ueda, T.; Tohda, H.; Chikazumi, N.; Eckstein, F.; Watanabe, K. Phosphorothioate-containing RNAs show mRNA activity in the prokaryotic translation systems in vitro. Nucleic Acids Res. 1991, 19, 547–552. [Google Scholar] [CrossRef][Green Version]
- Renders, M.; Emmerechts, G.; Rozenski, J.; Krecmerova, M.; Holy, A.; Herdewijn, P. Enzymatic synthesis of phosphonomethyl oligonucleotides by therminator polymerase. Angew. Chem. Int. Ed. Engl. 2007, 46, 2501–2504. [Google Scholar] [CrossRef]
- Renders, M.; Lievrouw, R.; Krecmerová, M.; Holý, A.; Herdewijn, P. Enzymatic Polymerization of Phosphonate Nucleosides. ChemBioChem 2008, 9, 2883–2888. [Google Scholar] [CrossRef]
- Liu, C.; Cozens, C.; Jaziri, F.; Rozenski, J.; Marechal, A.; Dumbre, S.; Pezo, V.; Marliere, P.; Pinheiro, V.B.; Groaz, E.; et al. Phosphonomethyl Oligonucleotides as Backbone-Modified Artificial Genetic Polymers. J. Am. Chem. Soc. 2018, 140, 6690–6699. [Google Scholar] [CrossRef]
- Johnson, J.A.; Lu, Y.Y.; Van Deventer, J.A.; Tirrell, D.A. Residue-specific incorporation of non-canonical amino acids into proteins: Recent developments and applications. Curr. Opin. Chem. Biol. 2010, 14, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Lercher, L.; Spicer, C.D.; Davis, B.G. Designing logical codon reassignment—Expanding the chemistry in biology. Chem. Sci. 2015, 6, 50–69. [Google Scholar] [CrossRef] [PubMed]
- Naowarojna, N.; Cheng, R.; Lopez, J.; Wong, C.; Qiao, L.; Liu, P. Chemical modifications of proteins and their applications in metalloenzyme studies. Synth. Syst. Biotechnol. 2021, 6, 32–49. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, P.; Van Deventer, J.A.; Link, A.J.; Tirrell, D.A. Introduction of an aliphatic ketone into recombinant proteins in a bacterial strain that overexpresses an editing-impaired leucyl-tRNA synthetase. Chembiochem 2009, 10, 2188–2190. [Google Scholar] [CrossRef]
- Gan, R.; Perez, J.G.; Carlson, E.D.; Ntai, I.; Isaacs, F.J.; Kelleher, N.L.; Jewett, M.C. Translation system engineering in Escherichia coli enhances non-canonical amino acid incorporation into proteins. Biotechnol. Bioeng. 2017, 114, 1074–1086. [Google Scholar] [CrossRef]
- Bryson, D.I.; Fan, C.; Guo, L.T.; Miller, C.; Soll, D.; Liu, D.R. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol. 2017, 13, 1253–1260. [Google Scholar] [CrossRef]
- Rackham, O.; Chin, J.W. A network of orthogonal ribosome x mRNA pairs. Nat. Chem. Biol. 2005, 1, 159–166. [Google Scholar] [CrossRef]
- Hui, A.; de Boer, H.A. Specialized ribosome system: Preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Natl. Acad. Sci. USA 1987, 84, 4762–4766. [Google Scholar] [CrossRef]
- Orelle, C.; Carlson, E.D.; Szal, T.; Florin, T.; Jewett, M.C.; Mankin, A.S. Protein synthesis by ribosomes with tethered subunits. Nature 2015, 524, 119–124. [Google Scholar] [CrossRef]
- Carlson, E.D.; d’Aquino, A.E.; Kim, D.S.; Fulk, E.M.; Hoang, K.; Szal, T.; Mankin, A.S.; Jewett, M.C. Engineered ribosomes with tethered subunits for expanding biological function. Nat. Commun. 2019, 10, 3920. [Google Scholar] [CrossRef]
- Schmied, W.H.; Tnimov, Z.; Uttamapinant, C.; Rae, C.D.; Fried, S.D.; Chin, J.W. Controlling orthogonal ribosome subunit interactions enables evolution of new function. Nature 2018, 564, 444–448. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, P.; Luo, X. Recent Technologies for Genetic Code Expansion and their Implications on Synthetic Biology Applications. J. Mol. Biol. 2022, 434, 167382. [Google Scholar] [CrossRef]
- Hohsaka, T.; Ashizuka, Y.; Murakami, H.; Sisido, M. Five-base codons for incorporation of nonnatural amino acids into proteins. Nucleic Acids Res. 2001, 29, 3646–3651. [Google Scholar] [CrossRef]
- Wang, K.; Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 2007, 25, 770–777. [Google Scholar] [CrossRef]
- Neumann, H.; Wang, K.; Davis, L.; Garcia-Alai, M.; Chin, J.W. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 2010, 464, 441–444. [Google Scholar] [CrossRef]
- Mukai, T.; Hayashi, A.; Iraha, F.; Sato, A.; Ohtake, K.; Yokoyama, S.; Sakamoto, K. Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 2010, 38, 8188–8195. [Google Scholar] [CrossRef]
- Kuru, E.; Maattala, R.M.; Noguera, K.; Stork, D.A.; Narasimhan, K.; Rittichier, J.; Wiegand, D.; Church, G.M. Release Factor Inhibiting Antimicrobial Peptides Improve Nonstandard Amino Acid Incorporation in Wild-type Bacterial Cells. ACS Chem. Biol. 2020, 15, 1852–1861. [Google Scholar] [CrossRef]
- Ravikumar, Y.; Nadarajan, S.P.; Yoo, T.H.; Lee, C.S.; Yun, H. Unnatural amino acid mutagenesis-based enzyme engineering. Trends Biotechnol. 2015, 33, 462–470. [Google Scholar] [CrossRef]
- Deepankumar, K.; Nadarajan, S.P.; Mathew, S.; Lee, S.-G.; Yoo, T.H.; Hong, E.Y.; Kim, B.-G.; Yun, H. Engineering Transaminase for Stability Enhancement and Site-Specific Immobilization through Multiple Noncanonical Amino Acids Incorporation. ChemCatChem 2015, 7, 417–421. [Google Scholar] [CrossRef]
- Cui, Z.; Johnston, W.A.; Alexandrov, K. Cell-Free Approach for Non-canonical Amino Acids Incorporation Into Polypeptides. Front. Bioeng. Biotechnol. 2020, 8, 1031. [Google Scholar] [CrossRef]
- Cui, Z.; Mureev, S.; Polinkovsky, M.E.; Tnimov, Z.; Guo, Z.; Durek, T.; Jones, A.; Alexandrov, K. Combining Sense and Nonsense Codon Reassignment for Site-Selective Protein Modification with Unnatural Amino Acids. ACS Synth. Biol. 2017, 6, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Drienovská, I.; Roelfes, G. Expanding the enzyme universe with genetically encoded unnatural amino acids. Nat. Catal. 2020, 3, 193–202. [Google Scholar] [CrossRef]
- Pagar, A.D.; Jeon, H.; Khobragade, T.P.; Sarak, S.; Giri, P.; Lim, S.; Yoo, T.H.; Ko, B.J.; Yun, H. Non-Canonical Amino Acid-Based Engineering of (R)-Amine Transaminase. Front. Chem. 2022, 10, 839636. [Google Scholar] [CrossRef] [PubMed]
- Deepankumar, K.; Shon, M.; Nadarajan, S.P.; Shin, G.; Mathew, S.; Ayyadurai, N.; Kim, B.-G.; Choi, S.-H.; Lee, S.-H.; Yun, H. Enhancing Thermostability and Organic Solvent Tolerance of ω-Transaminase through Global Incorporation of Fluorotyrosine. Adv. Synth. Catal. 2014, 356, 993–998. [Google Scholar] [CrossRef]
- Wilkinson, H.C.; Dalby, P.A. Fine-tuning the activity and stability of an evolved enzyme active-site through noncanonical amino-acids. FEBS J. 2021, 288, 1935–1955. [Google Scholar] [CrossRef]
- Jackson, J.C.; Duffy, S.P.; Hess, K.R.; Mehl, R.A. Improving nature’s enzyme active site with genetically encoded unnatural amino acids. J. Am. Chem. Soc. 2006, 128, 11124–11127. [Google Scholar] [CrossRef]
- Xiao, H.; Nasertorabi, F.; Choi, S.H.; Han, G.W.; Reed, S.A.; Stevens, R.C.; Schultz, P.G. Exploring the potential impact of an expanded genetic code on protein function. Proc. Natl. Acad. Sci. USA 2015, 112, 6961–6966. [Google Scholar] [CrossRef]
- Dominguez, M.A., Jr.; Thornton, K.C.; Melendez, M.G.; Dupureur, C.M. Differential effects of isomeric incorporation of fluorophenylalanines into PvuII endonuclease. Proteins 2001, 45, 55–61. [Google Scholar] [CrossRef]
- Smith, M.T.; Wu, J.C.; Varner, C.T.; Bundy, B.C. Enhanced protein stability through minimally invasive, direct, covalent, and site-specific immobilization. Biotechnol. Prog. 2013, 29, 247–254. [Google Scholar] [CrossRef]
- Cirino, P.C.; Tang, Y.; Takahashi, K.; Tirrell, D.A.; Arnold, F.H. Global incorporation of norleucine in place of methionine in cytochrome P450 BM-3 heme domain increases peroxygenase activity. Biotechnol. Bioeng. 2003, 83, 729–734. [Google Scholar] [CrossRef]
- Hoesl, M.G.; Acevedo-Rocha, C.G.; Nehring, S.; Royter, M.; Wolschner, C.; Wiltschi, B.; Budisa, N.; Antranikian, G. Lipase Congeners Designed by Genetic Code Engineering. ChemCatChem 2011, 3, 213–221. [Google Scholar] [CrossRef]
- Windle, C.L.; Simmons, K.J.; Ault, J.R.; Trinh, C.H.; Nelson, A.; Pearson, A.R.; Berry, A. Extending enzyme molecular recognition with an expanded amino acid alphabet. Proc. Natl. Acad. Sci. USA 2017, 114, 2610–2615. [Google Scholar] [CrossRef]
- Ugwumba, I.N.; Ozawa, K.; Xu, Z.-Q.; Ely, F.; Foo, J.-L.; Herlt, A.J.; Coppin, C.; Brown, S.; Taylor, M.C.; Ollis, D.L.; et al. Improving a Natural Enzyme Activity through Incorporation of Unnatural Amino Acids. J. Am. Chem. Soc. 2011, 133, 326–333. [Google Scholar] [CrossRef]
- Morikubo, N.; Fukuda, Y.; Ohtake, K.; Shinya, N.; Kiga, D.; Sakamoto, K.; Asanuma, M.; Hirota, H.; Yokoyama, S.; Hoshino, T. Cation−π Interaction in the Polyolefin Cyclization Cascade Uncovered by Incorporating Unnatural Amino Acids into the Catalytic Sites of Squalene Cyclase. J. Am. Chem. Soc. 2006, 128, 13184–13194. [Google Scholar] [CrossRef]
- Liu, X.; Yu, Y.; Hu, C.; Zhang, W.; Lu, Y.; Wang, J. Significant increase of oxidase activity through the genetic incorporation of a tyrosine-histidine cross-link in a myoglobin model of heme-copper oxidase. Angew. Chem. Int. Ed. Engl. 2012, 51, 4312–4316. [Google Scholar] [CrossRef]
- Zhou, Q.; Hu, M.; Zhang, W.; Jiang, L.; Perrett, S.; Zhou, J.; Wang, J. Probing the function of the Tyr-Cys cross-link in metalloenzymes by the genetic incorporation of 3-methylthiotyrosine. Angew. Chem. Int. Ed. Engl. 2013, 52, 1203–1207. [Google Scholar] [CrossRef]
- Lee, H.S.; Schultz, P.G. Biosynthesis of a Site-Specific DNA Cleaving Protein. J. Am. Chem. Soc. 2008, 130, 13194–13195. [Google Scholar] [CrossRef]
- Burke, A.J.; Lovelock, S.L.; Frese, A.; Crawshaw, R.; Ortmayer, M.; Dunstan, M.; Levy, C.; Green, A.P. Design and evolution of an enzyme with a non-canonical organocatalytic mechanism. Nature 2019, 570, 219–223. [Google Scholar] [CrossRef]
- Baker, P.J.; Montclare, J.K. Enhanced refoldability and thermoactivity of fluorinated phosphotriesterase. Chembiochem 2011, 12, 1845–1848. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Luo, X.; Li, J.; Reed, S.A.; Xiao, H.; Young, T.S.; Schultz, P.G. Enhancing protein stability with extended disulfide bonds. Proc. Natl. Acad. Sci. USA 2016, 113, 5910–5915. [Google Scholar] [CrossRef]
- Li, J.C.; Liu, T.; Wang, Y.; Mehta, A.P.; Schultz, P.G. Enhancing Protein Stability with Genetically Encoded Noncanonical Amino Acids. J. Am. Chem. Soc. 2018, 140, 15997–16000. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Rocha, C.G.; Hoesl, M.G.; Nehring, S.; Royter, M.; Wolschner, C.; Wiltschi, B.; Antranikian, G.; Budisa, N. Non-canonical amino acids as a useful synthetic biological tool for lipase-catalysed reactions in hostile environments. Catal. Sci. Technol. 2013, 3, 1198–1201. [Google Scholar] [CrossRef]
- Ohtake, K.; Yamaguchi, A.; Mukai, T.; Kashimura, H.; Hirano, N.; Haruki, M.; Kohashi, S.; Yamagishi, K.; Murayama, K.; Tomabechi, Y.; et al. Protein stabilization utilizing a redefined codon. Sci. Rep. 2015, 5, 9762. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Yang, X.; Lu, Z.; Liu, N.; Chen, Y. The “gate keeper” role of Trp222 determines the enantiopreference of diketoreductase toward 2-chloro-1-phenylethanone. PLoS ONE 2014, 9, e103792. [Google Scholar] [CrossRef]
- Drienovska, I.; Rioz-Martinez, A.; Draksharapu, A.; Roelfes, G. Novel artificial metalloenzymes by in vivo incorporation of metal-binding unnatural amino acids. Chem. Sci. 2015, 6, 770–776. [Google Scholar] [CrossRef]
- Kolev, J.N.; Zaengle, J.M.; Ravikumar, R.; Fasan, R. Enhancing the efficiency and regioselectivity of P450 oxidation catalysts by unnatural amino acid mutagenesis. Chembiochem 2014, 15, 1001–1010. [Google Scholar] [CrossRef]
- Ge, Y.; Fan, X.; Chen, P.R. A genetically encoded multifunctional unnatural amino acid for versatile protein manipulations in living cells. Chem. Sci. 2016, 7, 7055–7060. [Google Scholar] [CrossRef]
- Li, J.; Yu, J.; Zhao, J.; Wang, J.; Zheng, S.; Lin, S.; Chen, L.; Yang, M.; Jia, S.; Zhang, X.; et al. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem. 2014, 6, 352–361. [Google Scholar] [CrossRef]
- Zheng, S.; Fan, X.; Wang, J.; Zhao, J.; Chen, P.R. Dissection of Kinase Isoforms through Orthogonal and Chemical Inducible Signaling Cascades. Chembiochem 2017, 18, 1593–1598. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Mahesh, M.; Elsasser, S.J.; Hancock, S.M.; Uttamapinant, C.; Chin, J.W. Genetic encoding of photocaged cysteine allows photoactivation of TEV protease in live mammalian cells. J. Am. Chem. Soc. 2014, 136, 2240–2243. [Google Scholar] [CrossRef]
- Arbely, E.; Torres-Kolbus, J.; Deiters, A.; Chin, J.W. Photocontrol of tyrosine phosphorylation in mammalian cells via genetic encoding of photocaged tyrosine. J. Am. Chem. Soc. 2012, 134, 11912–11915. [Google Scholar] [CrossRef]
- Tian, R.; Liu, Y.; Cao, Y.; Zhang, Z.; Li, J.; Liu, L.; Du, G.; Chen, J. Titrating bacterial growth and chemical biosynthesis for efficient N-acetylglucosamine and N-acetylneuraminic acid bioproduction. Nat. Commun. 2020, 11, 5078. [Google Scholar] [CrossRef]
- Rovner, A.J.; Haimovich, A.D.; Katz, S.R.; Li, Z.; Grome, M.W.; Gassaway, B.M.; Amiram, M.; Patel, J.R.; Gallagher, R.R.; Rinehart, J.; et al. Recoded organisms engineered to depend on synthetic amino acids. Nature 2015, 518, 89–93. [Google Scholar] [CrossRef]
- Gan, F.; Liu, R.; Wang, F.; Schultz, P.G. Functional Replacement of Histidine in Proteins To Generate Noncanonical Amino Acid Dependent Organisms. J. Am. Chem. Soc. 2018, 140, 3829–3832. [Google Scholar] [CrossRef]
- Bessa-Neto, D.; Beliu, G.; Kuhlemann, A.; Pecoraro, V.; Doose, S.; Retailleau, N.; Chevrier, N.; Perrais, D.; Sauer, M.; Choquet, D. Bioorthogonal labeling of transmembrane proteins with non-canonical amino acids unveils masked epitopes in live neurons. Nat. Commun. 2021, 12, 6715. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Cai, W.; Tan, L.; Yu, Y.; Han, B.; Li, Y.; Xie, Y.; Su, Y.; Luo, X.; et al. Expanding the Structural Diversity of Protein Building Blocks with Noncanonical Amino Acids Biosynthesized from Aromatic Thiols. Angew. Chem. Int. Ed. Engl. 2021, 60, 10040–10048. [Google Scholar] [CrossRef]
- Mehl, R.A.; Anderson, J.C.; Santoro, S.W.; Wang, L.; Martin, A.B.; King, D.S.; Horn, D.M.; Schultz, P.G. Generation of a Bacterium with a 21 Amino Acid Genetic Code. J. Am. Chem. Soc. 2003, 125, 935–939. [Google Scholar] [CrossRef]
- Zhang, M.S.; Brunner, S.F.; Huguenin-Dezot, N.; Liang, A.D.; Schmied, W.H.; Rogerson, D.T.; Chin, J.W. Biosynthesis and genetic encoding of phosphothreonine through parallel selection and deep sequencing. Nat. Methods 2017, 14, 729–736. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, J.; Wang, L.; Tian, Z.; Cardenas, A.; Fang, X.; Chatterjee, A.; Xiao, H. Creation of Bacterial cells with 5-Hydroxytryptophan as a 21(st) Amino Acid Building Block. Chem 2020, 6, 2717–2727. [Google Scholar] [CrossRef]
- Roy, A.; Madden, C.; Ghirlanda, G. Photo-induced hydrogen production in a helical peptide incorporating a [FeFe] hydrogenase active site mimic. Chem. Commun. 2012, 48, 9816–9818. [Google Scholar] [CrossRef]
- Jang, W.D.; Kim, G.B.; Kim, Y.; Lee, S.Y. Applications of artificial intelligence to enzyme and pathway design for metabolic engineering. Curr. Opin. Biotechnol. 2022, 73, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Malik, S.; Gupta, A.; Srivastava, K.R. Revolutionizing enzyme engineering through artificial intelligence and machine learning. Emerg. Top. Life Sci. 2021, 5, 113–125. [Google Scholar] [PubMed]
- Choudhary, N.; Bharti, R.; Sharma, R. Role of artificial intelligence in chemistry. Mater. Today Proc. 2022, 48, 1527–1533. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; He, L.; Dong, S.; Xuan, J.; Cui, Q.; Feng, Y. Artificial Small Molecules as Cofactors and Biomacromolecular Building Blocks in Synthetic Biology: Design, Synthesis, Applications, and Challenges. Molecules 2023, 28, 5850. https://doi.org/10.3390/molecules28155850
Liu F, He L, Dong S, Xuan J, Cui Q, Feng Y. Artificial Small Molecules as Cofactors and Biomacromolecular Building Blocks in Synthetic Biology: Design, Synthesis, Applications, and Challenges. Molecules. 2023; 28(15):5850. https://doi.org/10.3390/molecules28155850
Chicago/Turabian StyleLiu, Fenghua, Lingling He, Sheng Dong, Jinsong Xuan, Qiu Cui, and Yingang Feng. 2023. "Artificial Small Molecules as Cofactors and Biomacromolecular Building Blocks in Synthetic Biology: Design, Synthesis, Applications, and Challenges" Molecules 28, no. 15: 5850. https://doi.org/10.3390/molecules28155850
APA StyleLiu, F., He, L., Dong, S., Xuan, J., Cui, Q., & Feng, Y. (2023). Artificial Small Molecules as Cofactors and Biomacromolecular Building Blocks in Synthetic Biology: Design, Synthesis, Applications, and Challenges. Molecules, 28(15), 5850. https://doi.org/10.3390/molecules28155850